
De Novo Transcriptome Sequencing Analysis of cDNA Library and Large-Scale Unigene Assembly in Japanese Red Pine (Pinus densiflora)

Abstract
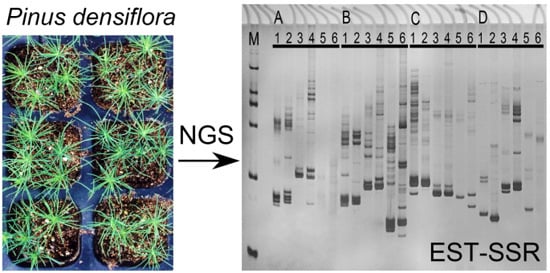
:
1. Introduction
2. Results and Discussion
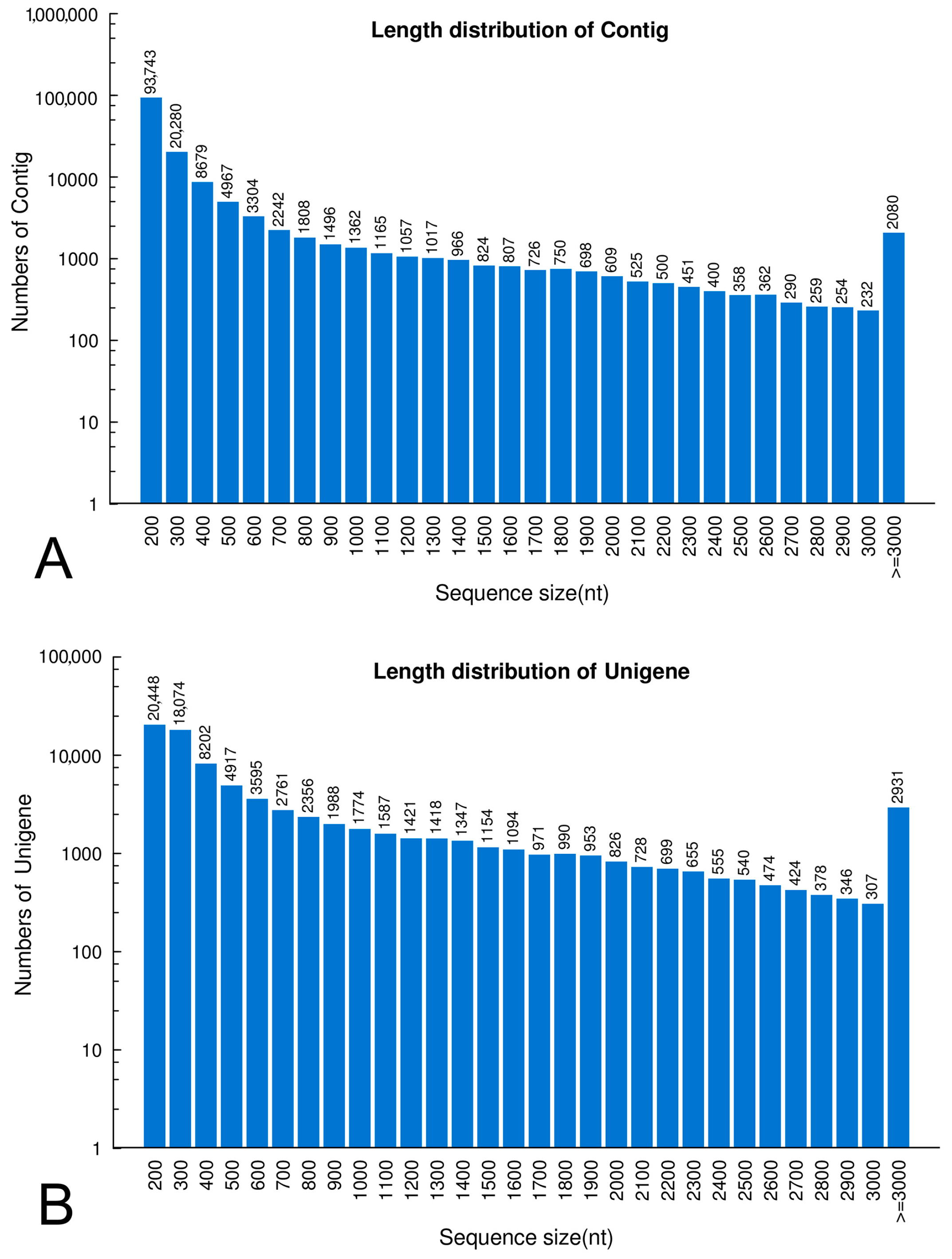
2.1. Transcriptome Sequencing and De Novo Assembly

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Map to P. taeda | Reads Number | Percentage |
|---|---|---|
| Total reads | 51,924,158 | 100.00% |
| Total mapped reads | 11,633,342 | 22.40% |
| Perfect match | 1,824,138 | 3.51% |
| Unique match | 10,889,229 | 20.97% |
| Multi-position match | 744,113 | 1.43% |
| Total unmapped reads | 40,290,816 | 77.60% |
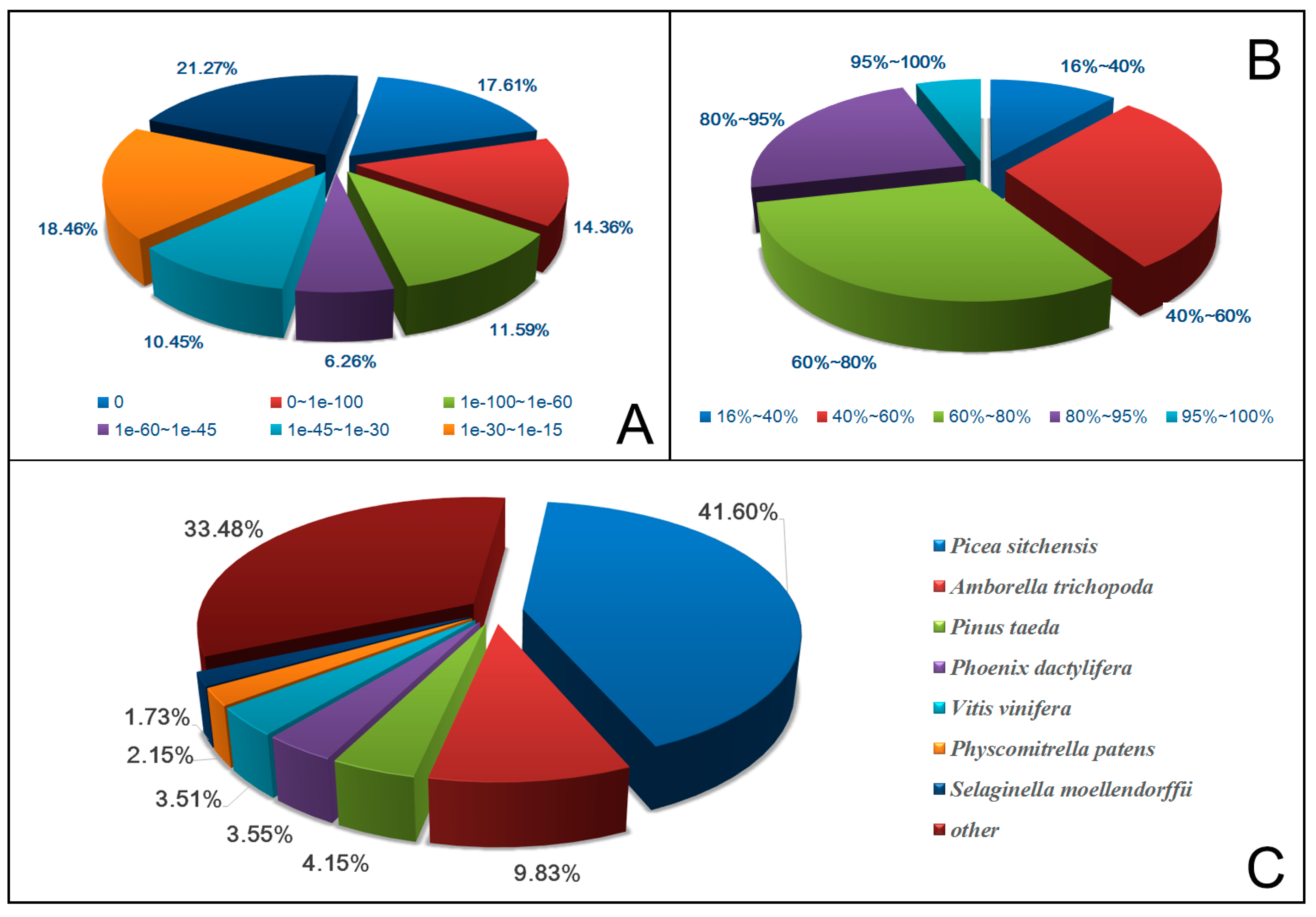
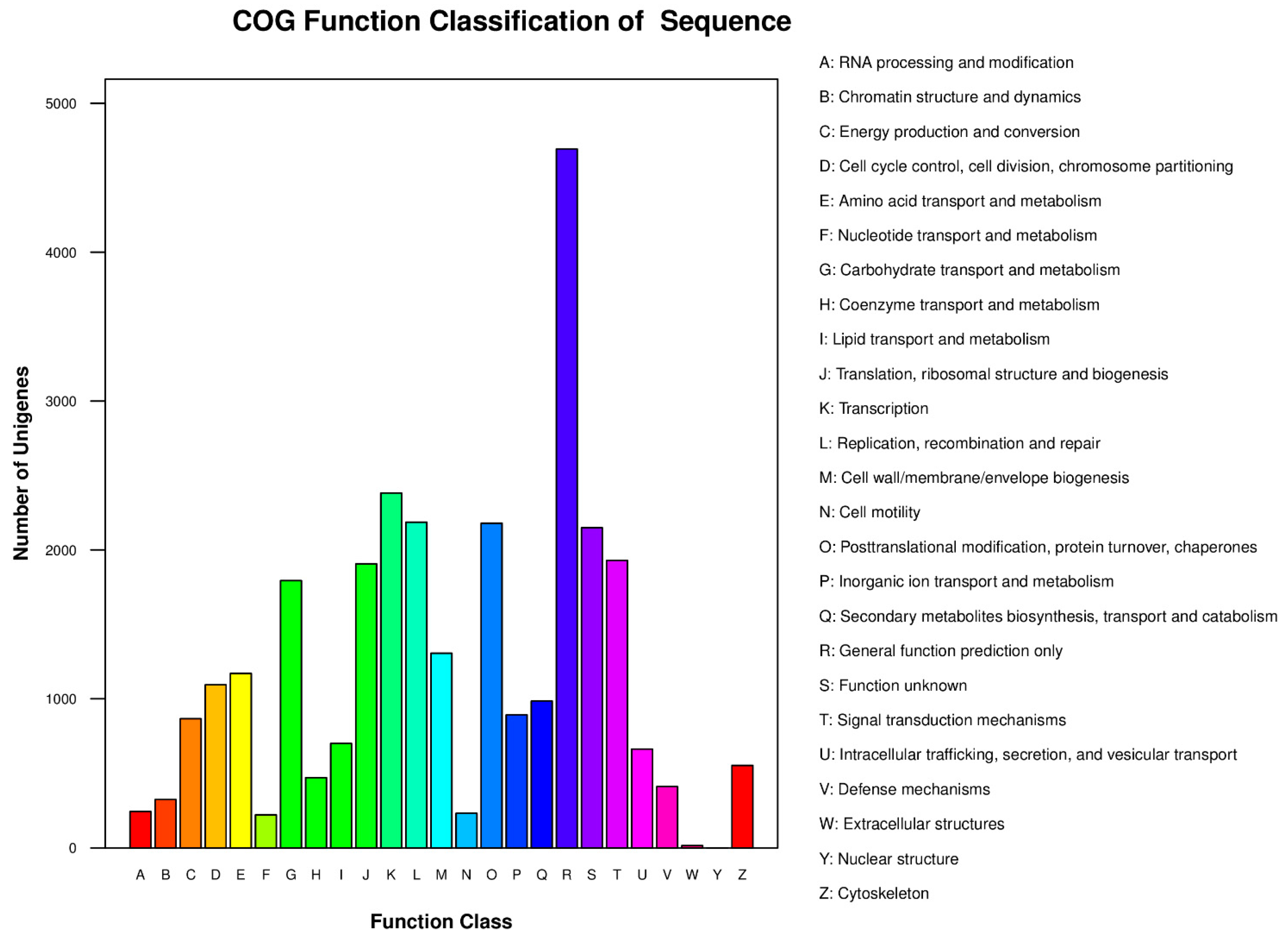
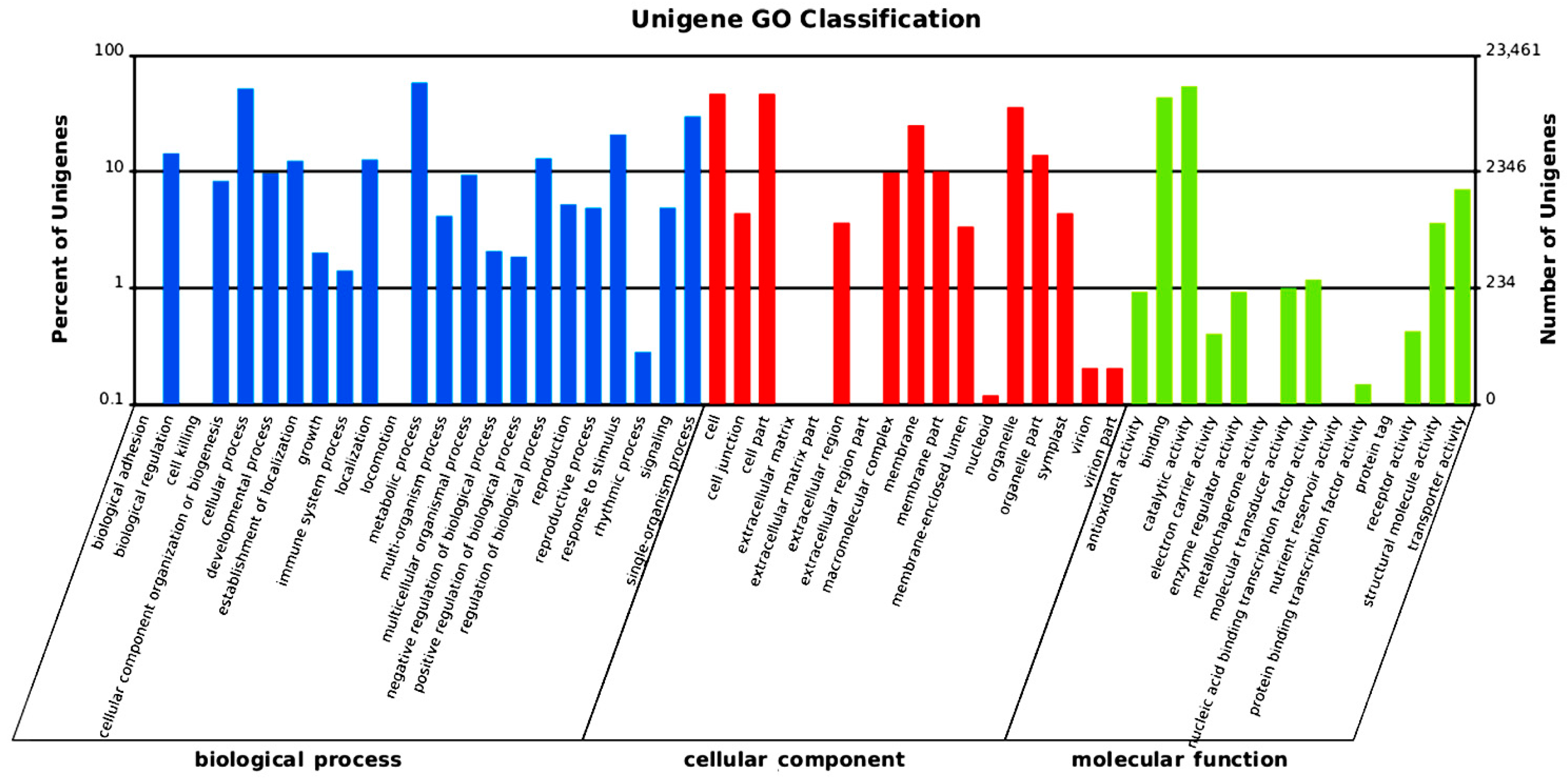
2.2. Gene Annotation and Analysis
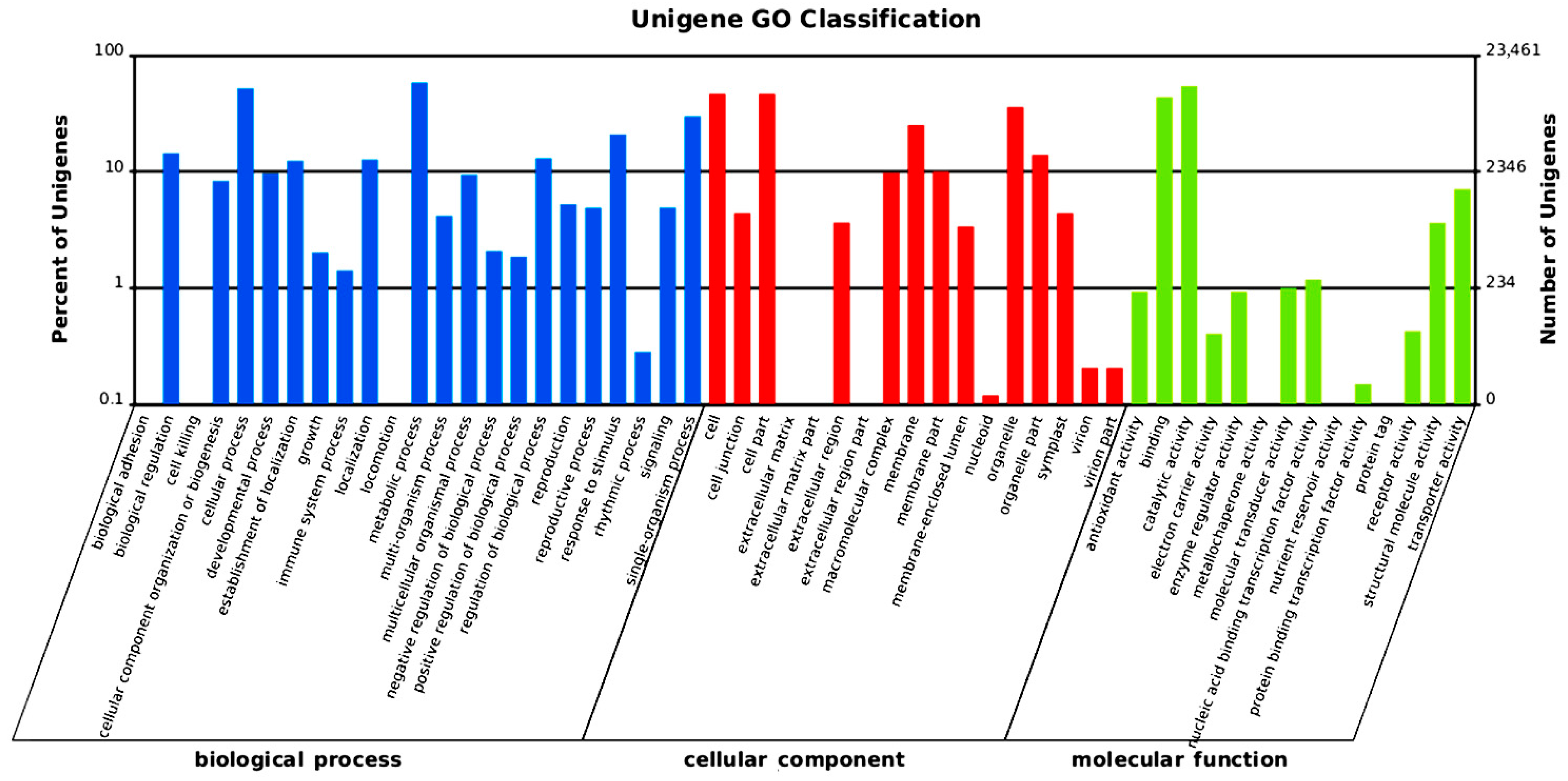
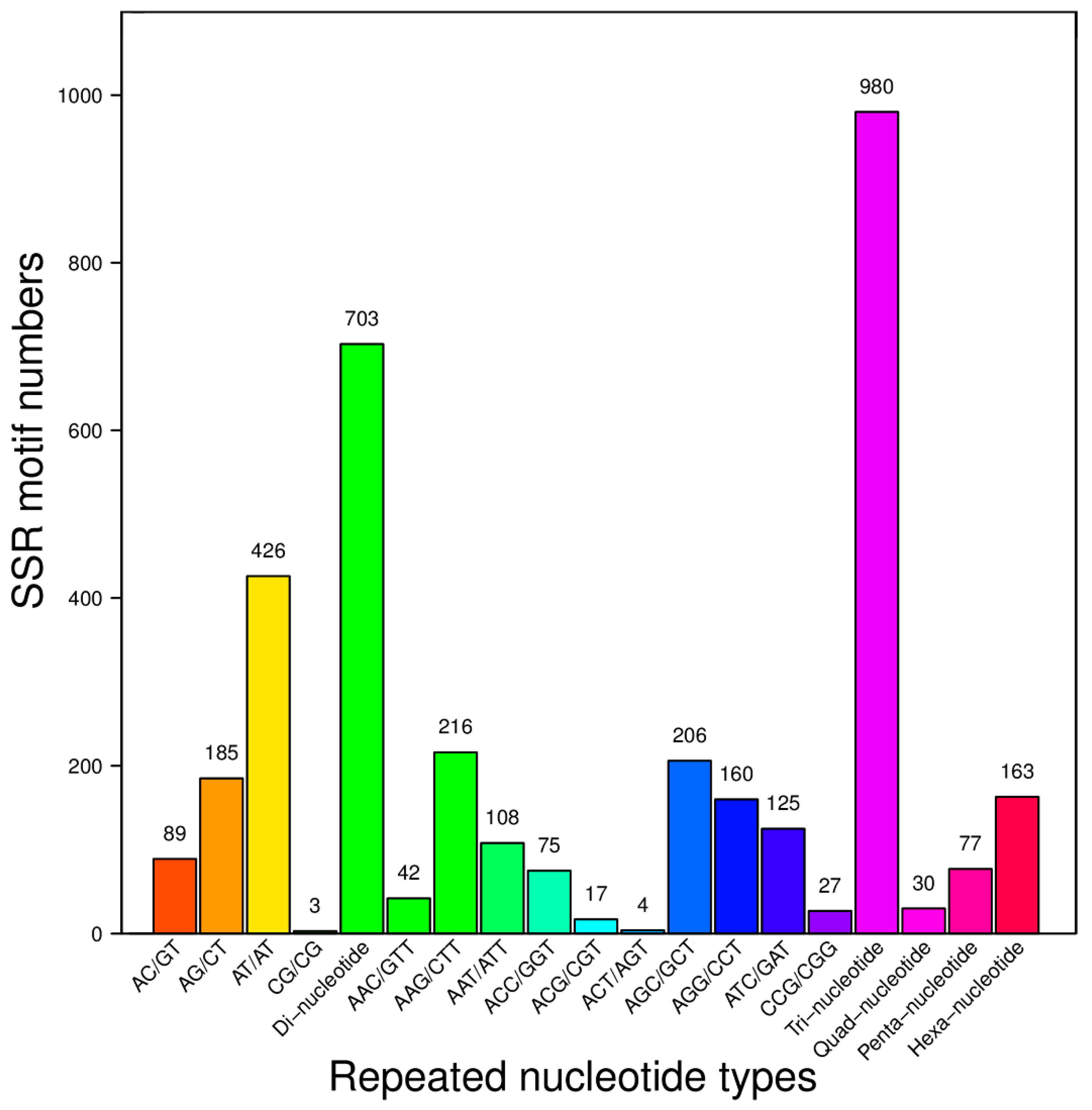
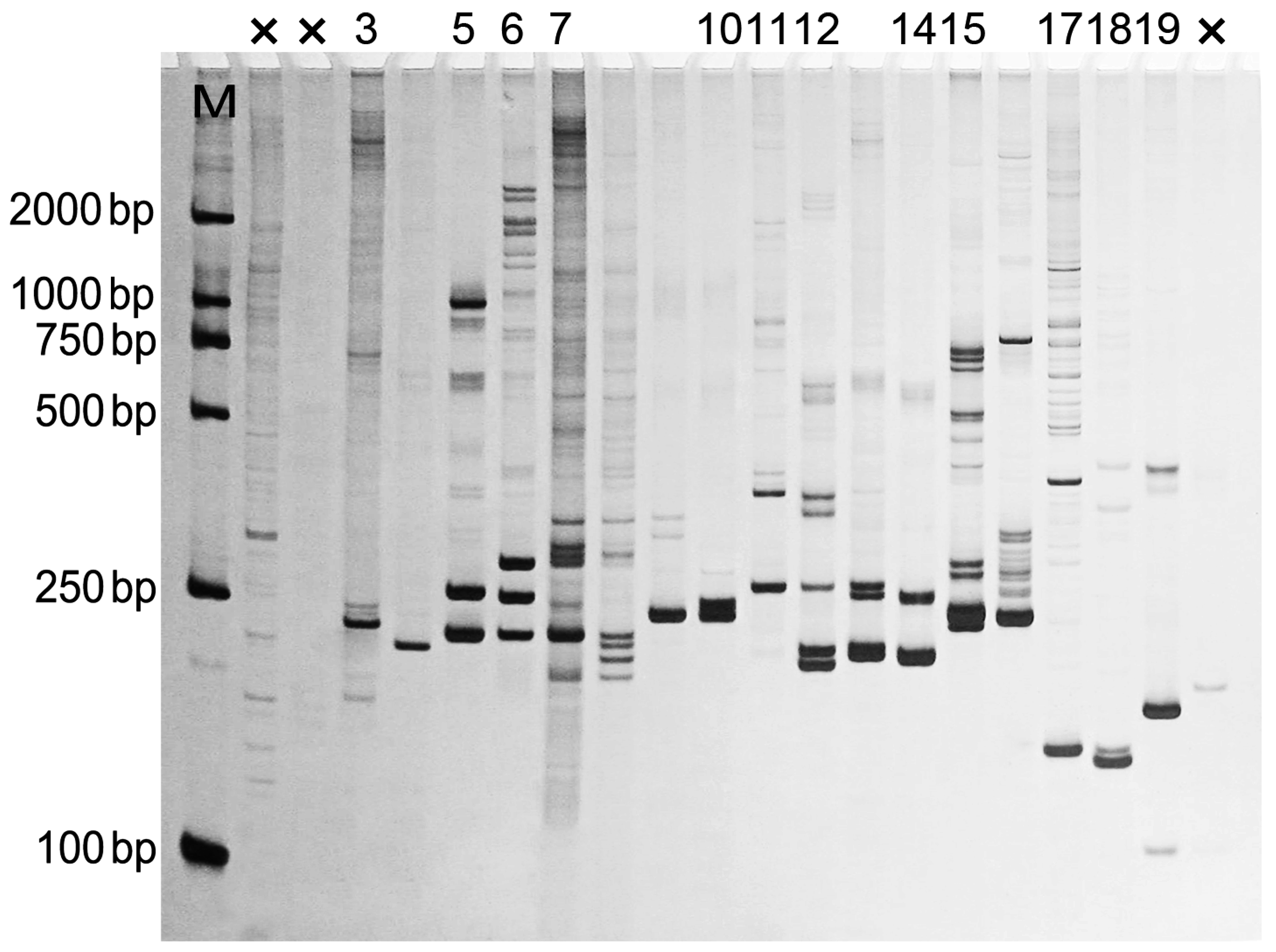
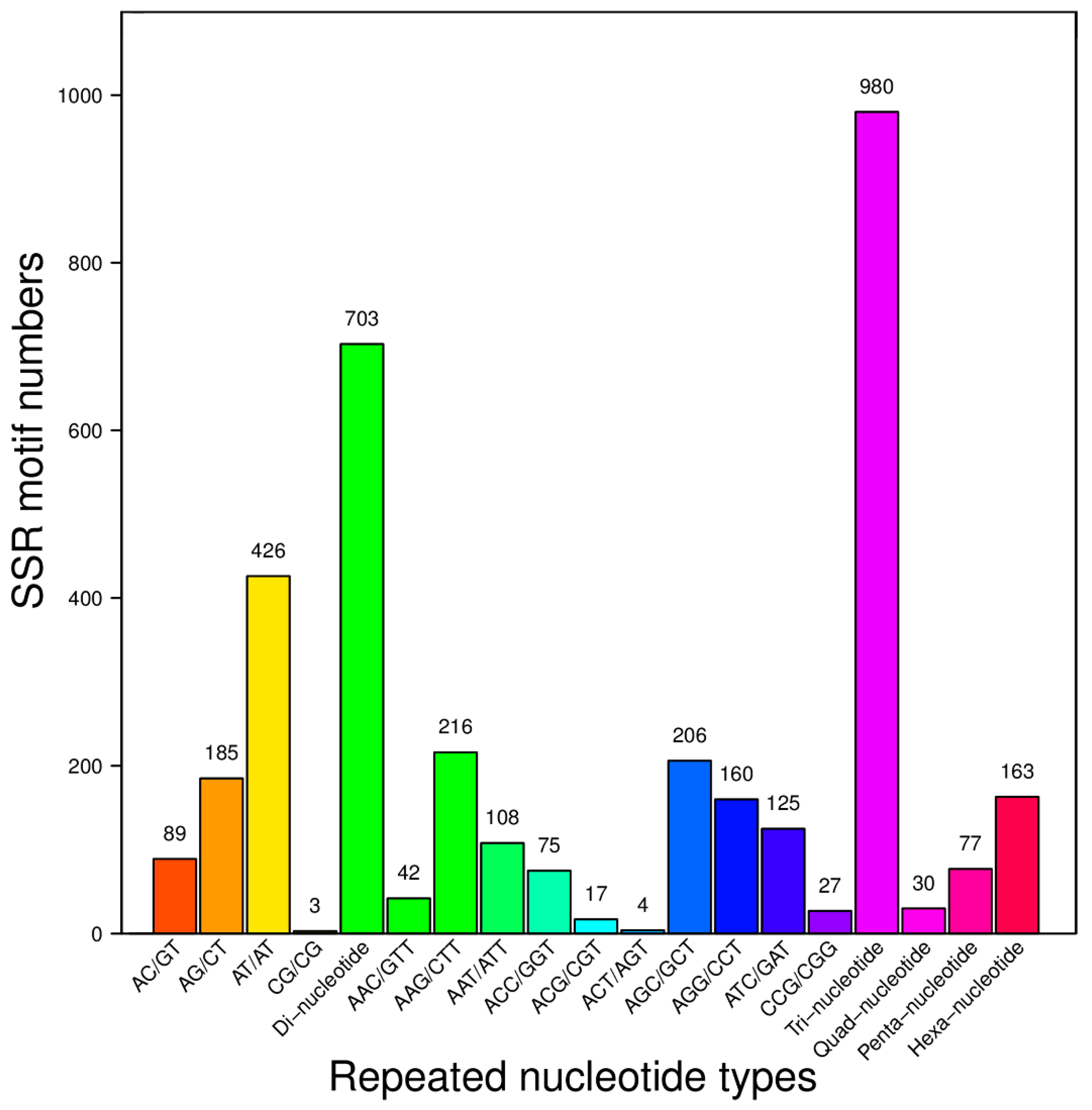
2.3. SSR Motifs Characterization and SSR Markers Development
| Number of Repeats | Di-Nucleotide Repeats | Tri-Nucleotide Repeats | Quad-Nucleotide Repeat | Penta-Nucleotide Repeats | Hexa-Nucleotide Repeats |
|---|---|---|---|---|---|
| 4 | - | - | - | 65 | 142 |
| 5 | - | 683 | 29 | 12 | 10 |
| 6 | 298 | 192 | 1 | 0 | 4 |
| 7 | 142 | 86 | 0 | 0 | 3 |
| 8 | 105 | 16 | 0 | 0 | 0 |
| 9 | 56 | 1 | 0 | 0 | 2 |
| 10 | 58 | 0 | 0 | 0 | 0 |
| 11 | 41 | 1 | 0 | 0 | 1 |
| 12 | 3 | 0 | 0 | 0 | 0 |
| 13 | 0 | 0 | 0 | 0 | 1 |
| 14 | 0 | 0 | 0 | 0 | 0 |
| 15 | 0 | 0 | 0 | 0 | 0 |
| 16 | 0 | 0 | 0 | 0 | 0 |
| 17 | 0 | 0 | 0 | 0 | 0 |
| 18 | 0 | 1 | 0 | 0 | 0 |
| 19 | 0 | 0 | 0 | 0 | 0 |
| 20 | 0 | 0 | 0 | 0 | 0 |
| 22 | 0 | 0 | 0 | 0 | 0 |
| 23 | 0 | 0 | 0 | 0 | 0 |
| SubTotal | 703 | 980 | 30 | 77 | 163 |
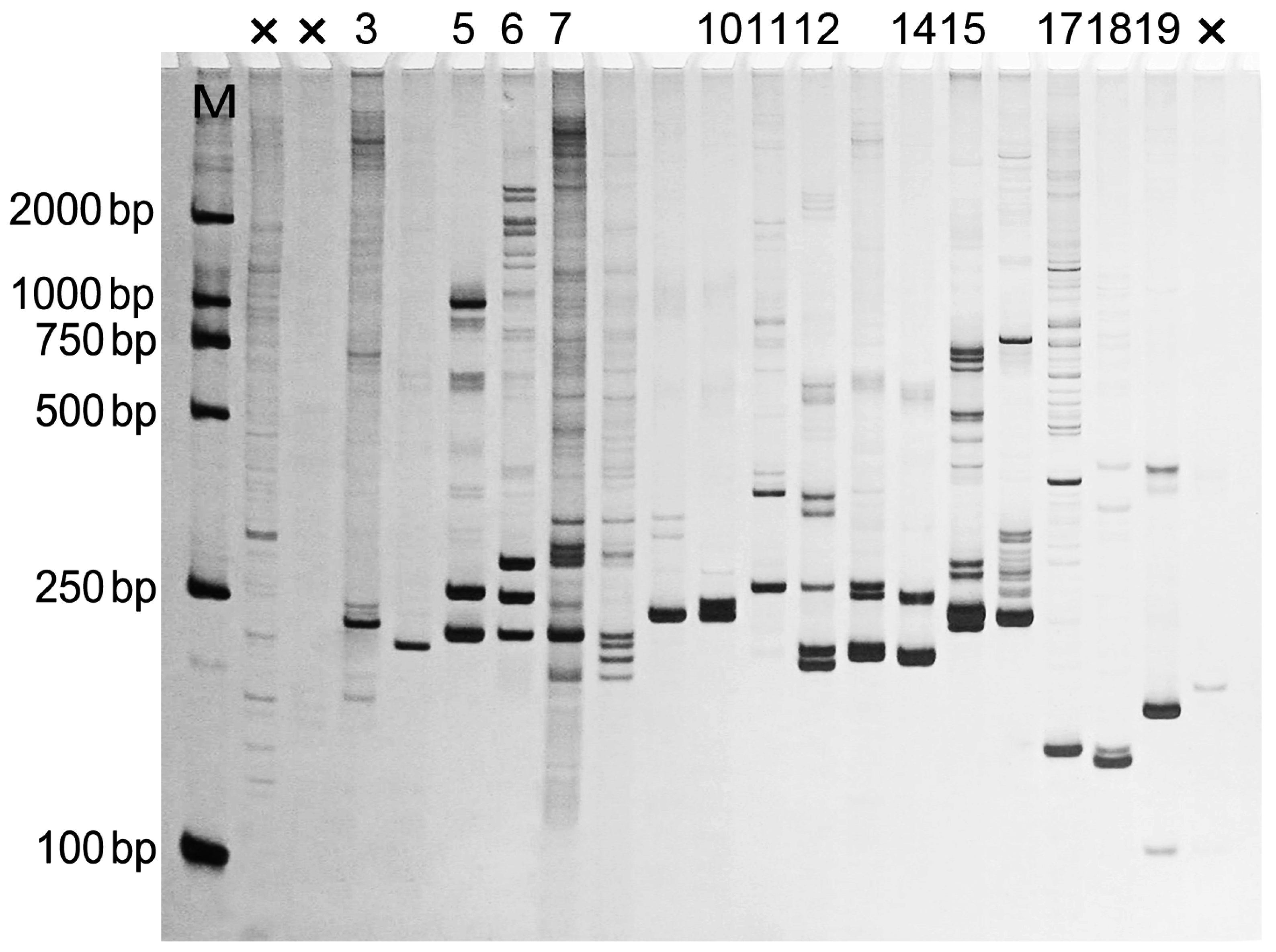
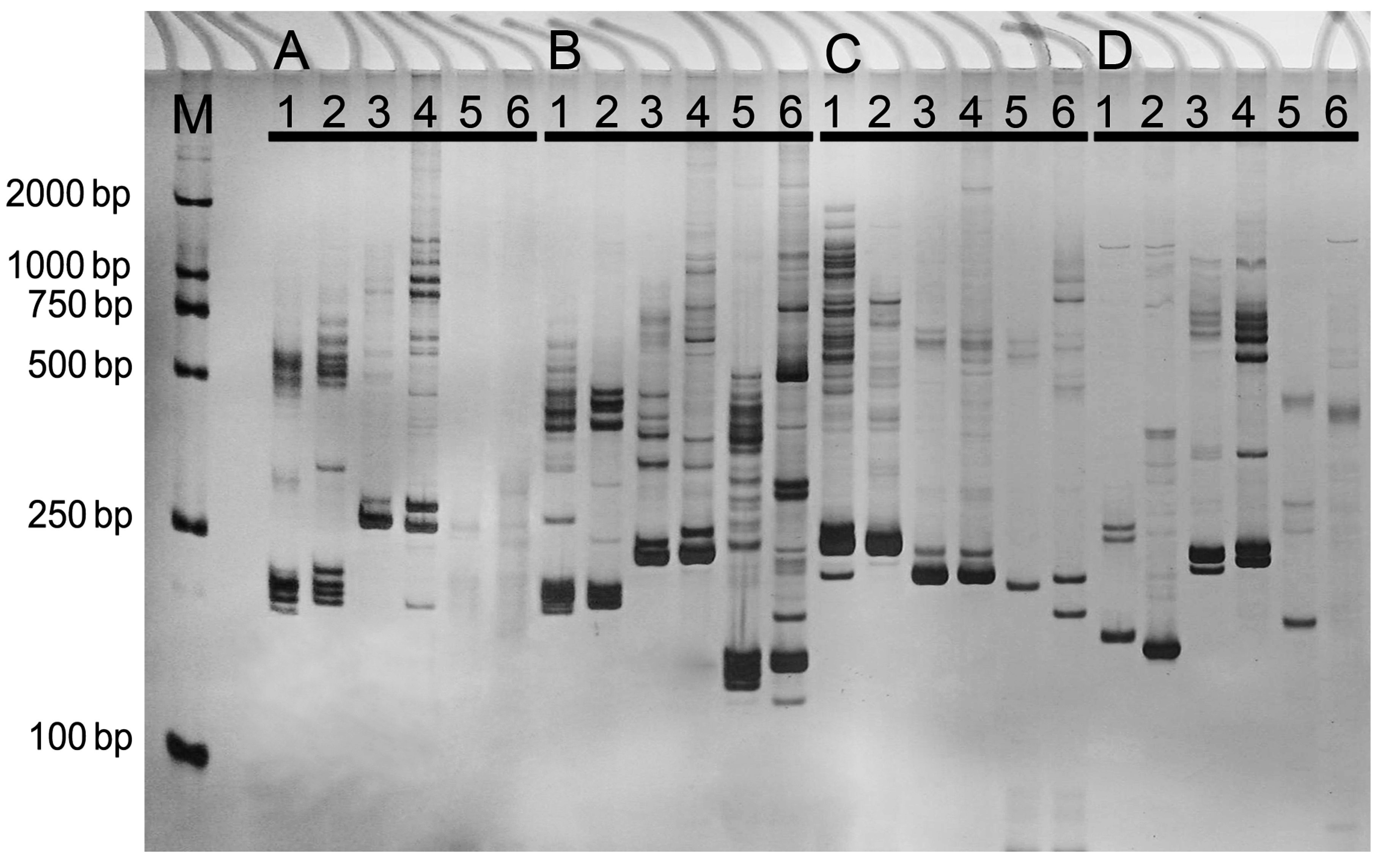
3. Experimental Section
3.1. Plant Material and RNA Extraction
3.2. Illumina Sequencing
3.3. Data Output and De Novo Assembly
3.4. Structural and Functional Annotation
3.5. SSR Loci Identification and Primer Pairs Design
3.6. EST-SSR Screening
4. Conclusions
Supplementary Materials
Author Contributions
Conflicts of Interest
References
- Prager, E.M.; Fowler, D.P.; Wilson, A.C. Rates of evolution in conifers (Pinaceae). Evolution 1976, 30, 637–649. [Google Scholar] [CrossRef]
- Lian, C.; Miwa, M.; Hogetsu, T. Outcrossing and paternity analysis of Pinus densiflora (Japanese red pine) by microsatellite polymorphism. Heredity 2001, 87, 88–98. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.B.; Kim, S.M.; Kang, M.K.; Kuzuyama, T.; Lee, J.K.; Park, S.C.; Shin, S.C.; Kim, S.U. Regulation of resin acid synthesis in Pinus densiflora by differential transcription of genes encoding multiple 1-deoxy-d-xylulose 5-phosphate synthase and 1-hydroxy-2-methyl-2-(E)-butenyl 4-diphosphate reductase genes. Tree Physiol. 2009, 29, 737–749. [Google Scholar] [CrossRef] [PubMed]
- Lian, C.; Narimatsu, M.; Nara, K.; Hogetsu, T. Tricholoma matsutake in a natural Pinus densiflora forest: Correspondence between above- and below-ground genets, association with multiple host trees and alteration of existing ectomycorrhizal communities. New Phytol. 2006, 171, 825–836. [Google Scholar] [CrossRef] [PubMed]
- Ju, Y.; Pan, J.; Wang, X.; Zhang, H. Detection of Bursaphelenchus xylophilus infection in Pinus massoniana from hyperspectral data. Nematology 2014, 16, 1197–1207. [Google Scholar] [CrossRef]
- Cavagnaro, P.F.; Chung, S.M.; Manin, S.; Yildiz, M.; Ali, A.; Alessandro, M.S.; Iorizzo, M.; Senalik, D.A.; Simon, P.W. Microsatellite isolation and marker development in carrot-genomic distribution, linkage mapping, genetic diversity analysis and marker transferability across Apiaceae. BMC Genom. 2011, 12, 386. [Google Scholar] [CrossRef] [PubMed]
- Powell, W.; Machray, G.C.; Provan, J. Polymorphism revealed by simple sequence repeats. Trends Plant Sci. 1996, 1, 215–222. [Google Scholar] [CrossRef]
- Wang, H.; Qi, X.; Gao, R.; Wang, J.; Dong, B.; Jiang, J.; Chen, S.; Guan, Z.; Fang, W.; Liao, Y.; Chen, F. Microsatellite polymorphism among Chrysanthemum sp. polyploids: The influence of whole genome duplication. Sci. Rep. 2014, 4, 6730. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Ravel, C.; Bernard, M.; Balfourier, F.; Leroy, P.; Feuillet, C.; Sourdille, P. Transferable bread wheat EST-SSRs can be useful for phylogenetic studies among the Triticeae species. Theor. Appl. Genet. 2006, 113, 407–418. [Google Scholar] [CrossRef] [PubMed]
- Ellis, J.; Burke, J. EST-SSRs as a resource for population genetic analyses. Heredity 2007, 99, 125–132. [Google Scholar] [CrossRef] [PubMed]
- Vendramin, E.; Dettori, M.T.; Giovinazzi, J.; Micali, S.; Quarta, R.; Verde, I. A set of EST-SSRs isolated from peach fruit transcriptome and their transportability across Prunus species. Mol. Ecol. Notes 2007, 7, 307–310. [Google Scholar] [CrossRef]
- Wei, W.; Qi, X.; Wang, L.; Zhang, Y.; Hua, W.; Li, D.; Lv, H.; Zhang, X. Characterization of the sesame (Sesamum indicum L.) global transcriptome using Illumina paired-end sequencing and development of EST-SSR markers. BMC Genom. 2011, 12, 451. [Google Scholar] [CrossRef] [PubMed]
- Meyer, E.; Aglyamova, G.V.; Wang, S.; Buchanan-Carter, J.; Abrego, D.; Colbourne, J.K.; Willis, B.L.; Matz, M.V. Sequencing and de novo analysis of a coral larval transcriptome using 454 GSFlx. BMC Genom. 2009, 10, 219. [Google Scholar] [CrossRef] [PubMed]
- Parchman, T.L.; Geist, K.S.; Grahnen, J.A.; Benkman, C.W.; Buerkle, C.A. Transcriptome sequencing in an ecologically important tree species: Assembly, annotation, and marker discovery. BMC Genom. 2010, 11, 180. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Jiang, J.; Chen, S.; Qi, X.; Peng, H.; Li, P.; Song, A.; Guan, Z.; Fang, W.; Liao, Y. Next-generation sequencing of the Chrysanthemum nankingense (Asteraceae) transcriptome permits large-scale unigene assembly and SSR marker discovery. PLoS ONE 2013, 8, e62293. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.J.; Song, S.H.; Wang, W.Q.; Song, S.Q. De novo assembly and characterization of germinating lettuce seed transcriptome using Illumina paired-end sequencing. Plant Physiol. Biochem. 2015, 96, 154–162. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Wang, J.; Jiang, J.; Chen, S.; Guan, Z.; Liao, Y.; Chen, F. Reference genes for normalizing transcription in diploid and tetraploid Arabidopsis. Sci. Rep. 2014, 4, 6781. [Google Scholar] [CrossRef] [PubMed]
- Zimin, A.; Stevens, K.A.; Crepeau, M.W.; Holtz-Morris, A.; Koriabine, M.; Marçais, G.; Puiu, D.; Roberts, M.; Wegrzyn, J.L.; de Jong, P.J. Sequencing and assembly of the 22-Gb loblolly pine genome. Genetics 2014, 196, 875–890. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Xu, C.; Lin, X.; Cui, B.; Wu, R.; Pang, X. De novo assembly and characterization of the fruit transcriptome of Chinese jujube (Ziziphus jujuba Mill.) using 454 pyrosequencing and the development of novel tri-nucleotide SSR markers. PLoS ONE 2014, 8, e106438. [Google Scholar] [CrossRef] [PubMed]
- Xiao, L.; Zhang, L.; Yang, G.; Zhu, H.; He, Y. Transcriptome of protoplasts reprogrammed into stem cells in Physcomitrella patens. PLoS ONE 2012, 7, e35961. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.-M.; Zhao, L.; Larson-Rabin, Z.; Li, D.-Z.; Guo, Z.H. De novo sequencing and characterization of the floral transcriptome of Dendrocalamus latiflorus (Poaceae: Bambusoideae). PLoS ONE 2012, 7, e42082. [Google Scholar] [CrossRef] [PubMed]
- Wegrzyn, J.L.; Liechty, J.D.; Stevens, K.A.; Wu, L.-S.; Loopstra, C.A.; Vasquez-Gross, H.A.; Dougherty, W.M.; Lin, B.Y.; Zieve, J.J.; Martínez-García, P.J. Unique features of the loblolly pine (Pinus taeda L.) megagenome revealed through sequence annotation. Genetics 2014, 196, 891–909. [Google Scholar] [CrossRef] [PubMed]
- Lalonde, E.; Ha, K.C.; Wang, Z.; Bemmo, A.; Kleinman, C.L.; Kwan, T.; Pastinen, T.; Majewski, J. RNA sequencing reveals the role of splicing polymorphisms in regulating human gene expression. Genome Res. 2011, 21, 545–554. [Google Scholar] [CrossRef] [PubMed]
- Sultan, M.; Schulz, M.H.; Richard, H.; Magen, A.; Klingenhoff, A.; Scherf, M.; Seifert, M.; Borodina, T.; Soldatov, A.; Parkhomchuk, D.; et al. A global view of gene activity and alternative splicing by deep sequencing of the human transcriptome. Science 2008, 321, 956–960. [Google Scholar] [CrossRef] [PubMed]
- Trick, M.; Long, Y.; Meng, J.; Bancroft, I. Single nucleotide polymorphism (SNP) discovery in the polyploid Brassica napus using Solexa transcriptome sequencing. Plant Biotechnol. J. 2009, 7, 334–346. [Google Scholar] [CrossRef] [PubMed]
- Guo, S.; Liu, J.; Zheng, Y.; Huang, M.; Zhang, H.; Gong, G.; He, H.; Ren, Y.; Zhong, S.; Fei, Z.; et al. Characterization of transcriptome dynamics during watermelon fruit development: Sequencing, assembly, annotation and gene expression profiles. BMC Genom. 2011, 12, 454. [Google Scholar] [CrossRef] [PubMed]
- Song, Q.; Shi, J.; Singh, S.; Fickus, E.; Costa, J.; Lewis, J.; Gill, B.; Ward, R.; Cregan, P. Development and mapping of microsatellite (SSR) markers in wheat. Theor. Appl. Genet. 2005, 110, 550–560. [Google Scholar] [CrossRef] [PubMed]
- Lian, C.; Miwa, M.; Hogetsu, T. Isolation and characterization of microsatellite loci from the Japanese red pine, Pinus densiflora. Mol. Ecol. 2000, 9, 1186–1188. [Google Scholar] [CrossRef] [PubMed]
- Choudhary, S.; Sethy, N.K.; Shokeen, B.; Bhatia, S. Development of chickpea EST-SSR markers and analysis of allelic variation across related species. Theor. Appl. Genet. 2009, 118, 591–608. [Google Scholar] [CrossRef] [PubMed]
- Saha, M.C.; Mian, M.A.; Eujayl, I.; Zwonitzer, J.C.; Wang, L.; May, G.D. Tall fescue EST-SSR markers with transferability across several grass species. Theor. Appl. Genet. 2004, 109, 783–791. [Google Scholar] [CrossRef] [PubMed]
- Varshney, R.K.; Graner, A.; Sorrells, M.E. Genic microsatellite markers in plants: Features and applications. Trends Biotechnol. 2005, 23, 48–55. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Bansal, R.; Gopalakrishna, T. Development and characterization of genic SSR markers for mungbean (Vigna radiata (L.) Wilczek). Euphytica 2014, 195, 245–258. [Google Scholar] [CrossRef]
- Scoles, G.; Gupta, S.; Prasad, M. Development and characterization of genic SSR markers in Medicago truncatula and their transferability in leguminous and non-leguminous species. Genom. Res. Counc. Can. 2009, 52, 761–771. [Google Scholar]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef] [PubMed]
- Cameron, M.; Williams, H.E.; Cannane, A. Improved gapped alignment in BLAST. IEEE ACM Trans. Comput. Biol. 2004, 1, 116–129. [Google Scholar] [CrossRef] [PubMed]
- Iseli, C.; Jongeneel, C.V.; Bucher, P. ESTScan: A program for detecting, evaluating, and reconstructing potential coding regions in EST sequences. ISMB 1999, 99, 138–148. [Google Scholar]
- Wang, Z.; Fang, B.; Chen, J.; Zhang, X.; Luo, Z.; Huang, L.; Chen, X.; Li, Y. De novo assembly and characterization of root transcriptome using Illumina paired-end sequencing and development of cSSR markers in sweet potato (Ipomoea batatas). BMC Genom. 2010, 11, 726. [Google Scholar] [CrossRef] [PubMed]
- Conesa, A.; Gotz, S.; Garcia-Gomez, J.M.; Terol, J.; Talon, M.; Robles, M. Blast2GO: A universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 2005, 21, 3674–3676. [Google Scholar] [CrossRef] [PubMed]
- Ye, J.; Fang, L.; Zheng, H.; Zhang, Y.; Chen, J.; Zhang, Z.; Wang, J.; Li, S.; Li, R.; Bolund, L. WEGO: A web tool for plotting GO annotations. Nucleic Acids Res. 2006, 34, W293–W297. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Ge, X.; Sun, M. Modified CTAB protocol using a silica matrix for isolation of plant genomic DNA. BioTechniques 2000, 28, 432–434. [Google Scholar] [PubMed]
- Wang, H.; Dong, B.; Jiang, J.; Fang, W.; Guan, Z.; Liao, Y.; Chen, S.; Chen, F. Characterization of in vitro haploid and doubled haploid Chrysanthemum morifolium plants via unfertilized ovule culture for phenotypical traits and DNA methylation pattern. Front. Plant Sci. 2014, 5, 738. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, L.; Zhang, S.; Lian, C. De Novo Transcriptome Sequencing Analysis of cDNA Library and Large-Scale Unigene Assembly in Japanese Red Pine (Pinus densiflora). Int. J. Mol. Sci. 2015, 16, 29047-29059. https://doi.org/10.3390/ijms161226139
Liu L, Zhang S, Lian C. De Novo Transcriptome Sequencing Analysis of cDNA Library and Large-Scale Unigene Assembly in Japanese Red Pine (Pinus densiflora). International Journal of Molecular Sciences. 2015; 16(12):29047-29059. https://doi.org/10.3390/ijms161226139
Chicago/Turabian StyleLiu, Le, Shijie Zhang, and Chunlan Lian. 2015. "De Novo Transcriptome Sequencing Analysis of cDNA Library and Large-Scale Unigene Assembly in Japanese Red Pine (Pinus densiflora)" International Journal of Molecular Sciences 16, no. 12: 29047-29059. https://doi.org/10.3390/ijms161226139
APA StyleLiu, L., Zhang, S., & Lian, C. (2015). De Novo Transcriptome Sequencing Analysis of cDNA Library and Large-Scale Unigene Assembly in Japanese Red Pine (Pinus densiflora). International Journal of Molecular Sciences, 16(12), 29047-29059. https://doi.org/10.3390/ijms161226139