
Systemically Administered, Target Organ-Specific Therapies for Regenerative Medicine

Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Local vs. Systemic Drug Delivery in Regenerative Medicine
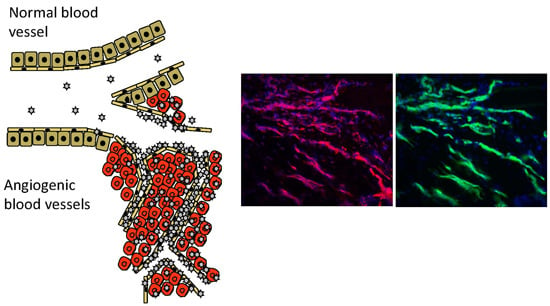
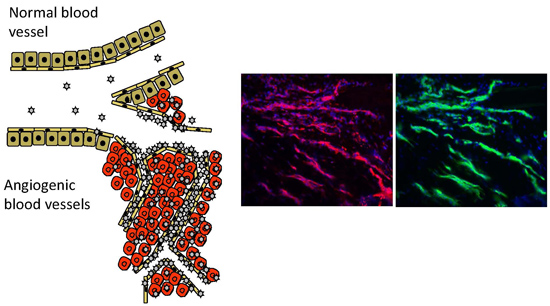
2. Vascular Heterogeneity—“Zip Codes” in Vasculature
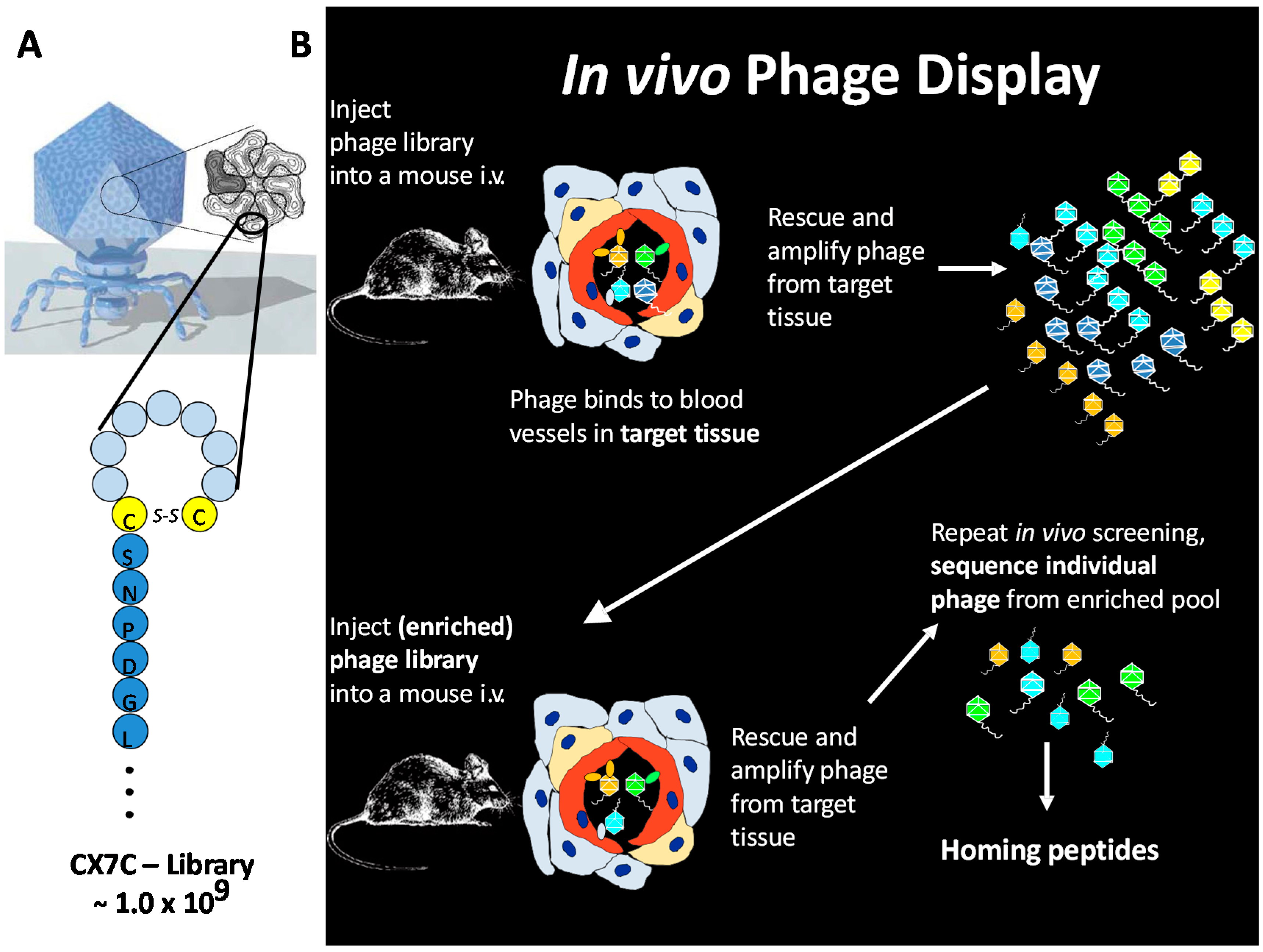
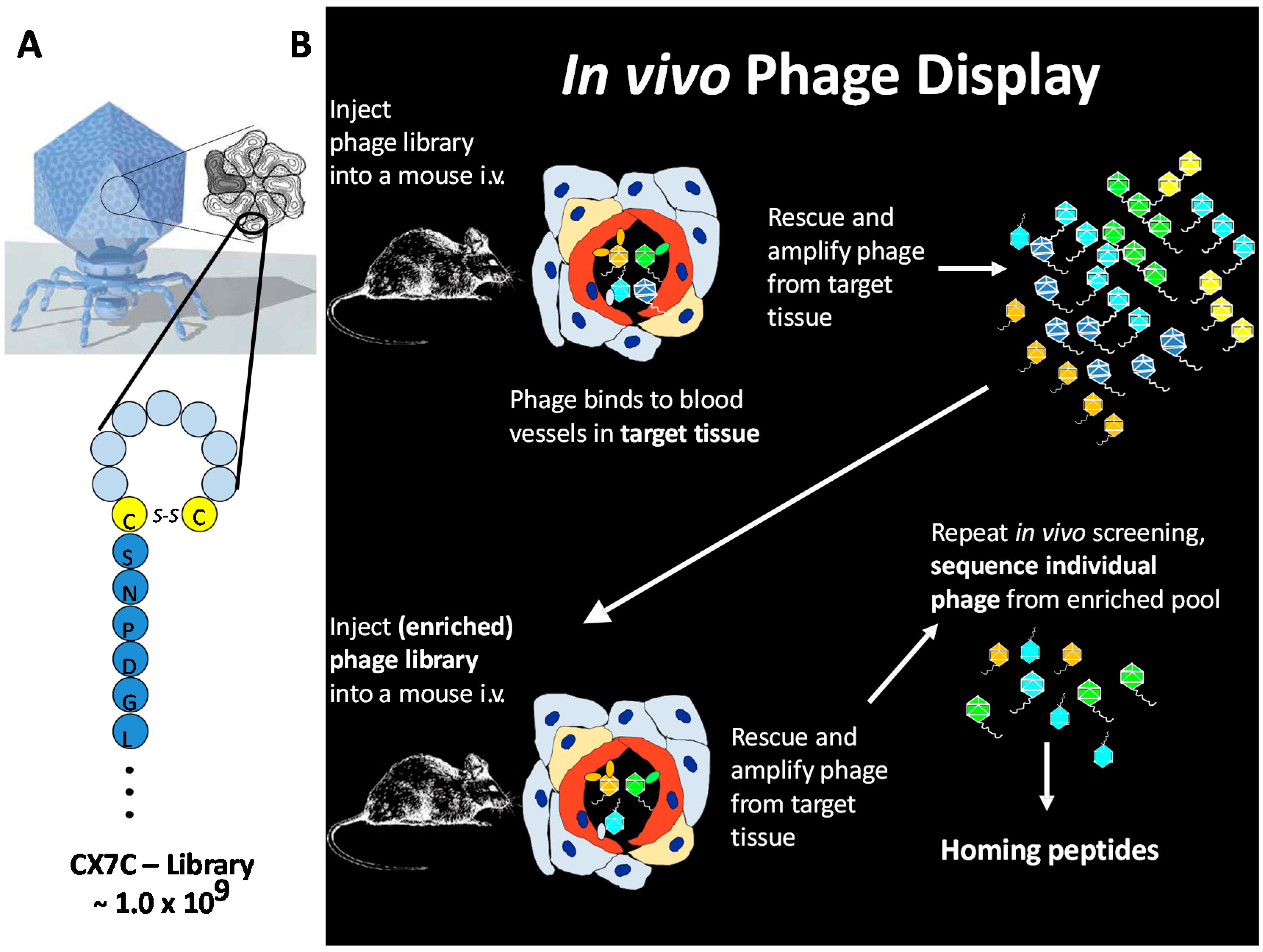
3. In Vivo Phage Display
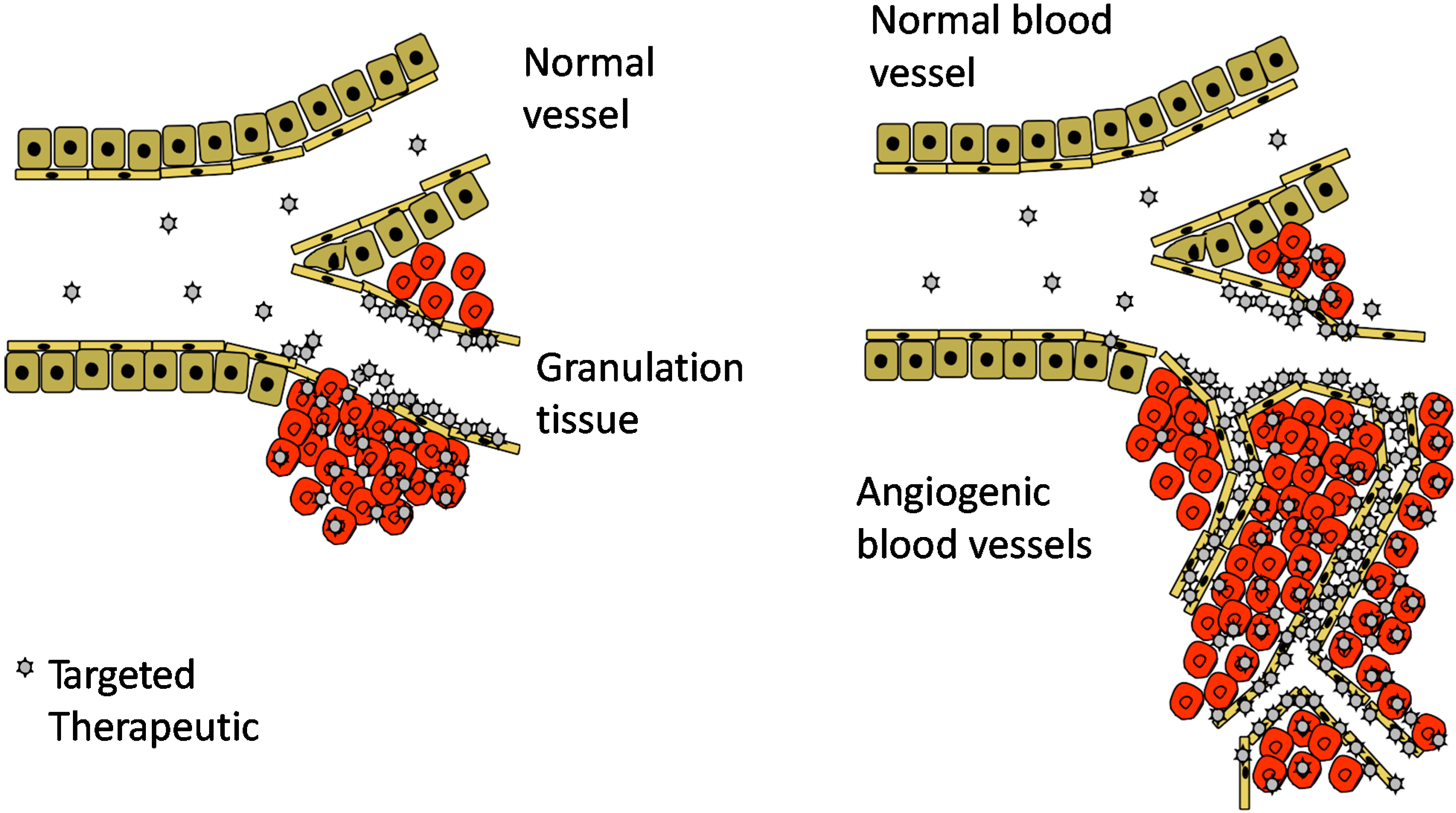
4. Angiogenesis—An Opportunity for Vascular Targeting in Regenerative Medicine
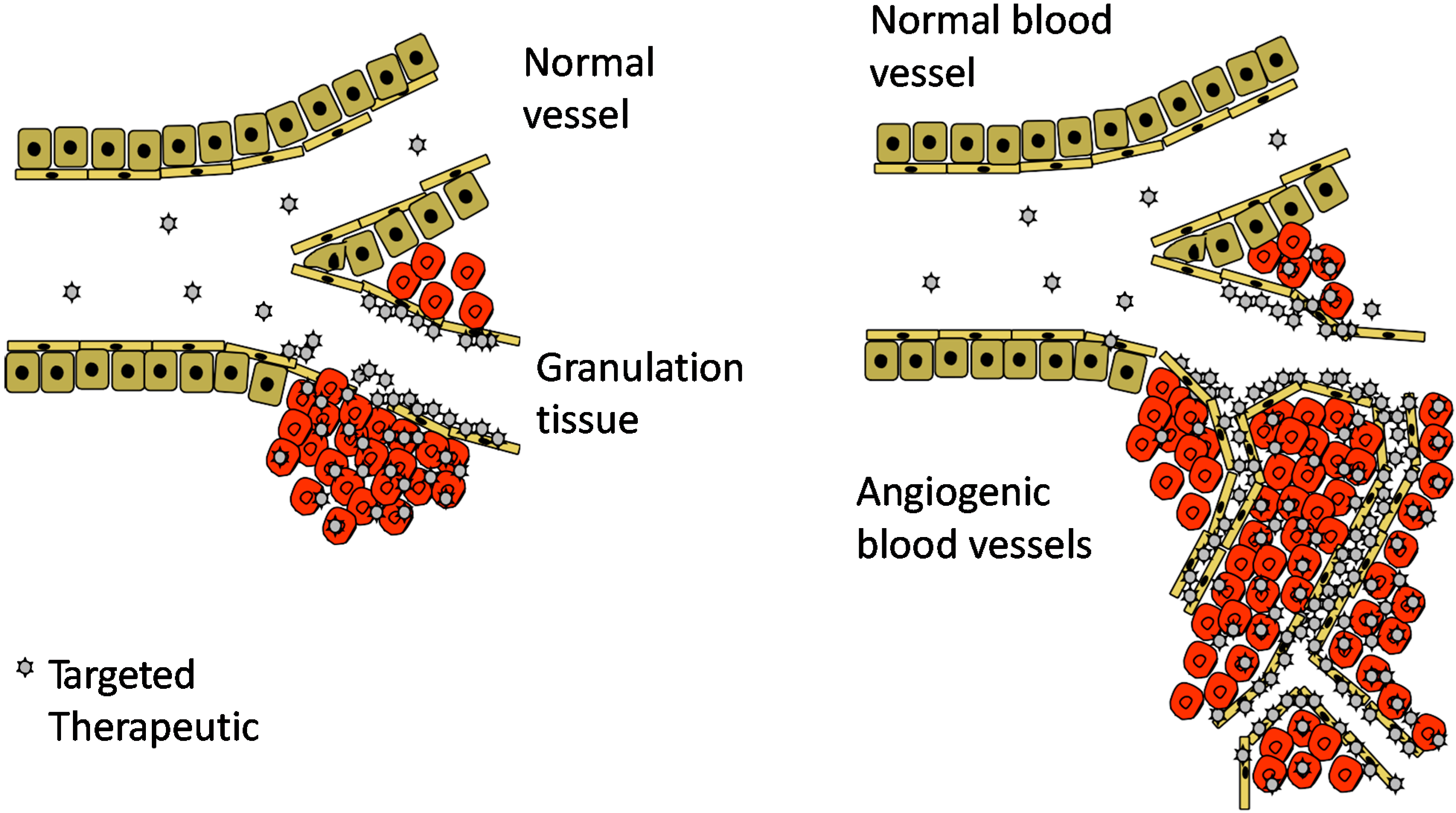
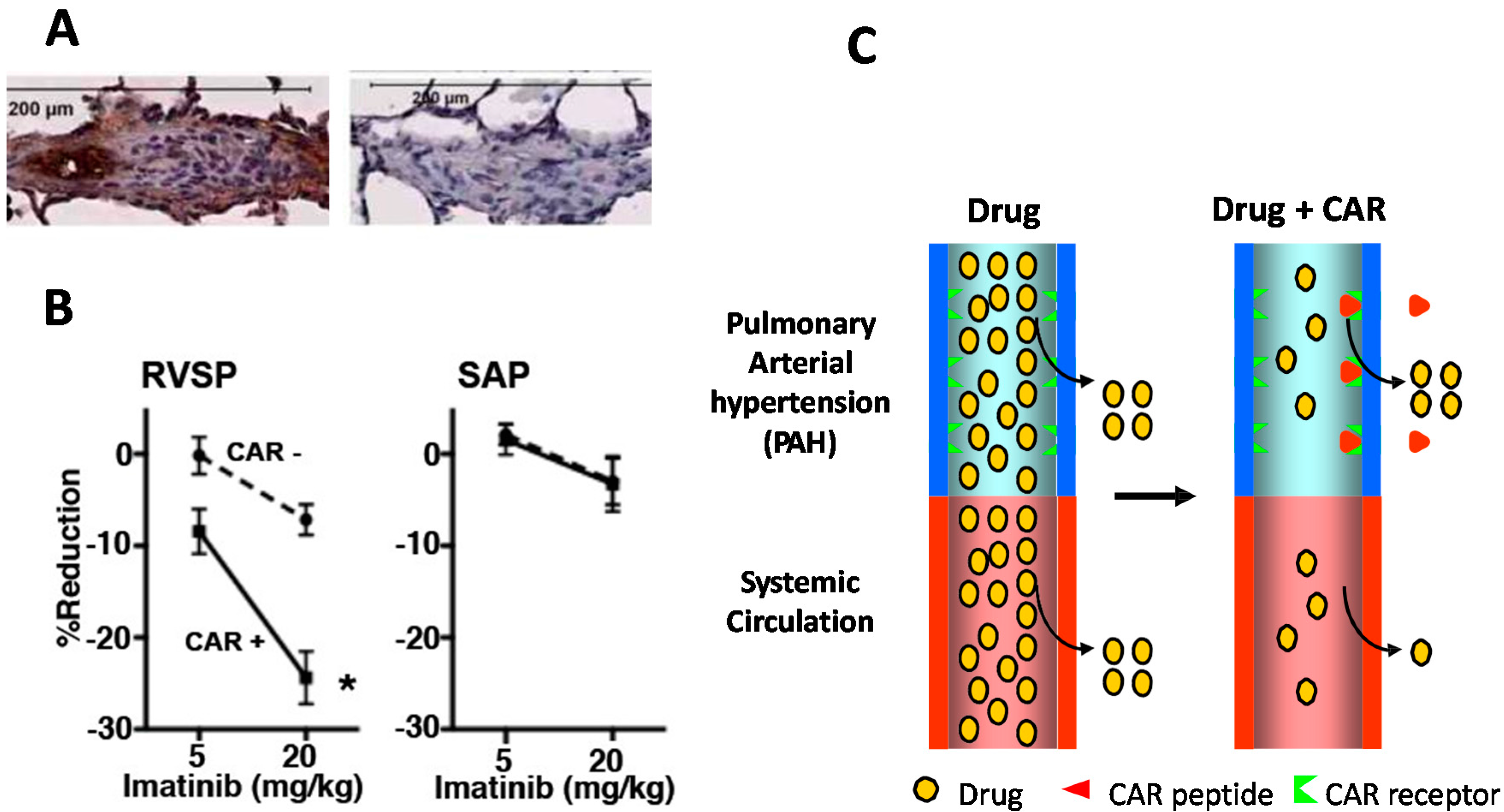
5. CRK and CAR Vascular Homing Peptides for Regenerative Medicine
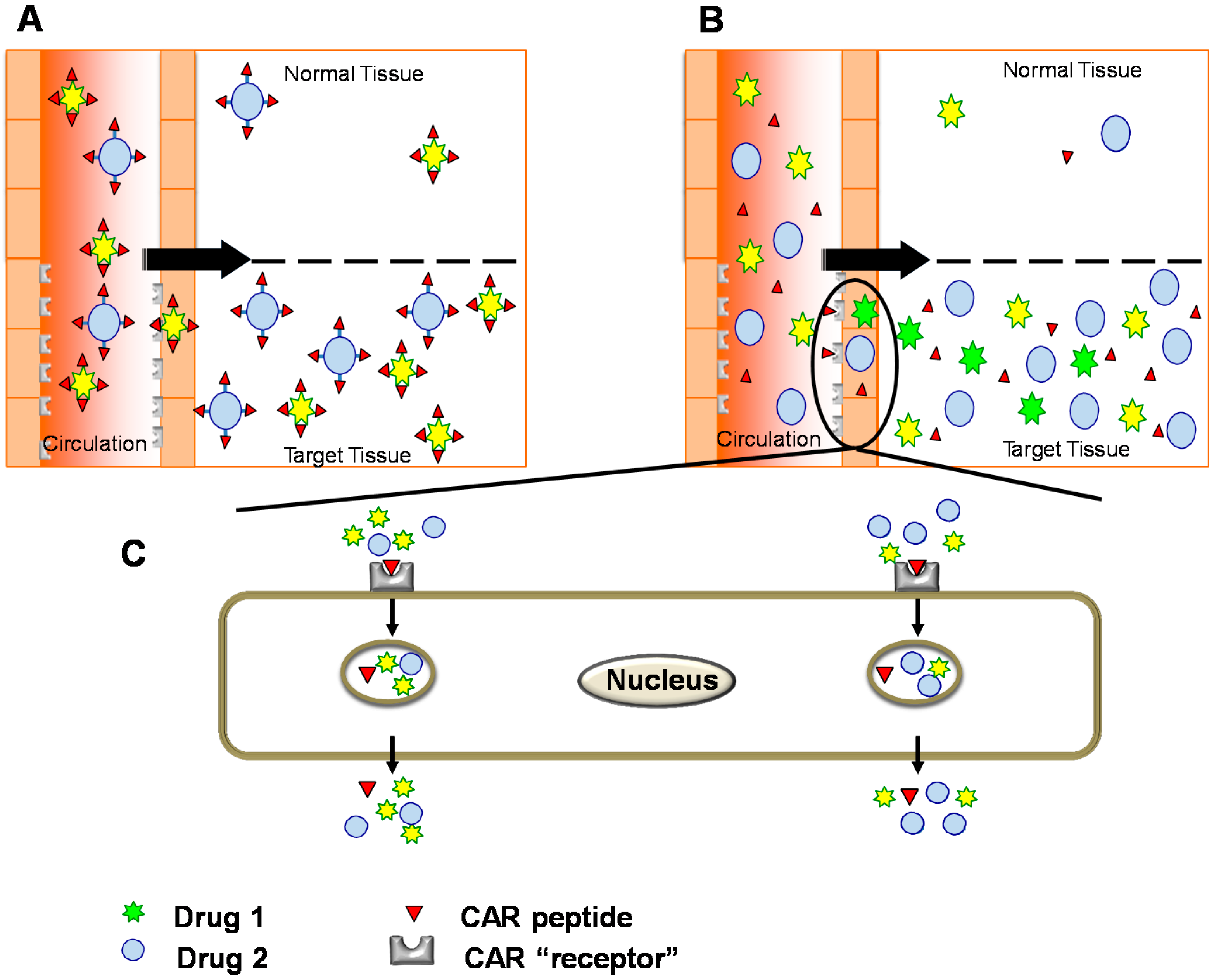
6. Conjugated Delivery
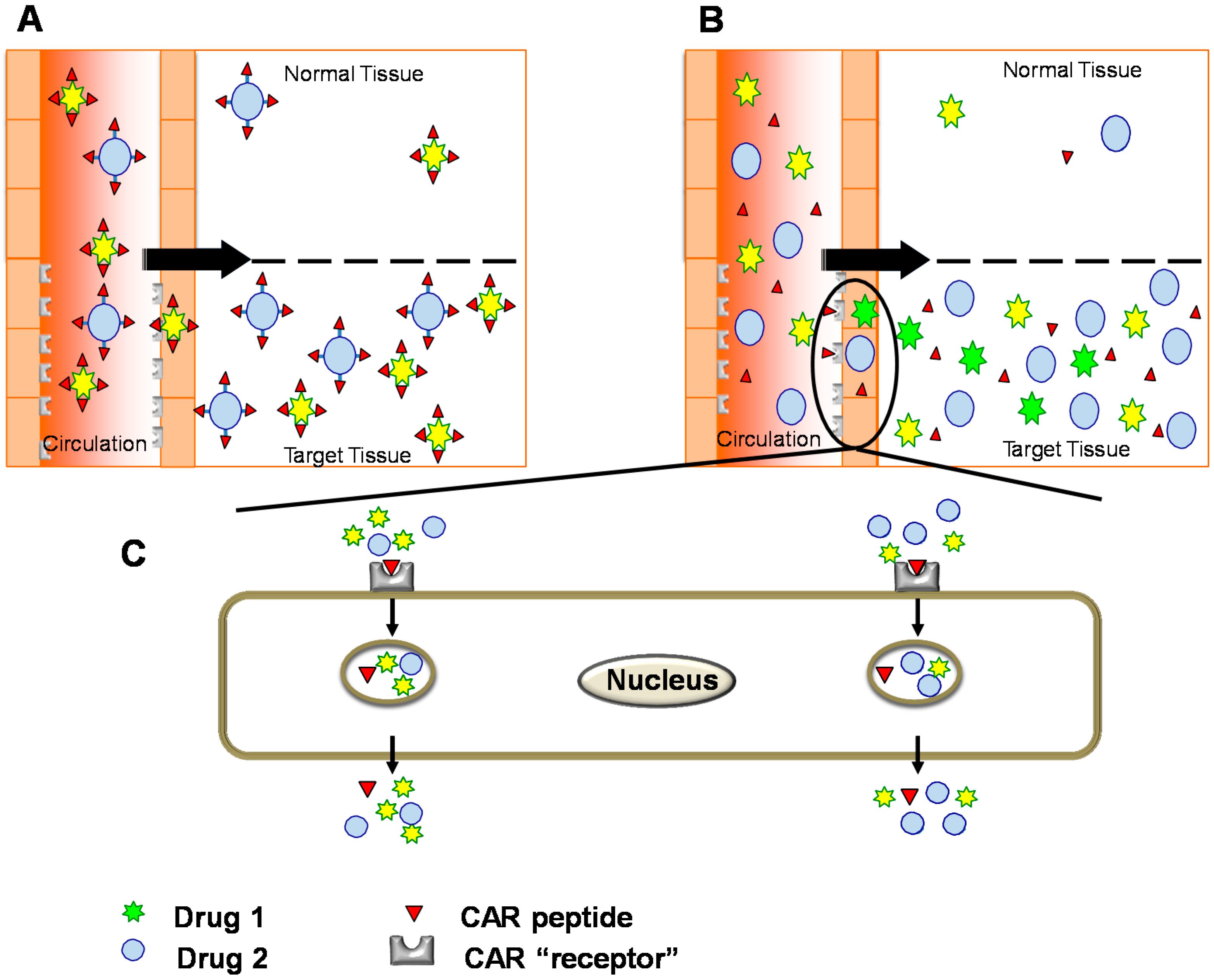
7. Bystander Effect
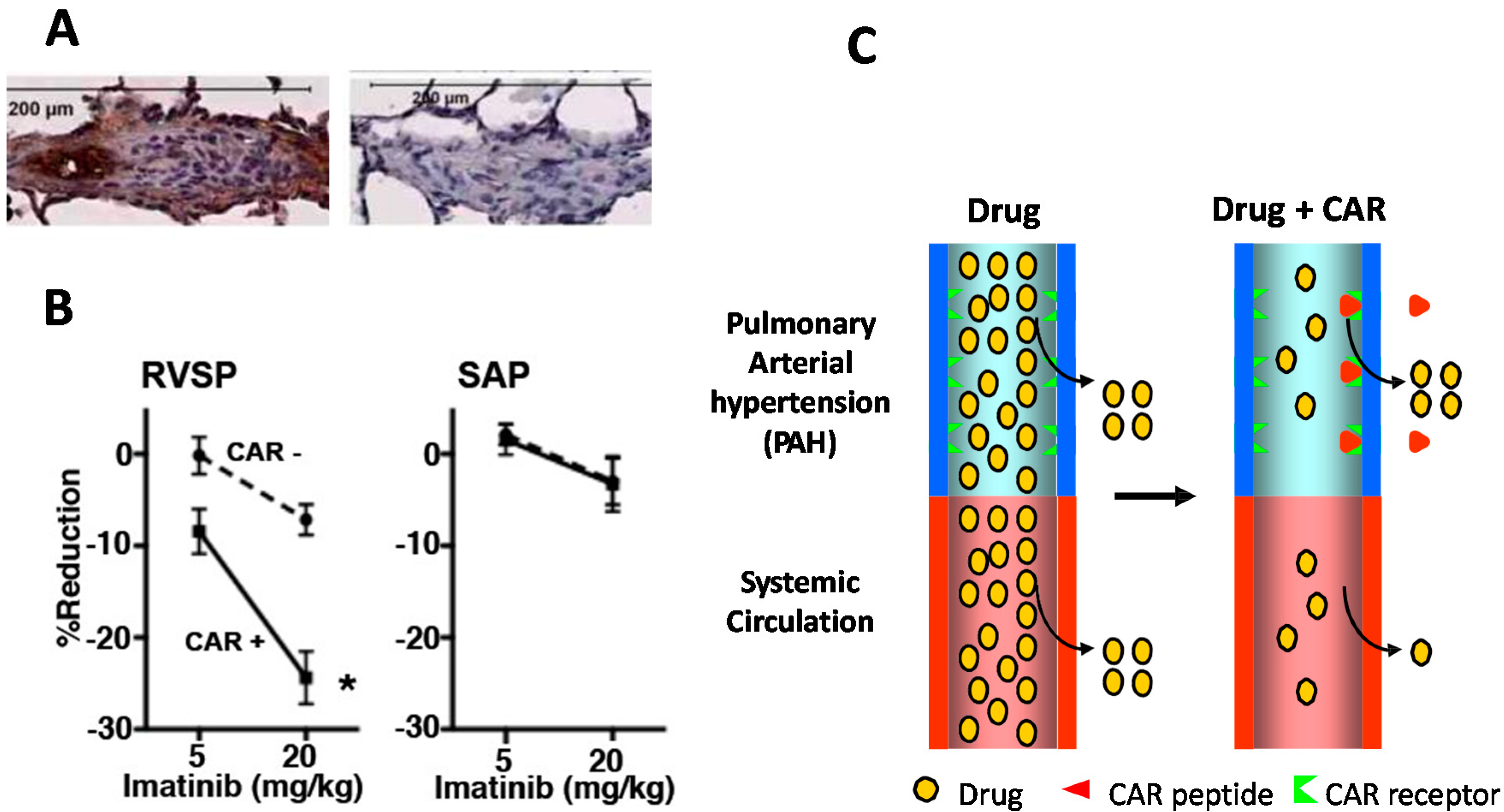
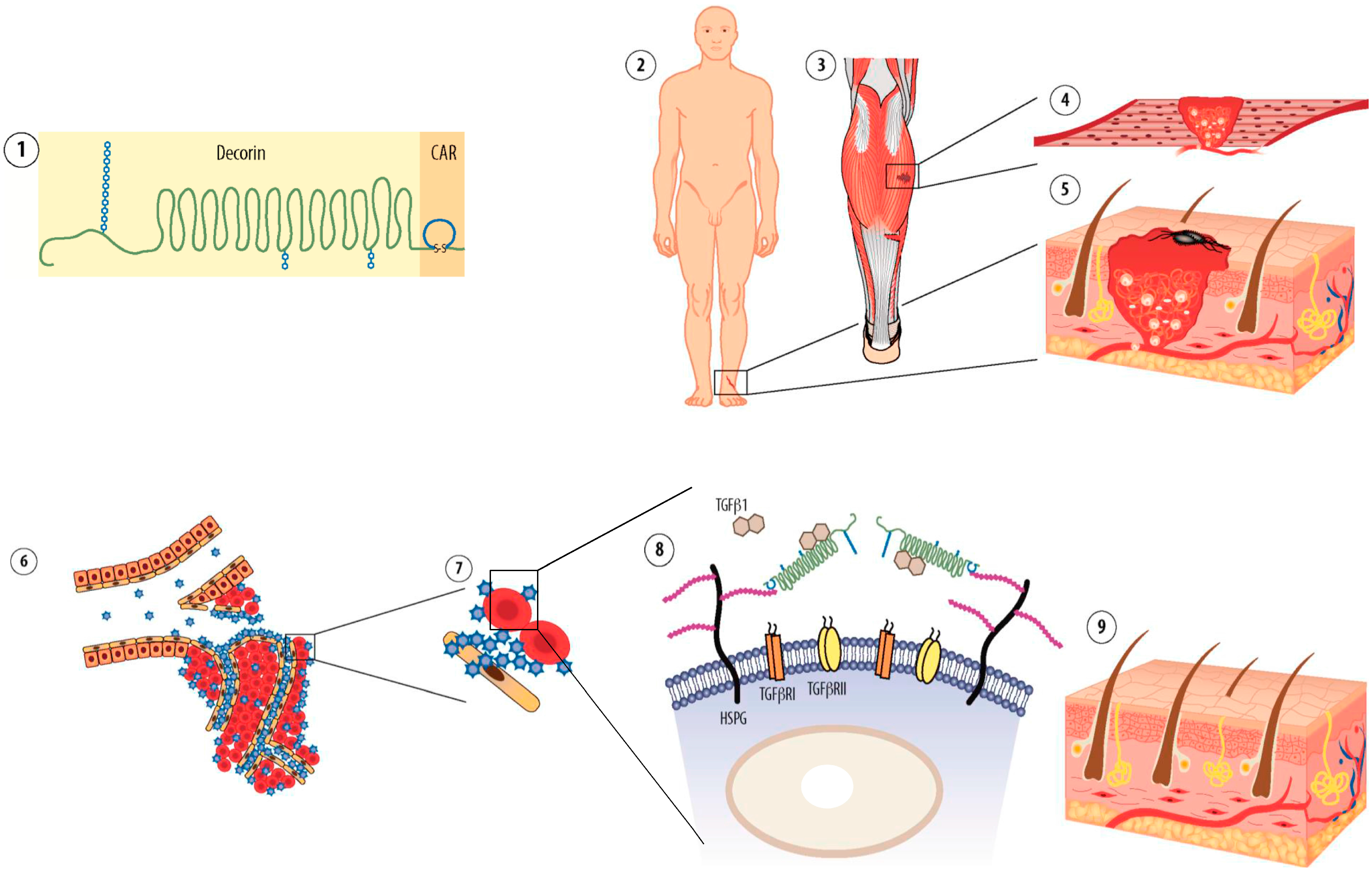
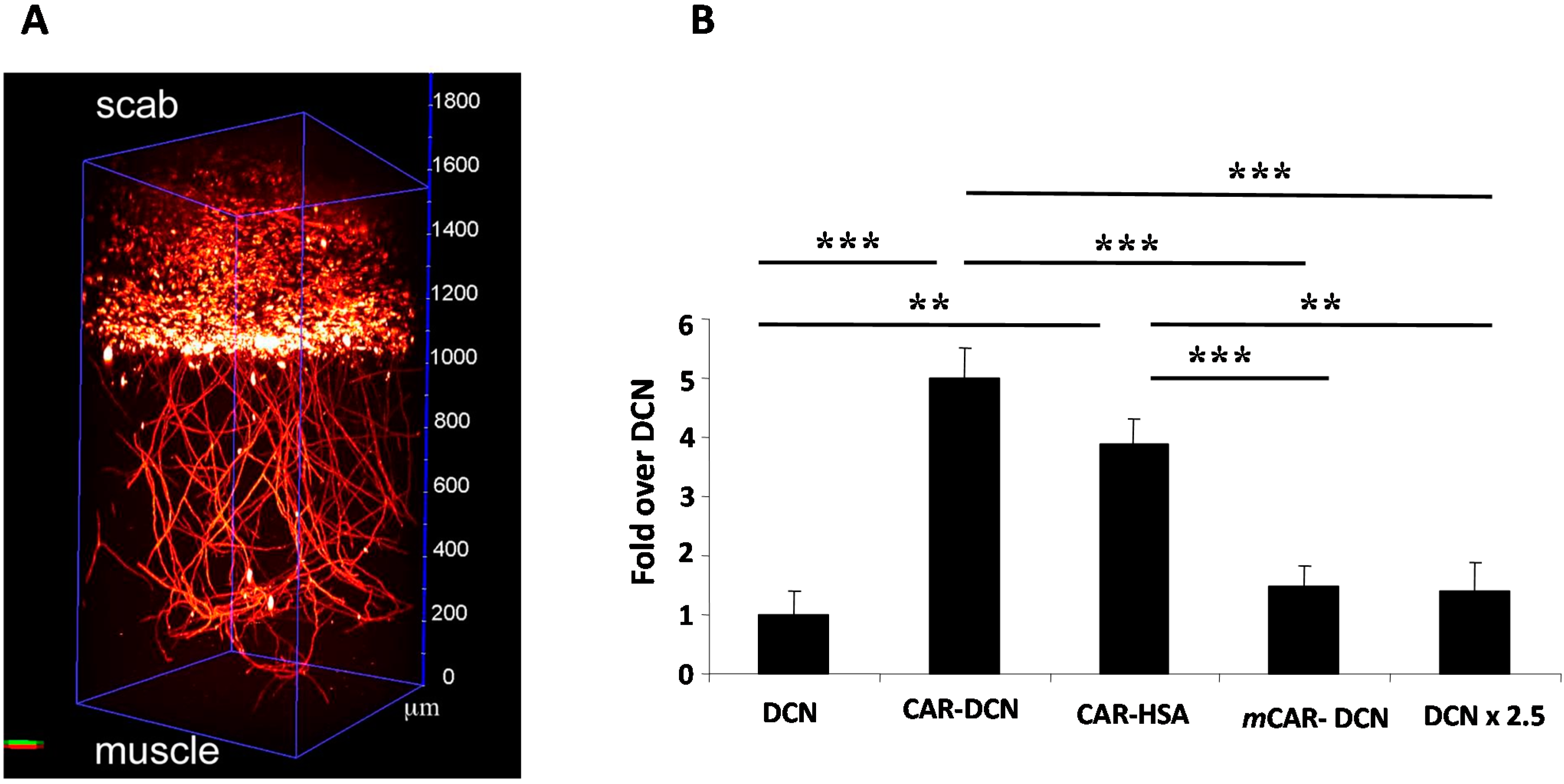
8. CAR-Decorin—Target Organ-Specific Systemically Administered Anti-Fibrotic Molecule
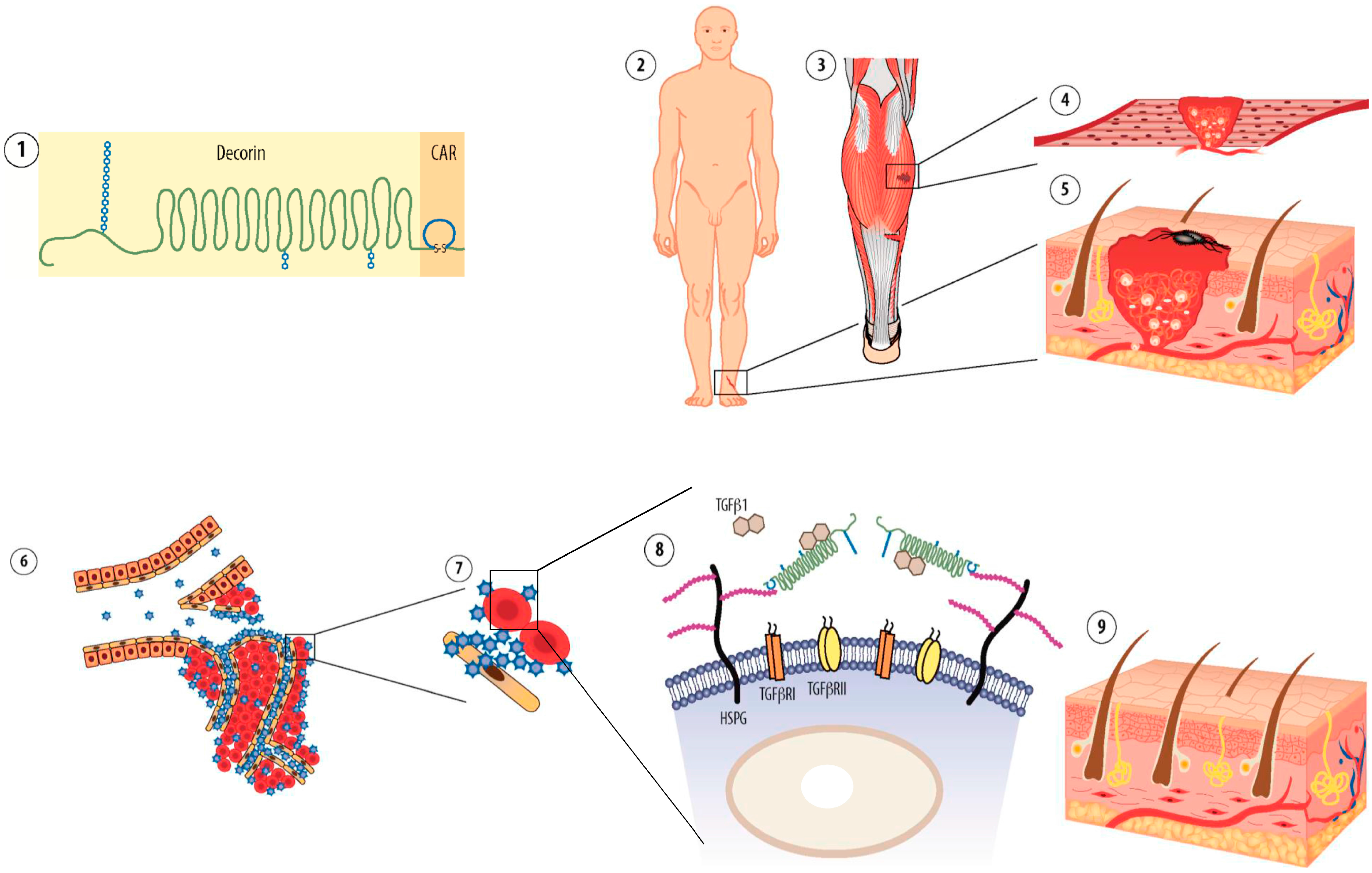
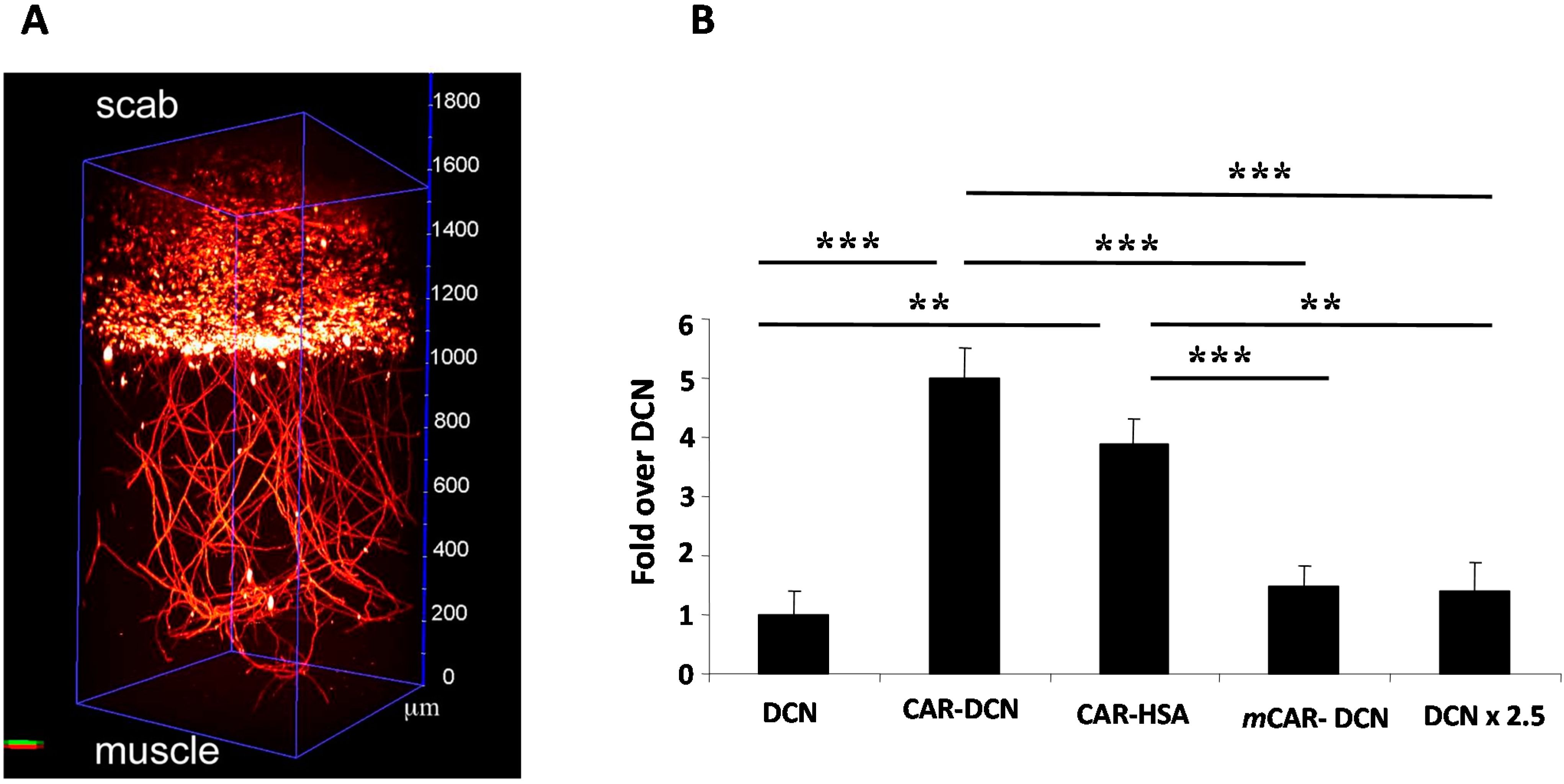
9. Future Perspectives
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Eming, S.A.; Martin, P.; Tomic-Canic, M. Wound repair and regeneration: Mechanisms, signaling, and translation. Sci. Transl. Med. 2014, 6, 265sr6. [Google Scholar] [CrossRef] [PubMed]
- Järvinen, T.A.; Järvinen, T.L.; Kääriäinen, M.; Kalimo, H.; Järvinen, M. Muscle injuries: Biology and treatment. Am. J. Sports Med. 2005, 33, 745–764. [Google Scholar] [CrossRef] [PubMed]
- Gurtner, G.C.; Werner, S.; Barrandon, Y.; Longaker, M.T. Wound repair and regeneration. Nature 2008, 453, 314–321. [Google Scholar] [CrossRef] [PubMed]
- Martino, M.M.; Briquez, P.S.; Maruyama, K.; Hubbell, J.A. Extracellular matrix-inspired growth factor delivery systems for bone regeneration. Adv. Drug Deliv. Rev. 2015. [Google Scholar] [CrossRef] [PubMed]
- Geer, D.J.; Swartz, D.D.; Andreadis, S.T. Biomimetic delivery of keratinocyte growth factor upon cellular demand for accelerated wound healing in vitro and in vivo. Am. J. Pathol. 2005, 167, 1575–1586. [Google Scholar] [CrossRef]
- Martino, M.M.; Briquez, P.S.; Guc, E.; Tortelli, F.; Kilarski, W.W.; Metzger, S.; Rice, J.J.; Kuhn, G.A.; Müller, R.; Swartz, M.A.; et al. Growth factors engineered for super-affinity to the extracellular matrix enhance tissue healing. Science 2014, 343, 885–888. [Google Scholar] [CrossRef] [PubMed]
- Arap, W.; Pasqualini, R.; Ruoslahti, E. Cancer treatment by targeted drug delivery to tumor vasculature in a mouse model. Science 1998, 279, 377–380. [Google Scholar] [CrossRef] [PubMed]
- Hussain, S.; Rodriguez-Fernandez, M.; Braun, G.B.; Doyle, F.J.; Ruoslahti, E. Quantity and accessibility for specific targeting of receptors in tumours. Sci. Rep. 2014, 4, 5232. [Google Scholar] [CrossRef] [PubMed]
- Ruoslahti, E.; Rajotte, D. An address system in the vasculature of normal tissues and tumors. Annu. Rev. Immunol. 2000, 18, 813–827. [Google Scholar] [CrossRef] [PubMed]
- AlDeghaither, D.; Smaglo, B.G.; Weiner, L.M. Beyond peptides and mAbs—Current status and future perspectives for biotherapeutics with novel constructs. J. Clin. Pharmacol. 2015, 55 (Suppl. 3), S4–S20. [Google Scholar] [CrossRef] [PubMed]
- Ruoslahti, E.; Bhatia, S.N.; Sailor, M.J. Targeting of drugs and nanoparticles to tumors. J. Cell Biol. 2010, 188, 759–768. [Google Scholar] [CrossRef] [PubMed]
- Teesalu, T.; Sugahara, K.N.; Ruoslahti, E. Tumor-penetrating peptides. Front. Oncol. 2013, 3, 216. [Google Scholar] [CrossRef] [PubMed]
- Huang, R.; Ma, H.; Guo, Y.; Liu, S.; Kuang, Y.; Shao, K.; Li, J.; Liu, Y.; Han, L.; Huang, S.; et al. Angiopep-conjugated nanoparticles for targeted long-term gene therapy of Parkinson’s disease. Pharm. Res. 2013, 30, 2549–2559. [Google Scholar] [CrossRef] [PubMed]
- Pasqualini, R.; Ruoslahti, E. Organ targeting in vivo using phage display peptide libraries. Nature 1996, 380, 364–366. [Google Scholar] [CrossRef] [PubMed]
- Nolan, D.J.; Ginsberg, M.; Israely, E.; Palikuqi, B.; Poulos, M.G.; James, D.; Ding, B.-S.; Schachterle, W.; Liu, Y.; Rosenwaks, Z.; et al. Molecular signatures of tissue-specific microvascular endothelial cell heterogeneity in organ maintenance and regeneration. Dev. Cell 2013, 26, 204–219. [Google Scholar] [CrossRef] [PubMed]
- Oh, P.; Borgstrom, P.; Witkiewicz, H.; Li, Y.; Borgstrom, B.J.; Chrastina, A.; Iwata, K.; Zinn, K.R.; Baldwin, R.; Testa, J.E.; et al. Live dynamic imaging of caveolae pumping targeted antibody rapidly and specifically across endothelium in the lung. Nat. Biotechnol. 2007, 25, 327–337. [Google Scholar] [CrossRef] [PubMed]
- Oh, P.; Testa, J.E.; Borgstrom, P.; Witkiewicz, H.; Li, Y.; Schnitzer, J.E. In vivo proteomic imaging analysis of caveolae reveals pumping system to penetrate solid tumors. Nat. Med. 2014, 20, 1062–1068. [Google Scholar] [CrossRef] [PubMed]
- Rajotte, D.; Arap, W.; Hagedorn, M.; Koivunen, E.; Pasqualini, R.; Ruoslahti, E. Molecular heterogeneity of the vascular endothelium revealed by in vivo phage display. J. Clin. Investig. 1998, 102, 430–437. [Google Scholar] [CrossRef] [PubMed]
- Ruoslahti, E. Vascular zip codes in angiogenesis and metastasis. Biochem. Soc. Trans. 2004, 32, 397–402. [Google Scholar] [CrossRef] [PubMed]
- Järvinen, T.A.; Ruoslahti, E. Molecular changes in the vasculature of injured tissues. Am. J. Pathol. 2007, 171, 702–711. [Google Scholar] [CrossRef] [PubMed]
- De Francesco, F.; Tirino, V.; Desiderio, V.; Ferraro, G.; D’Andrea, F.; Giuliano, M.; Libondi, G.; Pirozzi, G.; de Rosa, A.; Papaccio, G. Human CD34/CD90 ASCs are capable of growing as sphere clusters, producing high levels of VEGF and forming capillaries. PLoS ONE 2009, 4, e6537. [Google Scholar] [CrossRef] [PubMed]
- Teesalu, T.; Sugahara, K.N.; Ruoslahti, E. Mapping of vascular ZIP codes by phage display. Methods Enzymol. 2012, 503, 35–56. [Google Scholar] [PubMed]
- Järvinen, T.A. Design of target-seeking antifibrotic compounds. Methods Enzymol. 2012, 509, 243–261. [Google Scholar] [PubMed]
- Zhang, L.; Hoffman, J.A.; Ruoslahti, E. Molecular profiling of heart endothelial cells. Circulation 2005, 112, 1601–1611. [Google Scholar] [CrossRef] [PubMed]
- Whitney, M.A.; Crisp, J.L.; Nguyen, L.T.; Friedman, B.; Gross, L.A.; Steinbach, P.; Tsien, R.Y.; Nguyen, Q.T. Fluorescent peptides highlight peripheral nerves during surgery in mice. Nat. Biotechnol. 2011, 29, 352–356. [Google Scholar] [CrossRef] [PubMed]
- Schafer, M.; Werner, S. Cancer as an overhealing wound: An old hypothesis revisited. Nat. Rev. Mol. Cell Biol. 2008, 9, 628–638. [Google Scholar] [CrossRef] [PubMed]
- Papaccio, G. Insulitis and islet microvasculature in type 1 diabetes. Histol. Histopathol. 1993, 8, 751–759. [Google Scholar] [PubMed]
- Järvinen, M. Healing of a crush injury in rat striated muscle. 3. A micro-angiographical study of the effect of early mobilization and immobilization on capillary ingrowth. Acta Pathol. Microbiol. Scand. Sect. A Pathol. 1976, 84, 85–94. [Google Scholar]
- Järvinen, T.A.; Ruoslahti, E. Targeted antiscarring therapy for tissue injuries. Adv. Wound Care 2013, 2, 50–54. [Google Scholar] [CrossRef] [PubMed]
- Yanez, C.O.; Morales, A.R.; Yue, X.; Urakami, T.; Komatsu, M.; Järvinen, T.A.; Belfield, K.D. Deep vascular imaging in wounds by two-photon fluorescence microscopy. PLoS ONE 2013, 8, e67559. [Google Scholar] [CrossRef] [PubMed]
- Agemy, L.; Sugahara, K.N.; Kotamraju, V.R.; Gujraty, K.; Girard, O.M.; Kono, Y.; Mattrey, R.F.; Park, J.-H.; Sailor, M.J.; Jimenez, A.I.; et al. Nanoparticle-induced vascular blockade in human prostate cancer. Blood 2010, 116, 2847–2856. [Google Scholar] [CrossRef] [PubMed]
- Doll, F.; Schwager, K.; Hemmerle, T.; Neri, D. Murine analogues of etanercept and of F8-IL10 inhibit the progression of collagen-induced arthritis in the mouse. Arthritis Res. Ther. 2013, 15, R138. [Google Scholar] [CrossRef] [PubMed]
- Franz, M.; Doll, F.; Grun, K.; Richter, P.; Kose, N.; Ziffels, B.; Schubert, H.; Figulla, H.R.; Jung, C.; Gummert, J.; et al. Targeted delivery of interleukin-10 to chronic cardiac allograft rejection using a human antibody specific to the extra domain A of fibronectin. Int. J. Cardiol. 2015, 195, 311–322. [Google Scholar] [CrossRef] [PubMed]
- Galeazzi, M.; Bazzichi, L.; Sebastiani, G.D.; Neri, D.; Garcia, E.; Ravenni, N.; Giovannoni, L.; Wilton, J.; Bardelli, M.; Baldi, C.; et al. A phase IB clinical trial with Dekavil (F8-IL10), an immunoregulatory “armed antibody” for the treatment of rheumatoid arthritis, used in combination wiIh methotrexate. Isr. Med. Assoc. J. 2014, 16, 666. [Google Scholar] [PubMed]
- Schwager, K.; Kaspar, M.; Bootz, F.; Marcolongo, R.; Paresce, E.; Neri, D.; Trachsel, E. Preclinical characterization of DEKAVIL (F8-IL10), a novel clinical-stage immunocytokine which inhibits the progression of collagen-induced arthritis. Arthritis Res. Ther. 2009, 11, R142. [Google Scholar] [CrossRef] [PubMed]
- Corti, A.; Curnis, F. Tumor vasculature targeting through NGR peptide-based drug delivery systems. Curr. Pharm. Biotechnol. 2011, 12, 1128–1134. [Google Scholar] [CrossRef] [PubMed]
- Calcinotto, A.; Grioni, M.; Jachetti, E.; Curnis, F.; Mondino, A.; Parmiani, G.; Corti, A.; Bellone, M. Targeting TNF-α to neoangiogenic vessels enhances lymphocyte infiltration in tumors and increases the therapeutic potential of immunotherapy. J. Immunol. 2012, 188, 2687–2694. [Google Scholar] [CrossRef]
- Kean, T.J.; Duesler, L.; Young, R.G.; Dadabayev, A.; Olenyik, A.; Penn, M.; Wagner, J.; Fink, D.J.; Caplan, A.I.; Dennis, J.E. Development of a peptide-targeted, myocardial ischemia-homing, mesenchymal stem cell. J. Drug Target. 2012, 20, 23–32. [Google Scholar] [CrossRef] [PubMed]
- Järvinen, T.A.; Ruoslahti, E. Target-seeking antifibrotic compound enhances wound healing and suppresses scar formation in mice. Proc. Natl. Acad. Sci. USA 2010, 107, 21671–21676. [Google Scholar] [CrossRef] [PubMed]
- Ma, W.; Cai, S.; Du, J.; Tan, Y.; Chen, H.; Guo, Z.; Hu, H.; Fang, R.; Cai, S. SDF-1/54-DCN: A novel recombinant chimera with dual inhibitory effects on proliferation and chemotaxis of tumor cells. Biol. Pharm. Bull. 2008, 31, 1086–1091. [Google Scholar] [CrossRef] [PubMed]
- Kean, T.J.; Lin, P.; Caplan, A.I.; Dennis, J.E. MSCs: Delivery routes and engraftment, cell-targeting strategies, and immune modulation. Stem Cells Int. 2013, 2013, 732742. [Google Scholar] [CrossRef] [PubMed]
- Toba, M.; Alzoubi, A.; O’Neill, K.; Abe, K.; Urakami, T.; Komatsu, M.; Alvarez, D.; Järvinen, T.A.H.; Mann, D.; Ruoslahti, E.; et al. A novel vascular homing peptide strategy to selectively enhance pulmonary drug efficacy in pulmonary arterial hypertension. Am. J. Pathol. 2014, 184, 369–375. [Google Scholar] [CrossRef] [PubMed]
- Sugahara, K.N.; Teesalu, T.; Karmali, P.P.; Kotamraju, V.R.; Agemy, L.; Girard, O.M.; Hanahan, D.; Mattrey, R.F.; Ruoslahti, E. Tissue-penetrating delivery of compounds and nanoparticles into tumors. Cancer Cell 2009, 16, 510–520. [Google Scholar] [CrossRef] [PubMed]
- Teesalu, T.; Sugahara, K.N.; Kotamraju, V.R.; Ruoslahti, E. C-end rule peptides mediate neuropilin-1-dependent cell, vascular, and tissue penetration. Proc. Natl. Acad. Sci. USA 2009, 106, 16157–16162. [Google Scholar] [CrossRef] [PubMed]
- Sugahara, K.N.; Teesalu, T.; Karmali, P.P.; Kotamraju, V.R.; Agemy, L.; Greenwald, D.R.; Ruoslahti, E. Coadministration of a tumor-penetrating peptide enhances the efficacy of cancer drugs. Science 2010, 328, 1031–1035. [Google Scholar] [CrossRef] [PubMed]
- Ruoslahti, E. The RGD story: A personal account. Matrix Biol. 2003, 22, 459–465. [Google Scholar] [CrossRef]
- Urakami, T.; Jarvinen, T.A.; Toba, M.; Sawada, J.; Ambalavanan, N.; Mann, D.; McMurtry, I.; Oka, M.; Ruoslahti, E.; Komatsu, M. Peptide-directed highly selective targeting of pulmonary arterial hypertension. Am. J. Pathol. 2011, 178, 2489–2495. [Google Scholar] [CrossRef] [PubMed]
- Ruoslahti, E.; Yamaguchi, Y. Proteoglycans as modulators of growth factor activities. Cell 1991, 64, 867–869. [Google Scholar] [CrossRef]
- Border, W.A.; Ruoslahti, E. Transforming growth factor-β in disease: The dark side of tissue repair. J. Clin. Investig. 1992, 90, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Järvinen, T.A.H.; Prince, S. Decorin—A growth factor antagonist for tumor growth inhibition. BioMed Res. Int. 2015, in press. [Google Scholar]
- Border, W.A.; Noble, N.A.; Yamamoto, T.; Harper, J.R.; Yamaguchi, Y.; Pierschbacher, M.D.; Ruoslahti, E. Natural inhibitor of transforming growth factor-β protects against scarring in experimental kidney disease. Nature 1992, 360, 361–364. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, Y.; Ruoslahti, E. Expression of human proteoglycan in Chinese hamster ovary cells inhibits cell proliferation. Nature 1988, 336, 244–246. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, Y.; Mann, D.M.; Ruoslahti, E. Negative regulation of transforming growth factor-β by the proteoglycan decorin. Nature 1990, 346, 281–284. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Li, Y.; Shen, W.; Qiao, C.; Ambrosio, F.; Lavasani, M.; Nozaki, M.; Branca, M.F.; Huard, J. Relationships between transforming growth factor-β1, myostatin, and decorin: Implications for skeletal muscle fibrosis. J. Biol. Chem. 2007, 282, 25852–25863. [Google Scholar] [CrossRef] [PubMed]
- Vial, C.; Gutierrez, J.; Santander, C.; Cabrera, D.; Brandan, E. Decorin interacts with connective tissue growth factor (CTGF)/CCN2 by LRR12 inhibiting its biological activity. J. Biol. Chem. 2011, 286, 24242–24252. [Google Scholar] [CrossRef] [PubMed]
- Andrianifahanana, M.; Wilkes, M.C.; Gupta, S.K.; Rahimi, R.A.; Repellin, C.E.; Edens, M.; Wittenberger, J.; Yin, X.; Maidl, E.; Becker, J.; et al. Profibrotic TGF-β responses require the cooperative action of PDGF and ErbB receptor tyrosine kinases. FASEB J. 2013, 27, 4444–4454. [Google Scholar] [CrossRef] [PubMed]
- Järvinen, T.A.H.; Ruoslahti, E. Uusi lääkeaine estää arven muodostusta. Duodecim 2011, 127, 146–147. (In Finnish) [Google Scholar]
- Nahar, K.; Absar, S.; Gupta, N.; Kotamraju, V.R.; McMurtry, I.F.; Oka, M.; Komatsu, M.; Nozik-Grayck, E.; Ahsan, F. Peptide-coated liposomal fasudil enhances site specific vasodilation in pulmonary arterial hypertension. Mol. Pharm. 2014, 11, 4374–4384. [Google Scholar] [CrossRef] [PubMed]
- Gupta, N.; Patel, B.; Nahar, K.; Ahsan, F. Cell permeable peptide conjugated nanoerythrosomes of fasudil prolong pulmonary arterial vasodilation in PAH rats. Eur. J. Pharm. Biopharm. 2014, 88, 1046–1055. [Google Scholar] [CrossRef] [PubMed]
- Gupta, N.; Ibrahim, H.M.; Ahsan, F. Peptide-micelle hybrids containing fasudil for targeted delivery to the pulmonary arteries and arterioles to treat pulmonary arterial hypertension. J. Pharm. Sci. 2014, 103, 3743–3753. [Google Scholar] [CrossRef] [PubMed]
- Gupta, N.; Al-Saikhan, F.I.; Patel, B.; Rashid, J.; Ahsan, F. Fasudil and SOD packaged in peptide-studded-liposomes: Properties, pharmacokinetics and ex vivo targeting to isolated perfused rat lungs. Int. J. Pharm. 2015, 488, 33–43. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Järvinen, T.A.H.; May, U.; Prince, S. Systemically Administered, Target Organ-Specific Therapies for Regenerative Medicine. Int. J. Mol. Sci. 2015, 16, 23556-23571. https://doi.org/10.3390/ijms161023556
Järvinen TAH, May U, Prince S. Systemically Administered, Target Organ-Specific Therapies for Regenerative Medicine. International Journal of Molecular Sciences. 2015; 16(10):23556-23571. https://doi.org/10.3390/ijms161023556
Chicago/Turabian StyleJärvinen, Tero A. H., Ulrike May, and Stuart Prince. 2015. "Systemically Administered, Target Organ-Specific Therapies for Regenerative Medicine" International Journal of Molecular Sciences 16, no. 10: 23556-23571. https://doi.org/10.3390/ijms161023556
APA StyleJärvinen, T. A. H., May, U., & Prince, S. (2015). Systemically Administered, Target Organ-Specific Therapies for Regenerative Medicine. International Journal of Molecular Sciences, 16(10), 23556-23571. https://doi.org/10.3390/ijms161023556