
MicroRNA (miRNA) Signaling in the Human CNS in Sporadic Alzheimer’s Disease (AD)-Novel and Unique Pathological Features

Abstract
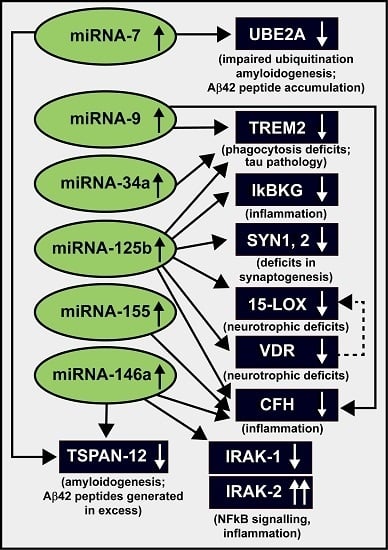
:
1. Introduction
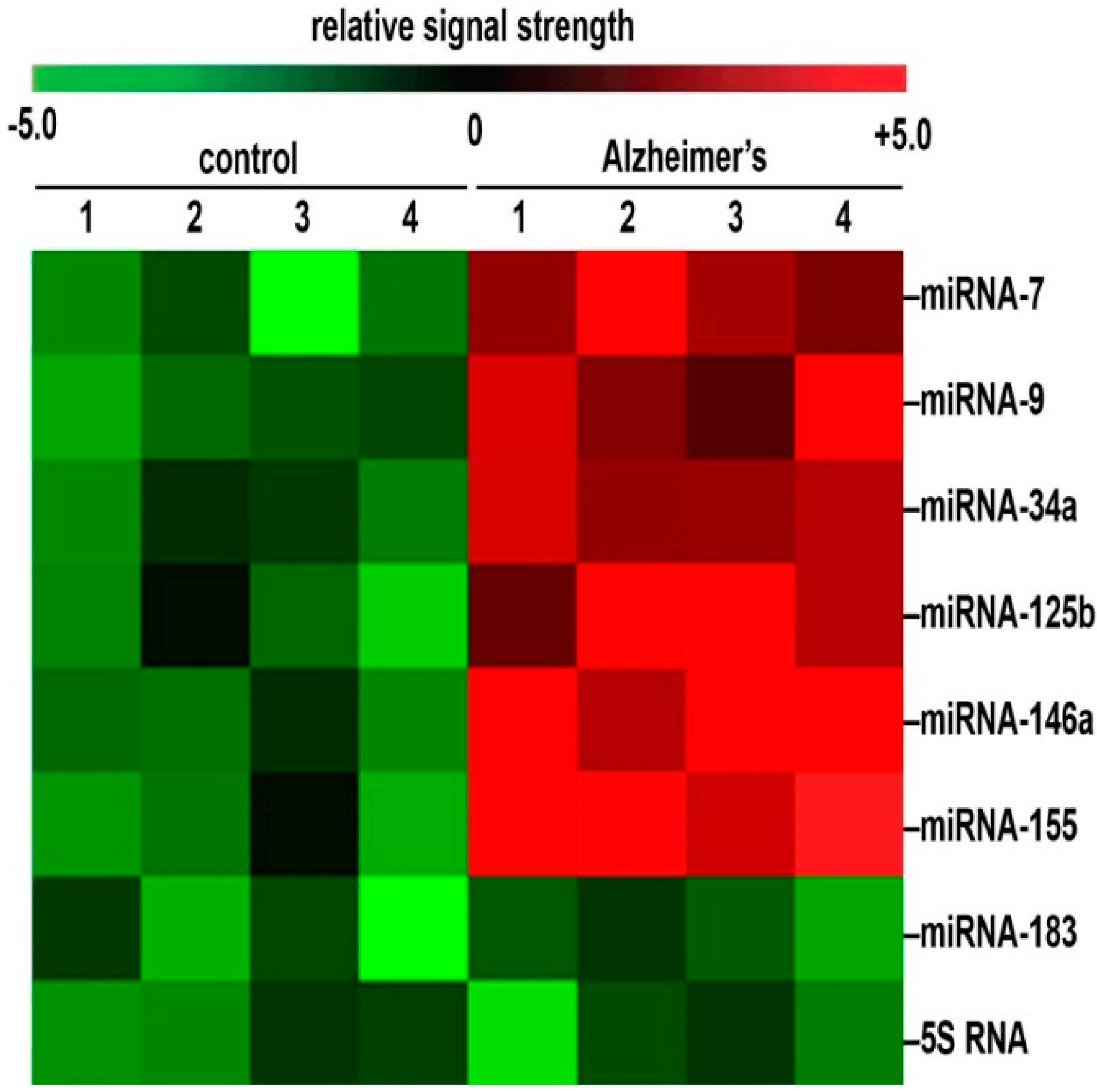
2. The High Selection Pressure and High Density of Genetic Information in Human Brain miRNAs
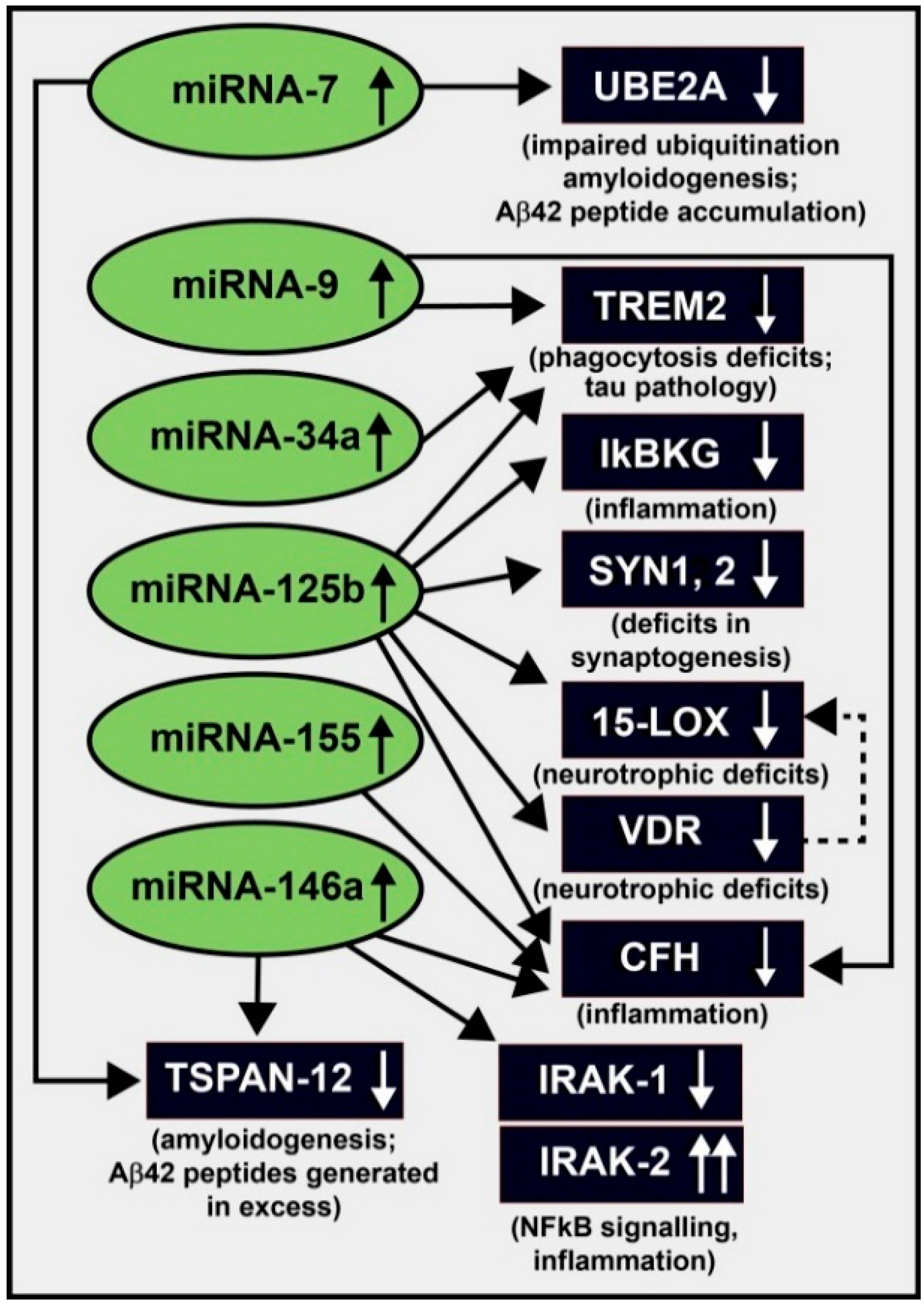
3. Up-Regulation of a Small Family of Six miRNAs Explains Many of the Neuropathological Deficits Seen in Sporadic Alzheimer’s Disease (AD)
4. NF-κB Relevance and miRNA Signaling in AD
5. NF-κB Inhibition versus Anti-miRNA (AM) Therapeutic Approaches
6. Summary
7. Concluding Remarks

{kind=link}
{kind=link}
{kind=link}
| miRNA (up-Regulated) | Target mRNA (down-Regulated) | Energy of Association EA (kcal/mol) | Consequence of Interaction (down-Regulated mRNA) | References |
|---|---|---|---|---|
| miRNA-7 | UBE2A, TSPAN-12 | −2.2 to −22.9 | amyloidogenesis | [32,81] |
| miRNA-9 | CFH, TREM2 | −22.2 | inflammation, innate-immune signaling, phagocytosis | [22,32,70,71,72,73,74,75,77] |
| miRNA-34a | TREM2 | −25.2 | phagocytosis; Aβ42 peptide clearance | [22,32,64,71,72,73,74,75,81] |
| miRNA-125b | 15-LOX, CFH, IκBKG, SYN-2, TREM2, VDR | −21.7 to −29.5 | inflammation, neurotrophism, phagocytosis, synaptogenesis | [22,32,64,68,71,72,73,74,75,81] |
| miRNA-146a | CFH, IRAK-1, TSPAN-12 | −24.5 to −26.4 | amyloidogenesis, inflammation, innate-immune signaling, NF-κB signaling | [32,39,63,64,81] |
| miRNA-155 | CFH | −26.1 | inflammation, innate-immune signaling | [32,33,39,64,81] |
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Afonso-Grunz, F.; Müller, S. Principles of miRNA–mRNA interactions: Beyond sequence complementarity. Cell. Mol. Life Sci. 2015, 72, 3127–3141. [Google Scholar] [CrossRef] [PubMed]
- Hill, J.M.; Lukiw, W.J. MicroRNA (miRNA)-mediated pathogenetic signaling in Alzheimer’s disease (AD). Neurochem. Res. 2015, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Roth, W.; Hecker, D.; Fava, E. Systems biology approaches to the study of biological networks underlying Alzheimer’s disease: Role of miRNAs. Methods Mol. Biol. 2016, 1303, 349–377. [Google Scholar] [PubMed]
- Zhao, Y.; Bhattacharjee, S.; Dua, P.; Alexandrov, P.N.; Lukiw, W.J. MicroRNA-based biomarkers and the diagnosis of Alzheimer’s disease. Front. Neurol. 2015, 6, 162. [Google Scholar] [CrossRef] [PubMed]
- Pogue, A.I.; Clement, C.; Hill, J.M.; Lukiw, W.J. Evolution of microRNA (miRNA) structure and function in plants and animals: Relevance to aging and disease. J. Aging Sci. 2014. [Google Scholar] [CrossRef]
- Burmistrova, O.A.; Goltsov, A.Y.; Abramova, L.I.; Kaleda, V.G.; Orlova, V.A.; Rogaev, E.I. MicroRNA in schizophrenia: Genetic and expression analysis of miR-130b (22q11). Biochemistry 2007, 72, 578–582. [Google Scholar] [CrossRef] [PubMed]
- Lukiw, W.J.; Andreeva, T.V.; Grigorenko, A.P.; Rogaev, E.I. Studying microRNA function and dysfunction in Alzheimer’s disease. Front. Genet. 2013, 3, 327–333. [Google Scholar] [CrossRef] [PubMed]
- Femminella, G.D.; Ferrara, N.; Rengo, G. The emerging role of microRNAs in Alzheimer’s disease. Front. Physiol. 2015, 6, 40. [Google Scholar] [CrossRef] [PubMed]
- Qiu, L.; Zhang, W.; Tan, E.K.; Zeng, L. Deciphering the function and regulation of microRNAs in Alzheimer’s disease and Parkinson’s disease. ACS Chem. Neurosci. 2014, 5, 884–894. [Google Scholar] [CrossRef] [PubMed]
- Tan, L.; Yu, J.T.; Tan, L. Causes and consequences of microRNA dysregulation in neurodegenerative diseases. Mol. Neurobiol. 2015, 51, 1249–1262. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Qin, C. General hallmarks of microRNAs in brain evolution and development. RNA Biol. 2015, 12, 701–708. [Google Scholar] [CrossRef] [PubMed]
- Sethi, P.; Lukiw, W.J. Micro-RNA abundance and stability in human brain: Specific alterations in Alzheimer’s disease temporal lobe neocortex. Neurosci. Lett. 2009, 459, 100–104. [Google Scholar] [CrossRef] [PubMed]
- Rüegger, S.; Großhans, H. MicroRNA turnover: When, how, and why. Trends Biochem. Sci. 2012, 37, 436–446. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Qin, Y.W.; Brewer, G.; Jing, Q. MicroRNA degradation and turnover: Regulating the regulators. Wiley Interdiscip. Rev. RNA. 2012, 3, 593–600. [Google Scholar] [CrossRef] [PubMed]
- Lukiw, W.J. Evolution and complexity of micro RNA in the human brain. Front. Genet. 2012, 3, 166. [Google Scholar] [CrossRef] [PubMed]
- MicroRNA Discovery and Profiling. Available online: www.lcsciences.com/microRNA (accessed on 7 December 2015).
- Lugli, G.; Cohen, A.M.; Bennett, D.A.; Shah, R.C.; Fields, C.J.; Hernandez, A.G.; Smalheiser, N.R. Plasma exosomal miRNAs in persons with and without Alzheimer disease: Altered expression and prospects for biomarkers. PLoS ONE 2015, 10, e0139233. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Ingolia, N.T.; Weissman, J.S.; Bartel, D.P. Mammalian microRNAs predominantly act to decrease target mRNA levels. Nature 2010, 466, 835–840. [Google Scholar] [CrossRef] [PubMed]
- Cloonan, N. Re-thinking miRNA-mRNA interactions: Intertwining issues confound target discovery. Bioessays 2015, 37, 379–388. [Google Scholar] [CrossRef] [PubMed]
- Cipolla, G.A. A non-canonical landscape of the microRNA system. Front. Genet. 2014, 5, 337. [Google Scholar] [CrossRef] [PubMed]
- Mukherji, S.; Ebert, M.S.; Zheng, G.X.; Tsang, J.S.; Sharp, P.A.; van Oudenaarden, A. MicroRNAs can generate thresholds in target gene expression. Nat. Genet. 2011, 43, 854–859. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharjee, S.; Zhao, Y.; Dua, P.; Rogaev, E.I.; Lukiw, W.J. MicroRNA-34a-mediated down-regulation of the microglial-enriched triggering receptor and phagocytosis-sensor TREM2 in age-related macular degeneration. PLoS ONE 2016, in press. [Google Scholar]
- Arteaga-Vázquez, M.; Caballero-Pérez, J.; Vielle-Calzada, J.P. A family of microRNAs present in plants and animals. Plant. Cell 2006, 18, 3355–3369. [Google Scholar] [CrossRef] [PubMed]
- Pogue, A.I.; Hill, J.M.; Lukiw, W.J. MicroRNA (miRNA): Sequence and stability, viroid-like properties, and disease association in the CNS. Brain Res. 2014, 1584, 73–79. [Google Scholar] [CrossRef] [PubMed]
- Hill, J.M.; Zhao, Y.; Bhattacharjee, S.; Lukiw, W.J. miRNAs and viroids utilize common strategies in genetic signal transfer. Front. Mol. Neurosci. 2014, 7, 10. [Google Scholar] [CrossRef] [PubMed]
- De Toro, J.; Herschlik, L.; Waldner, C.; Mongini, C. Emerging roles of exosomes in normal and pathological conditions: New insights for diagnosis and therapeutic applications. Front. Immunol. 2015, 6, 203. [Google Scholar] [CrossRef] [PubMed]
- Properzi, F.; Ferroni, E.; Poleggi, A.; Vinci, R. The regulation of exosome function in the CNS: Implications for neurodegeneration. Swiss Med. Wkly. 2015, 145, w14204. [Google Scholar] [CrossRef] [PubMed]
- Andreeva, K.; Cooper, N.G. MicroRNAs in the neural retina. Int. J. Genom. 2014, 2014, 165897. [Google Scholar] [CrossRef] [PubMed]
- Jimenez-Mateos, E.M. Role of microRNAs in innate neuroprotection mechanisms due to preconditioning of the brain. Front. Neurosci. 2015, 9, 118. [Google Scholar] [CrossRef] [PubMed]
- Cogoni, C.; Ruberti, F.; Barbato, C. MicroRNA landscape in Alzheimer’s disease. CNS Neurol. Disord. Drug Targets 2015, 14, 168–175. [Google Scholar] [CrossRef] [PubMed]
- Ohana, R.; Weiman-Kelman, B.; Raviv, S.; Tamm, E.R.; Pasmanik-Chor, M.; Rinon, A.; Netanely, D.; Shamir, R.; Solomon, A.S.; Ashery-Padan, R. MicroRNAs are essential for differentiation of the retinal pigmented epithelium and maturation of adjacent photoreceptors. Development 2015, 142, 2487–2498. [Google Scholar] [CrossRef] [PubMed]
- Lukiw, W.J.; Surjyadipta, B.; Dua, P.; Alexandrov, P.N. Common micro RNAs (miRNAs) target complement factor H (CFH) regulation in Alzheimer’s disease (AD) and in age-related macular degeneration (AMD). Int. J. Biochem. Mol. Biol. 2012, 3, 105–116. [Google Scholar] [PubMed]
- Devier, D.J.; Lovera, J.F.; Lukiw, W.J. Increase in NF-κB-sensitive miRNA-146a andmiRNA-155 in multiple sclerosis (MS) and pro-inflammatory neurodegeneration. Front. Mol. Neurosci. 2015, 8, 5. [Google Scholar] [CrossRef] [PubMed]
- Lukiw, W.J.; Dua, P.; Pogue, A.I.; Eicken, C.; Hill, J.M. Upregulation of micro RNA-146a (miRNA-146a), a marker for inflammatory neurodegeneration, in sporadic Creutzfeldt-Jakob disease (sCJD) and Gerstmann-Straussler-Scheinker (GSS) syndrome. J. Toxicol. Environ. Health A 2011, 74, 1460–1468. [Google Scholar] [CrossRef] [PubMed]
- Saba, R.; Medina, S.J.; Booth, S.A. A functional SNP catalog of overlapping miRNA-binding sites in genes implicated in prion disease and other neurodegenerative disorders. Hum. Mutat. 2014, 35, 1233–1248. [Google Scholar] [CrossRef] [PubMed]
- Majer, A.; Booth, S.A. Microdissection and transcriptional profiling: A window into the pathobiology of preclinical prion disease. Prion 2014, 8, 67–74. [Google Scholar] [CrossRef] [PubMed]
- Lukiw, W.J. Antagonism of NF-κB-up-regulated micro RNAs (miRNAs) in sporadic Alzheimer’s disease (AD)-anti-NF-κB vs. anti-miRNA strategies. Front. Genet. 2013, 4, 77. [Google Scholar] [CrossRef] [PubMed]
- Lukiw, W.J. NF-κB-regulated, proinflammatory miRNAs in Alzheimer’s disease. Alzheimers. Res. Ther. 2012, 4, 47. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.Y.; Alexandrov, P.N.; Pogue, A.I.; Zhao, Y.; Bhattacharjee, S.; Lukiw, W.J. miRNA-155 up-regulation and complement factor H (CFH) deficits in Down’s Syndrome. Neuroreport 2012, 23, 168–173. [Google Scholar] [CrossRef] [PubMed]
- Pogue, A.I.; Percy, M.E.; Cui, J.G.; Li, Y.Y.; Bhattacharjee, S.; Hill, J.M.; Kruck, T.P.; Zhao, Y.; Lukiw, W.J. Up-regulation of NF-κB-sensitive miRNA-125b and miRNA-146a in metal sulfate-stressed human astroglial (HAG) primary cell cultures. J. Inorg. Biochem. 2011, 105, 1434–1437. [Google Scholar] [CrossRef] [PubMed]
- Lukiw, W.J.; Cui, J.G.; Yuan, L.Y.; Bhattacharjee, P.S.; Corkern, M.; Clement, C.; Kammerman, E.M.; Ball, M.J.; Zhao, Y.; Sullivan, P.M.; et al. Acyclovir or Aβ42 peptides attenuate HSV-1-induced miRNA-146a levels in human primary brain cells. Neuroreport 2010, 21, 922–927. [Google Scholar] [CrossRef] [PubMed]
- Hill, J.M.; Clement, C.; Zhao, Y.; Lukiw, W.J. Induction of the pro-inflammatory NF-κB-sensitive miRNA-146a by human neurotrophic viruses. Front. Microbiol. 2015, 6, 43. [Google Scholar] [CrossRef] [PubMed]
- Hill, J.M.; Zhao, Y.; Clement, C.; Neumann, D.M.; Lukiw, W.J. HSV-1 infection of human brain cells induces miRNA-146a and Alzheimer-type inflammatory signaling. Neuroreport 2009, 20, 1500–1505. [Google Scholar] [CrossRef] [PubMed]
- Codocedo, J.F.; Inestrosa, N.C. Environmental control of microRNAs in the nervous system: Implications in plasticity and behavior. Neurosci. Biobehav. Rev. 2015. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Verma, S.; Mantri, S.; Berman, N.E.; Sandhir, R. Targeting microRNAs in prevention and treatment of neurodegenerative disorders. Drug Dev. Res. 2015, 76, 397–418. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.Z.; Ong, K.L.; Seeher, K.; Armstrong, N.J.; Thalamuthu, A.; Brodaty, H.; Sachdev, P.; Mather, K. Circulating microRNAs as biomarkers of Alzheimer’s disease: A systematic review. J. Alzheimers. Dis. 2015. [Google Scholar] [CrossRef] [PubMed]
- Karnati, H.K.; Panigrahi, M.K.; Gutti, R.K.; Greig, N.H.; Tamargo, I.A. miRNAs: Key players in neurodegenerative disorders and epilepsy. J. Alzheimers Dis. 2015, 48, 563–580. [Google Scholar] [CrossRef] [PubMed]
- Denzler, R.; Stoffel, M. The long, the short, and the unstructured: A unifying model of miRNA biogenesis. Mol. Cell 2015, 60, 4–6. [Google Scholar] [CrossRef] [PubMed]
- Fang, W.; Bartel, D.P. The menu of features that define primary microRNAs and enable de novo design of microRNA genes. Mol. Cell 2015, 60, 131–145. [Google Scholar] [CrossRef] [PubMed]
- Bartel, D.P. MicroRNAs: Target recognition and regulatory functions. Cell 2009, 136, 215–233. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Lukiw, W.J. Microbiome-generated amyloid and potential impact on amyloidogenesis in Alzheimer's disease (AD). J. Nat. Sci. 2015, 1, e138. [Google Scholar] [PubMed]
- Hill, J.M.; Pogue, A.I.; Lukiw, W.J. Pathogenic microRNAs common to brain and retinal degeneration; recent observations in Alzheimer’s disease and age-related macular degeneration. Front. Neurol. 2015, 6, 232. [Google Scholar] [CrossRef] [PubMed]
- Jiang, W.; Zhang, Y.; Meng, F.; Lian, B.; Chen, X.; Yu, X.; Dai, E.; Wang, S.; Liu, X.; Li, X.; et al. Identification of active transcription factor and miRNA regulatory pathways in Alzheimer’s disease. Bioinformatics 2013, 29, 2596–2602. [Google Scholar] [CrossRef] [PubMed]
- Hill, J.M.; Lukiw, W.J. Microbial-generated amyloids and Alzheimer's disease (AD). Front. Aging Neurosci. 2015, 7, 9. [Google Scholar] [CrossRef] [PubMed]
- Kiko, T.; Nakagawa, K.; Tsuduki, T.; Furukawa, K.; Arai, H.; Miyazawa, T. MicroRNAs in plasma and cerebrospinal fluid as potential markers for Alzheimer’s disease. J. Alzheimers Dis. 2014, 39, 253–259. [Google Scholar] [PubMed]
- Clement, C.; Hill, J.M.; Dua, P.; Culicchia, F.; Lukiw, W.J. Analysis of RNA from Alzheimer’s disease post-mortem brain tissues. Mol. Neurobiol. 2015, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Srinivasan, M.; Lahiri, D.K. NF-κB as a pivotal therapeutic target in the neurodegenerative pathologies of Alzheimer’s disease and multiple sclerosis. Expert Opin. Ther. Targets 2015, 19, 471–487. [Google Scholar] [CrossRef] [PubMed]
- Mihalas, A.B.; Meffert, M.K. IKK kinase assay for assessment of canonical NF-κB activation in neurons. Methods Mol. Biol. 2015, 1280, 61–74. [Google Scholar] [PubMed]
- Kaur, U.; Banerjee, P.; Bir, A.; Sinha, M.; Biswas, A.; Chakrabarti, S. Reactive oxygen species, redox signaling and neuroinflammation in Alzheimer’s disease: The NF-κB connection. Curr. Top. Med. Chem. 2015, 15, 446–457. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Jiang, L. Neuroinflammation in Alzheimer’s disease. Neuropsychiatr. Dis. Treat. 2015, 11, 243–256. [Google Scholar] [CrossRef] [PubMed]
- Engelmann, C.; Weih, F.; Haenold, R. Role of nuclear factor κB in central nervous system regeneration. Neural Regen. Res. 2014, 9, 707–711. [Google Scholar] [PubMed]
- Lanzillotta, A.; Porrini, V.; Bellucci, A.; Benarese, M.; Branca, C.; Parrella, E.; Spano, P.F.; Pizzi, M. NF-κB in innate neuroprotection and age-related neurodegenerative diseases. Front. Neurol. 2015, 6, 98. [Google Scholar] [CrossRef] [PubMed]
- Cui, J.G.; Li, Y.Y.; Zhao, Y.; Bhattacharjee, S.; Lukiw, W.J. Differential regulation of interleukin-1 receptor-associated kinase-1 (IRAK-1) and IRAK-2 by miRNA-146a and NF-κB in stressed human astroglial cells and in Alzheimer’s disease. J. Biol. Chem. 2010, 285, 38951–38960. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.Y.; Cui, J.G.; Dua, P.; Pogue, A.I.; Bhattacharjee, S.; Lukiw, W.J. Differential expression of miRNA-146a-regulated inflammatory genes in human primary neural, astroglial and microglial cells. Neurosci. Lett. 2011, 499, 109–113. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Bhattacharjee, S.; Jones, B.M.; Hill, J.M.; Clement, C.; Sambamurti, K.; Dua, P.; Lukiw, W.J. β-Amyloid precursor protein (βAPP) processing in Alzheimer’s disease (AD) and age-related macular degeneration (AMD). Mol. Neurobiol. 2015, 52, 533–544. [Google Scholar] [CrossRef] [PubMed]
- Boldin, M.P.; Baltimore, D. MicroRNAs, new effectors and regulators of NF-κB. Immunol. Rev. 2012, 246, 205–220. [Google Scholar] [CrossRef] [PubMed]
- Sen, R.; Smale, S.T. Selectivity of the NF-κB response. Cold Spring Harb. Perspect. Biol. 2010, 2, a000257. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Bhattacharjee, S.; Jones, B.M.; Hill, J.; Dua, P.; Lukiw, W.J. Regulation of neurotropic signaling by the inducible, NF-κB-sensitive miRNA-125b in Alzheimer’s disease and in primary human neuronal-glial cells. Mol. Neurobiol. 2014, 50, 97–106. [Google Scholar] [CrossRef] [PubMed]
- Lukiw, W.J.; Zhao, Y.; Cui, J.G. An NF-κB-sensitive micro RNA-146a-mediated inflammatory circuit in Alzheimer disease and in stressed human brain cells. J. Biol. Chem. 2008, 283, 31315–31322. [Google Scholar] [CrossRef] [PubMed]
- Lukiw, W.J. NF-κB-regulated micro RNAs (miRNAs) in primary human brain cells. Exp. Neurol. 2014, 235, 484–490. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharjee, S.; Zhao, Y.; Lukiw, W.J. Deficits in the miRNA-34a-regulated endogenous TREM2 phagocytosis sensor-receptor in Alzheimer’s disease (AD). Front. Aging Neurosci. 2014, 6, 116. [Google Scholar] [CrossRef] [PubMed]
- Jones, B.M.; Bhattacharjee, S.; Dua, P.; Hill, J.M.; Zhao, Y.; Lukiw, W.J. Regulating amyloidogenesis through the natural triggering receptor expressed inmyeloid/microglial cells 2 (TREM2). Front. Cell Neurosci. 2014, 8, 94. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Lukiw, W.J. TREM2 signaling, miRNA-34a and the extinction of phagocytosis. Front. Cell Neurosci. 2013, 7, 131. [Google Scholar] [CrossRef] [PubMed]
- Alexandrov, P.N.; Zhao, Y.; Jones, B.M.; Bhattacharjee, S.; Lukiw, W.J. Expression of the phagocytosis-essential protein TREM2 is down-regulated by an aluminum-inducedmiRNA-34a in a murine microglial cell line. J. Inorg. Biochem. 2013, 128, 267–269. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Bhattacharjee, S.; Jones, B.M.; Dua, P.; Alexandrov, P.N.; Hill, J.M.; Lukiw, W.J. Regulation of TREM2 expression by an NF-κB-sensitive miRNA-34a. Neuroreport 2013, 24, 318–323. [Google Scholar] [CrossRef] [PubMed]
- Lukiw, W.J.; Bazan, N.G. Strong nuclear factor-κB-DNA binding parallels cyclooxygenase-2 gene transcription in aging and in sporadic Alzheimer’s disease superior temporal lobe neocortex. J. Neurosci. Res. 1998, 53, 583–592. [Google Scholar] [CrossRef]
- Lukiw, W.J.; Alexandrov, P.N. Regulation of complement factor H (CFH) by multiple miRNAs in Alzheimer’s disease (AD) brain. Mol. Neurobiol. 2012, 46, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Fourier, A.; Portelius, E.; Zetterberg, H.; Blennow, K.; Quadrio, I.; Perret-Liaudet, A. Pre-analytical and analytical factors influencing Alzheimer’s disease cerebrospinal fluid biomarker variability. Clin. Chim. Acta 2015, 449, 9–15. [Google Scholar] [CrossRef] [PubMed]
- Alexandrov, P.N.; Dua, P.; Hill, J.M.; Bhattacharjee, S.; Zhao, Y.; Lukiw, W.J. microRNA(miRNA) speciation in Alzheimer’s disease (AD) cerebrospinal fluid (CSF) and extracellular fluid (ECF). Int. J. Biochem. Mol. Biol. 2012, 3, 365–373. [Google Scholar] [PubMed]
- Vos, S.J.; Visser, P.J.; Verhey, F.; Aalten, P.; Knol, D.; Ramakers, I.; Scheltens, P.; Rikkert, M.G.; Verbeek, M.M.; Teunissen, C.E. Variability of CSF Alzheimer’s disease biomarkers: Implications for clinical practice. PLoS ONE 2014, 9, e100784. [Google Scholar] [CrossRef] [PubMed]
- Lukiw, W.J. Variability in micro RNA (miRNA) abundance, speciation and complexity amongst different human populations and potential relevance to Alzheimer’s disease (AD). Front. Cell Neurosci. 2013, 7, 133. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Lukiw, W.J.; Louisiana State University. Personal communication, 2015.
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhao, Y.; Pogue, A.I.; Lukiw, W.J. MicroRNA (miRNA) Signaling in the Human CNS in Sporadic Alzheimer’s Disease (AD)-Novel and Unique Pathological Features. Int. J. Mol. Sci. 2015, 16, 30105-30116. https://doi.org/10.3390/ijms161226223
Zhao Y, Pogue AI, Lukiw WJ. MicroRNA (miRNA) Signaling in the Human CNS in Sporadic Alzheimer’s Disease (AD)-Novel and Unique Pathological Features. International Journal of Molecular Sciences. 2015; 16(12):30105-30116. https://doi.org/10.3390/ijms161226223
Chicago/Turabian StyleZhao, Yuhai, Aileen I. Pogue, and Walter J. Lukiw. 2015. "MicroRNA (miRNA) Signaling in the Human CNS in Sporadic Alzheimer’s Disease (AD)-Novel and Unique Pathological Features" International Journal of Molecular Sciences 16, no. 12: 30105-30116. https://doi.org/10.3390/ijms161226223
APA StyleZhao, Y., Pogue, A. I., & Lukiw, W. J. (2015). MicroRNA (miRNA) Signaling in the Human CNS in Sporadic Alzheimer’s Disease (AD)-Novel and Unique Pathological Features. International Journal of Molecular Sciences, 16(12), 30105-30116. https://doi.org/10.3390/ijms161226223