
Application of CRISPR/Cas9 Technology to HBV

Abstract
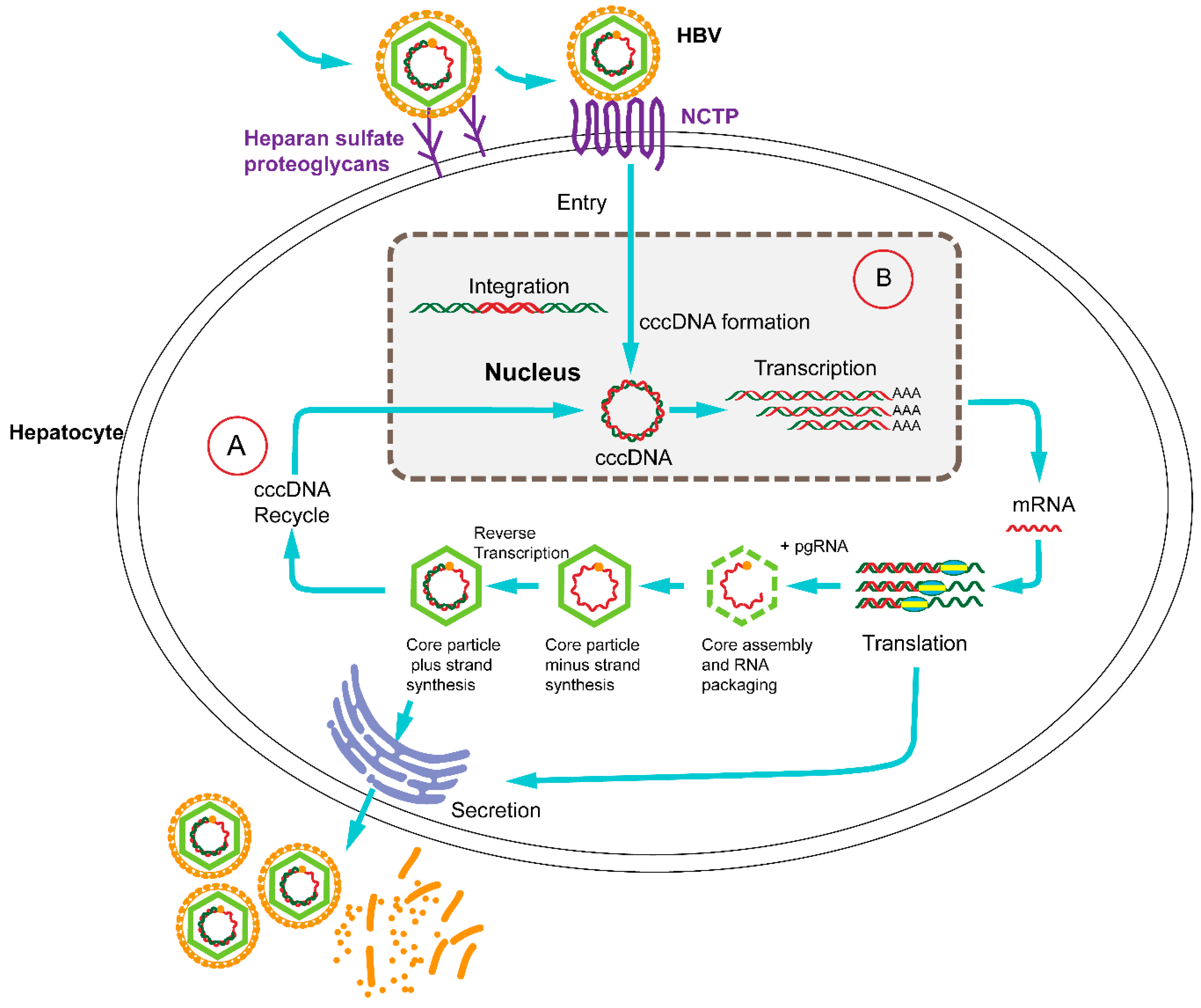
:1. Introduction
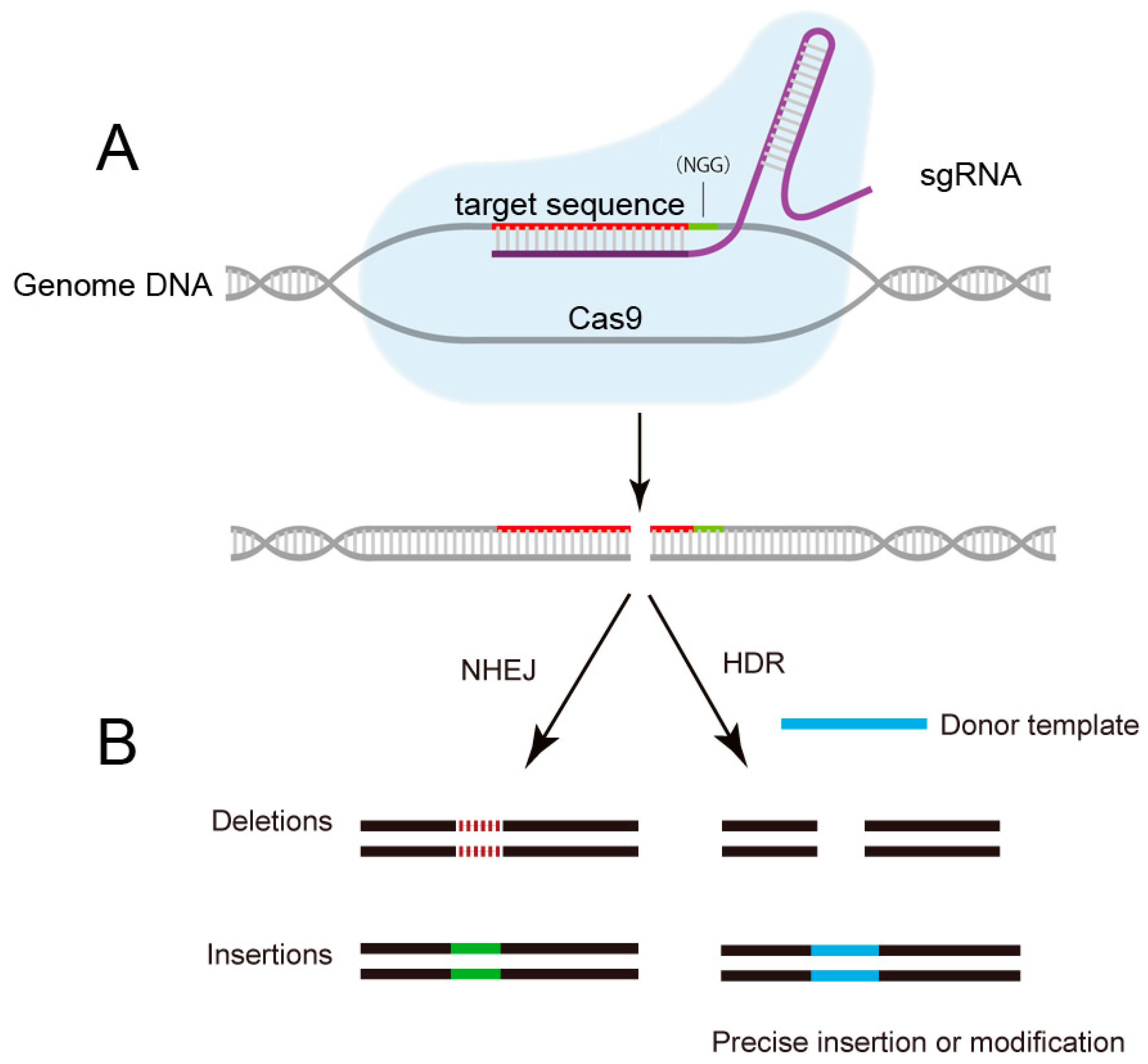
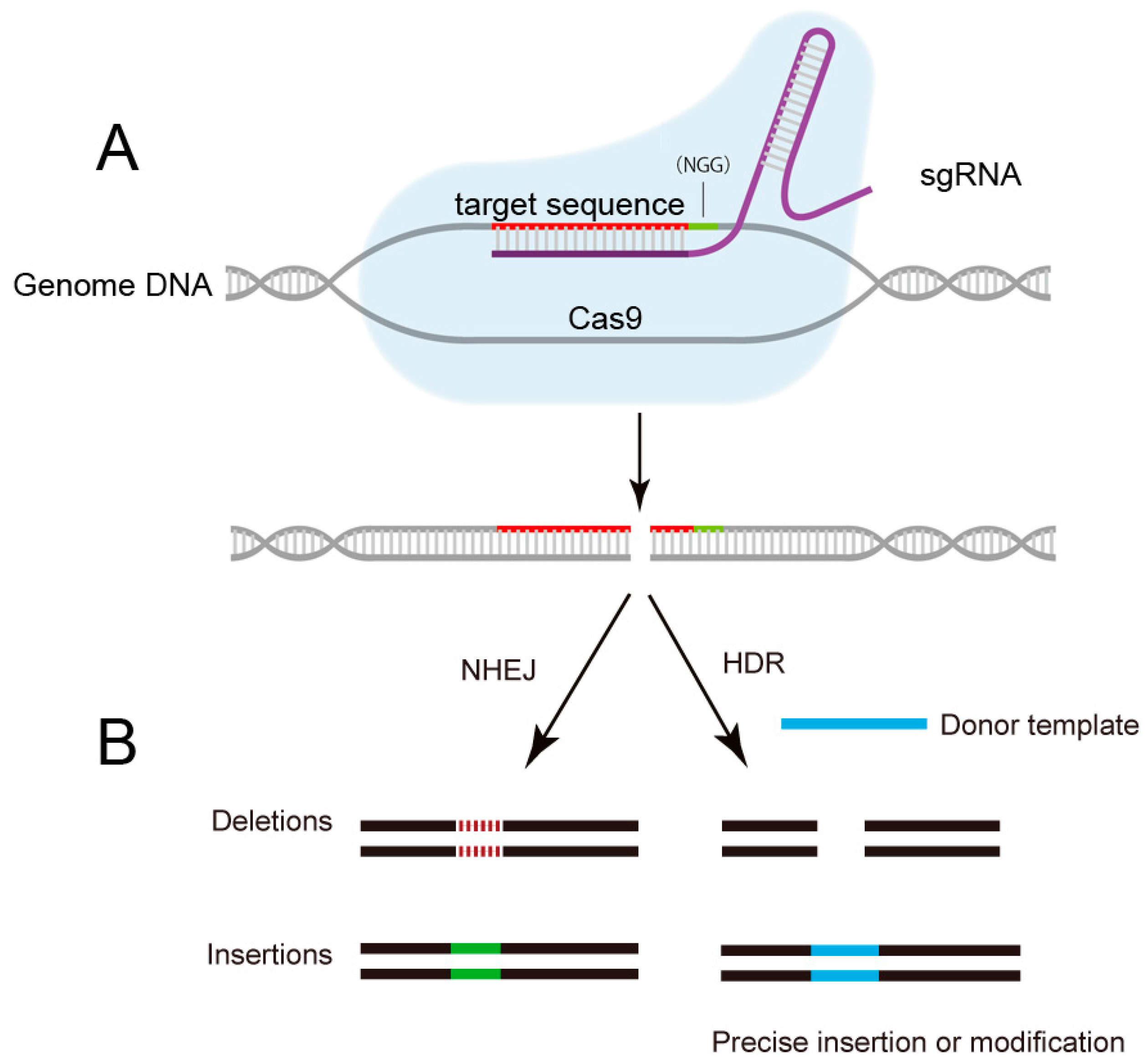
2. The Emergence of CRISPR/Cas9: From Bacterial Immune System to Versatile Genome Editing Tool
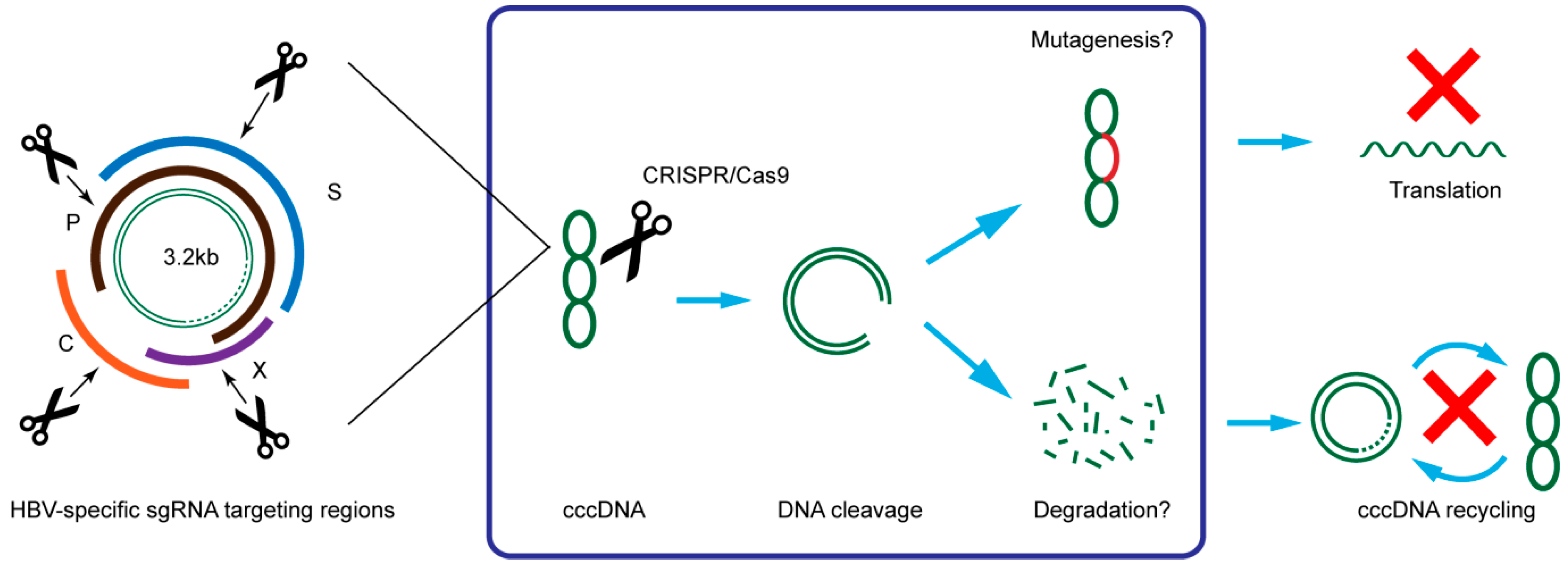
3. CRISPR/Cas9 Target HBV cccDNA and Inhibit HBV Replication

{kind=link}
{kind=link}
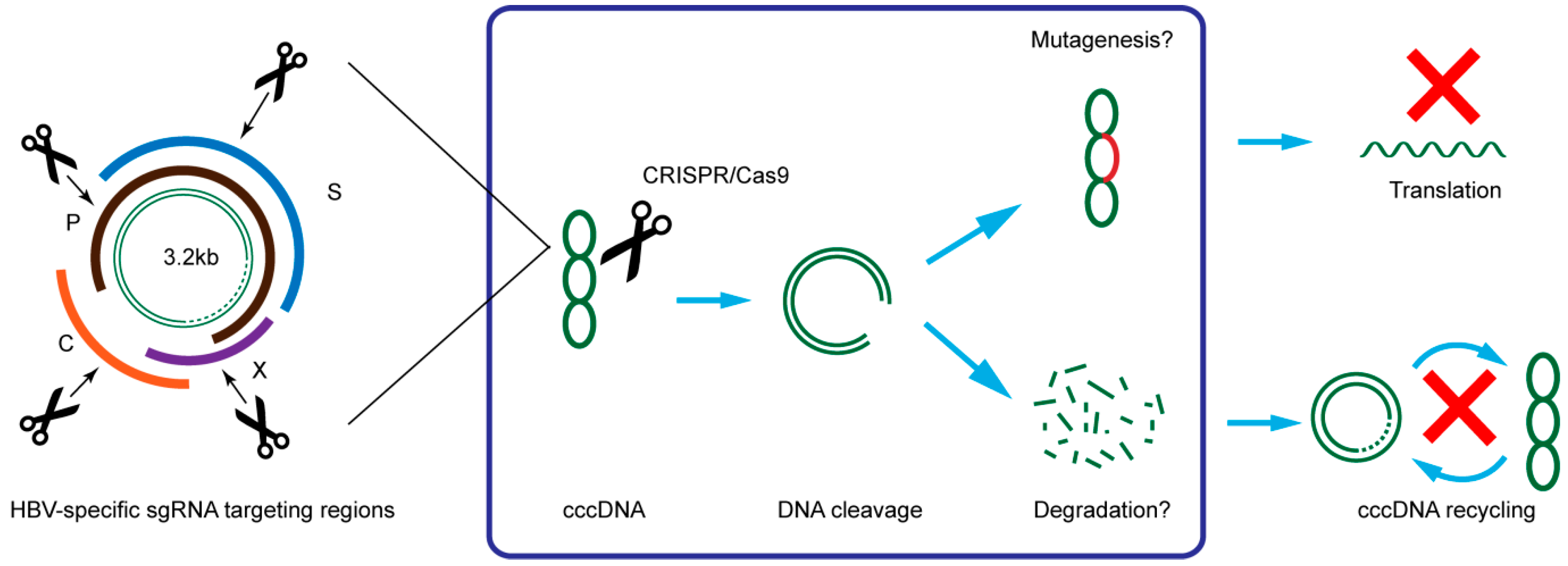{kind=link}
| Target | HBV Infection Model | Results | Reference |
|---|---|---|---|
| P1, S1, XCp, and PS2 ORFs | Huh7 | Reduction in HBsAg level in medium | Lin et al. [15] |
| HBV hydrodynamics-mouse model | Reduction in HBsAg level in serum | Lin et al. [15] | |
| P, S, and C ORFs | HepAD38 and HepaRG | Reduction in viral DNA and cccDNA levels. Reduction in HBsAg and HBeAg level in medium | Kennedy et al. [16] |
| ENII-CP/X and Pre-C ORFs | HepG2 with HBV receptor NTCP | Eight-fold inhibition of HBV infection | Seeger and Sohn [17] |
| P, S, X and C ORFs | HepG2 | Reduction of intracellular HBV replication intermediates and extracellular virion DNA | Liu et al. [18] |
| HBV hydrodynamics-mouse model | Reduction in HBsAg and HBeAg level in serum and the expression of HBcAg in liver | Liu et al. [18] | |
| P, S, X and C ORFs | HepG2.2.15 | Reduction in HBsAg level in medium and intracellular cccDNA | Zhen et al. [19] |
| HBV hydrodynamics-mouse model | Reduction in HBsAg level in serum | Zhen et al. [19] | |
| X/L and X ORFs | Huh7 | Reduction in HBsAg and HBeAg level in medium and intracellular cccDNA | Dong et al. [20] |
| HepG2.2.15 | Reduction in HBsAg level in medium | Dong et al. [20] | |
| HBV hydrodynamics-mouse model carrying cccDNA | Reduction in HBsAg and HBeAg level in serum and intrahepatic cccDNA | Dong et al. [20] | |
| P, S, X and C ORFs | HepG2 with HBV receptor NTCP | Reduction in HBsAg, HBV DNA, 3.5kb RNA and cccDNA levels in culture medium | Ramanan et al. [21] |
| HepG2.2.15 | Reduction in HBV DNA and cccDNA levels | Ramanan et al. [21] | |
| HBV hydrodynamics-mouse model | Reduction in HBsAg and viral DNA level in serum | Ramanan et al. [21] | |
| P, S, X and C ORFs | HuH-7 | Reduction in HBsAg and HBeAg level in medium | Wang et al. [22] |
| HepAD38 | Reduction in HBsAg, HBeAg, HBV DNA, and cccDNA levels in culture medium | Wang et al. [22] | |
| S and X ORFs | HepG2.2.15 and HepG2-H1.3 | Significant reduction in HBsAg level in medium | Karimova et al. [23] |
| HepG2 hNTCP | Significant reduction in HBsAg level in medium | Karimova et al. [23] |
4. The Limitations of the CRISPR/Cas9 Technology as a Novel Therapeutic for HBV
5. Potential Gene Editing Target for HBV Therapy with CRISPR/Cas9
6. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Ott, J.J.; Stevens, G.A.; Groeger, J.; Wiersma, S.T. Global epidemiology of hepatitis B virus infection: New estimates of age-specific HBsAg seroprevalence and endemicity. Vaccine 2012, 30, 2212–2219. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.M. Hepatitis B virus infection. N. Engl. J. Med. 1997, 337, 1733–1745. [Google Scholar] [CrossRef] [PubMed]
- Block, T.M.; Mehta, A.S.; Fimmel, C.J.; Jordan, R. Molecular viral oncology of hepatocellular carcinoma. Oncogene 2003, 22, 5093–5107. [Google Scholar] [CrossRef] [PubMed]
- Block, T.M.; Rawat, S.; Brosgart, C.L. Chronic hepatitis B: A wave of new therapies on the horizon. Antivir. Res. 2015, 121, 69–81. [Google Scholar] [CrossRef] [PubMed]
- Yuen, M.F.; Lai, C.L. Treatment of chronic hepatitis B: Evolution over two decades. J. Gastroenterol. Hepatol. 2011, 26, 138–143. [Google Scholar] [CrossRef] [PubMed]
- Zeisel, M.B.; Lucifora, J.; Mason, W.S.; Sureau, C.; Beck, J.; Levrero, M.; Kann, M.; Knolle, P.A.; Benkirane, M.; Durantel, D.; et al. Towards an HBV cure: State-of-the-ART and unresolved questions-report of the ANRS workshop on HBV cure. Gut 2015, 64, 1314–1326. [Google Scholar] [CrossRef] [PubMed]
- Shouval, D.; Lai, C.L.; Chang, T.T.; Cheinquer, H.; Martin, P.; Carosi, G.; Han, S.; Kaymakoglu, S.; Tamez, R.; Yang, J.; et al. Relapse of hepatitis B in HBeAg-negative chronic hepatitis B patients who discontinued successful entecavir treatment: The case for continuous antiviral therapy. J. Hepatol. 2009, 50, 289–295. [Google Scholar] [CrossRef] [PubMed]
- Jafri, S.M.; Lok, A.S. Antiviral therapy for chronic hepatitis B. Clin. Liver Dis. 2010, 14, 425–438. [Google Scholar] [CrossRef] [PubMed]
- Wursthorn, K.; Lutgehetmann, M.; Dandri, M.; Volz, T.; Buggisch, P.; Zollner, B.; Longerich, T.; Schirmacher, P.; Metzler, F.; Zankel, M.; et al. Peginterferon α-2b plus adefovir induce strong cccDNA decline and HBsAg reduction in patients with chronic hepatitis B. Hepatology 2006, 44, 675–684. [Google Scholar] [CrossRef] [PubMed]
- Shlomai, A.; Rice, C.M. Getting rid of a persistent troublemaker to cure hepatitis. Science 2014, 343, 1212–1213. [Google Scholar] [CrossRef] [PubMed]
- Ganem, D.; Varmus, H.E. The molecular biology of the hepatitis B viruses. Annu. Rev. Biochem. 1987, 56, 651–693. [Google Scholar] [CrossRef] [PubMed]
- Nassal, M. HBV cccDNA: Viral persistence reservoir and key obstacle for a cure of chronic hepatitis B. Gut 2015. [Google Scholar] [CrossRef] [PubMed]
- Cradick, T.J.; Keck, K.; Bradshaw, S.; Jamieson, A.C.; McCaffrey, A.P. Zinc-finger nucleases as a novel therapeutic strategy for targeting hepatitis B virus DNAs. Mol. Ther. 2010, 18, 947–954. [Google Scholar] [CrossRef] [PubMed]
- Bloom, K.; Ely, A.; Mussolino, C.; Cathomen, T.; Arbuthnot, P. Inactivation of hepatitis B virus replication in cultured cells and in vivo with engineered transcription activator-like effector nucleases. Mol. Ther. 2013, 21, 1889–1897. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.R.; Yang, H.C.; Kuo, Y.T.; Liu, C.J.; Yang, T.Y.; Sung, K.C.; Lin, Y.Y.; Wang, H.Y.; Wang, C.C.; Shen, Y.C.; et al. The crispr/cas9 system facilitates clearance of the intrahepatic hbv templates in vivo. Mol. Ther. Nucleic Acids 2014, 3. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, E.M.; Bassit, L.C.; Mueller, H.; Kornepati, A.V.; Bogerd, H.P.; Nie, T.; Chatterjee, P.; Javanbakht, H.; Schinazi, R.F.; Cullen, B.R. Suppression of hepatitis B virus DNA accumulation in chronically infected cells using a bacterial CRISPR/Cas RNA-guided DNA endonuclease. Virology 2015, 476, 196–205. [Google Scholar] [CrossRef] [PubMed]
- Seeger, C.; Sohn, J.A. Targeting hepatitis b virus with crispr/cas9. Mol. Ther. Nucleic Acids 2014, 3. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Hao, R.; Chen, S.; Guo, D.; Chen, Y. Inhibition of hepatitis B virus by the CRISPR/Cas9 system via targeting the conserved regions of the viral genome. J. Gen. Virol. 2015, 96, 2252–2261. [Google Scholar] [CrossRef] [PubMed]
- Zhen, S.; Hua, L.; Liu, Y.H.; Gao, L.C.; Fu, J.; Wan, D.Y.; Dong, L.H.; Song, H.F.; Gao, X. Harnessing the clustered regularly interspaced short palindromic repeat (CRISPR)/CRISPR-associated Cas9 system to disrupt the hepatitis B virus. Gene Ther. 2015, 22, 404–412. [Google Scholar] [CrossRef] [PubMed]
- Dong, C.; Qu, L.; Wang, H.; Wei, L.; Dong, Y.; Xiong, S. Targeting hepatitis B virus cccDNA by CRISPR/Cas9 nuclease efficiently inhibits viral replication. Antivir. Res. 2015, 118, 110–117. [Google Scholar] [CrossRef] [PubMed]
- Ramanan, V.; Shlomai, A.; Cox, D.B.; Schwartz, R.E.; Michailidis, E.; Bhatta, A.; Scott, D.A.; Zhang, F.; Rice, C.M.; Bhatia, S.N. CRISPR/Cas9 cleavage of viral DNA efficiently suppresses hepatitis B virus. Sci. Rep. 2015, 5. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Xu, Z.W.; Liu, S.; Zhang, R.Y.; Ding, S.L.; Xie, X.M.; Long, L.; Chen, X.M.; Zhuang, H.; Lu, F.M. Dual gRNAs guided CRISPR/Cas9 system inhibits hepatitis B virus replication. World J. Gastroenterol. 2015, 21, 9554–9565. [Google Scholar] [CrossRef] [PubMed]
- Karimova, M.; Beschorner, N.; Dammermann, W.; Chemnitz, J.; Indenbirken, D.; Bockmann, J.H.; Grundhoff, A.; Lüth, S.; Buchholz, F.; Schulze zur Wiesch, J.; et al. CRISPR/Cas9 nickase-mediated disruption of hepatitis B virus open reading frame S and X. Sci. Rep. 2015, 5. [Google Scholar] [CrossRef] [PubMed]
- Sander, J.D.; Joung, J.K. CRISPR-Cas systems for editing, regulating and targeting genomes. Nat. Biotechnol. 2014, 32, 347–355. [Google Scholar] [CrossRef] [PubMed]
- Barrangou, R.; Fremaux, C.; Deveau, H.; Richards, M.; Boyaval, P.; Moineau, S.; Romero, D.A.; Horvath, P. CRISPR provides acquired resistance against viruses in prokaryotes. Science 2007, 315, 1709–1712. [Google Scholar] [CrossRef] [PubMed]
- Deltcheva, E.; Chylinski, K.; Sharma, C.M.; Gonzales, K.; Chao, Y.; Pirzada, Z.A.; Eckert, M.R.; Vogel, J.; Charpentier, E. CRISPR RNA maturation by trans-encoded small RNA and host factor RNase III. Nature 2011, 471, 602–607. [Google Scholar] [CrossRef] [PubMed]
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 2012, 337, 816–821. [Google Scholar] [CrossRef] [PubMed]
- Mali, P.; Yang, L.; Esvelt, K.M.; Aach, J.; Guell, M.; DiCarlo, J.E.; Norville, J.E.; Church, G.M. RNA-guided human genome engineering via Cas9. Science 2013, 339, 823–826. [Google Scholar] [CrossRef] [PubMed]
- Jinek, M.; East, A.; Cheng, A.; Lin, S.; Ma, E.; Doudna, J. RNA-programmed genome editing in human cells. Elife 2013, 2. [Google Scholar] [CrossRef] [PubMed]
- Cong, L.; Ran, F.A.; Cox, D.; Lin, S.; Barretto, R.; Habib, N.; Hsu, P.D.; Wu, X.; Jiang, W.; Marraffini, L.A.; et al. Multiplex genome engineering using CRISPR/Cas systems. Science 2013, 339, 819–823. [Google Scholar] [CrossRef] [PubMed]
- Friedland, A.E.; Tzur, Y.B.; Esvelt, K.M.; Colaiacovo, M.P.; Church, G.M.; Calarco, J.A. Heritable genome editing in C. elegans via a CRISPR-Cas9 system. Nat. Methods 2013, 10, 741–743. [Google Scholar] [CrossRef] [PubMed]
- Hwang, W.Y.; Fu, Y.; Reyon, D.; Maeder, M.L.; Tsai, S.Q.; Sander, J.D.; Peterson, R.T.; Yeh, J.R.; Joung, J.K. Efficient genome editing in zebrafish using a CRISPR-Cas system. Nat. Biotechnol. 2013, 31, 227–229. [Google Scholar] [CrossRef] [PubMed]
- Port, F.; Chen, H.M.; Lee, T.; Bullock, S.L. Optimized CRISPR/Cas tools for efficient germline and somatic genome engineering in Drosophila. Proc. Natl. Acad. Sci. USA 2014, 111, E2967–E2976. [Google Scholar] [CrossRef] [PubMed]
- Jiang, W.; Bikard, D.; Cox, D.; Zhang, F.; Marraffini, L.A. RNA-guided editing of bacterial genomes using CRISPR-Cas systems. Nat. Biotechnol. 2013, 31, 233–239. [Google Scholar] [CrossRef] [PubMed]
- Feng, Z.; Zhang, B.; Ding, W.; Liu, X.; Yang, D.L.; Wei, P.; Cao, F.; Zhu, S.; Zhang, F.; Mao, Y.; et al. Efficient genome editing in plants using a CRISPR/Cas system. Cell Res. 2013, 23, 1229–1232. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Teng, F.; Li, T.; Zhou, Q. Simultaneous generation and germline transmission of multiple gene mutations in rat using CRISPR-Cas systems. Nat. Biotechnol. 2013, 31, 684–686. [Google Scholar] [CrossRef] [PubMed]
- Ganem, D.; Prince, A.M. Hepatitis B virus infection-natural history and clinical consequences. N. Engl. J. Med. 2004, 350, 1118–1129. [Google Scholar] [CrossRef] [PubMed]
- Dejean, A.; Lugassy, C.; Zafrani, S.; Tiollais, P.; Brechot, C. Detection of hepatitis B virus DNA in pancreas, kidney and skin of two human carriers of the virus. J. Gen. Virol. 1984, 65, 651–655. [Google Scholar] [CrossRef] [PubMed]
- Mason, A.; Wick, M.; White, H.; Perrillo, R. Hepatitis B virus replication in diverse cell types during chronic hepatitis B virus infection. Hepatology 1993, 18, 781–789. [Google Scholar] [CrossRef] [PubMed]
- Pontisso, P.; Poon, M.C.; Tiollais, P.; Brechot, C. Detection of hepatitis B virus DNA in mononuclear blood cells. Br. Med. J. (Clin. Res. Ed.) 1984, 288, 1563–1566. [Google Scholar] [CrossRef]
- Ran, F.A.; Cong, L.; Yan, W.X.; Scott, D.A.; Gootenberg, J.S.; Kriz, A.J.; Zetsche, B.; Shalem, O.; Wu, X.; Makarova, K.S.; et al. In vivo genome editing using Staphylococcus aureus Cas9. Nature 2015, 520, 186–191. [Google Scholar] [CrossRef] [PubMed]
- Truong, D.J.; Kuhner, K.; Kuhn, R.; Werfel, S.; Engelhardt, S.; Wurst, W.; Ortiz, O. Development of an intein-mediated split-Cas9 system for gene therapy. Nucleic Acids Res. 2015, 43, 6450–6458. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Foden, J.A.; Khayter, C.; Maeder, M.L.; Reyon, D.; Joung, J.K.; Sander, J.D. High-frequency off-target mutagenesis induced by CRISPR-Cas nucleases in human cells. Nat. Biotechnol. 2013, 31, 822–826. [Google Scholar] [CrossRef] [PubMed]
- Mali, P.; Aach, J.; Stranges, P.B.; Esvelt, K.M.; Moosburner, M.; Kosuri, S.; Yang, L.; Church, G.M. CAS9 transcriptional activators for target specificity screening and paired nickases for cooperative genome engineering. Nat. Biotechnol. 2013, 31, 833–838. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Sander, J.D.; Reyon, D.; Cascio, V.M.; Joung, J.K. Improving CRISPR-Cas nuclease specificity using truncated guide RNAs. Nat. Biotechnol. 2014, 32, 279–284. [Google Scholar] [CrossRef] [PubMed]
- Guilinger, J.P.; Thompson, D.B.; Liu, D.R. Fusion of catalytically inactive Cas9 to FokI nuclease improves the specificity of genome modification. Nat. Biotechnol. 2014, 32, 577–582. [Google Scholar] [CrossRef] [PubMed]
- Feitelson, M.A.; Lee, J. Hepatitis B virus integration, fragile sites, and hepatocarcinogenesis. Cancer Lett. 2007, 252, 157–170. [Google Scholar] [CrossRef] [PubMed]
- Schiffer, J.T.; Aubert, M.; Weber, N.D.; Mintzer, E.; Stone, D.; Jerome, K.R. Targeted DNA mutagenesis for the cure of chronic viral infections. J. Virol. 2012, 86, 8920–8936. [Google Scholar] [CrossRef] [PubMed]
- White, M.K.; Hu, W.; Khalili, K. The CRISPR/Cas9 genome editing methodology as a weapon against human viruses. Discov. Med. 2015, 19, 255–262. [Google Scholar] [PubMed]
- Perez, E.E.; Wang, J.; Miller, J.C.; Jouvenot, Y.; Kim, K.A.; Liu, O.; Wang, N.; Lee, G.; Bartsevich, V.V.; Lee, Y.L.; et al. Establishment of HIV-1 resistance in CD4+ T cells by genome editing using zinc-finger nucleases. Nat. Biotechnol. 2008, 26, 808–816. [Google Scholar] [CrossRef] [PubMed]
- Holt, N.; Wang, J.; Kim, K.; Friedman, G.; Wang, X.; Taupin, V.; Crooks, G.M.; Kohn, D.B.; Gregory, P.D.; Holmes, M.C.; et al. Human hematopoietic stem/progenitor cells modified by zinc-finger nucleases targeted to CCR5 control HIV-1 in vivo. Nat. Biotechnol. 2010, 28, 839–847. [Google Scholar] [CrossRef] [PubMed]
- Tebas, P.; Stein, D.; Tang, W.W.; Frank, I.; Wang, S.Q.; Lee, G.; Spratt, S.K.; Surosky, R.T.; Giedlin, M.A.; Nichol, G.; et al. Gene editing of CCR5 in autologous CD4 T cells of persons infected with HIV. N. Engl. J. Med. 2014, 370, 901–910. [Google Scholar] [CrossRef] [PubMed]
- Yan, H.; Zhong, G.; Xu, G.; He, W.; Jing, Z.; Gao, Z.; Huang, Y.; Qi, Y.; Peng, B.; Wang, H.; et al. Sodium taurocholate cotransporting polypeptide is a functional receptor for human hepatitis B and D virus. Elife 2012, 1. [Google Scholar] [CrossRef] [PubMed]
- Petersen, J.; Dandri, M.; Mier, W.; Lutgehetmann, M.; Volz, T.; von Weizsacker, F.; Haberkorn, U.; Fischer, L.; Pollok, J.M.; Erbes, B.; et al. Prevention of hepatitis B virus infection in vivo by entry inhibitors derived from the large envelope protein. Nat. Biotechnol. 2008, 26, 335–341. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lin, G.; Zhang, K.; Li, J. Application of CRISPR/Cas9 Technology to HBV. Int. J. Mol. Sci. 2015, 16, 26077-26086. https://doi.org/10.3390/ijms161125950
Lin G, Zhang K, Li J. Application of CRISPR/Cas9 Technology to HBV. International Journal of Molecular Sciences. 2015; 16(11):26077-26086. https://doi.org/10.3390/ijms161125950
Chicago/Turabian StyleLin, Guigao, Kuo Zhang, and Jinming Li. 2015. "Application of CRISPR/Cas9 Technology to HBV" International Journal of Molecular Sciences 16, no. 11: 26077-26086. https://doi.org/10.3390/ijms161125950
APA StyleLin, G., Zhang, K., & Li, J. (2015). Application of CRISPR/Cas9 Technology to HBV. International Journal of Molecular Sciences, 16(11), 26077-26086. https://doi.org/10.3390/ijms161125950