
An Overview of Predictors for Intrinsically Disordered Proteins over 2010–2014

Abstract
:1. Introduction
2. Predictors Developed in the Last Five Years

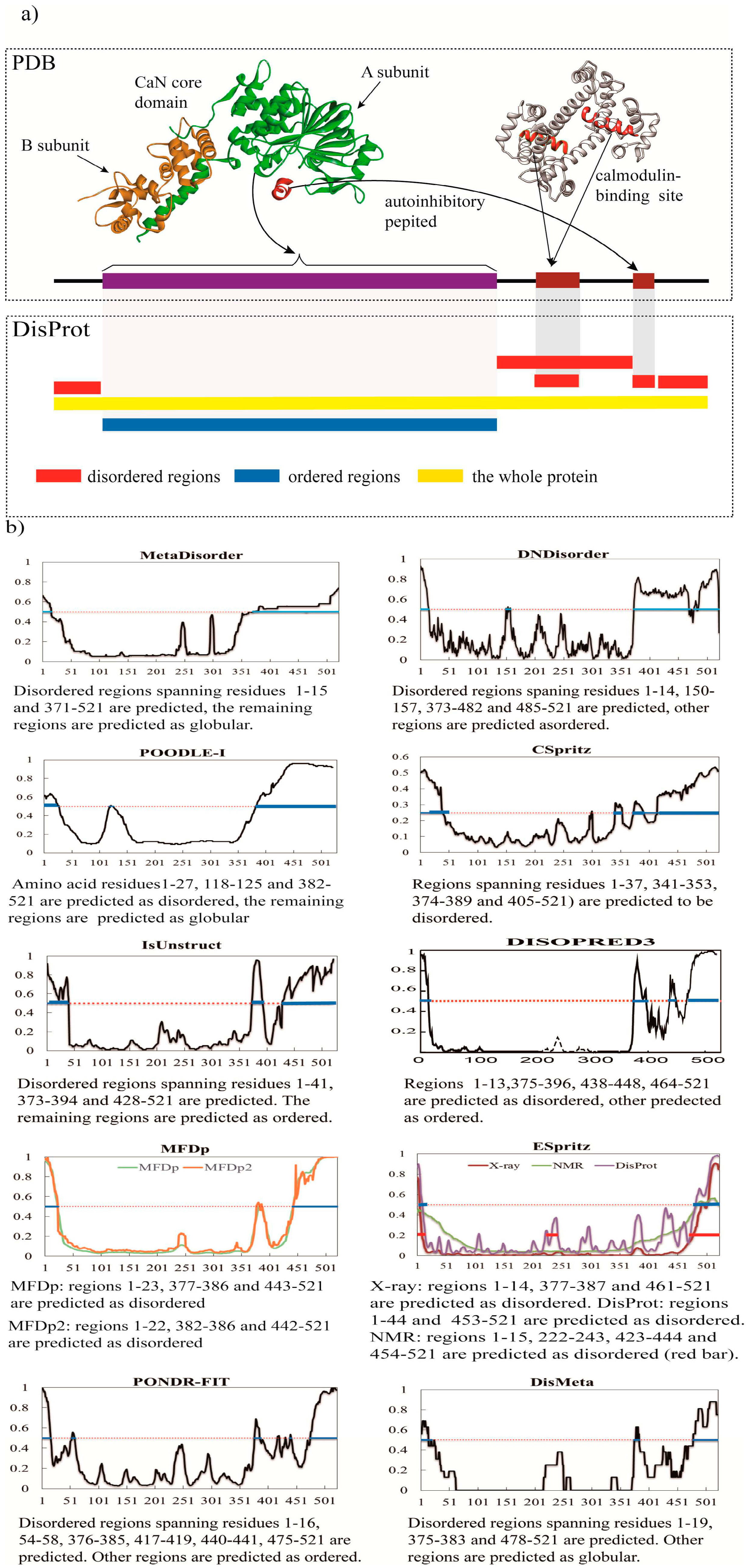{kind=link}
{kind=link}
| Name | Year | PSI-BLAST | Availability |
|---|---|---|---|
| MFDp [14] | 2010 | X | X |
| PONDR-FIT [15] | 2010 | X | X |
| SPA [16] | 2010 | ||
| DisCon [17] | 2011 | X | |
| POODLE-I [18] | 2011 | X | |
| Cspritz [19] | 2011 | X | X |
| MetaDisorder [20] | 2012 | X | X |
| Espritz [21] | 2012 | X | |
| SPINDE-D [22] | 2012 | X | |
| Dndisorder [23] | 2013 | X | X |
| IsUntruct [24] | 2013 | X | |
| MFDp2 [25] | 2013 | X | X |
| RAPID [26] | 2013 | ||
| PON-Diso [27] | 2014 | X | |
| DisMeta [28] | 2014 | X | X |
| DisPredict [29] | 2014 | ||
| DISOPRED3 [30] | 2014 | X | X |
2.1. Predictors Based on Machine Learning Classifiers
2.2. Methods Based on a Meta-Approach Which Combines Predictions from Multiple Base Predictors
2.3. Methods Based on the Physicochemical Properties
3. A Brief Comparative Assessment of the Recent Developed Predictors of IDPs
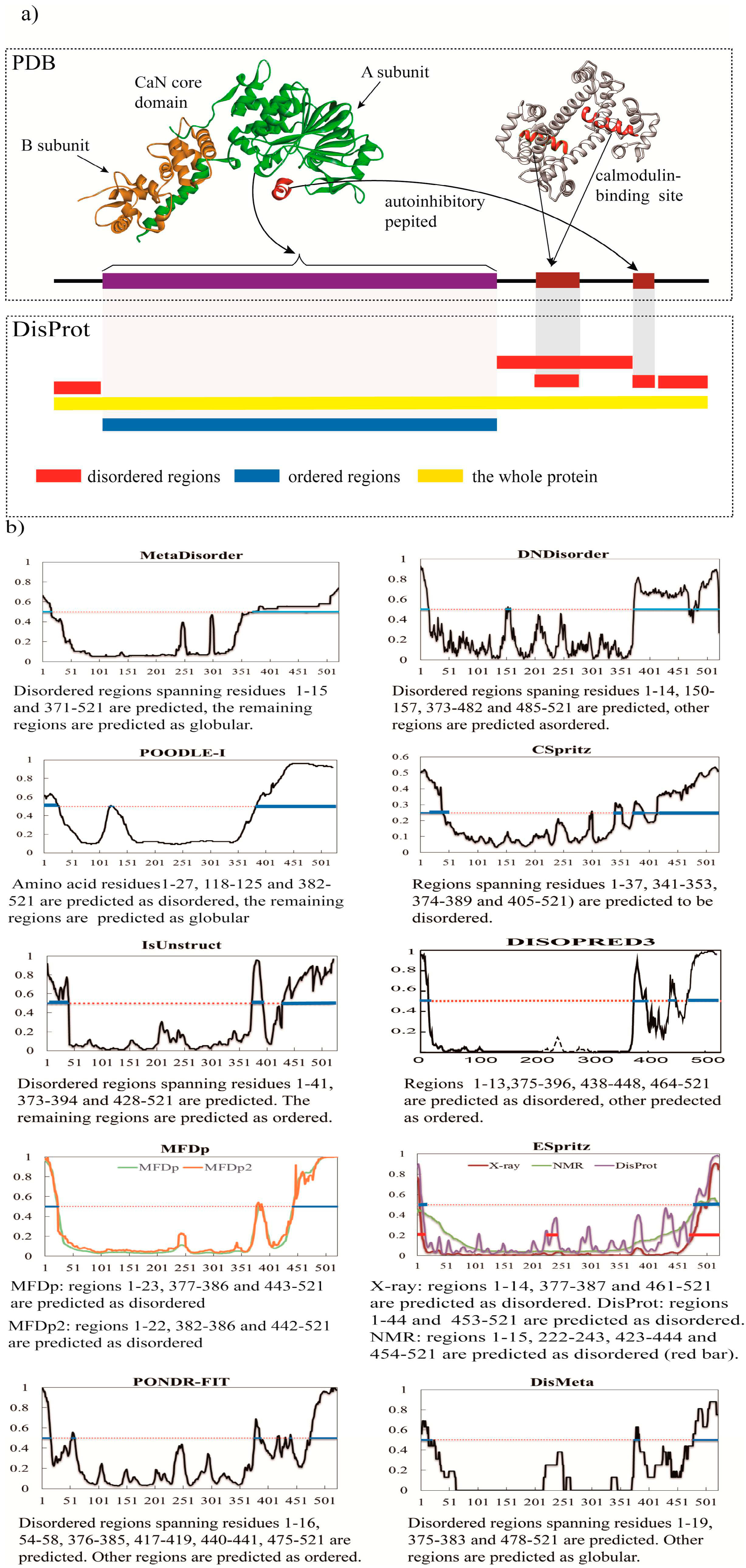
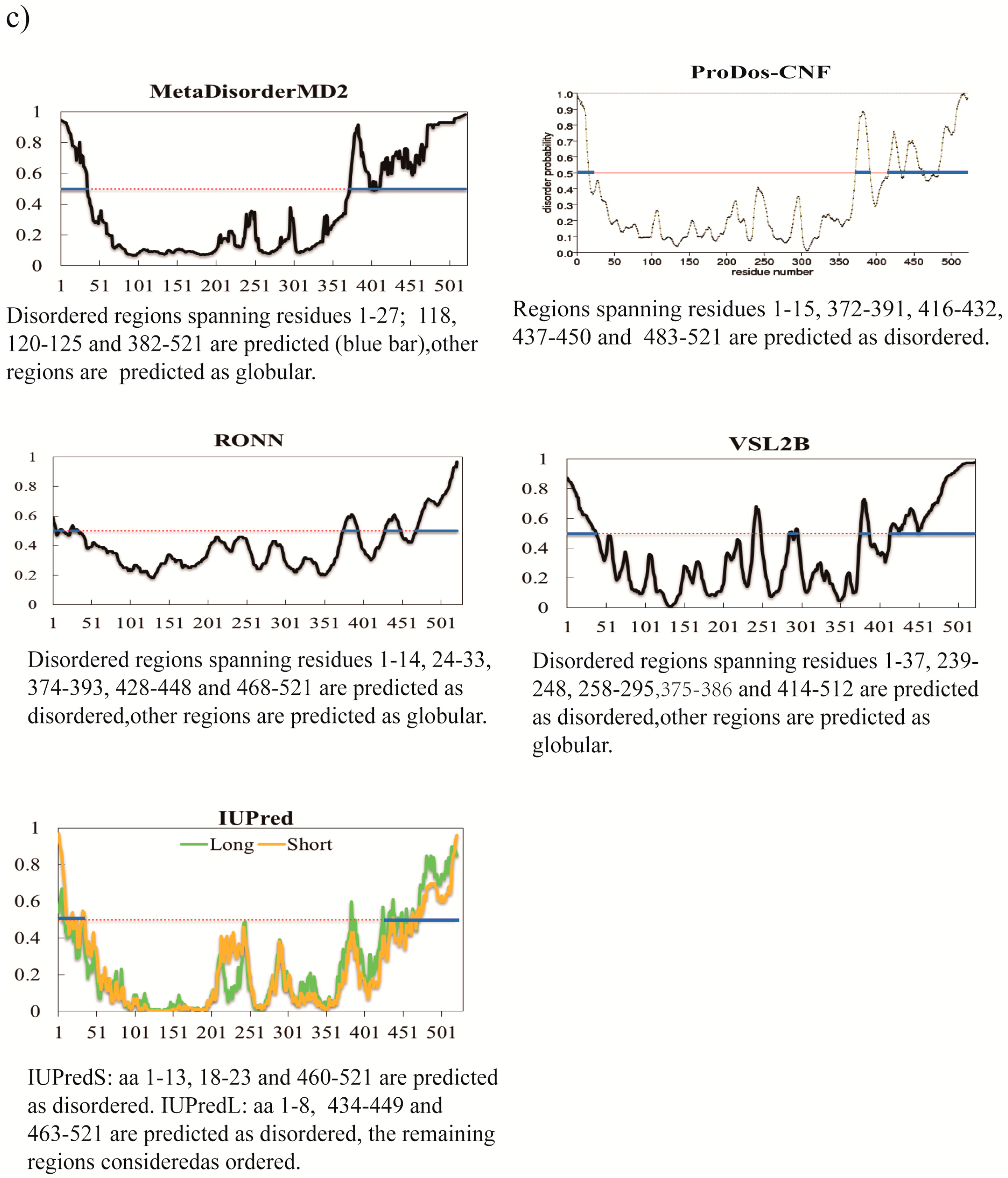
| Predictor | No. of Disordered Residues | Total % Disorder | ACC |
|---|---|---|---|
| Metadisorder | 166 | 0.32 | 0.9804 |
| Dndisorder | 160 | 0.31 | 0.9731 |
| MetaDisorderMD2 | 184 | 0.35 | 0.9559 |
| POODLE-I | 172 | 0.33 | 0.9539 |
| Cspritz | 173 | 0.33 | 0.9001 |
| IsUnstruct | 158 | 0.30 | 0.8848 |
| PrDos-CNF | 106 | 0.20 | 0.8752 |
| RONN | 115 | 0.22 | 0.8695 |
| Espritz_NMR | 144 | 0.28 | 0.8694 |
| DISOPRED3 | 102 | 0.20 | 0.8675 |
| MFDp | 112 | 0.21 | 0.8656 |
| MFDp2 | 107 | 0.21 | 0.8618 |
| VSL2B | 178 | 0.34 | 0.8580 |
| Espritz_X-ray | 85 | 0.16 | 0.8522 |
| IUPredL | 83 | 0.16 | 0.8503 |
| IUPredS | 81 | 0.16 | 0.8234 |
| PONDR-FIT | 84 | 0.16 | 0.8215 |
| DisMeta | 69 | 0.13 | 0.8157 |
| Espritz_DisProt | 113 | 0.22 | 0.7888 |
4. Concluding Remarks
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Ferron, F.; Longhi, S.; Canard, B.; Karlin, D. A practical overview of protein disorder prediction methods. Proteins 2006, 65, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Tompa, P.; Han, K.H. Intrinsically disordered proteins. Phys. Today 2012, 65, 64–65. [Google Scholar] [CrossRef]
- Fischer, E. Einfluss der Configuration auf die Wirkung der Enzyme. Berichte Deutschen Chemischen Gesellschaft 2006, 27, 2985–2993. [Google Scholar] [CrossRef]
- Uversky, V.N. Intrinsically disordered proteins from A to Z. Int. J. Biochem. Cell Biol. 2011, 43, 1090–1103. [Google Scholar] [CrossRef] [PubMed]
- Dunker, A.K.; Oldfield, C.J.; Meng, J.; Romero, P.; Yang, J.Y.; Chen, J.W.; Vacic, V.; Obradovic, Z.; Uversky, V.N. The unfoldomics decade: An update on intrinsically disordered proteins. BMC Genom. 2008, 9. [Google Scholar] [CrossRef] [PubMed]
- Fukuchi, S.; Hosoda, K.; Homma, K.; Gojobori, T.; Nishikawa, K. Binary classification of protein molecules into intrinsically disordered and ordered segments. BMC Struct. Biol. 2011, 11, 29. [Google Scholar] [CrossRef] [PubMed]
- Dunker, A.K.; Silman, I.; Uversky, V.N.; Sussman, J.L. Function and structure of inherently disordered proteins. Curr. Opin. Struct. Biol. 2008, 18, 756–764. [Google Scholar] [CrossRef] [PubMed]
- Tompa, P. Intrinsically disordered proteins: A 10-year recap. Trends Biochem. Sci. 2012, 37, 509–516. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N.; Oldfield, C.J.; Dunker, A.K. Intrinsically disordered proteins in human diseases: Introducing the D2 concept. Annu. Rev. Biophys. 2008, 37, 215–246. [Google Scholar] [CrossRef] [PubMed]
- Deng, X.; Eickholt, J.; Cheng, J. A comprehensive overview of computational protein disorder prediction methods. Mol. Biosyst. 2012, 8, 114–121. [Google Scholar] [CrossRef] [PubMed]
- Williams, R.J.P. The conformation properties of proteins in solution. Biol. Rev. 1979, 54, 389–437. [Google Scholar] [CrossRef] [PubMed]
- Han, P.; Zhang, X.; Feng, Z.P. Predicting disordered regions in proteins using the profiles of amino acid indices. BMC Bioinform. 2009, 10. [Google Scholar] [CrossRef] [PubMed]
- He, B.; Wang, K.J.; Liu, Y.L.; Xue, B.; Uversky, V.N.; Dunker, A.K. Predicting intrinsic disorder in proteins: An overview. Cell Res. 2009, 19, 929–949. [Google Scholar] [CrossRef] [PubMed]
- Multilayered Fusion-base Disorder predictor. Available online: http://biomine.ece.ualberta.ca/MFDp.html (accessed on 7 September 2015).
- Protein Disorder Predictors. Available online: www.disprot.org/predictors.php (accessed on 7 September 2015).
- Xue, B.; Hsu, W.-L.; Lee, J.-H.; Lu, H.; Dunker, A.K.; Uversky, V.N. SPA: Short peptide analyzer of intrinsic disorder status of short peptides. Genes Cells 2010, 15, 635–646. [Google Scholar] [CrossRef] [PubMed]
- Disorder Content Predictor. Available online: http://biomine.ece.ualberta.ca/DisCon/ (accessed on 7 September 2015).
- POODLE-I. Available online: http://mbs.cbrc.jp/poodle/poodle-i.html/ (accessed on 7 September 2015).
- Disorder Prediction with CSpritz. Available online: http://protein.bio.unipd.it/cspritz/ (accessed on 7 September 2015).
- GeneSilico MetaDisorder Service. Available online: http://iimcb.genesilico.pl/metadisorder/ (accessed on 7 September 2015).
- Espritz. Available online: http://protein.bio.unipdc.it/espritz/ (accessed on 7 September 2015).
- SPINDE-D. Available online: http://sparks.informatics.iupui.edu/ (accessed on 7 September 2015).
- DNdisorder. Available online: http://iris.rnet.missouri.edu/dndisorder/ (accessed on 7 September 2015).
- The BioInformatics Group. Available online: http://bioinfo.protres.ru/IsUnstruct/ (accessed on 7 September 2015).
- MFDp2 Webserver–Biomine. Available online: http://biomine.ece.ualberta.ca/MFDp2/ (accessed on 7 September 2015).
- RAPID: Regression-Based Accurate Prediction of Protein Intrinsic Disorder Content. Available online: http://biomine.ece.ualberta.ca/RAPID/ (accessed on 7 September 2015).
- PON-Diso. Available online: http://structure.bmc.lu.se/PON-Diso/ (accessed on 7 September 2015).
- Disorder Prediction Meta-Server. Available online: http://www-nmr.cabm.rutgers.edu/bioinformatics/disorder/ (accessed on 7 September 2015).
- DisPredict. Available online: http://cs.uno.edu/~tamjid/Software.html (accessed on 7 September 2015).
- UCL Department of Computer Science, The PSIPRED Protein Sequence Analysis Workbench. Available online: http://bioinf.cs.ucl.ac.uk/disopred (accessed on 7 September 2015).
- Walsh, I.; Martin, A.J.; di Domenico, T.; Tosatto, S.C. ESpritz: Accurate and fast prediction of protein disorder. Bioinformatics 2012, 28, 503–509. [Google Scholar] [CrossRef] [PubMed]
- Baldi, P.; Brunak, S.; Frasconi, P.; Soda, G.; Pollastri, G. Exploiting the past and the future in protein secondary structure prediction. Bioinformatics 1999, 15, 937–946. [Google Scholar] [CrossRef] [PubMed]
- Sickmeier, M.; Hamilton, J.A.; LeGall, T.; Vacic, V.; Cortese, M.S.; Tantos, A.; Szabo, B.; Tompa, P.; Chen, J.; Uversky, V.N. DisProt: the database of disordered proteins. Nucleic Acids Res. 2007, 35, 786–793. [Google Scholar] [CrossRef] [PubMed]
- Walsh, I.; Martin, A.J.M.; di Domenico, T.; Vullo, A.; Pollastri, G.; Tosatto, S.C.E. CSpritz: Accurate prediction of protein disorder segments with annotation for homology, secondary structure and linear motifs. Nucleic Acids Res. 2011, 39, 190–196. [Google Scholar] [CrossRef] [PubMed]
- Gibson, T.J. Cell regulation: Determined to signal discrete cooperation. Trends Biochem. Sci. 2009, 34, 471–482. [Google Scholar] [CrossRef] [PubMed]
- Diella, F.; Haslam, N.; Chica, C.; Budd, A.; Michael, S.; Brown, N.P.; Travé, G.; Gibson, T.J. Understanding eukaryotic linear motifs and their role in cell signaling and regulation. Front Biosci. 2008, 13, 6580–6603. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Faraggi, E.; Xue, B.; Dunker, A.K.; Uversky, V.N.; Zhou, Y. SPINE-D: Accurate Prediction of Short and Long Disordered Regions by a Single Neural-Network Based Method. J. Biomol. Struct. Dyn. 2012, 29, 799–813. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Faraggi, E.; Zhou, Y. Fluctuations of backbone torsion angles obtained from NMR-determined structures and their prediction. Protein 2010, 78, 3353–3362. [Google Scholar] [CrossRef] [PubMed]
- Monastyrskyy, B.; Fidelis, K.; Moult, J.; Tramontano, A.; Kryshtafovych, A. Evaluation of disorder predictions in CASP9. Proteins 2011, 79, 107–118. [Google Scholar] [CrossRef] [PubMed]
- Eickholt, J.; Cheng, J. DNdisorder: Predicting protein disorder using boosting and deep networks. BMC Bioinform. 2013, 14. [Google Scholar] [CrossRef] [PubMed]
- Hinton, G. A practical guide to training restricted Boltzmann machines. Momentum 2010, 9, 926. [Google Scholar]
- Medina, M.W.; Gao, F.; Naidoo, D.; Rudel, L.L.; Temel, R.E.; McDaniel, A.L.; Marshall, S.M.; Krauss, R.M. Coordinately regulated alternative splicing of genes involved in cholesterol biosynthesis and uptake. PLoS ONE 2011, 6, 604–607. [Google Scholar] [CrossRef] [PubMed]
- Deng, X.; Eickholt, J.; Cheng, J. PreDisorder: Ab initio sequence-based prediction of protein disordered regions. BMC Bioinform. 2009, 10. [Google Scholar] [CrossRef] [PubMed]
- Monastyrskyy, B.; Kryshtafovych, A.; Moult, J.; Tramontano, A.; Fidelis, K. Assessment of protein disorder region predictions in CASP10. Proteins 2014, 82, 127–137. [Google Scholar] [CrossRef] [PubMed]
- Iqbal, S.; Hoque, M.T. DisPredict: A Predictor of Disordered Protein from Sequence using RBF Kernel. Tech. Rep. 2014, 5, 211–218. [Google Scholar]
- Mizianty, M.J.; Stach, W.; Chen, K.; Kedarisetti, K.D.; Disfani, F.M.; Kurgan, L. Improved sequence-based prediction of disordered regions with multilayer fusion of multiple information sources. Bioinformatics 2010, 26, 489–496. [Google Scholar] [CrossRef]
- Yan, J.; Mizianty, M.J.; Filipow, P.L.; Uversky, V.N.; Kurgan, L. RAPID: Fast and accurate sequence-based prediction of intrinsic disorder content on proteomic scale. BBA Proteins Proteom. 2013, 1834, 1671–1680. [Google Scholar] [CrossRef] [PubMed]
- Mizianty, M.J.; Zhang, T.; Xue, B.; Zhou, Y.; Dunker, A.K.; Uversky, V.N.; Kurgan, L. In-silico prediction of disorder content using hybrid sequence representation. BMC Bioinform. 2011, 12. [Google Scholar] [CrossRef] [PubMed]
- Ward, J.J.; Sodhi, J.S.; McGuffin, L.J.; Buxton, B.F.; Jones, D.T. Prediction and functional analysis of native disorder in proteins from the three kingdoms of life. J. Mol. Biol. 2004, 337, 635–645. [Google Scholar] [CrossRef] [PubMed]
- Ward, J.J.; McGuffin, L.J.; Bryson, K.; Buxton, B.F.; Jones, D.T. The DISOPRED server for the prediction of protein disorder. Bioinformatics 2004, 20, 2138–2139. [Google Scholar] [CrossRef] [PubMed]
- Jones, D.T.; Cozzetto, D. DISOPRED3: Precise disordered region predictions with annotated protein-binding activity. Bioinformatics 2015, 31, 857–863. [Google Scholar] [CrossRef] [PubMed]
- Ali, H.; Urolagin, S.; Gurarslan, O.; Vihinen, M. Performance of Protein Disorder Prediction Programs on Amino Acid Substitutions. Hum. Mutat. 2014, 35, 794–804. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Madden, T.L.; Schaffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef] [PubMed]
- Breiman, L. Random forests. Mach. Learn. 2001, 45, 5–32. [Google Scholar] [CrossRef]
- Kawashima, S.; Kanehisa, M. AAindex: Amino acid index database. Nucleic Acids Res. 2000, 28, 374–374. [Google Scholar] [CrossRef] [PubMed]
- Romero, P.; Obradovic, Z.; Dunker, A.K. Sequence Data Analysis for Long Disordered Regions Prediction in the Calcineurin Family. Genome Inform. 1997, 8, 110–124. [Google Scholar]
- Xue, B.; Dunbrack, R.L.; Williams, R.W.; Dunker, A.K.; Uversky, V.N. PONDR-FIT: A meta-predictor of intrinsically disordered amino acids. Biochim. Biophys. Acta 2010, 1804, 996–1010. [Google Scholar] [CrossRef] [PubMed]
- Radivojac, P.; Obradović, Z.; Brown, C.J.; Dunker, A.K. Prediction of boundaries between intrinsically ordered and disordered protein regions. Pac. Symp. Biocomput. 2003, 8, 216–227. [Google Scholar]
- Romero, P.; Obradovic, Z.; Li, X.; Garner, E.C.; Brown, C.J.; Dunker, A.K. Sequence complexity of disordered protein. Proteins 2001, 42, 38–48. [Google Scholar] [CrossRef]
- Prilusky, J.; Felder, C.E.; Zeev-Ben-Mordehai, T.; Rydberg, E.H.; Man, O.; Beckmann, J.S.; Silman, I.; Sussman, J.L. FoldIndex©: A simple tool to predict whether a given protein sequence is intrinsically unfolded. Bioinformatics 2005, 21, 3435–3438. [Google Scholar] [CrossRef] [PubMed]
- Dosztányi, Z.; Csizmok, V.; Tompa, P.; Simon, I. IUPred: Web server for the prediction of intrinsically unstructured regions of proteins based on estimated energy content. Bioinformatics 2005, 21, 3433–3434. [Google Scholar] [CrossRef]
- Campen, A.; Williams, R.M.; Brown, C.J.; Meng, J.; Uversky, V.N.; Dunker, A.K. TOP-IDP-Scale: A New Amino Acid Scale Measuring Propensity for Intrinsic Disorder. Protein Pept. Lett. 2008, 15, 956–963. [Google Scholar] [CrossRef] [PubMed]
- Hirose, S.; Shimizu, K.; Noguchi, T. POODLE-I: Disordered Region Prediction by Integrating POODLE Series and Structural Information Predictors Based on a Workflow Approach. Silico Biol. 2011, 10, 185–191. [Google Scholar]
- Kozlowski, L.P.; Bujnicki, J.M. MetaDisorder: A meta-server for the prediction of intrinsic disorder in proteins. BMC Bioinform. 2012, 13. [Google Scholar] [CrossRef] [PubMed]
- Linding, R.; Jensen, L.J.; Diella, F.; Bork, P.; Gibson, T.J.; Russell, R.B. Protein disorder prediction: Implications for structural proteomics. Structure 2003, 11, 1453–1459. [Google Scholar] [CrossRef] [PubMed]
- Linding, R.; Russell, R.B.; Neduva, V.; Gibson, T.J. GlobPlot: Exploring protein sequences for globularity and disorder. Nucleic Acids Res. 2003, 31, 3701–3708. [Google Scholar] [CrossRef] [PubMed]
- Su, C.-T.; Chen, C.-Y.; Hsu, C.-M. iPDA: Integrated protein disorder analyzer. Nucleic Acids Res. 2007, 35, 465–472. [Google Scholar] [CrossRef] [PubMed]
- Fuxreiter, M.; Tompa, P.; Simon, I. Local structural disorder imparts plasticity on linear motifs. Bioinformatics 2007, 23, 950–956. [Google Scholar] [CrossRef]
- Shimizu, K.; Hirose, S.; Noguchi, T. POODLE-S: Web application for predicting protein disorder by using physicochemical features and reduced amino acid set of a position-specific scoring matrix. Bioinformatics 2007, 23, 2337–2338. [Google Scholar] [CrossRef]
- Hirose, S.; Shimizu, K.; Kanai, S.; Kuroda, Y.; Noguchi, T. POODLE-L: A two-level SVM prediction system for reliably predicting long disordered regions. Bioinformatics 2007, 23, 2046–2053. [Google Scholar] [CrossRef] [PubMed]
- Ishida, T.; Kinoshita, K. PrDOS: Prediction of disordered protein regions from amino acid sequence. Nucleic Acids Res. 2007, 35, 460–464. [Google Scholar] [CrossRef] [PubMed]
- Vullo, A.; Bortolami, O.; Pollastri, G.; Tosatto, S.C.E. Spritz: A server for the prediction of intrinsically disordered regions in protein sequences using kernel machines. Nucleic Acids Res. 2006, 34, 164–168. [Google Scholar] [CrossRef] [PubMed]
- Su, C.-T.; Chen, C.-Y.; Ou, Y.-Y. Protein disorder prediction by condensed PSSM considering propensity for order or disorder. BMC Bioinform. 2006, 7. [Google Scholar] [CrossRef]
- Yang, Z.R.; Thomson, R.; McNeil, P.; Esnouf, R.M. RONN: The bio-basis function neural network technique applied to the detection of natively disordered regions in proteins. Bioinformatics 2005, 21, 3369–3376. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.J.; Acton, T.B.; Montelione, G.T. DisMeta: A meta server for construct design and optimization. In Structural Genomics, 1st ed.; Chen, Y.W., Ed.; Humana Press: Totowa, NJ, USA, 2014; Volume 1091, pp. 3–16. [Google Scholar]
- Rost, B.; Yachdav, G.; Liu, J.F. The PredictProtein server. Nucleic Acids Res. 2004, 32, 321–326. [Google Scholar] [CrossRef] [PubMed]
- Emanuelsson, O.; Brunak, S.; von Heijne, G.; Nielsen, H. Locating proteins in the cell using TargetP, SignalP and related tools. Nat. Protoc. 2007, 2, 953–971. [Google Scholar] [CrossRef]
- Zhou, H.Y.; Zhou, Y.Q. Predicting the topology of transmembrane helical proteins using mean burial propensity and a hidden-Markov-model-based method. Protein Sci. 2003, 12, 1547–1555. [Google Scholar] [CrossRef] [PubMed]
- Wootton, J.C.; Federhen, S. Analysis of compositionally biased regions in sequence databases. Methods Enzymol. 1996, 266, 554–571. [Google Scholar] [PubMed]
- McGuffin, L.J. Intrinsic disorder prediction from the analysis of multiple protein fold recognition models. Bioinformatics 2008, 24, 1798–1804. [Google Scholar] [CrossRef] [PubMed]
- Mizianty, M.J.; Peng, Z.; Kurgan, L. MFDp2: Accurate predictor of disorder in proteins by fusion of disorder probabilities, content and profiles. IPDs 2013, 1, e24428. [Google Scholar] [CrossRef]
- Galzitskaya, O.; Garbuzynskiy, S.; Lobanov, M.Y. Prediction of natively unfolded regions in protein chains. Mol. Biol. 2006, 40, 298–304. [Google Scholar] [CrossRef]
- Lobanov, M.Y.; Sokolovskiy, I.V.; Galzitskaya, O.V. IsUnstruct: Prediction of the residue status to be ordered or disordered in the protein chain by a method based on the Ising model. J. Biomol. Struct. Dyn. 2013, 31, 1034–1043. [Google Scholar] [CrossRef] [PubMed]
- Dosztanyi, Z.; Csizmok, V.; Tompa, P.; Simon, I. The pairwise energy content estimated from amino acid composition discriminates between folded and intrinsically unstructured proteins. J. Mol. Biol. 2005, 347, 827–839. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N. Natively unfolded proteins: A point where biology waits for physics. Protein Sci. 2002, 11, 739–756. [Google Scholar] [CrossRef] [PubMed]
- Peng, Z.-L.; Kurgan, L. Comprehensive comparative assessment of in-silico predictors of disordered regions. Curr. Protein Pept. Sci. 2012, 13, 6–18. [Google Scholar] [CrossRef] [PubMed]
- Walsh, I.; Giollo, M.; di Domenico, T.; Ferrari, C.; Zimmermann, O.; Tosatto, S.C. Comprehensive large-scale assessment of intrinsic protein disorder. Bioinformatics 2015, 31, 201–208. [Google Scholar] [CrossRef] [PubMed]
- Dosztányi, Z.; Mészáros, B.; Simon, I. Bioinformatical approaches to characterize intrinsically disordered/unstructured proteins. Brief. Bioinform. 2010, 11, 225–243. [Google Scholar] [CrossRef] [PubMed]
- Obradovic, Z.; Peng, K.; Vucetic, S.; Radivojac, P.; Dunker, A.K. Exploiting heterogeneous sequence properties improves prediction of protein disorder. Proteins 2005, 61, 176–182. [Google Scholar] [CrossRef] [PubMed]
- Kissinger, C.R.; Parge, H.E.; Knighton, D.R.; Lewis, C.T.; Pelletier, L.A.; Tempczyk, A.; Kalish, V.J.; Tucker, K.D.; Showalter, R.E.; Moomaw, E.W.; et al. Crystal-structures of human calcineurin and the human FKBP12-FK506-calcineurin complex. Nature 1995, 378, 641–644. [Google Scholar] [CrossRef]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, J.; Feng, Y.; Wang, X.; Li, J.; Liu, W.; Rong, L.; Bao, J. An Overview of Predictors for Intrinsically Disordered Proteins over 2010–2014. Int. J. Mol. Sci. 2015, 16, 23446-23462. https://doi.org/10.3390/ijms161023446
Li J, Feng Y, Wang X, Li J, Liu W, Rong L, Bao J. An Overview of Predictors for Intrinsically Disordered Proteins over 2010–2014. International Journal of Molecular Sciences. 2015; 16(10):23446-23462. https://doi.org/10.3390/ijms161023446
Chicago/Turabian StyleLi, Jianzong, Yu Feng, Xiaoyun Wang, Jing Li, Wen Liu, Li Rong, and Jinku Bao. 2015. "An Overview of Predictors for Intrinsically Disordered Proteins over 2010–2014" International Journal of Molecular Sciences 16, no. 10: 23446-23462. https://doi.org/10.3390/ijms161023446
APA StyleLi, J., Feng, Y., Wang, X., Li, J., Liu, W., Rong, L., & Bao, J. (2015). An Overview of Predictors for Intrinsically Disordered Proteins over 2010–2014. International Journal of Molecular Sciences, 16(10), 23446-23462. https://doi.org/10.3390/ijms161023446