
Old Things New View: Ascorbic Acid Protects the Brain in Neurodegenerative Disorders

{kind=link}
{kind=link}
Abstract
:1. Introduction
2. General Aspects of Ascorbic Acid
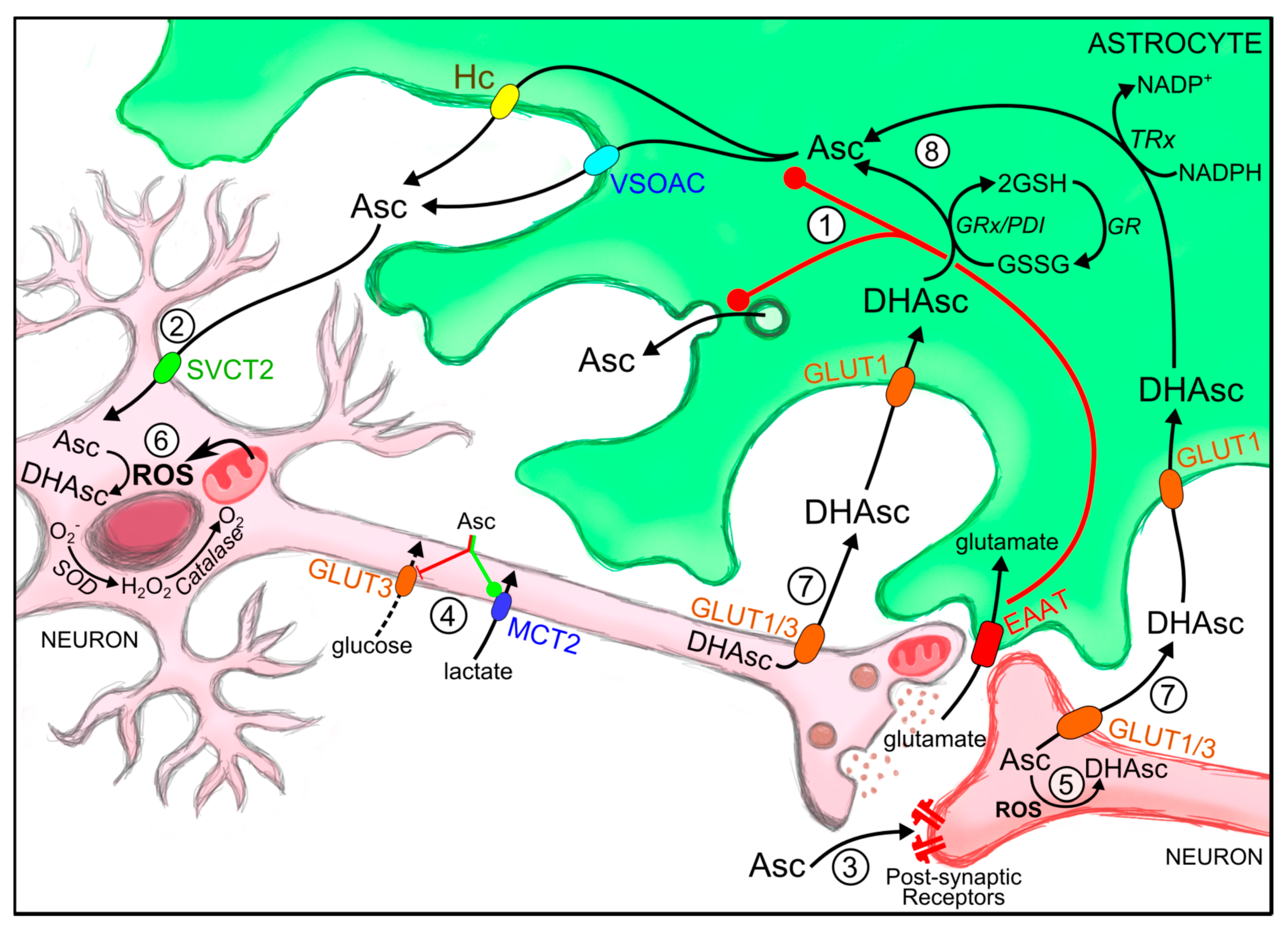
2.1. Ascorbic Acid Uptake and Distribution in the CNS
2.1.1. Ascorbic Acid Homeostasis and Synaptic Activity
2.1.2. Mechanisms for Astrocytic Ascorbic Acid Recycling and Efflux
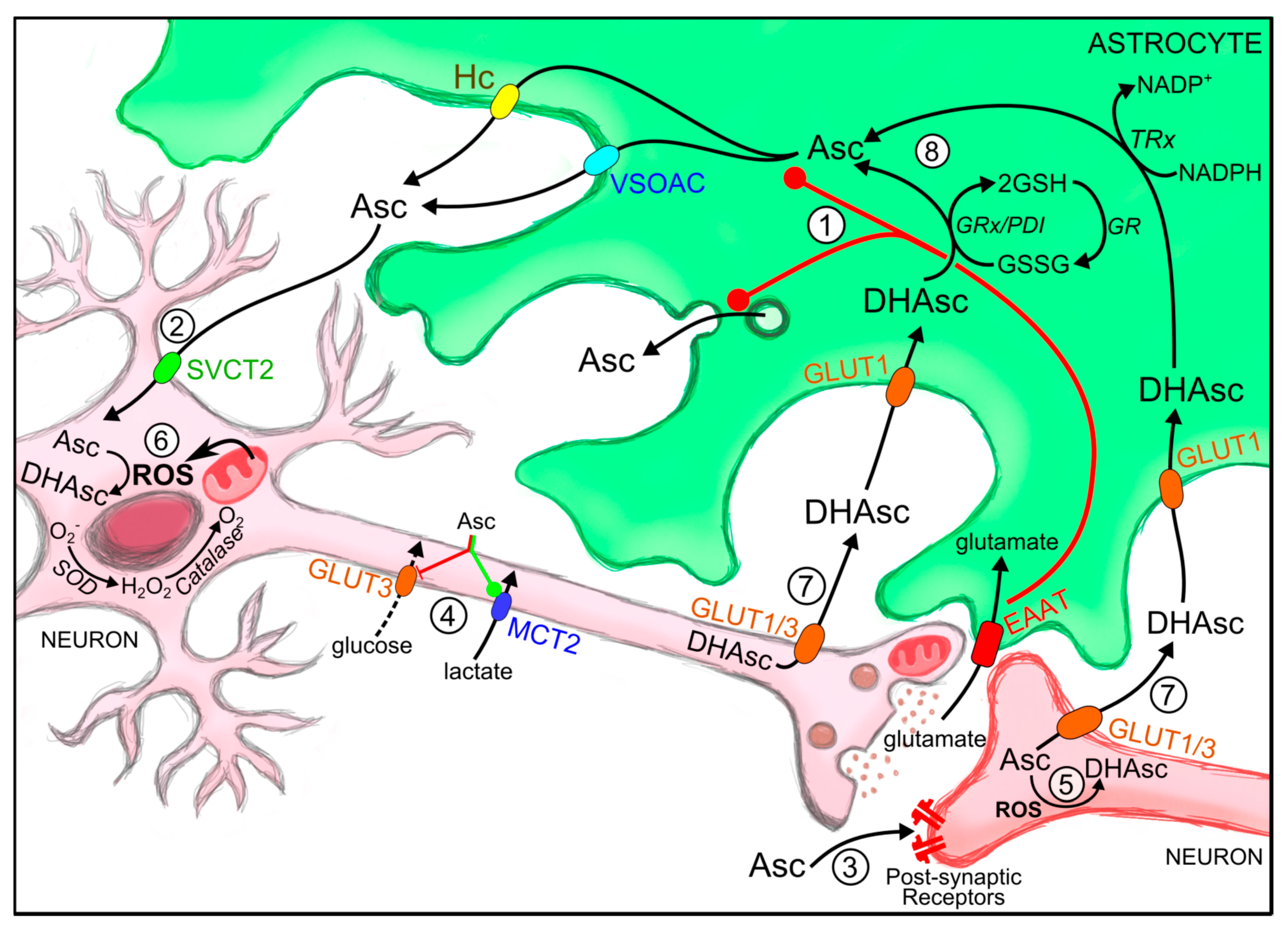
3. Roles of Ascorbic Acid at the CNS
3.1. Ascorbic Acid as a Neuromodulator of Synaptic Activity
3.2. Ascorbic Acid and the Ascorbic Acid Metabolic Switch
4. High Metabolism and ROS Production by Neurons
4.1. High Neuronal Metabolism during Synaptic Activity
4.2. ROS Production and the Ascorbic Acid Metabolic Switch
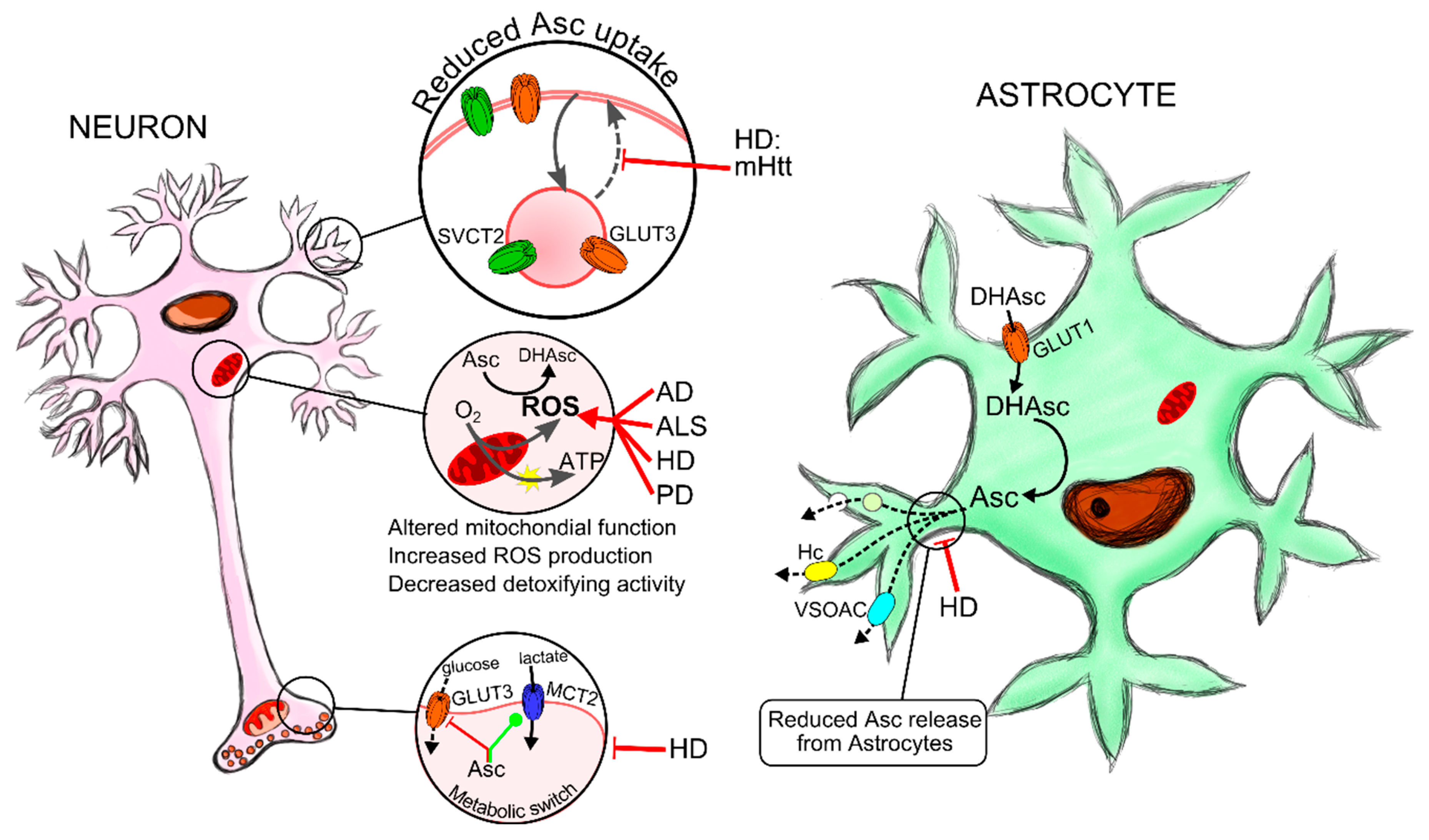
5. Oxidative Stress and Neurodegenerative Disorders
5.1. Redox Imbalance, Ascorbic Acid and Neurodegeneration
5.1.1. Amyotrophic Lateral Sclerosis
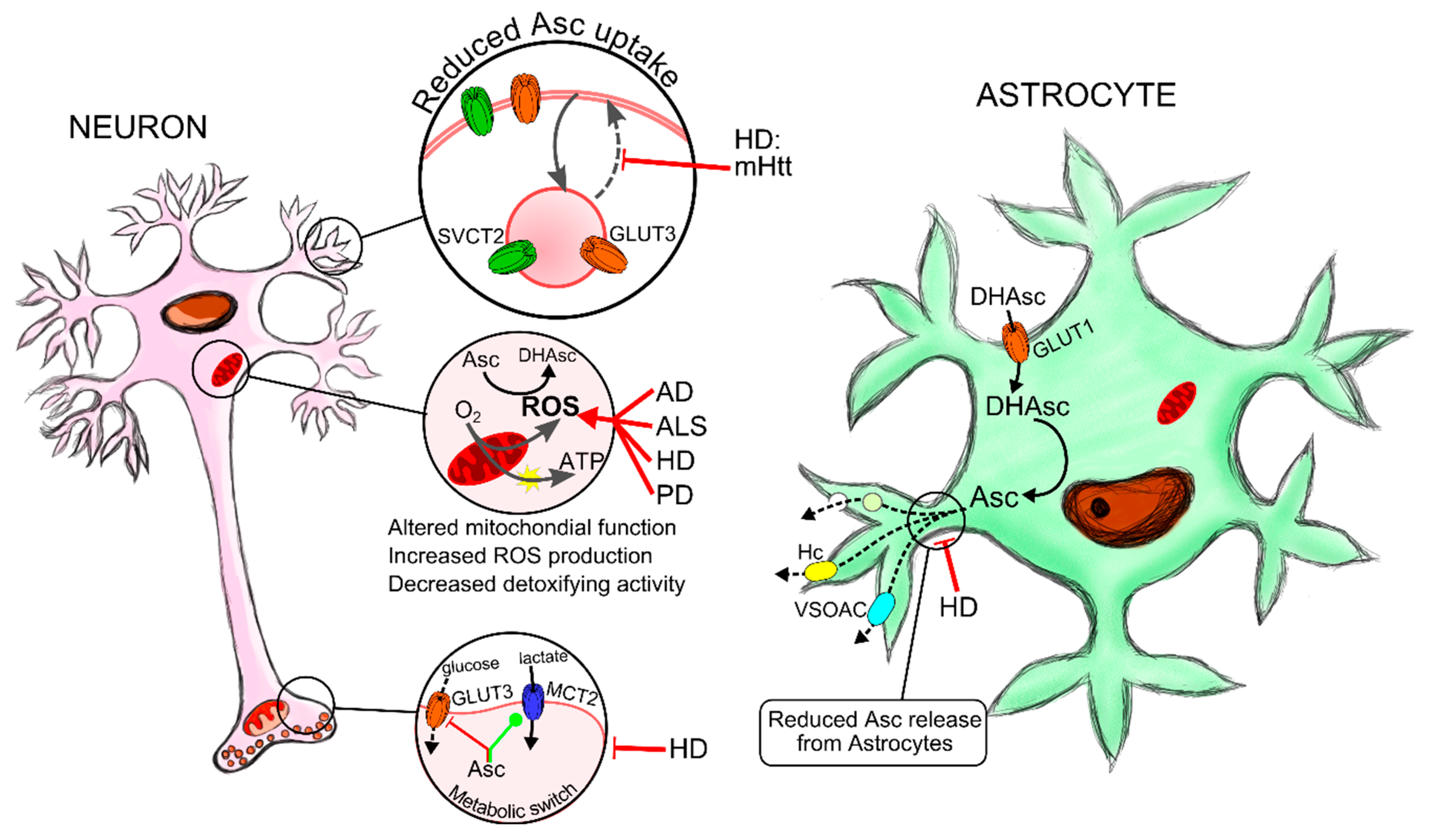
5.1.2. Alzheimer’s Disease
5.1.3. Huntington’s Disease
5.1.4. Parkinson’s Disease
6. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
References
- Du, J.; Cullen, J.J.; Buettner, G.R. Ascorbic acid: Chemistry, biology and the treatment of cancer. Biochim. Biophys. Acta 2012, 1826, 443–457. [Google Scholar] [CrossRef] [PubMed]
- Lachapelle, M.Y.; Drouin, G. Inactivation dates of the human and guinea pig vitamin C genes. Genetica 2011, 139, 199–207. [Google Scholar] [CrossRef] [PubMed]
- Nishikimi, M.; Kawai, T.; Yagi, K. Guinea pigs possess a highly mutated gene for l-gulono-γ-lactone oxidase, the key enzyme for l-ascorbic acid biosynthesis missing in this species. J. Biol. Chem. 1992, 267, 21967–21972. [Google Scholar] [PubMed]
- May, J.M. Vitamin c transport and its role in the central nervous system. Subcell. Biochem. 2012, 56, 85–103. [Google Scholar] [PubMed]
- Harrison, F.E.; May, J.M. Vitamin c function in the brain: Vital role of the ascorbate transporter SVCT2. Free Radic. Biol. Med. 2009, 46, 719–730. [Google Scholar] [CrossRef] [PubMed]
- Frei, B.; England, L.; Ames, B.N. Ascorbate is an outstanding antioxidant in human blood plasma. Proc. Natl. Acad. Sci. USA 1989, 86, 6377–6381. [Google Scholar] [CrossRef] [PubMed]
- Park, J.B.; Levine, M. Purification, cloning and expression of dehydroascorbic acid-reducing activity from human neutrophils: Identification as glutaredoxin. Biochem. J. 1996, 315, 931–938. [Google Scholar] [CrossRef] [PubMed]
- Himmelreich, U.; Drew, K.N.; Serianni, A.S.; Kuchel, P.W. 13C NMR studies of vitamin C transport and its redox cycling in human erythrocytes. Biochemistry 1998, 37, 7578–7588. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, T.; Casini, A.F.; Nishikimi, M. Molecular cloning and functional expression of rat liver glutathione-dependent dehydroascorbate reductase. J. Biol. Chem. 1998, 273, 28708–28712. [Google Scholar] [CrossRef] [PubMed]
- Castro, M.A.; Pozo, M.; Cortes, C.; Garcia Mde, L.; Concha, I.I.; Nualart, F. Intracellular ascorbic acid inhibits transport of glucose by neurons, but not by astrocytes. J. Neurochem. 2007, 102, 773–782. [Google Scholar] [CrossRef] [PubMed]
- Castro, M.A.; Angulo, C.; Brauchi, S.; Nualart, F.; Concha, I.I. Ascorbic acid participates in a general mechanism for concerted glucose transport inhibition and lactate transport stimulation. Pflugers Arch. 2008, 457, 519–528. [Google Scholar] [CrossRef] [PubMed]
- Castro, M.A.; Beltrán, F.A.; Brauchi, S.; Concha, I.I. A metabolic switch in brain: Glucose and lactate metabolism modulation by ascorbic acid. J. Neurochem. 2009, 110, 423–440. [Google Scholar] [CrossRef] [PubMed]
- Spector, R.; Lorenzo, A.V. Ascorbic acid homeostasis in the central nervous system. Am. J. Physiol. 1973, 225, 757–763. [Google Scholar] [PubMed]
- Attwell, D.; Laughlin, S.B. An energy budget for signaling in the grey matter of the brain. J. Cereb. Blood Flow Metab. 2001, 21, 1133–1145. [Google Scholar] [CrossRef] [PubMed]
- Alle, H.; Roth, A.; Geiger, J.R. Energy-efficient action potentials in hippocampal mossy fibers. Science 2009, 325, 1405–1408. [Google Scholar] [CrossRef] [PubMed]
- Wilson, J.X. Antioxidant defense of the brain: A role for astrocytes. Can. J. Physiol. Pharmacol. 1997, 75, 1149–1163. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, K.A.; Wywial, E.; Perez, V.I.; Lambert, A.J.; Edrey, Y.H.; Lewis, K.N.; Grimes, K.; Lindsey, M.L.; Brand, M.D.; Buffenstein, R. Walking the oxidative stress tightrope: A perspective from the naked mole-rat, the longest-living rodent. Curr. Pharm. Des. 2011, 17, 2290–2307. [Google Scholar] [CrossRef] [PubMed]
- Sahoo, A.; Chainy, G.B. Alterations in the activities of cerebral antioxidant enzymes of rat are related to aging. Int. J. Dev. Neurosci. 1997, 15, 939–948. [Google Scholar] [CrossRef]
- Ciani, E.; Groneng, L.; Voltattorni, M.; Rolseth, V.; Contestabile, A.; Paulsen, R.E. Inhibition of free radical production or free radical scavenging protects from the excitotoxic cell death mediated by glutamate in cultures of cerebellar granule neurons. Brain Res. 1996, 728, 1–6. [Google Scholar] [CrossRef]
- Heafield, M.T.; Fearn, S.; Steventon, G.B.; Waring, R.H.; Williams, A.C.; Sturman, S.G. Plasma cysteine and sulphate levels in patients with motor neurone, parkinson’s and alzheimer’s disease. Neurosci. Lett. 1990, 110, 216–220. [Google Scholar] [CrossRef]
- Halliwell, B. Oxidative stress and neurodegeneration: Where are we now? J. Neurochem. 2006, 97, 1634–1658. [Google Scholar] [CrossRef] [PubMed]
- Goswami, A.; Dikshit, P.; Mishra, A.; Mulherkar, S.; Nukina, N.; Jana, N.R. Oxidative stress promotes mutant huntingtin aggregation and mutant huntingtin-dependent cell death by mimicking proteasomal malfunction. Biochem. Biophys. Res. Commun. 2006, 342, 184–190. [Google Scholar] [CrossRef] [PubMed]
- Wyttenbach, A.; Sauvageot, O.; Carmichael, J.; Diaz-Latoud, C.; Arrigo, A.P.; Rubinsztein, D.C. Heat shock protein 27 prevents cellular polyglutamine toxicity and suppresses the increase of reactive oxygen species caused by huntingtin. Hum. Mol. Genet. 2002, 11, 1137–1151. [Google Scholar] [CrossRef] [PubMed]
- Browne, S.E.; Bowling, A.C.; MacGarvey, U.; Baik, M.J.; Berger, S.C.; Muqit, M.M.; Bird, E.D.; Beal, M.F. Oxidative damage and metabolic dysfunction in huntington’s disease: Selective vulnerability of the basal ganglia. Ann. Neurol. 1997, 41, 646–653. [Google Scholar] [CrossRef] [PubMed]
- Warner, T.A.; Kang, J.Q.; Kennard, J.A.; Harrison, F.E. Low brain ascorbic acid increases susceptibility to seizures in mouse models of decreased brain ascorbic acid transport and alzheimer's disease. Epilepsy Res. 2015, 110, 20–25. [Google Scholar] [CrossRef] [PubMed]
- Acuña, A.I.; Esparza, M.; Kramm, C.; Beltrán, F.A.; Parra, A.V.; Cepeda, C.; Toro, C.A.; Vidal, R.L.; Hetz, C.; Concha, I.I.; et al. A failure in energy metabolism and antioxidant uptake precede symptoms of huntington’s disease in mice. Nat. Commun. 2013, 4, 2917. [Google Scholar] [CrossRef] [PubMed]
- Mazziotta, J.C.; Phelps, M.E.; Pahl, J.J.; Huang, S.C.; Baxter, L.R.; Riege, W.H.; Hoffman, J.M.; Kuhl, D.E.; Lanto, A.B.; Wapenski, J.A.; et al. Reduced cerebral glucose metabolism in asymptomatic subjects at risk for huntington’s disease. N. Engl. J. Med. 1987, 316, 357–362. [Google Scholar] [CrossRef] [PubMed]
- Navarro, A.; Boveris, A. Brain mitochondrial dysfunction and oxidative damage in parkinson’s disease. J. Bioenerg. Biomembr. 2009, 41, 517–521. [Google Scholar] [CrossRef] [PubMed]
- Berger, T.M.; Polidori, M.C.; Dabbagh, A.; Evans, P.J.; Halliwell, B.; Morrow, J.D.; Roberts, L.J., 2nd; Frei, B. Antioxidant activity of vitamin C in iron-overloaded human plasma. J. Biol. Chem. 1997, 272, 15656–15660. [Google Scholar] [CrossRef] [PubMed]
- Kiuchi, K.; Nishikimi, M.; Yagi, K. Purification and characterization of l-gulonolactone oxidase from chicken kidney microsomes. Biochemistry 1982, 21, 5076–5082. [Google Scholar] [CrossRef] [PubMed]
- Grunewald, R.A. Ascorbic acid in the brain. Brain Res. Brain Res. Rev. 1993, 18, 123–133. [Google Scholar] [CrossRef]
- Harrison, F.E.; Bowman, G.L.; Polidori, M.C. Ascorbic acid and the brain: Rationale for the use against cognitive decline. Nutrients 2014, 6, 1752–1781. [Google Scholar] [CrossRef] [PubMed]
- Siliprandi, L.; Vanni, P.; Kessler, M.; Semenza, G. Na+-dependent, electroneutral l-ascorbate transport across brush border membrane vesicles from guinea pig small intestine. BBA Biomembr. 1979, 552, 129–142. [Google Scholar] [CrossRef]
- Levine, M.; Wang, Y.; Padayatty, S.J.; Morrow, J. A new recommended dietary allowance of vitamin C for healthy young women. Proc. Nat. Acad. Sci. USA 2001, 98, 9842–9846. [Google Scholar] [CrossRef] [PubMed]
- Carr, A.C.; Frei, B. Toward a new recommended dietary allowance for vitamin C based on antioxidant and health effects in humans. Am. J. Clin. Nutr. 1999, 69, 1086–1107. [Google Scholar] [PubMed]
- Deruelle, F.; Baron, B. Vitamin C: Is supplementation necessary for optimal health? J. Altern. Complement. Med. 2008, 14, 1291–1298. [Google Scholar] [CrossRef] [PubMed]
- Levine, G.N.; Frei, B.; Koulouris, S.N.; Gerhard, M.D.; Keaney, J.F., Jr.; Vita, J.A. Ascorbic acid reverses endothelial vasomotor dysfunction in patients with coronary artery disease. Circulation 1996, 93, 1107–1113. [Google Scholar] [CrossRef] [PubMed]
- Friedman, G.J.; Sherry, S.; Ralli, E.P. The mechanism of the excretion of vitamin c by the human kidney at low and normal plasma levels of ascorbic acid. J. Clin. Investig. 1940, 19, 685–689. [Google Scholar] [CrossRef] [PubMed]
- Faaland, C.A.; Race, J.E.; Ricken, G.; Warner, F.J.; Williams, W.J.; Holtzman, E.J. Molecular characterization of two novel transporters from human and mouse kidney and from LLC-PK1 cells reveals a novel conserved family that is homologous to bacterial and aspergillus nucleobase transporters. BBA Gene Struct. Expr. 1998, 1442, 353–360. [Google Scholar] [CrossRef]
- Daruwala, R.; Song, J.; Koh, W.S.; Rumsey, S.C.; Levine, M. Cloning and functional characterization of the human sodium-dependent vitamin C transporters HSVCT1 and HSVCT2. FEBS Lett. 1999, 460, 480–484. [Google Scholar] [CrossRef]
- Tsukaguchi, H.; Tokui, T.; Mackenzie, B.; Berger, U.V.; Chen, X.Z.; Wang, Y.; Brubaker, R.F.; Hediger, M.A. A family of mammalian Na+-dependent l-ascorbic acid transporters. Nature 1999, 399, 70–75. [Google Scholar] [PubMed]
- Castro, M.; Caprile, T.; Astuya, A.; Millan, C.; Reinicke, K.; Vera, J.C.; Vasquez, O.; Aguayo, L.G.; Nualart, F. High-affinity sodium-vitamin C co-transporters (SVCT) expression in embryonic mouse neurons. J. Neurochem. 2001, 78, 815–823. [Google Scholar] [CrossRef] [PubMed]
- Garcia Mde, L.; Salazar, K.; Millan, C.; Rodriguez, F.; Montecinos, H.; Caprile, T.; Silva, C.; Cortes, C.; Reinicke, K.; Vera, J.C.; et al. Sodium vitamin C cotransporter SVCT2 is expressed in hypothalamic glial cells. Glia 2005, 50, 32–47. [Google Scholar] [CrossRef] [PubMed]
- Takanaga, H.; Mackenzie, B.; Hediger, M.A. Sodium-dependent ascorbic acid transporter family SLC23. Pflugers Arch. 2004, 447, 677–682. [Google Scholar] [CrossRef] [PubMed]
- Wilson, J.X. Regulation of vitamin C transport. Annu. Rev. Nutr. 2005, 25, 105–125. [Google Scholar] [CrossRef] [PubMed]
- Lam, D.K.; Daniel, P.M. The influx of ascorbic acid into the rat’s brain. Quart. J. Exp. Physiol. 1986, 71, 483–489. [Google Scholar] [CrossRef]
- Angelow, S.; Haselbach, M.; Galla, H.J. Functional characterisation of the active ascorbic acid transport into cerebrospinal fluid using primary cultured choroid plexus cells. Brain Res. 2003, 988, 105–113. [Google Scholar] [CrossRef]
- Nualart, F.; Mack, L.; Garcia, A.; Cisternas, P.; Bongarzone, E.R.; Heitzer, M.; Jara, N.; Martinez, F.; Ferrada, L.; Espinoza, F.; et al. Vitamin C transporters, recycling and the bystander effect in the nervous system: SVCT2 versus gluts. J. Stem Cell Res. Ther. 2014, 4, 209. [Google Scholar] [CrossRef] [PubMed]
- Schenk, J.O.; Miller, E.; Gaddis, R.; Adams, R.N. Homeostatic control of ascorbate concentration in CNS extracellular fluid. Brain Res. 1982, 253, 353–356. [Google Scholar] [CrossRef]
- Agus, D.B.; Gambhir, S.S.; Pardridge, W.M.; Spielholz, C.; Baselga, J.; Vera, J.C.; Golde, D.W. Vitamin C crosses the blood-brain barrier in the oxidized form through the glucose transporters. J. Clin. Investig. 1997, 100, 2842–2848. [Google Scholar] [CrossRef] [PubMed]
- Taverna, R.D.; Langdon, R.G. Reversible association of cytochalasin B with the human erythrocyte membrane. Inhibition of glucose transport and the stoichiometry of cytochalasin binding. BBA Biomembr. 1973, 323, 207–219. [Google Scholar] [CrossRef]
- Agus, D.B.; Vera, J.C.; Golde, D.W. Stromal cell oxidation: A mechanism by which tumors obtain vitamin C. Cancer Res. 1999, 59, 4555–4558. [Google Scholar] [PubMed]
- Nualart, F.J.; Rivas, C.I.; Montecinos, V.P.; Godoy, A.S.; Guaiquil, V.H.; Golde, D.W.; Vera, J.C. Recycling of vitamin C by a bystander effect. J. Biol. Chem. 2003, 278, 10128–10133. [Google Scholar] [CrossRef] [PubMed]
- Rice, M.E. Ascorbate regulation and its neuroprotective role in the brain. Trends Neurosci. 2000, 23, 209–216. [Google Scholar] [CrossRef]
- Rice, M.E.; Russo-Menna, I. Differential compartmentalization of brain ascorbate and glutathione between neurons and glia. Neuroscience 1998, 82, 1213–1223. [Google Scholar] [CrossRef]
- Milby, K.; Oke, A.; Adams, R.N. Detailed mapping of ascorbate distribution in rat brain. Neurosci. Lett. 1982, 28, 169–174. [Google Scholar] [CrossRef]
- Stamford, J.A.; Kruk, Z.L.; Millar, J. Regional differences in extracellular ascorbic acid levels in the rat brain determined by high speed cyclic voltammetry. Brain Res. 1984, 299, 289–295. [Google Scholar] [CrossRef]
- Miele, M.; Fillenz, M. In vivo determination of extracellular brain ascorbate. J. Neurosci. Methods 1996, 70, 15–19. [Google Scholar] [CrossRef]
- Hediger, M.A. New view at C. Nat. Med. 2002, 8, 445–446. [Google Scholar] [CrossRef] [PubMed]
- Basse-Tomusk, A.; Rebec, G.V. Regional distribution of ascorbate and 3,4-dihydroxyphenylacetic acid (DOPAC) in rat striatum. Brain Res. 1991, 538, 29–35. [Google Scholar] [CrossRef]
- Ghasemzadeh, B.; Cammack, J.; Adams, R.N. Dynamic changes in extracellular fluid ascorbic acid monitored by in vivo electrochemistry. Brain Res. 1991, 547, 162–166. [Google Scholar] [CrossRef]
- O’Neill, R.D.; Fillenz, M.; Sundstrom, L.; Rawlins, J.N. Voltammetrically monitored brain ascorbate as an index of excitatory amino acid release in the unrestrained rat. Neurosci. Lett. 1984, 52, 227–233. [Google Scholar] [CrossRef]
- Boutelle, M.G.; Svensson, L.; Fillenz, M. Rapid changes in striatal ascorbate in response to tail-pinch monitored by constant potential voltammetry. Neuroscience 1989, 30, 11–17. [Google Scholar] [CrossRef]
- Carr, A.; Frei, B. Does vitamin C act as a pro-oxidant under physiological conditions? FASEB J. 1999, 13, 1007–1024. [Google Scholar] [PubMed]
- Maellaro, E.; del Bello, B.; Sugherini, L.; Santucci, A.; Comporti, M.; Casini, A.F. Purification and characterization of glutathione-dependent dehydroascorbate reductase from rat liver. Biochem. J. 1994, 301, 471–476. [Google Scholar] [CrossRef] [PubMed]
- Washburn, M.P.; Wells, W.W. Identification of the dehydroascorbic acid reductase and thioltransferase (glutaredoxin) activities of bovine erythrocyte glutathione peroxidase. Biochem. Biophys. Res. Commun. 1999, 257, 567–571. [Google Scholar] [CrossRef] [PubMed]
- Guaiquil, V.H.; Farber, C.M.; Golde, D.W.; Vera, J.C. Efficient transport and accumulation of vitamin C in Hl-60 cells depleted of glutathione. J. Biol. Chem. 1997, 272, 9915–9921. [Google Scholar] [PubMed]
- May, J.M.; Qu, Z.C.; Mendiratta, S. Protection and recycling of α-tocopherol in human erythrocytes by intracellular ascorbic acid. Arch. Biochem. Biophys. 1998, 349, 281–289. [Google Scholar] [CrossRef] [PubMed]
- Savini, I.; Duflot, S.; Avigliano, L. Dehydroascorbic acid uptake in a human keratinocyte cell line (HaCaT) is glutathione-independent. Biochem. J. 2000, 345(3), 665–672. [Google Scholar] [CrossRef] [PubMed]
- Nualart, F.; Salazar, K.; Oyarce, K.; Cisternas, P.; Jara, N.; Silva-Alvarez, C.; Pastor, P.; Martinez, F.; Garcia, A.; Garcia-Robles Mde, L.; et al. Typical and atypical stem cells in the brain, vitamin C effect and neuropathology. Biol. Res. 2012, 45, 243–256. [Google Scholar] [CrossRef] [PubMed]
- Rumsey, S.C.; Kwon, O.; Xu, G.W.; Burant, C.F.; Simpson, I.; Levine, M. Glucose transporter isoforms GLUT1 and GLUT3 transport dehydroascorbic acid. J. Biol. Chem. 1997, 272, 18982–18989. [Google Scholar] [CrossRef] [PubMed]
- Vera, J.C.; Rivas, C.I.; Fischbarg, J.; Golde, D.W. Mammalian facilitative hexose transporters mediate the transport of dehydroascorbic acid. Nature 1993, 364, 79–82. [Google Scholar] [CrossRef] [PubMed]
- Makar, T.K.; Nedergaard, M.; Preuss, A.; Gelbard, A.S.; Perumal, A.S.; Cooper, A.J. Vitamin E, ascorbate, glutathione, glutathione disulfide, and enzymes of glutathione metabolism in cultures of chick astrocytes and neurons: Evidence that astrocytes play an important role in antioxidative processes in the brain. J. Neurochem. 1994, 62, 45–53. [Google Scholar] [CrossRef] [PubMed]
- Desagher, S.; Glowinski, J.; Premont, J. Astrocytes protect neurons from hydrogen peroxide toxicity. J. Neurosci. 1996, 16, 2553–2562. [Google Scholar] [PubMed]
- Dringen, R.; Kussmaul, L.; Gutterer, J.M.; Hirrlinger, J.; Hamprecht, B. The glutathione system of peroxide detoxification is less efficient in neurons than in astroglial cells. J. Neurochem. 1999, 72, 2523–2530. [Google Scholar] [CrossRef] [PubMed]
- Rohl, C.; Armbrust, E.; Herbst, E.; Jess, A.; Gulden, M.; Maser, E.; Rimbach, G.; Bosch-Saadatmandi, C. Mechanisms involved in the modulation of astroglial resistance to oxidative stress induced by activated microglia: Antioxidative systems, peroxide elimination, radical generation, lipid peroxidation. Neurotox. Res. 2010, 17, 317–331. [Google Scholar] [CrossRef] [PubMed]
- Macco, R.; Pelizzoni, I.; Consonni, A.; Vitali, I.; Giacalone, G.; Martinelli Boneschi, F.; Codazzi, F.; Grohovaz, F.; Zacchetti, D. Astrocytes acquire resistance to iron-dependent oxidative stress upon proinflammatory activation. J. Neuroinflamm. 2013, 10, 130. [Google Scholar] [CrossRef] [PubMed]
- Almeida, A.; Almeida, J.; Bolanos, J.P.; Moncada, S. Different responses of astrocytes and neurons to nitric oxide: The role of glycolytically generated ATP in astrocyte protection. Proc. Natl. Acad. Sci. USA 2001, 98, 15294–15299. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Nogales, P.; Almeida, A.; Bolanos, J.P. Peroxynitrite protects neurons against nitric oxide-mediated apoptosis. A key role for glucose-6-phosphate dehydrogenase activity in neuroprotection. J. Biol. Chem. 2003, 278, 864–874. [Google Scholar] [CrossRef] [PubMed]
- Ben-Yoseph, O.; Boxer, P.A.; Ross, B.D. Assessment of the role of the glutathione and pentose phosphate pathways in the protection of primary cerebrocortical cultures from oxidative stress. J. Neurochem. 1996, 66, 2329–2337. [Google Scholar] [CrossRef] [PubMed]
- Lovatt, D.; Sonnewald, U.; Waagepetersen, H.S.; Schousboe, A.; He, W.; Lin, J.H.; Han, X.; Takano, T.; Wang, S.; Sim, F.J.; et al. The transcriptome and metabolic gene signature of protoplasmic astrocytes in the adult murine cortex. J. Neurosci. 2007, 27, 12255–12266. [Google Scholar] [CrossRef] [PubMed]
- Cahoy, J.D.; Emery, B.; Kaushal, A.; Foo, L.C.; Zamanian, J.L.; Christopherson, K.S.; Xing, Y.; Lubischer, J.L.; Krieg, P.A.; Krupenko, S.A.; et al. A transcriptome database for astrocytes, neurons, and oligodendrocytes: A new resource for understanding brain development and function. J. Neurosci. 2008, 28, 264–278. [Google Scholar] [CrossRef] [PubMed]
- Astuya, A.; Caprile, T.; Castro, M.; Salazar, K.; Garcia Mde, L.; Reinicke, K.; Rodriguez, F.; Vera, J.C.; Millan, C.; Ulloa, V.; et al. Vitamin C uptake and recycling among normal and tumor cells from the central nervous system. J. Neurosci. Res. 2005, 79, 146–156. [Google Scholar] [CrossRef] [PubMed]
- Yusa, T. Increased extracellular ascorbate release reflects glutamate re-uptake during the early stage of reperfusion after forebrain ischemia in rats. Brain Res. 2001, 897, 104–113. [Google Scholar] [CrossRef]
- Patterson, J.W.; Mastin, D.W. Some effects of dehydroascorbic acid on the central nervous system. Am. J. Physiol. 1951, 167, 119–126. [Google Scholar] [PubMed]
- Deponte, M. Glutathione catalysis and the reaction mechanisms of glutathione-dependent enzymes. BBA Gen. Subj. 2013, 1830, 3217–3266. [Google Scholar] [CrossRef] [PubMed]
- Winkler, B.S.; Orselli, S.M.; Rex, T.S. The redox couple between glutathione and ascorbic acid: A chemical and physiological perspective. Free Radic. Biol. Med. 1994, 17, 333–349. [Google Scholar] [CrossRef]
- Wells, W.W.; Xu, D.P.; Yang, Y.F.; Rocque, P.A. Mammalian thioltransferase (glutaredoxin) and protein disulfide isomerase have dehydroascorbate reductase activity. J. Biol. Chem. 1990, 265, 15361–15364. [Google Scholar] [PubMed]
- Wells, W.W.; Xu, D.P. Dehydroascorbate reduction. J. Bioenerg. Biomembr. 1994, 26, 369–377. [Google Scholar] [CrossRef] [PubMed]
- Linster, C.L.; van Schaftingen, E. Vitamin C. Biosynthesis, recycling and degradation in mammals. FEBS J. 2007, 274, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Board, P.G.; Coggan, M.; Chelvanayagam, G.; Easteal, S.; Jermiin, L.S.; Schulte, G.K.; Danley, D.E.; Hoth, L.R.; Griffor, M.C.; Kamath, A.V.; et al. Identification, characterization, and crystal structure of the ω class glutathione transferases. J. Biol. Chem. 2000, 275, 24798–24806. [Google Scholar] [CrossRef] [PubMed]
- Whitbread, A.K.; Masoumi, A.; Tetlow, N.; Schmuck, E.; Coggan, M.; Board, P.G. Characterization of the ω class of glutathione transferases. Methods Enzymol. 2005, 401, 78–99. [Google Scholar] [PubMed]
- Del Bello, B.; Maellaro, E.; Sugherini, L.; Santucci, A.; Comporti, M.; Casini, A.F. Purification of NADPH-dependent dehydroascorbate reductase from rat liver and its identification with 3 α-hydroxysteroid dehydrogenase. Biochem. J. 1994, 304, 385–390. [Google Scholar] [CrossRef] [PubMed]
- Mendiratta, S.; Qu, Z.C.; May, J.M. Enzyme-dependent ascorbate recycling in human erythrocytes: Role of thioredoxin reductase. Free Radic. Biol. Med. 1998, 25, 221–228. [Google Scholar] [CrossRef]
- Rose, R.C. Cerebral metabolism of oxidized ascorbate. Brain Res. 1993, 628, 49–55. [Google Scholar] [CrossRef]
- Dragan, M.; Dixon, S.J.; Jaworski, E.; Chan, T.S.; O’Brien, P.J.; Wilson, J.X. Coenzyme Q1 depletes NAD(P)H and impairs recycling of ascorbate in astrocytes. Brain Res. 2006, 1078, 9–18. [Google Scholar] [CrossRef] [PubMed]
- Siushansian, R.; Tao, L.; Dixon, S.J.; Wilson, J.X. Cerebral astrocytes transport ascorbic acid and dehydroascorbic acid through distinct mechanisms regulated by cyclic AMP. J. Neurochem. 1997, 68, 2378–2385. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, J.; Toku, K.; Zhang, B.; Ishihara, K.; Sakanaka, M.; Maeda, N. Astrocytes prevent neuronal death induced by reactive oxygen and nitrogen species. Glia 1999, 28, 85–96. [Google Scholar] [CrossRef]
- Gegg, M.E.; Beltran, B.; Salas-Pino, S.; Bolanos, J.P.; Clark, J.B.; Moncada, S.; Heales, S.J. Differential effect of nitric oxide on glutathione metabolism and mitochondrial function in astrocytes and neurones: Implications for neuroprotection/neurodegeneration? J. Neurochem. 2003, 86, 228–237. [Google Scholar] [CrossRef] [PubMed]
- Dringen, R.; Pfeiffer, B.; Hamprecht, B. Synthesis of the antioxidant glutathione in neurons: Supply by astrocytes of CysGly as precursor for neuronal glutathione. J. Neurosci. 1999, 19, 562–569. [Google Scholar] [PubMed]
- Langeveld, C.H.; Jongenelen, C.A.; Schepens, E.; Stoof, J.C.; Bast, A.; Drukarch, B. Cultured rat striatal and cortical astrocytes protect mesencephalic dopaminergic neurons against hydrogen peroxide toxicity independent of their effect on neuronal development. Neurosci. Lett. 1995, 192, 13–16. [Google Scholar] [CrossRef]
- Fujita, T.; Tozaki-Saitoh, H.; Inoue, K. P2y1 receptor signaling enhances neuroprotection by astrocytes against oxidative stress via il-6 release in hippocampal cultures. Glia 2009, 57, 244–257. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Vartiainen, N.E.; Ying, W.; Chan, P.H.; Koistinaho, J.; Swanson, R.A. Astrocytes protect neurons from nitric oxide toxicity by a glutathione-dependent mechanism. J. Neurochem. 2001, 77, 1601–1610. [Google Scholar] [CrossRef] [PubMed]
- Lucius, R.; Sievers, J. Postnatal retinal ganglion cells in vitro: Protection against reactive oxygen species (ROS)-induced axonal degeneration by cocultured astrocytes. Brain Res. 1996, 743, 56–62. [Google Scholar] [CrossRef]
- Wilson, J.X.; Peters, C.E.; Sitar, S.M.; Daoust, P.; Gelb, A.W. Glutamate stimulates ascorbate transport by astrocytes. Brain Res. 2000, 858, 61–66. [Google Scholar] [CrossRef]
- Selkirk, J.V.; Nottebaum, L.M.; Vana, A.M.; Verge, G.M.; Mackay, K.B.; Stiefel, T.H.; Naeve, G.S.; Pomeroy, J.E.; Petroski, R.E.; Moyer, J.; et al. Role of the GLT-1 subtype of glutamate transporter in glutamate homeostasis: The GLT-1-preferring inhibitor way-855 produces marginal neurotoxicity in the rat hippocampus. Eur. J. Neurosci. 2005, 21, 3217–3228. [Google Scholar] [CrossRef] [PubMed]
- Bak, L.K.; Schousboe, A.; Waagepetersen, H.S. The glutamate/gaba-glutamine cycle: Aspects of transport, neurotransmitter homeostasis and ammonia transfer. J. Neurochem. 2006, 98, 641–653. [Google Scholar] [CrossRef] [PubMed]
- Miele, M.; Boutelle, M.G.; Fillenz, M. The physiologically induced release of ascorbate in rat brain is dependent on impulse traffic, calcium influx and glutamate uptake. Neuroscience 1994, 62, 87–91. [Google Scholar] [CrossRef]
- Rebec, G.V.; Pierce, R.C. A vitamin as neuromodulator: Ascorbate release into the extracellular fluid of the brain regulates dopaminergic and glutamatergic transmission. Prog. Neurobiol. 1994, 43, 537–565. [Google Scholar] [CrossRef]
- Bell, J.A.; Beglan, C.L.; London, E.D. Interaction of ascorbic acid with the neurotoxic effects of NMDA and sodium nitroprusside. Life Sci. 1996, 58, 367–371. [Google Scholar] [CrossRef]
- Lane, D.J.; Lawen, A. The glutamate aspartate transporter (GLAST) mediates l-glutamate-stimulated ascorbate-release via swelling-activated anion channels in cultured neonatal rodent astrocytes. Cell Biochem. Biophys. 2013, 65, 107–119. [Google Scholar] [CrossRef] [PubMed]
- Cammack, J.; Ghasemzadeh, B.; Adams, R.N. The pharmacological profile of glutamate-evoked ascorbic acid efflux measured by in vivo electrochemistry. Brain Res. 1991, 565, 17–22. [Google Scholar] [CrossRef]
- Siushansian, R.; Dixon, S.J.; Wilson, J.X. Osmotic swelling stimulates ascorbate efflux from cerebral astrocytes. J. Neurochem. 1996, 66, 1227–1233. [Google Scholar] [CrossRef] [PubMed]
- Corti, A.; Casini, A.F.; Pompella, A. Cellular pathways for transport and efflux of ascorbate and dehydroascorbate. Arch. Biochem. Biophys. 2010, 500, 107–115. [Google Scholar] [CrossRef] [PubMed]
- Ekholm, A.; Katsura, K.; Kristian, T.; Liu, M.; Folbergrova, J.; Siesjo, B.K. Coupling of cellular energy state and ion homeostasis during recovery following brain ischemia. Brain Res. 1993, 604, 185–191. [Google Scholar] [CrossRef]
- Ahmad, S.; Evans, W.H. Post-translational integration and oligomerization of connexin 26 in plasma membranes and evidence of formation of membrane pores: Implications for the assembly of gap junctions. Biochem. J. 2002, 365, 693–699. [Google Scholar] [CrossRef] [PubMed]
- Locke, D.; Koreen, I.V.; Liu, J.Y.; Harris, A.L. Reversible pore block of connexin channels by cyclodextrins. J. Biol. Chem. 2004, 279, 22883–22892. [Google Scholar] [CrossRef] [PubMed]
- Ramundo-Orlando, A.; Serafino, A.; Schiavo, R.; Liberti, M.; d’Inzeo, G. Permeability changes of connexin32 hemi channels reconstituted in liposomes induced by extremely low frequency, low amplitude magnetic fields. BBA Biomembr. 2005, 1668, 33–40. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Von Zastrow, M.; Tritton, T.R.; Castle, J.D. Identification of l-ascorbic acid in secretion granules of the rat parotid gland. J. Biol. Chem. 1984, 259, 11746–11750. [Google Scholar] [PubMed]
- Kivirikko, K.I.; Prockop, D.J. Hydroxylation of proline in synthetic polypeptides with purified protocollagen hydroxylase. J. Biol. Chem. 1967, 242, 4007–4012. [Google Scholar] [PubMed]
- Kivirikko, K.I.; Prockop, D.J. Enzymatic hydroxylation of proline and lysine in protocollagen. Proc. Nat. Acad. Sci. USA 1967, 57, 782–789. [Google Scholar] [CrossRef] [PubMed]
- Kivirikko, K.I.; Prockop, D.J. Partial characterization of protocollagen from embryonic cartilage. Biochem. J. 1967, 102, 432–442. [Google Scholar] [CrossRef] [PubMed]
- Peterkofsky, B. Ascorbate requirement for hydroxylation and secretion of procollagen: Relationship to inhibition of collagen synthesis in scurvy. Am. J. Clin. Nutr. 1991, 54, 1135S–1140S. [Google Scholar] [PubMed]
- Padh, H. Cellular functions of ascorbic acid. Biochem. Cell Biol. 1990, 68, 1166–1173. [Google Scholar] [CrossRef] [PubMed]
- Levine, M.; Wang, Y.; Rumsey, S.C. Analysis of ascorbic acid and dehydroascorbic acid in biological samples. Methods Enzymol. 1999, 299, 65–76. [Google Scholar] [PubMed]
- Chatterjee, I.B.; Majumder, A.K.; Nandi, B.K.; Subramanian, N. Synthesis and some major functions of vitamin C in animals. Ann. N. Y. Acad. Sci. 1975, 258, 24–47. [Google Scholar] [CrossRef] [PubMed]
- Padayatty, S.J.; Levine, M. Vitamin C and coronary microcirculation. Circulation 2001, 103, E117. [Google Scholar] [CrossRef] [PubMed]
- Verrax, J.; Calderon, P.B. The controversial place of vitamin C in cancer treatment. Biochem. Pharmacol. 2008, 76, 1644–1652. [Google Scholar] [CrossRef] [PubMed]
- Traber, M.G.; Stevens, J.F. Vitamins C and E: Beneficial effects from a mechanistic perspective. Free Radic. Biol. Med. 2011, 51, 1000–1013. [Google Scholar] [CrossRef] [PubMed]
- Tveden-Nyborg, P.; Johansen, L.K.; Raida, Z.; Villumsen, C.K.; Larsen, J.O.; Lykkesfeldt, J. Vitamin C deficiency in early postnatal life impairs spatial memory and reduces the number of hippocampal neurons in guinea pigs. Am. J. Clin. Nutr. 2009, 90, 540–546. [Google Scholar] [CrossRef] [PubMed]
- Lykkesfeldt, J.; Trueba, G.P.; Poulsen, H.E.; Christen, S. Vitamin C deficiency in weanling guinea pigs: Differential expression of oxidative stress and DNA repair in liver and brain. Br. J. Nutr. 2007, 98, 1116–1119. [Google Scholar] [CrossRef] [PubMed]
- Kratzing, C.C.; Kelly, J.D.; Kratzing, J.E. Ascorbic acid in fetal rat brain. J. Neurochem. 1985, 44, 1623–1624. [Google Scholar] [CrossRef] [PubMed]
- Tveden-Nyborg, P.; Vogt, L.; Schjoldager, J.G.; Jeannet, N.; Hasselholt, S.; Paidi, M.D.; Christen, S.; Lykkesfeldt, J. Maternal vitamin C deficiency during pregnancy persistently impairs hippocampal neurogenesis in offspring of guinea pigs. PLoS ONE 2012, 7, e48488. [Google Scholar] [CrossRef] [PubMed]
- Sotiriou, S.; Gispert, S.; Cheng, J.; Wang, Y.; Chen, A.; Hoogstraten-Miller, S.; Miller, G.F.; Kwon, O.; Levine, M.; Guttentag, S.H.; et al. Ascorbic-acid transporter Slc23a1 is essential for vitamin C transport into the brain and for perinatal survival. Nat. Med. 2002, 8, 514–517. [Google Scholar] [CrossRef] [PubMed]
- Naseer, M.I.; Ullah, N.; Ullah, I.; Koh, P.O.; Lee, H.Y.; Park, M.S.; Kim, M.O. Vitamin C protects against ethanol and PTZ-induced apoptotic neurodegeneration in prenatal rat hippocampal neurons. Synapse 2011, 65, 562–571. [Google Scholar] [CrossRef] [PubMed]
- Niki, E. Action of ascorbic acid as a scavenger of active and stable oxygen radicals. Am. J. Clin. Nutr. 1991, 54, 1119S–1124S. [Google Scholar] [PubMed]
- Santos, I.M.; Tome Ada, R.; Saldanha, G.B.; Ferreira, P.M.; Militao, G.C.; Freitas, R.M. Oxidative stress in the hippocampus during experimental seizures can be ameliorated with the antioxidant ascorbic acid. Oxid. Med. Cell. Longev. 2009, 2, 214–221. [Google Scholar] [CrossRef] [PubMed]
- Qiu, S.; Li, L.; Weeber, E.J.; May, J.M. Ascorbate transport by primary cultured neurons and its role in neuronal function and protection against excitotoxicity. J. Neurosci. Res. 2007, 85, 1046–1056. [Google Scholar] [CrossRef] [PubMed]
- Coyle, J.T.; Puttfarcken, P. Oxidative stress, glutamate, and neurodegenerative disorders. Science 1993, 262, 689–695. [Google Scholar] [CrossRef] [PubMed]
- Lipton, S.A.; Rosenberg, P.A. Excitatory amino acids as a final common pathway for neurologic disorders. N. Engl. J. Med. 1994, 330, 613–622. [Google Scholar] [PubMed]
- Choi, D.W. Glutamate neurotoxicity and diseases of the nervous system. Neuron 1988, 1, 623–634. [Google Scholar] [CrossRef]
- Todd, R.D.; Bauer, P.A. Ascorbate modulates 5-[3H]hydroxytryptamine binding to central 5-HT3 sites in bovine frontal cortex. J. Neurochem. 1988, 50, 1505–1512. [Google Scholar] [CrossRef] [PubMed]
- Majewska, M.D.; Bell, J.A. Ascorbic acid protects neurons from injury induced by glutamate and NMDA. Neuroreport 1990, 1, 194–196. [Google Scholar] [CrossRef] [PubMed]
- Majewska, M.D.; Bell, J.A.; London, E.D. Regulation of the NMDA receptor by redox phenomena: Inhibitory role of ascorbate. Brain Res. 1990, 537, 328–332. [Google Scholar] [CrossRef]
- Levine, M.; Morita, K.; Heldman, E.; Pollard, H.B. Ascorbic acid regulation of norepinephrine biosynthesis in isolated chromaffin granules from bovine adrenal medulla. J. Biol. Chem. 1985, 260, 15598–15603. [Google Scholar] [PubMed]
- Levine, M.; Morita, K.; Pollard, H. Enhancement of norepinephrine biosynthesis by ascorbic acid in cultured bovine chromaffin cells. J. Biol. Chem. 1985, 260, 12942–12947. [Google Scholar] [PubMed]
- Paterson, I.A.; Hertz, L. Sodium-independent transport of noradrenaline in mouse and rat astrocytes in primary culture. J. Neurosci. Res. 1989, 23, 71–77. [Google Scholar] [CrossRef] [PubMed]
- Atlante, A.; Gagliardi, S.; Minervini, G.M.; Ciotti, M.T.; Marra, E.; Calissano, P. Glutamate neurotoxicity in rat cerebellar granule cells: A major role for xanthine oxidase in oxygen radical formation. J. Neurochem. 1997, 68, 2038–2045. [Google Scholar] [CrossRef] [PubMed]
- Kuo, C.H.; Hata, F.; Yoshida, H.; Yamatodani, A.; Wada, H. Effect of ascorbic acid on release of acetylcholine from synaptic vesicles prepared from different species of animals and release of noradrenaline from synaptic vesicles of rat brain. Life Sci. 1979, 24, 911–915. [Google Scholar] [CrossRef]
- Fan, S.F.; Yazulla, S. Modulation of voltage-dependent k+ currents (Ik(v)) in retinal bipolar cells by ascorbate is mediated by dopamine d1 receptors. Vis. Neurosci. 1999, 16, 923–931. [Google Scholar] [CrossRef] [PubMed]
- Nelson, M.T.; Joksovic, P.M.; Su, P.; Kang, H.W.; Van Deusen, A.; Baumgart, J.P.; David, L.S.; Snutch, T.P.; Barrett, P.Q.; Lee, J.H.; et al. Molecular mechanisms of subtype-specific inhibition of neuronal t-type calcium channels by ascorbate. J. Neurosci. 2007, 27, 12577–12583. [Google Scholar] [CrossRef] [PubMed]
- Calero, C.I.; Vickers, E.; Moraga Cid, G.; Aguayo, L.G.; von Gersdorff, H.; Calvo, D.J. Allosteric modulation of retinal GABA receptors by ascorbic acid. J. Neurosci. 2011, 31, 9672–9682. [Google Scholar] [CrossRef] [PubMed]
- Eldridge, C.F.; Bunge, M.B.; Bunge, R.P.; Wood, P.M. Differentiation of axon-related schwann cells in vitro. I. Ascorbic acid regulates basal lamina assembly and myelin formation. J. Cell Biol. 1987, 105, 1023–1034. [Google Scholar] [CrossRef] [PubMed]
- Araque, A.; Parpura, V.; Sanzgiri, R.P.; Haydon, P.G. Tripartite synapses: Glia, the unacknowledged partner. Trends Neurosci. 1999, 22, 208–215. [Google Scholar] [CrossRef]
- Pellerin, L.; Magistretti, P.J. Glutamate uptake into astrocytes stimulates aerobic glycolysis: A mechanism coupling neuronal activity to glucose utilization. Proc. Natl. Acad. Sci. USA 1994, 91, 10625–10629. [Google Scholar] [CrossRef] [PubMed]
- Demestre, M.; Boutelle, M.; Fillenz, M. Stimulated release of lactate in freely moving rats is dependent on the uptake of glutamate. J. Physiol. 1997, 499, 825–832. [Google Scholar] [CrossRef] [PubMed]
- Magistretti, P.J.; Pellerin, L. The astrocyte-mediated coupling between synaptic activity and energy metabolism operates through volume transmission. Prog. Brain Res. 2000, 125, 229–240. [Google Scholar] [PubMed]
- Allaman, I.; Gavillet, M.; Belanger, M.; Laroche, T.; Viertl, D.; Lashuel, H.A.; Magistretti, P.J. Amyloid-β aggregates cause alterations of astrocytic metabolic phenotype: Impact on neuronal viability. J. Neurosci. 2010, 30, 3326–3338. [Google Scholar] [CrossRef] [PubMed]
- Bittar, P.G.; Charnay, Y.; Pellerin, L.; Bouras, C.; Magistretti, P.J. Selective distribution of lactate dehydrogenase isoenzymes in neurons and astrocytes of human brain. J. Cereb. Blood Flow Metab. 1996, 16, 1079–1089. [Google Scholar] [CrossRef] [PubMed]
- Laughton, J.D.; Charnay, Y.; Belloir, B.; Pellerin, L.; Magistretti, P.J.; Bouras, C. Differential messenger RNA distribution of lactate dehydrogenase LDH-1 and LDH-5 isoforms in the rat brain. Neuroscience 2000, 96, 619–625. [Google Scholar] [CrossRef]
- Pellerin, L.; Bergersen, L.H.; Halestrap, A.P.; Pierre, K. Cellular and subcellular distribution of monocarboxylate transporters in cultured brain cells and in the adult brain. J. Neurosci. Res. 2005, 79, 55–64. [Google Scholar] [CrossRef] [PubMed]
- Pierre, K.; Pellerin, L. Monocarboxylate transporters in the central nervous system: Distribution, regulation and function. J. Neurochem. 2005, 94, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Chih, C.P.; Roberts, E.L., Jr. Energy substrates for neurons during neural activity: A critical review of the astrocyte-neuron lactate shuttle hypothesis. J. Cereb. Blood Flow Metab. 2003, 23, 1263–1281. [Google Scholar] [CrossRef] [PubMed]
- Dienel, G.A.; Wang, R.Y.; Cruz, N.F. Generalized sensory stimulation of conscious rats increases labeling of oxidative pathways of glucose metabolism when the brain glucose-oxygen uptake ratio rises. J. Cereb. Blood Flow Metab. 2002, 22, 1490–1502. [Google Scholar] [CrossRef] [PubMed]
- Gjedde, A. Cerebral blood flow change in arterial hypoxemia is consistent with negligible oxygen tension in brain mitochondria. Neuroimage 2002, 17, 1876–1881. [Google Scholar] [PubMed]
- Hertz, L. The astrocyte-neuron lactate shuttle: A challenge of a challenge. J. Cereb. Blood Flow Metab. 2004, 24, 1241–1248. [Google Scholar] [CrossRef] [PubMed]
- Hertz, L.; Kala, G. Energy metabolism in brain cells: Effects of elevated ammonia concentrations. Metab. Brain Dis. 2007, 22, 199–218. [Google Scholar] [CrossRef] [PubMed]
- Mangia, S.; Garreffa, G.; Bianciardi, M.; Giove, F.; di Salle, F.; Maraviglia, B. The aerobic brain: Lactate decrease at the onset of neural activity. Neuroscience 2003, 118, 7–10. [Google Scholar] [CrossRef]
- Mangia, S.; Giove, F.; Bianciardi, M.; di Salle, F.; Garreffa, G.; Maraviglia, B. Issues concerning the construction of a metabolic model for neuronal activation. J. Neurosci Res. 2003, 71, 463–467. [Google Scholar] [CrossRef] [PubMed]
- Mangia, S.; Simpson, I.A.; Vannucci, S.J.; Carruthers, A. The in vivo neuron-to-astrocyte lactate shuttle in human brain: Evidence from modeling of measured lactate levels during visual stimulation. J. Neurochem. 2009, 109 (Suppl. 1), 55–62. [Google Scholar] [CrossRef] [PubMed]
- Beltrán, F.A.; Acuña, A.I.; Miro, M.P.; Angulo, C.; Concha, I.I.; Castro, M.A. Ascorbic acid-dependent GLUT3 inhibition is a critical step for switching neuronal metabolism. J. Cell. Physiol. 2011, 226, 3286–3294. [Google Scholar] [CrossRef] [PubMed]
- Covarrubias-Pinto, A.; Moll, P.; Solis-Maldonado, M.; Acuña, A.I.; Riveros, A.; Paz Miro, M.; Papic, E.; Beltrán, F.A.; Cepeda, C.; Concha, I.I.; et al. Beyond the redox imbalance: Oxidative stress contributes to an impaired GLUT3 modulation in huntington’s disease. Free Radic. Biol. Med. 2015. [Google Scholar] [CrossRef] [PubMed]
- Mink, J.W.; Blumenschine, R.J.; Adams, D.B. Ratio of central nervous system to body metabolism in vertebrates: Its constancy and functional basis. Am. J. Physiol. 1981, 241, R203–R212. [Google Scholar] [PubMed]
- Abeles, M. Corticonics: Neural Circuits of the Cerebral Cortex; Cambridge University Press: New York, NY, USA, 1991; p. 280. [Google Scholar]
- Braitenberg, V.; Schüz, A. Cortex: Statistics and Geometry of Neuronal Connectivity; Springer: Berlin, Germany, 1998. [Google Scholar]
- Sibson, N.R.; Dhankhar, A.; Mason, G.F.; Rothman, D.L.; Behar, K.L.; Shulman, R.G. Stoichiometric coupling of brain glucose metabolism and glutamatergic neuronal activity. Proc. Natl. Acad. Sci. USA 1998, 95, 316–321. [Google Scholar] [CrossRef] [PubMed]
- Sokoloff, L.; Reivich, M.; Kennedy, C.; Des Rosiers, M.H.; Patlak, C.S.; Pettigrew, K.D.; Sakurada, O.; Shinohara, M. The [14C]deoxyglucose method for the measurement of local cerebral glucose utilization: Theory, procedure, and normal values in the conscious and anesthetized albino rat. J. Neurochem. 1977, 28, 897–916. [Google Scholar] [CrossRef] [PubMed]
- Giove, F.; Mangia, S.; Bianciardi, M.; Garreffa, G.; di Salle, F.; Morrone, R.; Maraviglia, B. The physiology and metabolism of neuronal activation: In vivo studies by NMR and other methods. Magn. Reson. Imaging 2003, 21, 1283–1293. [Google Scholar] [CrossRef] [PubMed]
- Boado, R.J.; Pardridge, W.M. Measurement of blood-brain barrier glut1 glucose transporter and actin mRNA by a quantitative polymerase chain reaction assay. J. Neurochem. 1994, 62, 2085–2090. [Google Scholar] [CrossRef] [PubMed]
- Bolz, S.; Farrell, C.L.; Dietz, K.; Wolburg, H. Subcellular distribution of glucose transporter (GLUT-1) during development of the blood-brain barrier in rats. Cell Tissue Res. 1996, 284, 355–365. [Google Scholar] [CrossRef] [PubMed]
- Nualart, F.; Godoy, A.; Reinicke, K. Expression of the hexose transporters GLUT1 and GLUT2 during the early development of the human brain. Brain Res. 1999, 824, 97–104. [Google Scholar] [CrossRef]
- Maher, F.; Vannucci, S.; Takeda, J.; Simpson, I.A. Expression of mouse-GLUT3 and human-GLUT3 glucose transporter proteins in brain. Biochem. Biophys. Res. Commun. 1992, 182, 703–711. [Google Scholar] [CrossRef]
- Lowry, O.H.; Passonneau, J.V. The relationships between substrates and enzymes of glycolysis in brain. J. Biol. Chem. 1964, 239, 31–42. [Google Scholar] [PubMed]
- Beltrán, F.A.; Acuña, A.I.; Miró, M.P.; Castro, M. Brain energy metabolism in health and disease. In Neuroscience—Dealing with Frontiers; Contreras, C.M., Ed. InTech: Rijeka, Croatia, 2012; p. 32. [Google Scholar]
- Kety, S.S. The general metabolism of the brain in vivo. In The Metabolism of the Nervous System; Richter, D., Ed.; Elsevier: Philadelphia, PA, USA, 1957; p. 17. [Google Scholar]
- Sokoloff, L. Quantitative measurements of cerebral blood flow in man. Methods Med. Res. 1960, 8, 253–261. [Google Scholar] [PubMed]
- Schurr, A.; West, C.A.; Rigor, B.M. Lactate-supported synaptic function in the rat hippocampal slice preparation. Science 1988, 240, 1326–1328. [Google Scholar] [CrossRef] [PubMed]
- Izumi, Y.; Ishii, K.; Katsuki, H.; Benz, A.M.; Zorumski, C.F. β-Hydroxybutyrate fuels synaptic function during development. Histological and physiological evidence in rat hippocampal slices. J. Clin. Investing. 1998, 101, 1121–1132. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Wyss, M.T.; Jolivet, R.; Buck, A.; Magistretti, P.J.; Weber, B. In vivo evidence for lactate as a neuronal energy source. J. Neurosci. 2011, 31, 7477–7485. [Google Scholar] [CrossRef] [PubMed]
- Chinopoulos, C.; Adam-Vizi, V. Mitochondria deficient in complex I activity are depolarized by hydrogen peroxide in nerve terminals: Relevance to parkinson’s disease. J. Neurochem. 2001, 76, 302–306. [Google Scholar] [CrossRef] [PubMed]
- Dawson, V.L.; Dawson, T.M.; London, E.D.; Bredt, D.S.; Snyder, S.H. Nitric oxide mediates glutamate neurotoxicity in primary cortical cultures. Proc. Natl. Acad. Sci. USA 1991, 88, 6368–6371. [Google Scholar] [CrossRef] [PubMed]
- Lafon-Cazal, M.; Pietri, S.; Culcasi, M.; Bockaert, J. NMDA-dependent superoxide production and neurotoxicity. Nature 1993, 364, 535–537. [Google Scholar] [CrossRef] [PubMed]
- Bondy, S.C.; Lee, D.K. Oxidative stress induced by glutamate receptor agonists. Brain Res. 1993, 610, 229–233. [Google Scholar] [CrossRef]
- Vesce, S.; Kirk, L.; Nicholls, D.G. Relationships between superoxide levels and delayed calcium deregulation in cultured cerebellar granule cells exposed continuously to glutamate. J. Neurochem. 2004, 90, 683–693. [Google Scholar] [CrossRef] [PubMed]
- Kahlert, S.; Zundorf, G.; Reiser, G. Glutamate-mediated influx of extracellular Ca2+ is coupled with reactive oxygen species generation in cultured hippocampal neurons but not in astrocytes. J. Neurosci. Res. 2005, 79, 262–271. [Google Scholar] [CrossRef] [PubMed]
- Neymotin, A.; Petri, S.; Calingasan, N.Y.; Wille, E.; Schafer, P.; Stewart, C.; Hensley, K.; Beal, M.F.; Kiaei, M. Lenalidomide (revlimid) administration at symptom onset is neuroprotective in a mouse model of amyotrophic lateral sclerosis. Exp. Neurol. 2009, 220, 191–197. [Google Scholar] [CrossRef] [PubMed]
- Shaw, P.J. Molecular and cellular pathways of neurodegeneration in motor neurone disease. J. Neurol. Neurosurg. Psychiatry 2005, 76, 1046–1057. [Google Scholar] [CrossRef] [PubMed]
- Rosen, D.R.; Siddique, T.; Patterson, D.; Figlewicz, D.A.; Sapp, P.; Hentati, A.; Donaldson, D.; Goto, J.; O’Regan, J.P.; Deng, H.X.; et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 1993, 362, 59–62. [Google Scholar] [CrossRef] [PubMed]
- Lin, M.T.; Beal, M.F. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature 2006, 443, 787–795. [Google Scholar] [CrossRef] [PubMed]
- Mattiazzi, M.; D’Aurelio, M.; Gajewski, C.D.; Martushova, K.; Kiaei, M.; Beal, M.F.; Manfredi, G. Mutated human sod1 causes dysfunction of oxidative phosphorylation in mitochondria of transgenic mice. J. Biol. Chem. 2002, 277, 29626–29633. [Google Scholar] [CrossRef] [PubMed]
- Orth, M.; Schapira, A.H. Mitochondria and degenerative disorders. Am. J. Med. Genet. 2001, 106, 27–36. [Google Scholar] [CrossRef] [PubMed]
- Wenk, G.L. Neuropathologic changes in alzheimer’s disease. J. Clin. Psychiatry 2003, 64, 7–10. [Google Scholar] [PubMed]
- Duyckaerts, C.; Delatour, B.; Potier, M.C. Classification and basic pathology of alzheimer disease. Acta Neuropathol. 2009, 118, 5–36. [Google Scholar] [CrossRef] [PubMed]
- Anandatheerthavarada, H.K.; Devi, L. Amyloid precursor protein and mitochondrial dysfunction in alzheimer’s disease. Neuroscientist 2007, 13, 626–638. [Google Scholar] [CrossRef] [PubMed]
- Gu, M.; Gash, M.T.; Mann, V.M.; Javoy-Agid, F.; Cooper, J.M.; Schapira, A.H. Mitochondrial defect in huntington’s disease caudate nucleus. Ann. Neurol. 1996, 39, 385–389. [Google Scholar] [CrossRef] [PubMed]
- Moreira, P.I.; Santos, M.S.; Oliveira, C.R. Alzheimer’s disease: A lesson from mitochondrial dysfunction. Antioxid. Redox Signal. 2007, 9, 1621–1630. [Google Scholar] [CrossRef] [PubMed]
- Swerdlow, R.H.; Khan, S.M. The alzheimer’s disease mitochondrial cascade hypothesis: An update. Exp. Neurol. 2009, 218, 308–315. [Google Scholar] [CrossRef] [PubMed]
- Gabuzda, D.; Busciglio, J.; Chen, L.B.; Matsudaira, P.; Yankner, B.A. Inhibition of energy metabolism alters the processing of amyloid precursor protein and induces a potentially amyloidogenic derivative. J. Biol. Chem. 1994, 269, 13623–13628. [Google Scholar] [PubMed]
- Apelt, J.; Bigl, M.; Wunderlich, P.; Schliebs, R. Aging-related increase in oxidative stress correlates with developmental pattern of β-secretase activity and β-amyloid plaque formation in transgenic TG2576 mice with alzheimer-like pathology. Int. J. Dev. Neurosci. 2004, 22, 475–484. [Google Scholar] [CrossRef] [PubMed]
- Hefti, F.; Goure, W.F.; Jerecic, J.; Iverson, K.S.; Walicke, P.A.; Krafft, G.A. The case for soluble Aβ oligomers as a drug target in alzheimer’s disease. Trends Pharmacol. Sci. 2013, 34, 261–266. [Google Scholar] [CrossRef] [PubMed]
- Sanmartin, C.D.; Adasme, T.; Hidalgo, C.; Paula-Lima, A.C. The antioxidant N-acetylcysteine prevents the mitochondrial fragmentation induced by soluble amyloid-β peptide oligomers. Neurodegener. Dis. 2012, 10, 34–37. [Google Scholar] [CrossRef] [PubMed]
- Bush, A.I.; Masters, C.L.; Tanzi, R.E. Copper, β-amyloid, and alzheimer’s disease: Tapping a sensitive connection. Proc. Natl. Acad. Sci. USA 2003, 100, 11193–11194. [Google Scholar] [CrossRef] [PubMed]
- Ma, T.; Hoeffer, C.A.; Wong, H.; Massaad, C.A.; Zhou, P.; Iadecola, C.; Murphy, M.P.; Pautler, R.G.; Klann, E. Amyloid β-induced impairments in hippocampal synaptic plasticity are rescued by decreasing mitochondrial superoxide. J. Neurosci. 2011, 31, 5589–5595. [Google Scholar] [CrossRef] [PubMed]
- Harrison, F.E.; Hosseini, A.H.; McDonald, M.P.; May, J.M. Vitamin C reduces spatial learning deficits in middle-aged and very old app/psen1 transgenic and wild-type mice. Pharmacol. Biochem. Behav. 2009, 93, 443–450. [Google Scholar] [CrossRef] [PubMed]
- Kennard, J.A.; Harrison, F.E. Intravenous ascorbate improves spatial memory in middle-aged APP/PSEN1 and wild type mice. Behav. Brain Res. 2014, 264, 34–42. [Google Scholar] [CrossRef] [PubMed]
- Murakami, K.; Murata, N.; Ozawa, Y.; Kinoshita, N.; Irie, K.; Shirasawa, T.; Shimizu, T. Vitamin C restores behavioral deficits and amyloid-β oligomerization without affecting plaque formation in a mouse model of alzheimer’s disease. J. Alzheimer’s Dis. 2011, 26, 7–18. [Google Scholar]
- Kook, S.Y.; Lee, K.M.; Kim, Y.; Cha, M.Y.; Kang, S.; Baik, S.H.; Lee, H.; Park, R.; Mook-Jung, I. High-dose of vitamin C supplementation reduces amyloid plaque burden and ameliorates pathological changes in the brain of 5XFAD mice. Cell Death Dis. 2014, 5, e1083. [Google Scholar] [CrossRef] [PubMed]
- Charlton, K.E.; Rabinowitz, T.L.; Geffen, L.N.; Dhansay, M.A. Lowered plasma vitamin C, but not vitamin e, concentrations in dementia patients. J. Nutr. Health Aging 2004, 8, 99–107. [Google Scholar] [PubMed]
- Riviere, S.; Birlouez-Aragon, I.; Nourhashemi, F.; Vellas, B. Low plasma vitamin C in alzheimer patients despite an adequate diet. Int. J. Geriatr. Psychiatry 1998, 13, 749–754. [Google Scholar] [CrossRef]
- Zandi, P.P.; Anthony, J.C.; Khachaturian, A.S.; Stone, S.V.; Gustafson, D.; Tschanz, J.T.; Norton, M.C.; Welsh-Bohmer, K.A.; Breitner, J.C.; Cache County Study, G. Reduced risk of alzheimer disease in users of antioxidant vitamin supplements: The cache county study. Arch. Neurol. 2004, 61, 82–88. [Google Scholar] [CrossRef] [PubMed]
- DiFiglia, M. Excitotoxic injury of the neostriatum: A model for huntington’s disease. Trends Neurosci. 1990, 13, 286–289. [Google Scholar] [CrossRef]
- Twelvetrees, A.E.; Yuen, E.Y.; Arancibia-Carcamo, I.L.; MacAskill, A.F.; Rostaing, P.; Lumb, M.J.; Humbert, S.; Triller, A.; Saudou, F.; Yan, Z.; et al. Delivery of GABAARs to synapses is mediated by HAP1-KIF5 and disrupted by mutant huntingtin. Neuron 2010, 65, 53–65. [Google Scholar] [CrossRef] [PubMed]
- Caviston, J.P.; Holzbaur, E.L. Huntingtin as an essential integrator of intracellular vesicular trafficking. Trends Cell Biol. 2009, 19, 147–155. [Google Scholar] [CrossRef] [PubMed]
- Bamford, K.A.; Caine, E.D.; Kido, D.K.; Cox, C.; Shoulson, I. A prospective evaluation of cognitive decline in early huntington’s disease: Functional and radiographic correlates. Neurology 1995, 45, 1867–1873. [Google Scholar] [CrossRef] [PubMed]
- Brandt, J.; Folstein, S.E.; Folstein, M.F. Differential cognitive impairment in alzheimer’s disease and huntington’s disease. Ann. Neurol. 1988, 23, 555–561. [Google Scholar] [CrossRef] [PubMed]
- Shiwach, R. Psychopathology in huntington’s disease patients. Acta Psychiatr. Scand. 1994, 90, 241–246. [Google Scholar] [CrossRef] [PubMed]
- Bruyn, G.W.; von Wolferen, W.J. Pathogenesis of huntington’s chorea. Lancet 1973, 1, 1382. [Google Scholar] [CrossRef]
- Hersch, S.M.; Gevorkian, S.; Marder, K.; Moskowitz, C.; Feigin, A.; Cox, M.; Como, P.; Zimmerman, C.; Lin, M.; Zhang, L.; et al. Creatine in huntington disease is safe, tolerable, bioavailable in brain and reduces serum 8OH2’dG. Neurology 2006, 66, 250–252. [Google Scholar] [CrossRef] [PubMed]
- Browne, S.E.; Ferrante, R.J.; Beal, M.F. Oxidative stress in huntington’s disease. Brain Pathol. 1999, 9, 147–163. [Google Scholar] [CrossRef] [PubMed]
- Browne, S.E.; Beal, M.F. Oxidative damage in huntington’s disease pathogenesis. Antioxid. Redox Signal. 2006, 8, 2061–2073. [Google Scholar] [CrossRef] [PubMed]
- Sorolla, M.A.; Reverter-Branchat, G.; Tamarit, J.; Ferrer, I.; Ros, J.; Cabiscol, E. Proteomic and oxidative stress analysis in human brain samples of huntington disease. Free Radic. Biol. Med. 2008, 45, 667–678. [Google Scholar] [CrossRef] [PubMed]
- Santamaria, A.; Perez-Severiano, F.; Rodriguez-Martinez, E.; Maldonado, P.D.; Pedraza-Chaverri, J.; Rios, C.; Segovia, J. Comparative analysis of superoxide dismutase activity between acute pharmacological models and a transgenic mouse model of huntington’s disease. Neurochem. Res. 2001, 26, 419–424. [Google Scholar] [CrossRef] [PubMed]
- Gines, S.; Seong, I.S.; Fossale, E.; Ivanova, E.; Trettel, F.; Gusella, J.F.; Wheeler, V.C.; Persichetti, F.; MacDonald, M.E. Specific progressive camp reduction implicates energy deficit in presymptomatic huntington’s disease knock-in mice. Hum. Mol. Genet. 2003, 12, 497–508. [Google Scholar] [CrossRef] [PubMed]
- Kuwert, T.; Lange, H.W.; Boecker, H.; Titz, H.; Herzog, H.; Aulich, A.; Wang, B.C.; Nayak, U.; Feinendegen, L.E. Striatal glucose consumption in chorea-free subjects at risk of huntington’s disease. J. Neurol. 1993, 241, 31–36. [Google Scholar] [CrossRef] [PubMed]
- Leenders, K.L.; Frackowiak, R.S.; Quinn, N.; Marsden, C.D. Brain energy metabolism and dopaminergic function in huntington’s disease measured in vivo using positron emission tomography. Mov. Disord. 1986, 1, 69–77. [Google Scholar] [CrossRef] [PubMed]
- Powers, W.J.; Videen, T.O.; Markham, J.; McGee-Minnich, L.; Antenor-Dorsey, J.V.; Hershey, T.; Perlmutter, J.S. Selective defect of in vivo glycolysis in early huntington’s disease striatum. Proc. Natl. Acad. Sci. USA 2007, 104, 2945–2949. [Google Scholar] [CrossRef] [PubMed]
- Benchoua, A.; Trioulier, Y.; Zala, D.; Gaillard, M.C.; Lefort, N.; Dufour, N.; Saudou, F.; Elalouf, J.M.; Hirsch, E.; Hantraye, P.; et al. Involvement of mitochondrial complex II defects in neuronal death produced by N-terminus fragment of mutated huntingtin. Mol. Biol. Cell 2006, 17, 1652–1663. [Google Scholar] [CrossRef] [PubMed]
- Brennan, W.A., Jr.; Bird, E.D.; Aprille, J.R. Regional mitochondrial respiratory activity in huntington’s disease brain. J. Neurochem. 1985, 44, 1948–1950. [Google Scholar] [CrossRef] [PubMed]
- Browne, S.E. Mitochondria and huntington’s disease pathogenesis: Insight from genetic and chemical models. Ann. N. Y. Acad. Sci. 2008, 1147, 358–382. [Google Scholar] [CrossRef] [PubMed]
- Lim, D.; Fedrizzi, L.; Tartari, M.; Zuccato, C.; Cattaneo, E.; Brini, M.; Carafoli, E. Calcium homeostasis and mitochondrial dysfunction in striatal neurons of huntington disease. J. Biol. Chem. 2008, 283, 5780–5789. [Google Scholar] [CrossRef] [PubMed]
- Milakovic, T.; Johnson, G.V. Mitochondrial respiration and ATP production are significantly impaired in striatal cells expressing mutant huntingtin. J. Biol. Chem. 2005, 280, 30773–30782. [Google Scholar] [CrossRef] [PubMed]
- Tabrizi, S.J.; Cleeter, M.W.; Xuereb, J.; Taanman, J.W.; Cooper, J.M.; Schapira, A.H. Biochemical abnormalities and excitotoxicity in huntington’s disease brain. Ann. Neurol. 1999, 45, 25–32. [Google Scholar] [CrossRef]
- Damiano, M.; Galvan, L.; Deglon, N.; Brouillet, E. Mitochondria in huntington’s disease. BBA Mol. Basis Dis. 2010, 1802, 52–61. [Google Scholar] [CrossRef] [PubMed]
- Parker, W.D., Jr.; Boyson, S.J.; Luder, A.S.; Parks, J.K. Evidence for a defect in NADH: Ubiquinone oxidoreductase (complex I) in huntington’s disease. Neurology 1990, 40, 1231–1234. [Google Scholar] [CrossRef] [PubMed]
- Brouillet, E.; Conde, F.; Beal, M.F.; Hantraye, P. Replicating huntington’s disease phenotype in experimental animals. Prog. Neurobiol. 1999, 59, 427–468. [Google Scholar] [CrossRef]
- Ruan, Q.; Lesort, M.; MacDonald, M.E.; Johnson, G.V. Striatal cells from mutant huntingtin knock-in mice are selectively vulnerable to mitochondrial complex II inhibitor-induced cell death through a non-apoptotic pathway. Hum. Mol. Genet. 2004, 13, 669–681. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Choo, Y.S.; Johnson, G.V.; MacDonald, M.; Detloff, P.J.; Lesort, M. Mutant huntingtin directly increases susceptibility of mitochondria to the calcium-induced permeability transition and cytochrome c release. Hum. Mol. Genet. 2004, 13, 1407–1420. [Google Scholar] [CrossRef] [PubMed]
- Panov, A.V.; Gutekunst, C.A.; Leavitt, B.R.; Hayden, M.R.; Burke, J.R.; Strittmatter, W.J.; Greenamyre, J.T. Early mitochondrial calcium defects in huntington’s disease are a direct effect of polyglutamines. Nat. Neurosci. 2002, 5, 731–736. [Google Scholar] [CrossRef] [PubMed]
- Petrasch-Parwez, E.; Nguyen, H.P.; Lobbecke-Schumacher, M.; Habbes, H.W.; Wieczorek, S.; Riess, O.; Andres, K.H.; Dermietzel, R.; von Horsten, S. Cellular and subcellular localization of huntingtin [corrected] aggregates in the brain of a rat transgenic for huntington disease. J. Comp. Neurol. 2007, 501, 716–730. [Google Scholar] [CrossRef] [PubMed]
- Cui, L.; Jeong, H.; Borovecki, F.; Parkhurst, C.N.; Tanese, N.; Krainc, D. Transcriptional repression of PGC-1α by mutant huntingtin leads to mitochondrial dysfunction and neurodegeneration. Cell 2006, 127, 59–69. [Google Scholar] [CrossRef] [PubMed]
- McGill, J.K.; Beal, M.F. PGC-1α, a new therapeutic target in huntington’s disease? Cell 2006, 127, 465–468. [Google Scholar] [CrossRef] [PubMed]
- St-Pierre, J.; Drori, S.; Uldry, M.; Silvaggi, J.M.; Rhee, J.; Jager, S.; Handschin, C.; Zheng, K.; Lin, J.; Yang, W.; et al. Suppression of reactive oxygen species and neurodegeneration by the PGC-1 transcriptional coactivators. Cell 2006, 127, 397–408. [Google Scholar] [CrossRef] [PubMed]
- Rebec, G.V.; Barton, S.J.; Marseilles, A.M.; Collins, K. Ascorbate treatment attenuates the huntington behavioral phenotype in mice. Neuroreport 2003, 14, 1263–1265. [Google Scholar] [CrossRef] [PubMed]
- Dorner, J.L.; Miller, B.R.; Klein, E.L.; Murphy-Nakhnikian, A.; Andrews, R.L.; Barton, S.J.; Rebec, G.V. Corticostriatal dysfunction underlies diminished striatal ascorbate release in the R6/2 mouse model of huntington’s disease. Brain Res. 2009, 1290, 111–120. [Google Scholar] [CrossRef] [PubMed]
- Rebec, G.V.; Conroy, S.K.; Barton, S.J. Hyperactive striatal neurons in symptomatic huntington R6/2 mice: Variations with behavioral state and repeated ascorbate treatment. Neuroscience 2006, 137, 327–336. [Google Scholar] [CrossRef] [PubMed]
- Hirsch, E.C.; Jenner, P.; Przedborski, S. Pathogenesis of parkinson’s disease. Mov. Disord. 2013, 28, 24–30. [Google Scholar] [CrossRef] [PubMed]
- Braak, H.; del Tredici, K.; Rub, U.; de Vos, R.A.; Jansen Steur, E.N.; Braak, E. Staging of brain pathology related to sporadic parkinson’s disease. Neurobiol. Aging 2003, 24, 197–211. [Google Scholar] [CrossRef]
- Booij, J.; Tissingh, G.; Boer, G.J.; Speelman, J.D.; Stoof, J.C.; Janssen, A.G.; Wolters, E.C.; van Royen, E.A. [123I]FP-CIT SPECT shows a pronounced decline of striatal dopamine transporter labelling in early and advanced parkinson’s disease. J. Neurol. Neurosurg. Psychiatry 1997, 62, 133–140. [Google Scholar] [CrossRef] [PubMed]
- Iranzo, A.; Lomena, F.; Stockner, H.; Valldeoriola, F.; Vilaseca, I.; Salamero, M.; Molinuevo, J.L.; Serradell, M.; Duch, J.; Pavia, J.; et al. Decreased striatal dopamine transporter uptake and substantia nigra hyperechogenicity as risk markers of synucleinopathy in patients with idiopathic rapid-eye-movement sleep behaviour disorder: A prospective study [corrected]. Lancet Neurol. 2010, 9, 1070–1077. [Google Scholar] [CrossRef]
- Kaufman, M.J.; Madras, B.K. Severe depletion of cocaine recognition sites associated with the dopamine transporter in parkinson’s-diseased striatum. Synapse 1991, 9, 43–49. [Google Scholar] [CrossRef] [PubMed]
- Biskup, S.; Gerlach, M.; Kupsch, A.; Reichmann, H.; Riederer, P.; Vieregge, P.; Wullner, U.; Gasser, T. Genes associated with parkinson syndrome. J. Neurol. 2008, 255 5, 8–17. [Google Scholar] [CrossRef] [PubMed]
- Schapira, A.H. Mitochondrial dysfunction in neurodegenerative diseases. Neurochem. Res. 2008, 33, 2502–2509. [Google Scholar] [CrossRef] [PubMed]
- Henchcliffe, C.; Beal, M.F. Mitochondrial biology and oxidative stress in parkinson disease pathogenesis. Nat. Clin. Pract. Neurol. 2008, 4, 600–609. [Google Scholar] [CrossRef] [PubMed]
- Bindoff, L.A.; Birch-Machin, M.; Cartlidge, N.E.; Parker, W.D., Jr.; Turnbull, D.M. Mitochondrial function in Parkinson’s disease. Lancet 1989, 2, 49. [Google Scholar] [CrossRef]
- Mizuno, Y.; Ohta, S.; Tanaka, M.; Takamiya, S.; Suzuki, K.; Sato, T.; Oya, H.; Ozawa, T.; Kagawa, Y. Deficiencies in complex I subunits of the respiratory chain in parkinson’s disease. Biochem. Biophys. Res. Commun. 1989, 163, 1450–1455. [Google Scholar] [CrossRef]
- Schapira, A.H.; Cooper, J.M.; Dexter, D.; Jenner, P.; Clark, J.B.; Marsden, C.D. Mitochondrial complex I deficiency in parkinson’s disease. Lancet 1989, 1, 1269. [Google Scholar] [CrossRef]
- Xiang, W.; Schlachetzki, J.C.; Helling, S.; Bussmann, J.C.; Berlinghof, M.; Schaffer, T.E.; Marcus, K.; Winkler, J.; Klucken, J.; Becker, C.M. Oxidative stress-induced posttranslational modifications of α-synuclein: Specific modification of α-synuclein by 4-hydroxy-2-nonenal increases dopaminergic toxicity. Mol. Cell. Neurosci. 2013, 54, 71–83. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.; Jyoti, S.; Naz, F.; Shakya, B.; Rahul; Afzal, M.; Siddique, Y.H. Effect of l-ascorbic acid on the climbing ability and protein levels in the brain of drosophila model of Parkinson’s disease. Int. J. Neurosci. 2012, 122, 704–709. [Google Scholar] [CrossRef] [PubMed]
- Ballaz, S.; Morales, I.; Rodriguez, M.; Obeso, J.A. Ascorbate prevents cell death from prolonged exposure to glutamate in an in vitro model of human dopaminergic neurons. J. Neurosci. Res. 2013, 91, 1609–1617. [Google Scholar] [CrossRef] [PubMed]
- Foy, C.J.; Passmore, A.P.; Vahidassr, M.D.; Young, I.S.; Lawson, J.T. Plasma chain-breaking antioxidants in Alzheimer’s disease, vascular dementia and parkinson’s disease. QJM 1999, 92, 39–45. [Google Scholar] [CrossRef] [PubMed]
- Sohmiya, M.; Tanaka, M.; Tak, N.W.; Yanagisawa, M.; Tanino, Y.; Suzuki, Y.; Okamoto, K.; Yamamoto, Y. Redox status of plasma coenzyme Q10 indicates elevated systemic oxidative stress in parkinson’s disease. J. Neurol. Sci. 2004, 223, 161–166. [Google Scholar] [CrossRef] [PubMed]
- Ide, K.; Yamada, H.; Umegaki, K.; Mizuno, K.; Kawakami, N.; Hagiwara, Y.; Matsumoto, M.; Yoshida, H.; Kim, K.; Shiosaki, E.; et al. Lymphocyte vitamin C levels as potential biomarker for progression of parkinson’s disease. Nutrition 2015, 31, 406–408. [Google Scholar] [CrossRef] [PubMed]
- Fitzgerald, K.C.; O’Reilly, E.J.; Fondell, E.; Falcone, G.J.; McCullough, M.L.; Park, Y.; Kolonel, L.N.; Ascherio, A. Intakes of vitamin C and carotenoids and risk of amyotrophic lateral sclerosis: Pooled results from 5 cohort studies. Ann. Neurol. 2013, 73, 236–245. [Google Scholar] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Covarrubias-Pinto, A.; Acuña, A.I.; Beltrán, F.A.; Torres-Díaz, L.; Castro, M.A. Old Things New View: Ascorbic Acid Protects the Brain in Neurodegenerative Disorders. Int. J. Mol. Sci. 2015, 16, 28194-28217. https://doi.org/10.3390/ijms161226095
Covarrubias-Pinto A, Acuña AI, Beltrán FA, Torres-Díaz L, Castro MA. Old Things New View: Ascorbic Acid Protects the Brain in Neurodegenerative Disorders. International Journal of Molecular Sciences. 2015; 16(12):28194-28217. https://doi.org/10.3390/ijms161226095
Chicago/Turabian StyleCovarrubias-Pinto, Adriana, Aníbal Ignacio Acuña, Felipe Andrés Beltrán, Leandro Torres-Díaz, and Maite Aintzane Castro. 2015. "Old Things New View: Ascorbic Acid Protects the Brain in Neurodegenerative Disorders" International Journal of Molecular Sciences 16, no. 12: 28194-28217. https://doi.org/10.3390/ijms161226095
APA StyleCovarrubias-Pinto, A., Acuña, A. I., Beltrán, F. A., Torres-Díaz, L., & Castro, M. A. (2015). Old Things New View: Ascorbic Acid Protects the Brain in Neurodegenerative Disorders. International Journal of Molecular Sciences, 16(12), 28194-28217. https://doi.org/10.3390/ijms161226095