
Mutational Spectrum Analysis of Neurodegenerative Diseases and Its Pathogenic Implication

Abstract
:1. Introduction
2. Results and Discussion
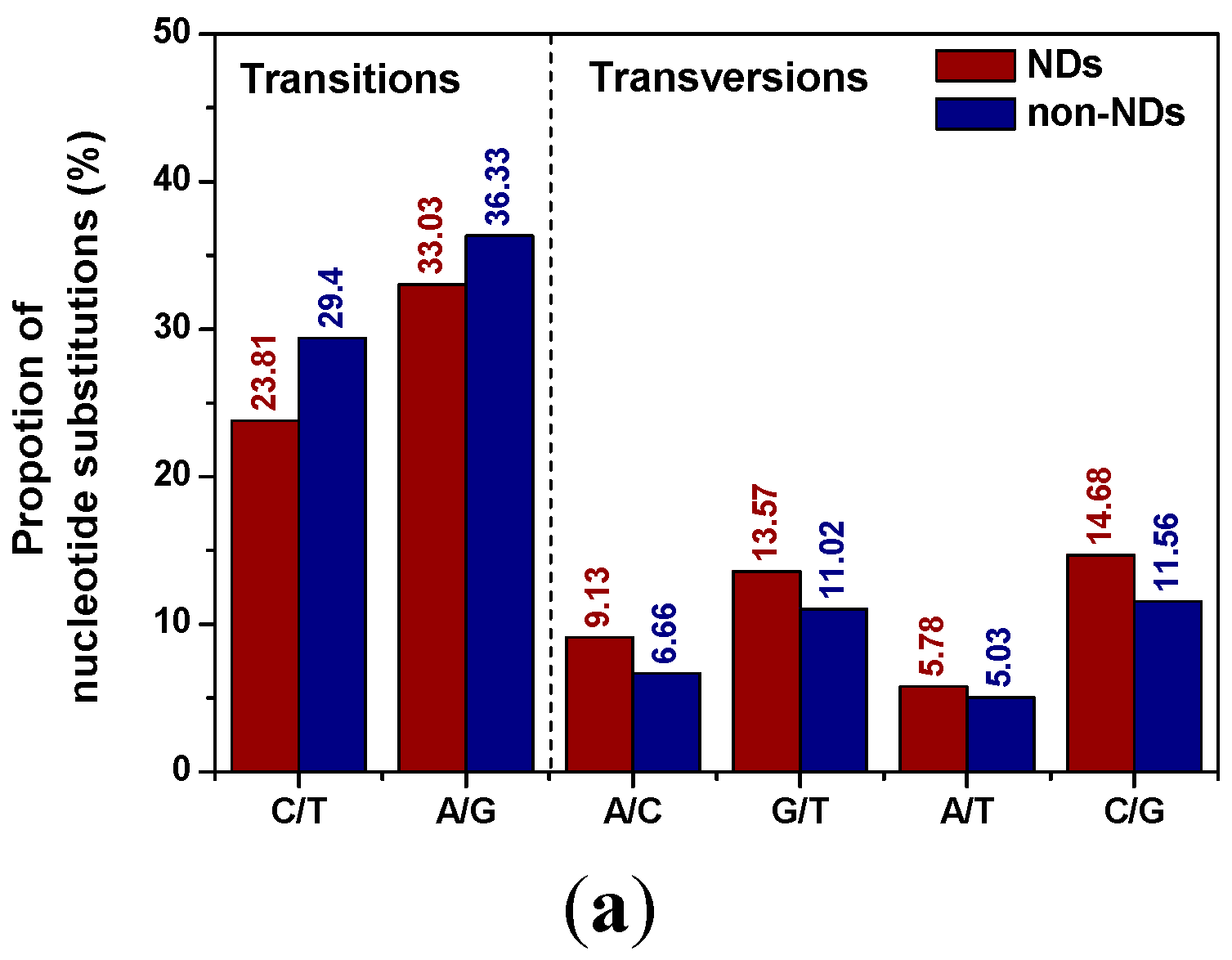
2.1. Distribution Patterns of Pucleotide Substitutions

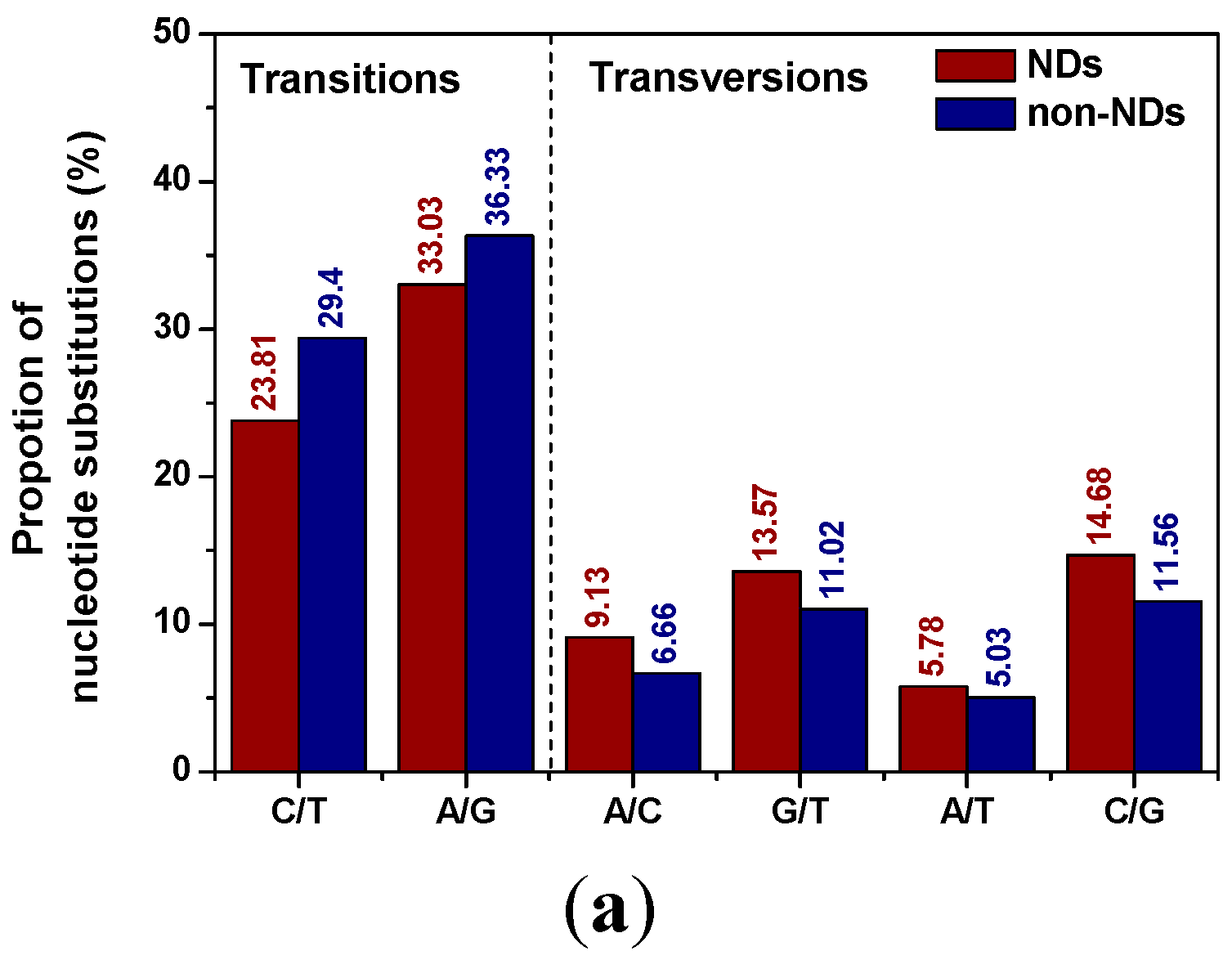{kind=link}
{kind=link}
| From | To | ||||
|---|---|---|---|---|---|
| A | T | C | G | Total | |
| A | ― | 3.56 (2.09) | 5.01 (2.76) | 10.12 (9.19) | 18.69 (14.04) |
| T | 2.22 (2.94) | ― | 8.90 (10.35) | 4.78 (4.08) | 15.90 (17.37) |
| C | 4.12 (3.90) | 14.91 (19.05) | ― | 6.23 (4.77) | 25.26 (27.72) |
| G | 22.91 (27.14) | 8.79 (6.94) | 8.45 (6.79) | ― | 40.15 (40.87) |
| Total | 29.25 (33.98) | 27.26 (28.06) | 22.36 (19.91) | 21.13 (18.04) | ― |
2.2. Position Distribution Patterns of Nucleotide Substitution
| Position | NDs | Non-NDs | ||||
|---|---|---|---|---|---|---|
| Ts (%) | Tv (%) | Total (%) | Ts (%) | Tv (%) | Total (%) | |
| First | 26.97 | 16.75 | 43.72 | 29.79 | 12.24 | 42.03 |
| Second | 29.04 | 20.24 | 49.28 | 35.46 | 17.32 | 52.78 |
| Third | 0.67 | 6.34 | 7.01 | 0.47 | 4.72 | 5.19 |
| Total | 56.68 | 43.32 | 100 | 65.72 | 34.28 | 100 |
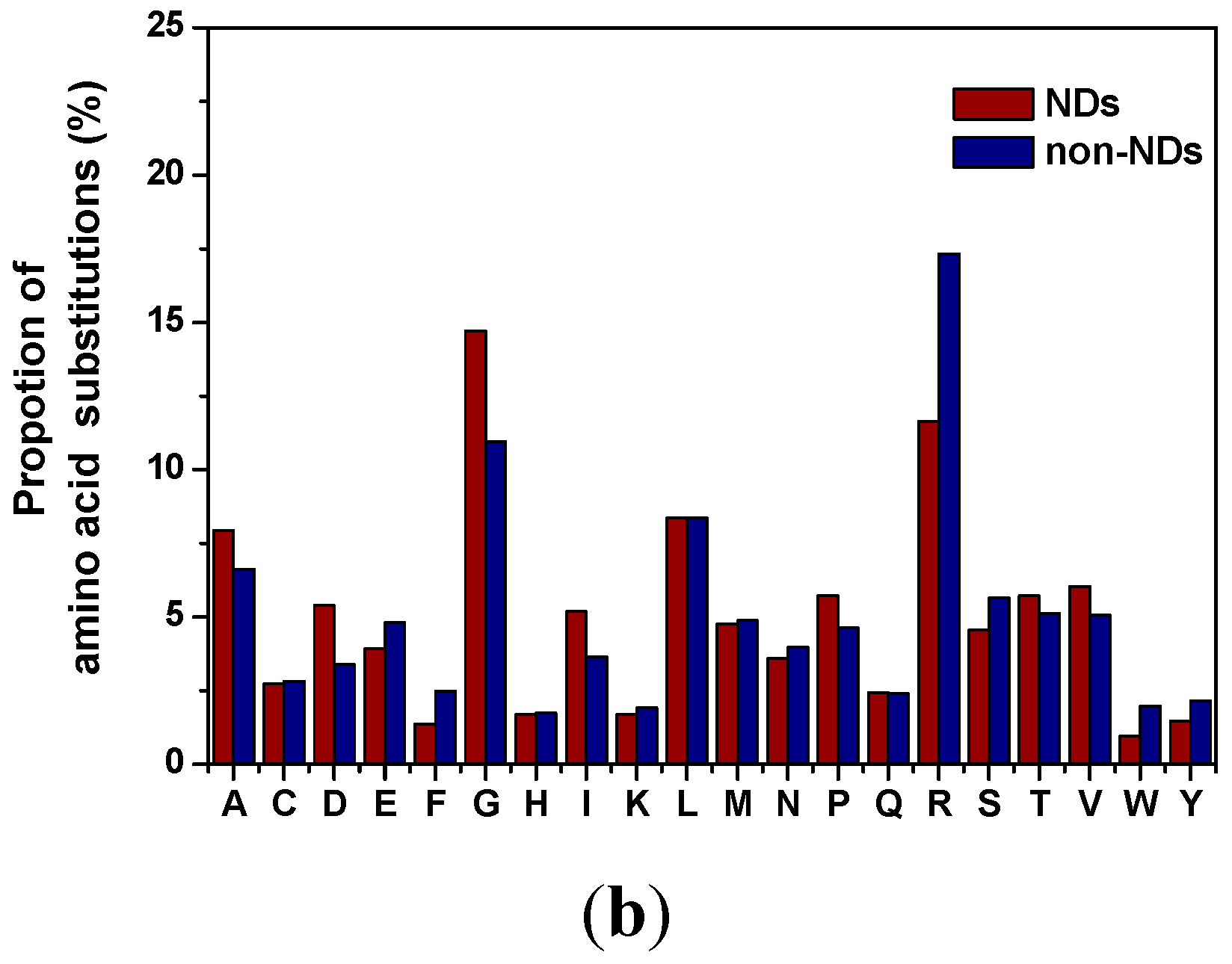
2.3. Distribution of Amino Acid Substitutions
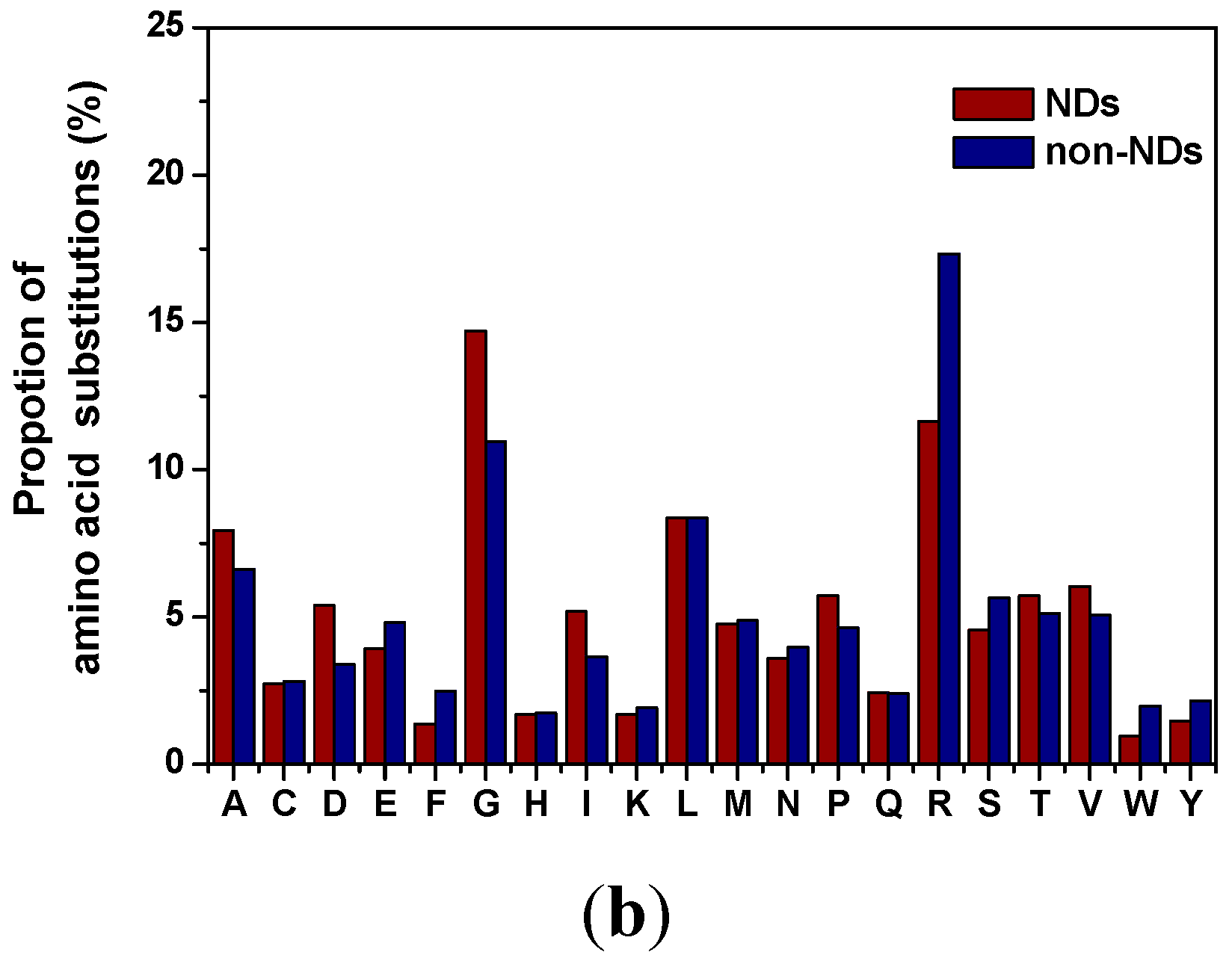
3. Experimental Section
4. Conclusion
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Krawczak, M.; Cooper, D.N. The human gene mutation database. Trends Genet. 1997, 13, 121–122. [Google Scholar] [CrossRef]
- Wang, Z.; Moult, J. SNPs, protein structure, and disease. Hum. Mutat. 2001, 17, 263–270. [Google Scholar] [CrossRef] [PubMed]
- Naya, H.; Romero, H.; Zavala, A.; Alvarez, B.; Musto, H. Aerobiosis increases the genomic guanine plus cytosine content (GC%) in prokaryotes. J. Mol. Evol. 2002, 55, 260–264. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Xiong, S.; Xie, C.; Markesbery, W.R.; Lovell, M.A. Increased oxidative damage in nuclear and mitochondrial DNA in Alzheimer’s disease. J. Neurochem. 2005, 93, 953–962. [Google Scholar] [CrossRef] [PubMed]
- Nunomura, A.; Perry, G.; Aliev, G.; Hirai, K.; Takeda, A.; Balraj, E.K.; Jones, P.K.; Ghanbari, H.; Wataya, T.; Shimohama, S.; et al. Oxidative damage is the earliest event in Alzheimer disease. J. Neuropathol. Exp. Neurol. 2001, 60, 759–767. [Google Scholar] [PubMed]
- Zhang, J.; Perry, G.; Smith, M.A.; Robertson, D.; Olson, S.J.; Graham, D.G.; Montine, T.J. Parkinson’s disease is associated with oxidative damage to cytoplasmic DNA and RNA in substantia nigra neurons. Am. J. Pathol. 1999, 154, 1423–1429. [Google Scholar] [CrossRef]
- Nakabeppu, Y.; Tsuchimoto, D.; Yamaguchi, H.; Sakumi, K. Oxidative damage in nucleic acids and Parkinson's disease. J. Neurosci. Res. 2007, 85, 919–934. [Google Scholar] [CrossRef] [PubMed]
- Shimura-Miura, H.; Hattori, N.; Kang, D.; Miyako, K.; Nakabeppu, Y.; Mizuno, Y. Increased 8-oxo-dGTPase in the mitochondria of substantia nigral neurons in Parkinson’s disease. Ann. Neurol. 1999, 46, 920–924. [Google Scholar] [CrossRef]
- Polidori, M.C.; Mecocci, P.; Browne, S.E.; Senin, U.; Beal, M.F. Oxidative damage to mitochondrial DNA in Huntington’s disease parietal cortex. Neurosci. Lett. 1999, 272, 53–56. [Google Scholar] [CrossRef]
- Kikuchi, H.; Furuta, A.; Nishioka, K.; Suzuki, S.O.; Nakabeppu, Y.; Iwaki, T. Impairment of mitochondrial DNA repair enzymes against accumulation of 8-oxo-guanine in the spinal motor neurons of amyotrophic lateral sclerosis. Acta Neuropathol. 2002, 103, 408–414. [Google Scholar] [CrossRef] [PubMed]
- Vitkup, D.; Sander, C.; Church, G.M. The amino-acid mutational spectrum of human genetic disease. Genome Biol. 2003, 4, 72. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.; Vihinen, M. Spectrum of disease-causing mutations in protein secondary structures. BMC Struct. Biol. 2007, 7, 56. [Google Scholar] [CrossRef] [PubMed]
- Ollila, J.; Lappalainen, I.; Vihinen, M. Sequence specificity in CpG mutation hotspots. FEBS Lett. 1996, 396, 119–122. [Google Scholar] [CrossRef]
- Cruts, M.; Theuns, J.; van Broeckhoven, C. Locus-specific mutation databases for neurodegenerative brain diseases. Hum. Mutat. 2012, 33, 1340–1344. [Google Scholar] [CrossRef] [PubMed]
- Nuytemans, K.; Theuns, J.; van Broeckhoven, C. Genetic etiology of Parkinson’s disease associated with mutations in the SNCA, PARK2, PINK1, PARK7 and LRRK2 genes: An update. Hum. Mutat. 2010, 31, 763–780. [Google Scholar] [CrossRef] [PubMed]
- Kenney, S.M.; Cox, D.W. Sequence variation database for the Wilson disease copper transporter, ATP7B. Hum. Mutat. 2007, 28, 1171–1177. [Google Scholar] [CrossRef] [PubMed]
- Davies, L.P.; Macintyre, G.; Cox, D.W. New Mutations in the Wilson Disease Gene, ATP7B: Implications for molecular testing. Genet. Test. 2008, 12, 139–145. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, M.; Takahashi, Y.; Koike, A.; Fukuda, Y.; Goto, J.; Tsuji, S. A mutation database for amyotrophic lateral sclerosis. Hum. Mutat. 2010, 31, 1003–1010. [Google Scholar] [CrossRef] [PubMed]
- Known Point Variations in Human Prion Gene Coding Region. Available online: http://www.mad-cow.org/prion_point_mutations.html (accessed on 15 August 2014).
- UniProtKB. Available online: http://ebi4.uniprot.org/uniprot/P42858 (accessed on 15 August 2014).
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shen, L.; Ji, H.-F. Mutational Spectrum Analysis of Neurodegenerative Diseases and Its Pathogenic Implication. Int. J. Mol. Sci. 2015, 16, 24295-24301. https://doi.org/10.3390/ijms161024295
Shen L, Ji H-F. Mutational Spectrum Analysis of Neurodegenerative Diseases and Its Pathogenic Implication. International Journal of Molecular Sciences. 2015; 16(10):24295-24301. https://doi.org/10.3390/ijms161024295
Chicago/Turabian StyleShen, Liang, and Hong-Fang Ji. 2015. "Mutational Spectrum Analysis of Neurodegenerative Diseases and Its Pathogenic Implication" International Journal of Molecular Sciences 16, no. 10: 24295-24301. https://doi.org/10.3390/ijms161024295
APA StyleShen, L., & Ji, H.-F. (2015). Mutational Spectrum Analysis of Neurodegenerative Diseases and Its Pathogenic Implication. International Journal of Molecular Sciences, 16(10), 24295-24301. https://doi.org/10.3390/ijms161024295