
Cytokine-Modulating Strategies and Newer Cytokine Targets for Arthritis Therapy

Abstract
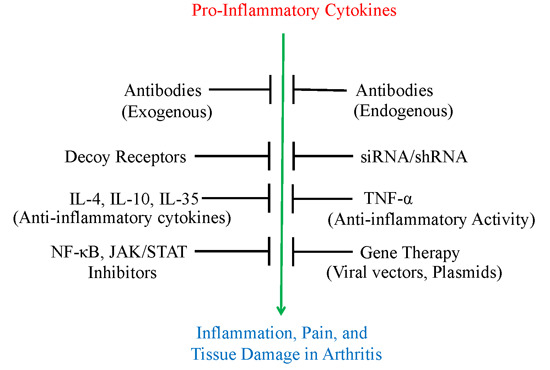
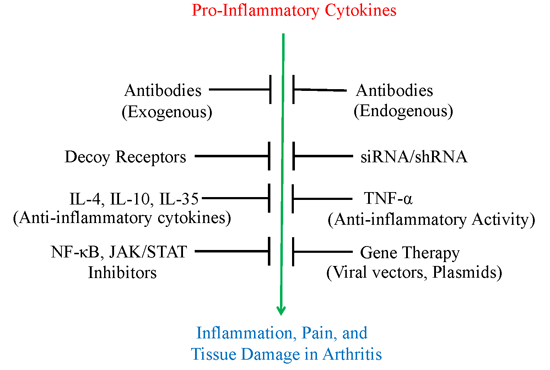
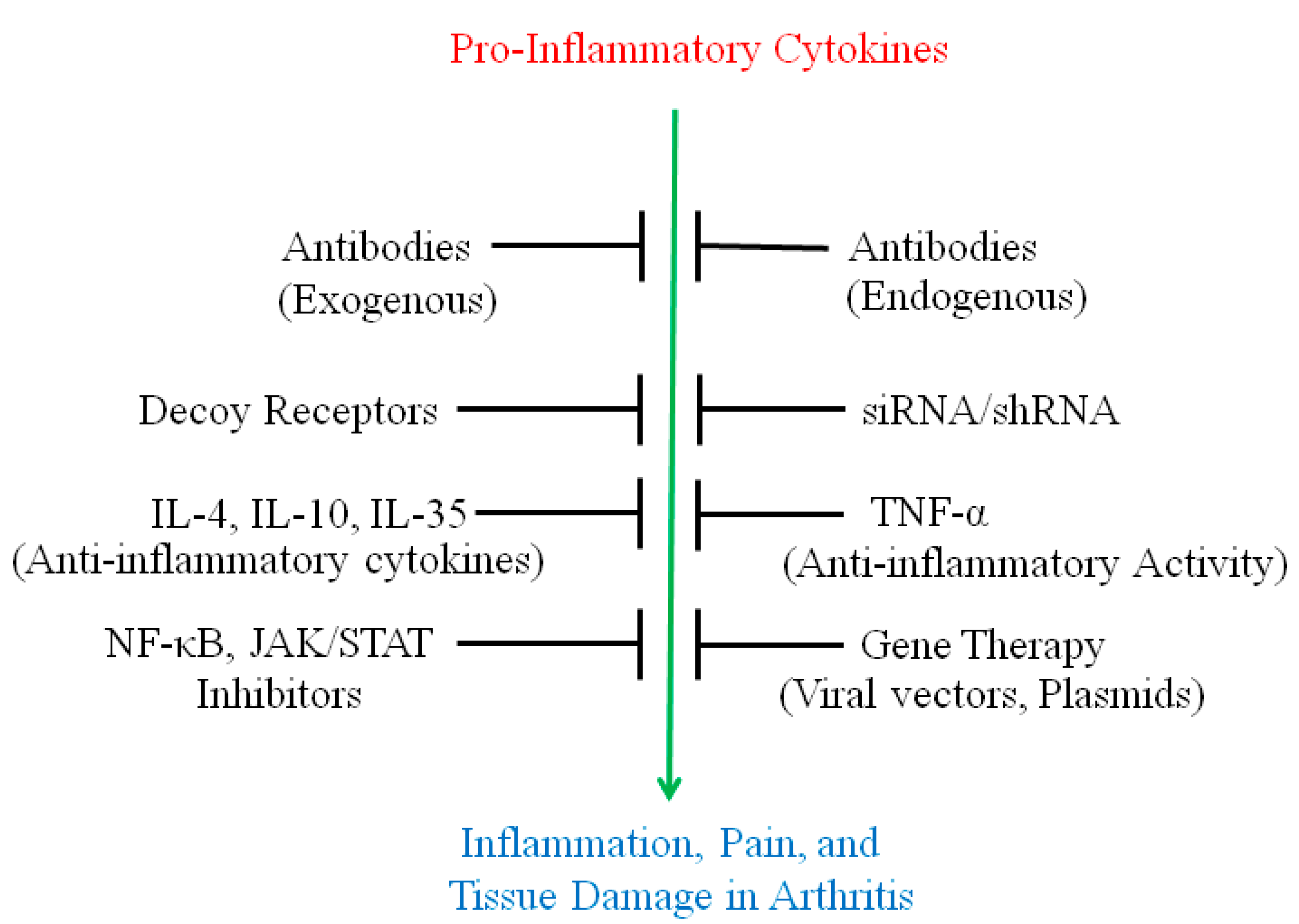
:
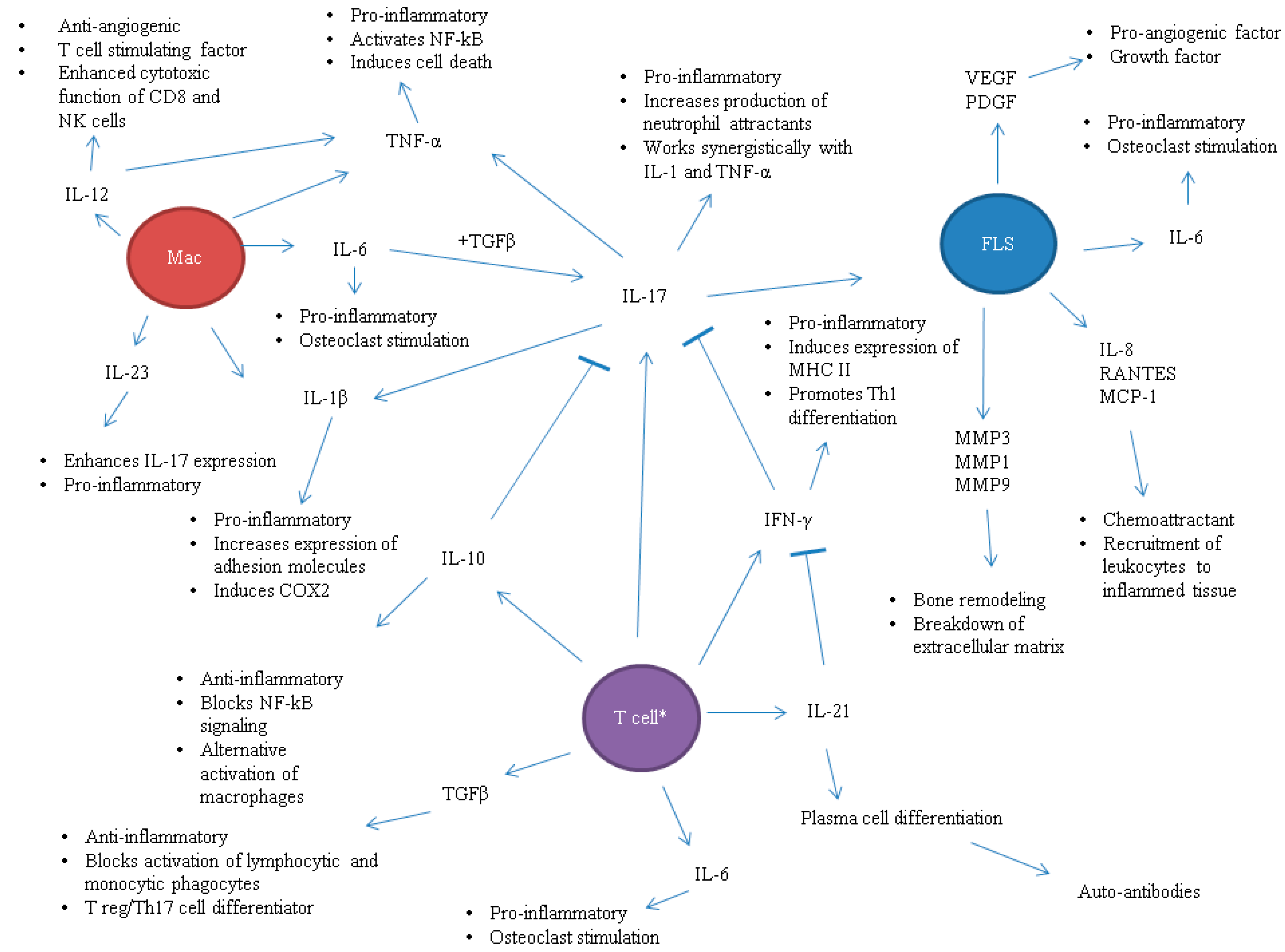
1. Introduction


{kind=link}
{kind=link}
{kind=link}
| Target | Agent | Type | Mechanism of Action | Ref. |
|---|---|---|---|---|
| IL-1 | Anakinra a | Recombinant IL-1Rα | Prevents binding of IL-1β to IL-1Rα | [19] |
| Anti-IL-1β c | Human IgG1 monoclonal antibody (mAb) | Binds tightly to IL-1β and neutralizes it | [20] | |
| Canakinumab b | Human IgG1 mAb | Neutralizes the activity of IL-1β | [21] | |
| Gevokizumab b | Humanized IgG2 mAb | Neutralizes the activity of IL-1β | [22] | |
| LY2189102 b | Humanized IgG4 mAb | Neutralizes the activity of IL-1β | [23] | |
| Rilonacept b | Ligand-binding domain of IL-1RI and human IgG1 fusion protein | Attaches to and neutralizes circulating IL-1β before it can bind its receptor | [24] | |
| IL-6 | MAB406 c | mAb | Blocks IL-6 signaling | [ 25,26] |
| MRA b | Humanized mAb | Inhibits IL-6 signaling | [27] | |
| Tocilizumab a | Humanized mAb | Binds to IL-6Rα chain and blocks IL-6 signaling | [28,29] | |
| TNFα | Infliximab a | Recombinant IgG1 mAb | Binds to TNFα and prevents it from binding to its receptor | [30,31] |
| Adalimumab a | Recombinant IgG1 mAb | Binds to TNFα and prevents it from activating TNF receptors | [32] | |
| Etanercept a | Extracellular domain of TNF receptor II (p75) and the Fc portion of IgG1 fusion protein | Functions as a decoy receptor to TNFα | [33] | |
| Golimumab b | Human mAb | Neutralizes TNFα bioactivity | [34] | |
| IL-17 | Secukinumab b | Human IgG1k mAb | Selectively binds and neutralizes IL-17A | [35,36] |
| Ixekizumab b | Humanized IgG4 mAb | Neutralizes IL-17A | [37] | |
| Brodalumab b | Human anti-IL17RA mAb | Inhibits the activity of IL-17 | [38] | |
| IL-12/IL-23 | Ustekinumab b | Human IgG1κ mAb | Blocks the biologic activity of IL-12 and IL-23 through their common p40 subunit by inhibiting their receptors | [39] |
| Guselkumab b | Human mAb | An IL-23p19-targeted mAb | [40] |
| Target Cytokine | Vector | Mode of Gene Transfer | Model | Ref. |
|---|---|---|---|---|
| IL-1β | rAAV-IL-1Ra | In vivo | Mouse | [43] |
| TNF-α | sTNFR plasmid electrotransfer | In vivo | Mouse | [46,47] |
| IFN-β | Adenovirus vector | Intra-articular | Rat | [49] |
| IL-4 | Adenoviral vector | In vivo | Mouse | [50] |
| IL-10 | Lentivirus using inflammation promoter switches Saa3 and Mmp13 | Intra-articular | Mouse | [51] |
| IL-18 | Adenoviral vector | In vivo | Mouse | [52] |
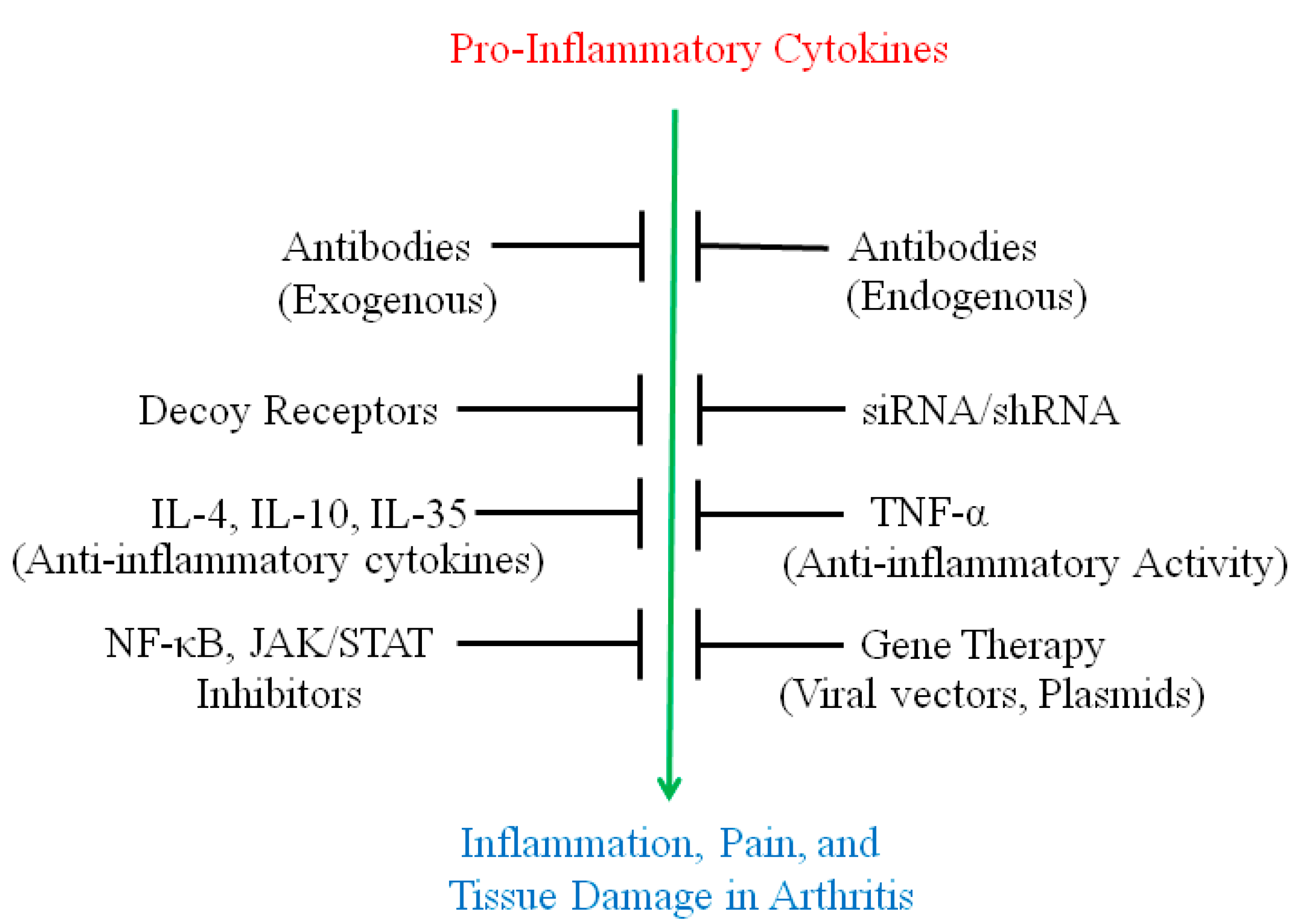
2. Use of Antibodies and Decoy Receptors against Pro-Inflammatory Cytokines/Cytokine Receptors for the Treatment of Arthritis
3. The Use of Small Molecule Inhibitors of Cytokine Production for Controlling Inflammation
4. Gene Therapy for Modulating Cytokine Response to Control Arthritis
4.1. IL-1β
4.2. TNFα
4.3. IL-18
4.4. IFN-β
4.5. IL-4 and IL-10
4.6. Small Interfering RNA (SiRNA)/Short Hairpin RNA (ShRNA)-Induced Cytokine Modulation for Arthritis Therapy
5. The Relatively Newer Cytokines (IL-32, IL-34 and IL-35) and Their Roles in Inflammation and Autoimmune Arthritis
5.1. IL-32
5.2. IL-34
5.3. IL-35
6. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Astry, B.; Harberts, E.; Moudgil, K.D. A cytokine-centric view of the pathogenesis and treatment of autoimmune arthritis. J. Interferon Cytokine Res. 2011, 31, 927–940. [Google Scholar] [CrossRef] [PubMed]
- Clavel, G.; Thiolat, A.; Boissier, M.C. Interleukin newcomers creating new numbers in rheumatology: IL-34 to IL-38. Jt. Bone Spine 2013, 80, 449–453. [Google Scholar] [CrossRef]
- Leng, R.X.; Pan, H.F.; Tao, J.H.; Ye, D.Q. IL-19, IL-20 and IL-24: Potential therapeutic targets for autoimmune diseases. Expert Opin. Ther. Targets 2011, 15, 119–126. [Google Scholar] [CrossRef] [PubMed]
- Garlanda, C.; Dinarello, C.A.; Mantovani, A. The interleukin-1 family: Back to the future. Immunity 2013, 39, 1003–1018. [Google Scholar] [CrossRef] [PubMed]
- Gaffen, S.L.; Jain, R.; Garg, A.V.; Cua, D.J. The IL-23–IL-17 immune axis: From mechanisms to therapeutic testing. Nat. Rev. Immunol. 2014, 14, 585–600. [Google Scholar] [CrossRef] [PubMed]
- Tang, C.; Chen, S.; Qian, H.; Huang, W. Interleukin-23: As a drug target for autoimmune inflammatory diseases. Immunology 2012, 135, 112–124. [Google Scholar] [CrossRef] [PubMed]
- Bettelli, E.; Korn, T.; Kuchroo, V.K. Th17: The third member of the effector T cell trilogy. Curr. Opin. Immunol. 2007, 19, 652–657. [Google Scholar] [CrossRef] [PubMed]
- Rajaiah, R.; Puttabyatappa, M.; Polumuri, S.K.; Moudgil, K.D. Interleukin-27 and interferon-γ are involved in regulation of autoimmune arthritis. J. Biol. Chem. 2011, 286, 2817–2825. [Google Scholar] [CrossRef] [PubMed]
- Adamopoulos, I.E.; Pflanz, S. The emerging role of interleukin 27 in inflammatory arthritis and bone destruction. Cytokine Growth Factor Rev. 2013, 24, 115–121. [Google Scholar] [CrossRef] [PubMed]
- Clark, I.A. How TNF was recognized as a key mechanism of disease. Cytokine Growth Factor Rev. 2007, 18, 335–343. [Google Scholar] [CrossRef] [PubMed]
- Walsh, L.J.; Trinchieri, G.; Waldorf, H.A.; Whitaker, D.; Murphy, G.F. Human dermal mast cells contain and release tumor necrosis factor alpha, which induces endothelial leukocyte adhesion molecule 1. Proc. Natl. Acad. Sci. USA 1991, 88, 4220–4224. [Google Scholar] [CrossRef] [PubMed]
- Wajant, H.; Pfizenmaier, K.; Scheurich, P. Tumor necrosis factor signaling. Cell Death Differ. 2003, 10, 45–65. [Google Scholar] [CrossRef] [PubMed]
- Locksley, R.M.; Killeen, N.; Lenardo, M.J. The TNF and TNF receptor superfamilies: Integrating mammalian biology. Cell 2001, 104, 487–501. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Goeddel, D.V. TNF-R1 signaling: A beautiful pathway. Science 2002, 296, 1634–1635. [Google Scholar] [CrossRef] [PubMed]
- Kant, S.; Swat, W.; Zhang, S.; Zhang, Z.-Y.; Neel, B.G.; Flavell, R.A.; Davis, R.J. TNF-stimulated MAP kinase activation mediated by a RHO family gtpase signaling pathway. Genes Dev. 2011, 25, 2069–2078. [Google Scholar] [CrossRef] [PubMed]
- Hot, A.; Miossec, P. Effects of interleukin (IL)-17a and IL-17F in human rheumatoid arthritis synoviocytes. Ann. Rheum. Dis. 2011, 70, 727–732. [Google Scholar] [CrossRef] [PubMed]
- Shabgah, A.G.; Fattahi, E.; Shahneh, F.Z. Interleukin-17 in human inflammatory diseases. Postep. Dermatol. Alergol. 2014, 31, 256–261. [Google Scholar] [CrossRef]
- Sarkar, S.; Justa, S.; Brucks, M.; Endres, J.; Fox, D.A.; Zhou, X.; Alnaimat, F.; Whitaker, B.; Wheeler, J.C.; Jones, B.H.; et al. Interleukin (IL)-17a, f and af in inflammation: A study in collagen-induced arthritis and rheumatoid arthritis. Clin. Exp. Immunol. 2014, 177, 652–661. [Google Scholar] [CrossRef] [PubMed]
- Mertens, M.; Singh, J.A. Anakinra for rheumatoid arthritis: A systematic review. J. Rheumatol. 2009, 36, 1118–1125. [Google Scholar] [CrossRef] [PubMed]
- Goh, A.X.; Bertin-Maghit, S.; Ping Yeo, S.; Ho, A.W.; Derks, H.; Mortellaro, A.; Wang, C.I. A novel human anti-interleukin-1beta neutralizing monoclonal antibody showing in vivo efficacy. MAbs 2014, 6, 765–773. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, A.; Tannenbaum, S.; Rordorf, C.; Lowe, P.J.; Floch, D.; Gram, H.; Roy, S. Pharmacokinetic and pharmacodynamic properties of canakinumab, a human anti-interleukin-1β monoclonal antibody. Clin. Pharmacokinet. 2012, 51, e1–e18. [Google Scholar] [CrossRef] [PubMed]
- Geiler, J.; McDermott, M.F. Gevokizumab, an anti-IL-1β MAB for the potential treatment of type 1 and 2 diabetes, rheumatoid arthritis and cardiovascular disease. Curr. Opin. Mol. Ther. 2010, 12, 755–769. [Google Scholar] [PubMed]
- Bihorel, S.; Fiedler-Kelly, J.; Ludwig, E.; Sloan-Lancaster, J.; Raddad, E. Population pharmacokinetic modeling of ly2189102 after multiple intravenous and subcutaneous administrations. AAPS J. 2014, 16, 1009–1017. [Google Scholar] [CrossRef] [PubMed]
- McDermott, M.F. Rilonacept in the treatment of chronic inflammatory disorders. Drugs Today 2009, 45, 423–430. [Google Scholar] [CrossRef] [PubMed]
- Liang, B.; Gardner, D.B.; Griswold, D.E.; Bugelski, P.J.; Song, X.Y. Anti-interleukin-6 monoclonal antibody inhibits autoimmune responses in a murine model of systemic lupus erythematosus. Immunology 2006, 119, 296–305. [Google Scholar] [CrossRef] [PubMed]
- LaSpina, M.; Tripathi, S.; Gatto, L.A.; Bruch, D.; Maier, K.G.; Kittur, D.S. An interleukin-6-neutralizing antibody prevents cyclosporine-induced nephrotoxicity in mice. J. Surg. Res. 2008, 148, 121–125. [Google Scholar] [CrossRef] [PubMed]
- Straub, R.H.; Harle, P.; Yamana, S.; Matsuda, T.; Takasugi, K.; Kishimoto, T.; Nishimoto, N. Anti-interleukin-6 receptor antibody therapy favors adrenal androgen secretion in patients with rheumatoid arthritis: A randomized, double-blind, placebo-controlled study. Arthritis Rheumatol. 2006, 54, 1778–1785. [Google Scholar] [CrossRef]
- Semerano, L.; Thiolat, A.; Minichiello, E.; Clavel, G.; Bessis, N.; Boissier, M.C. Targeting IL-6 for the treatment of rheumatoid arthritis: Phase ii investigational drugs. Expert Opin. Investig. Drugs 2014, 23, 979–999. [Google Scholar] [CrossRef] [PubMed]
- Nishimoto, N.; Kishimoto, T.; Yoshizaki, K. Anti-interleukin 6 receptor antibody treatment in rheumatic disease. Ann. Rheum. Dis. 2000, 59 (Suppl. S1), i21–i27. [Google Scholar] [CrossRef]
- Lou, H.; Zong, Y.; Ge, Y.R.; Cheng, J.W.; Wei, R.L. Efficacy and tolerability of latanoprost compared with timolol in the treatment of patients with chronic angle-closure glaucoma. Curr. Med. Res. Opin. 2014, 30, 1367–1373. [Google Scholar] [CrossRef] [PubMed]
- St Clair, E.W.; van der Heijde, D.M.; Smolen, J.S.; Maini, R.N.; Bathon, J.M.; Emery, P.; Keystone, E.; Schiff, M.; Kalden, J.R.; Wang, B.; et al. Combination of infliximab and methotrexate therapy for early rheumatoid arthritis: A randomized, controlled trial. Arthritis Rheumatol. 2004, 50, 3432–3443. [Google Scholar] [CrossRef]
- Wiens, A.; Correr, C.J.; Venson, R.; Otuki, M.F.; Pontarolo, R. A systematic review and meta-analysis of the efficacy and safety of adalimumab for treating rheumatoid arthritis. Rheumatol. Int. 2010, 30, 1063–1070. [Google Scholar] [CrossRef] [PubMed]
- Moreland, L.W.; Baumgartner, S.W.; Schiff, M.H.; Tindall, E.A.; Fleischmann, R.M.; Weaver, A.L.; Ettlinger, R.E.; Cohen, S.; Koopman, W.J.; Mohler, K.; et al. Treatment of rheumatoid arthritis with a recombinant human tumor necrosis factor receptor (p75)-fc fusion protein. N. Engl. J. Med. 1997, 337, 141–147. [Google Scholar] [CrossRef] [PubMed]
- Keystone, E.C.; Genovese, M.C.; Klareskog, L.; Hsia, E.C.; Hall, S.T.; Miranda, P.C.; Pazdur, J.; Bae, S.C.; Palmer, W.; Zrubek, J.; et al. Golimumab, a human antibody to tumour necrosis factor α given by monthly subcutaneous injections, in active rheumatoid arthritis despite methotrexate therapy: The GO-FORWARD study. Ann. Rheum. Dis. 2009, 68, 789–796. [Google Scholar] [CrossRef] [PubMed]
- Langley, R.G.; Elewski, B.E.; Lebwohl, M.; Reich, K.; Griffiths, C.E.; Papp, K.; Puig, L.; Nakagawa, H.; Spelman, L.; Sigurgeirsson, B.; et al. Secukinumab in plaque psoriasis—Results of two phase 3 trials. N. Engl. J. Med. 2014, 371, 326–338. [Google Scholar] [CrossRef] [PubMed]
- Patel, D.D.; Lee, D.M.; Kolbinger, F.; Antoni, C. Effect of IL-17a blockade with secukinumab in autoimmune diseases. Ann. Rheum. Dis. 2013, 72 (Suppl. S2), ii116–ii123. [Google Scholar] [CrossRef]
- Leonardi, C.; Matheson, R.; Zachariae, C.; Cameron, G.; Li, L.; Edson-Heredia, E.; Braun, D.; Banerjee, S. Anti-interleukin-17 monoclonal antibody ixekizumab in chronic plaque psoriasis. N. Engl. J. Med. 2012, 366, 1190–1199. [Google Scholar] [CrossRef] [PubMed]
- Mease, P.J.; Genovese, M.C.; Greenwald, M.W.; Ritchlin, C.T.; Beaulieu, A.D.; Deodhar, A.; Newmark, R.; Feng, J.; Erondu, N.; Nirula, A. Brodalumab, an anti-IL17RA monoclonal antibody, in psoriatic arthritis. N. Engl. J. Med. 2014, 370, 2295–2306. [Google Scholar] [CrossRef]
- Sandborn, W.J.; Gasink, C.; Gao, L.L.; Blank, M.A.; Johanns, J.; Guzzo, C.; Sands, B.E.; Hanauer, S.B.; Targan, S.; Rutgeerts, P.; et al. Ustekinumab induction and maintenance therapy in refractory crohn’s disease. N. Engl. J. Med. 2012, 367, 1519–1528. [Google Scholar] [CrossRef] [PubMed]
- Sofen, H.; Smith, S.; Matheson, R.T.; Leonardi, C.L.; Calderon, C.; Brodmerkel, C.; Li, K.; Campbell, K.; Marciniak, S.J., Jr.; Wasfi, Y.; et al. Guselkumab (an IL-23-specific mab) demonstrates clinical and molecular response in patients with moderate-to-severe psoriasis. J. Allergy Clin. Immunol. 2014, 133, 1032–1040. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, Y.; Tanaka, T. Interleukin 6 and rheumatoid arthritis. BioMed Res. Int. 2014, 2014. [Google Scholar] [CrossRef] [PubMed]
- Calabrese, L.H.; Rose-John, S. IL-6 biology: Implications for clinical targeting in rheumatic disease. Nat. Rev. 2014, 10, 720–727. [Google Scholar]
- Pan, R.Y.; Chen, S.L.; Xiao, X.; Liu, D.W.; Peng, H.J.; Tsao, Y.P. Therapy and prevention of arthritis by recombinant adeno-associated virus vector with delivery of interleukin-1 receptor antagonist. Arthritis Rheumatol. 2000, 43, 289–297. [Google Scholar] [CrossRef]
- Evans, C.H.; Ghivizzani, S.C.; Herndon, J.H.; Wasko, M.C.; Reinecke, J.; Wehling, P.; Robbins, P.D. Clinical trials in the gene therapy of arthritis. Clin. Orthop. Relat. Res. 2000, 379, S300–S307. [Google Scholar] [CrossRef]
- Wehling, P.; Reinecke, J.; Baltzer, A.W.; Granrath, M.; Schulitz, K.P.; Schultz, C.; Krauspe, R.; Whiteside, T.W.; Elder, E.; Ghivizzani, S.C.; et al. Clinical responses to gene therapy in joints of two subjects with rheumatoid arthritis. Hum. Gene Ther. 2009, 20, 97–101. [Google Scholar] [CrossRef] [PubMed]
- Bloquel, C.; Bessis, N.; Boissier, M.C.; Scherman, D.; Bigey, P. Gene therapy of collagen-induced arthritis by electrotransfer of human tumor necrosis factor-α soluble receptor I variants. Hum. Gene Ther. 2004, 15, 189–201. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.M.; Ho, S.H.; Hahn, W.; Jeong, J.G.; Park, E.J.; Lee, H.J.; Yu, S.S.; Lee, C.S.; Lee, Y.W.; Kim, S. Electro-gene therapy of collagen-induced arthritis by using an expression plasmid for the soluble p75 tumor necrosis factor receptor-Fc fusion protein. Gene Ther. 2003, 10, 1216–1224. [Google Scholar] [CrossRef] [PubMed]
- Mease, P.J.; Hobbs, K.; Chalmers, A.; El-Gabalawy, H.; Bookman, A.; Keystone, E.; Furst, D.E.; Anklesaria, P.; Heald, A.E. Local delivery of a recombinant adenoassociated vector containing a tumour necrosis factor alpha antagonist gene in inflammatory arthritis: A phase 1 dose-escalation safety and tolerability study. Ann. Rheum. Dis. 2009, 68, 1247–1254. [Google Scholar] [CrossRef] [PubMed]
- Adriaansen, J.; Kuhlman, R.R.; van Holten, J.; Kaynor, C.; Vervoordeldonk, M.J.; Tak, P.P. Intraarticular interferon-beta gene therapy ameliorates adjuvant arthritis in rats. Hum. Gene Ther. 2006, 17, 985–996. [Google Scholar] [CrossRef] [PubMed]
- Woods, J.M.; Tokuhira, M.; Berry, J.C.; Katschke, K.J., Jr.; Kurata, H.; Damergis, J.A., Jr.; Arai, K.; Koch, A.E. Interleukin-4 adenoviral gene therapy reduces production of inflammatory cytokines and prostaglandin e2 by rheumatoid arthritis synovium ex vivo. J. Investig. Med. 1999, 47, 285–292. [Google Scholar] [PubMed]
- Vermeij, E.A.; Broeren, M.G.; Bennink, M.B.; Arntz, O.J.; Gjertsson, I.; van Lent P, L.E.M.; van den Berg, W.B.; Koenders, M.I.; van de Loo, F.A. Disease-regulated local IL-10 gene therapy diminishes synovitis and cartilage proteoglycan depletion in experimental arthritis. Ann. Rheum. Dis. 2014. [Google Scholar] [CrossRef]
- Smeets, R.L.; van de Loo, F.A.; Arntz, O.J.; Bennink, M.B.; Joosten, L.A.; van den Berg, W.B. Adenoviral delivery of IL-18 binding protein c ameliorates collagen-induced arthritis in mice. Gene Ther. 2003, 10, 1004–1011. [Google Scholar] [CrossRef] [PubMed]
- Brennan, F.M.; McInnes, I.B. Evidence that cytokines play a role in rheumatoid arthritis. J. Clin. Investig. 2008, 118, 3537–3545. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.P.; Hasan, S.; Sharma, S.; Nagra, S.; Yamaguchi, D.T.; Wong, D.; Bh, H.; Hossain, A. Th17 cells in inflammation and autoimmunity. Autoimmun. Rev. 2014, 13, 1174–1181. [Google Scholar] [CrossRef] [PubMed]
- Shetty, A.; Hanson, R.; Korsten, P.; Shawagfeh, M.; Arami, S.; Volkov, S.; Vila, O.; Swedler, W.; Shunaigat, A.N.; Smadi, S.; et al. Tocilizumab in the treatment of rheumatoid arthritis and beyond. Drug Des. Dev. Ther. 2014, 8, 349–364. [Google Scholar]
- Alghasham, A.; Rasheed, Z. Therapeutic targets for rheumatoid arthritis: Progress and promises. Autoimmunity 2014, 47, 77–94. [Google Scholar] [CrossRef] [PubMed]
- Bongartz, T.; Sutton, A.J.; Sweeting, M.J.; Buchan, I.; Matteson, E.L.; Montori, V. Anti-TNF antibody therapy in rheumatoid arthritis and the risk of serious infections and malignancies: Systematic review and meta-analysis of rare harmful effects in randomized controlled trials. JAMA 2006, 295, 2275–2285. [Google Scholar] [CrossRef]
- Cantini, F.; Niccoli, L.; Goletti, D. Tuberculosis risk in patients treated with non-anti-tumor necrosis factor-alpha (TNF-α) targeted biologics and recently licensed TNF-α inhibitors: Data from clinical trials and national registries. J. Rheumatol. Suppl. 2014, 91, 56–64. [Google Scholar] [CrossRef] [PubMed]
- Choy, E.H.; Kavanaugh, A.F.; Jones, S.A. The problem of choice: Current biologic agents and future prospects in RA. Nat. Rev. Rheumatol. 2013, 9, 154–163. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.Y.; Chi, H.H.; Rajaiah, R.; Moudgil, K.D. Exogenous tumour necrosis factor alpha induces suppression of autoimmune arthritis. Arthritis Res. Ther. 2008, 10, R38. [Google Scholar] [CrossRef] [PubMed]
- Williams, R.O. Paradoxical effects of tumour necrosis factor-α in adjuvant-induced arthritis. Arthritis Res. Ther. 2008, 10, 113. [Google Scholar] [CrossRef] [PubMed]
- Elicabe, R.J.; Cargnelutti, E.; Serer, M.I.; Stege, P.W.; Valdez, S.R.; Toscano, M.A.; Rabinovich, G.A.; Di Genaro, M.S. Lack of TNFR p55 results in heightened expression of IFN-γ and IL-17 during the development of reactive arthritis. J. Immunol. 2010, 185, 4485–4495. [Google Scholar] [CrossRef] [PubMed]
- Masli, S.; Turpie, B. Anti-inflammatory effects of tumour necrosis factor (TNF)-α are mediated via TNF-R2 (p75) in tolerogenic transforming growth factor-β-treated antigen-presenting cells. Immunology 2009, 127, 62–72. [Google Scholar] [CrossRef] [PubMed]
- Zakharova, M.; Ziegler, H.K. Paradoxical anti-inflammatory actions of TNF-α: Inhibition of IL-12 and IL-23 via TNF receptor 1 in macrophages and dendritic cells. J. Immunol. 2005, 175, 5024–5033. [Google Scholar] [CrossRef] [PubMed]
- Zagury, D.; Burny, A.; Gallo, R.C. Toward a new generation of vaccines: The anti-cytokine therapeutic vaccines. Proc. Natl. Acad. Sci. USA 2001, 98, 8024–8029. [Google Scholar] [CrossRef] [PubMed]
- Spohn, G.; Keller, I.; Beck, M.; Grest, P.; Jennings, G.T.; Bachmann, M.F. Active immunization with IL-1 displayed on virus-like particles protects from autoimmune arthritis. Eur. J. Immunol. 2008, 38, 877–887. [Google Scholar] [CrossRef]
- Bertin-Maghit, S.M.; Capini, C.J.; Bessis, N.; Chomilier, J.; Muller, S.; Abbas, A.; Autin, L.; Spadoni, J.L.; Rappaport, J.; Therwath, A.; et al. Improvement of collagen-induced arthritis by active immunization against murine IL-1β peptides designed by molecular modelling. Vaccine 2005, 23, 4228–4235. [Google Scholar] [CrossRef] [PubMed]
- Dalum, I.; Butler, D.M.; Jensen, M.R.; Hindersson, P.; Steinaa, L.; Waterston, A.M.; Grell, S.N.; Feldmann, M.; Elsner, H.I.; Mouritsen, S. Therapeutic antibodies elicited by immunization against TNF-α. Nat. Biotechnol. 1999, 17, 666–669. [Google Scholar] [CrossRef] [PubMed]
- Salomon, I.; Netzer, N.; Wildbaum, G.; Schif-Zuck, S.; Maor, G.; Karin, N. Targeting the function of IFN-γ-inducible protein 10 suppresses ongoing adjuvant arthritis. J. Immunol. 2002, 169, 2685–2693. [Google Scholar] [CrossRef]
- Ratsimandresy, R.A.; Duvallet, E.; Assier, E.; Semerano, L.; Delavallee, L.; Bessis, N.; Zagury, J.F.; Boissier, M.C. Active immunization against IL-23p19 improves experimental arthritis. Vaccine 2011, 29, 9329–9336. [Google Scholar] [CrossRef]
- Hilton, S.W.D.J. Inhibitors of cytokine signal transduction. J. Biol. Chem. 2004, 279, 821–824. [Google Scholar] [CrossRef] [PubMed]
- Gaestel, M.; Mengel, A.; Bothe, U.; Asadullah, K. Protein kinases as small molecule inhibitor targets in inflammation. Curr. Med. Chem. 2007, 14, 2214–2234. [Google Scholar] [CrossRef] [PubMed]
- Milici, A.J.; Kudlacz, E.M.; Audoly, L.; Zwillich, S.; Changelian, P. Cartilage preservation by inhibition of janus kinase 3 in two rodent models of rheumatoid arthritis. Arthritis Res. Ther. 2008, 10, R14. [Google Scholar] [CrossRef] [PubMed]
- Lundquist, L.M.; Cole, S.W.; Sikes, M.L. Efficacy and safety of tofacitinib for treatment of rheumatoid arthritis. World J. Orthop. 2014, 5, 504–511. [Google Scholar] [CrossRef] [PubMed]
- Maiga, M.; Lun, S.; Guo, H.; Winglee, K.; Ammerman, N.C.; Bishai, W.R. Risk of tuberculosis reactivation with tofacitinib (cp-690550). J. Infect. Dis. 2012, 205, 1705–1708. [Google Scholar] [CrossRef] [PubMed]
- Venkatesha, S.H.; Berman, B.M.; Moudgil, K.D. Herbal medicinal products target defined biochemical and molecular mediators of inflammatory autoimmune arthritis. Bioorg. Med. Chem. 2011, 19, 21–29. [Google Scholar] [CrossRef] [PubMed]
- Nanjundaiah, S.M.; Venkatesha, S.H.; Yu, H.; Tong, L.; Stains, J.P.; Moudgil, K.D. Celastrus and its bioactive celastrol protect against bone damage in autoimmune arthritis by modulating osteoimmune cross-talk. J. Biol. Chem. 2012, 287, 22216–22226. [Google Scholar] [CrossRef] [PubMed]
- Venkatesha, S.H.; Astry, B.; Nanjundaiah, S.M.; Yu, H.; Moudgil, K.D. Suppression of autoimmune arthritis by celastrus-derived celastrol through modulation of pro-inflammatory chemokines. Bioorg. Med. Chem. 2012, 20, 5229–5234. [Google Scholar] [CrossRef] [PubMed]
- Matsunaga, N.; Tsuchimori, N.; Matsumoto, T.; Ii, M. Tak-242 (resatorvid), a small-molecule inhibitor of toll-like receptor (TLR) 4 signaling, binds selectively to TLR4 and interferes with interactions between TLR4 and its adaptor molecules. Mol. Pharmacol. 2010, 79, 34–41. [Google Scholar] [CrossRef] [PubMed]
- McCormack, W.J.; Parker, A.E.; O’Neill, L.A. Toll-like receptors and nod-like receptors in rheumatic diseases. Arthritis Res. Ther. 2009, 11, 243. [Google Scholar] [CrossRef] [PubMed]
- Evans, C.H.; Ghivizzani, S.C.; Robbins, P.D. Getting arthritis gene therapy into the clinic. Nat. Rev. 2011, 7, 244–249. [Google Scholar]
- Jorgensen, C.; Apparailly, F. Prospects for gene therapy in inflammatory arthritis. Best Pract. Res. 2010, 24, 541–552. [Google Scholar] [CrossRef]
- Nakasato, M.; Nonomura, Y.; Miyasaka, N.; Kohsaka, H. The gene delivery system for rheumatoid synovium. Jpn. J. Clin. Immunol. 2008, 31, 17–22. [Google Scholar] [CrossRef]
- Traister, R.S.; Hirsch, R. Gene therapy for arthritis. Mod. Rheumatol. 2008, 18, 2–14. [Google Scholar] [CrossRef] [PubMed]
- Woods, A.M.; Thompson, S.J.; Wooley, P.H.; Panayi, G.; Klavinskis, L.S. Immune modulation of collagen-induced arthritis by intranasal cytokine gene delivery: A model for the therapy of rheumatoid arthritis. Arthritis Rheumatol. 2005, 52, 3761–3771. [Google Scholar] [CrossRef]
- Schmidt-Weber, C.B.; Pohlers, D.; Siegling, A.; Schadlich, H.; Buchner, E.; Volk, H.D.; Palombo-Kinne, E.; Emmrich, F.; Kinne, R.W. Cytokine gene activation in synovial membrane, regional lymph nodes, and spleen during the course of rat adjuvant arthritis. Cell Immunol. 1999, 195, 53–65. [Google Scholar] [CrossRef] [PubMed]
- Ji, J.D.; Lee, W.J. Interleukin-18 gene polymorphisms and rheumatoid arthritis: A meta-analysis. Gene 2013, 523, 27–32. [Google Scholar] [CrossRef]
- Zhang, W.; Cong, X.L.; Qin, Y.H.; He, Z.W.; He, D.Y.; Dai, S.M. IL-18 upregulates the production of key regulators of osteoclastogenesis from fibroblast-like synoviocytes in rheumatoid arthritis. Inflammation 2013, 36, 103–109. [Google Scholar] [CrossRef] [PubMed]
- Tak, P.P. IFN-β in rheumatoid arthritis. Front. Biosci. 2004, 9, 3242–3247. [Google Scholar] [CrossRef]
- Lee, S.J.; Lee, A.; Hwang, S.R.; Park, J.S.; Jang, J.; Huh, M.S.; Jo, D.G.; Yoon, S.Y.; Byun, Y.; Kim, S.H.; et al. TNF-α gene silencing using polymerized sirna/thiolated glycol chitosan nanoparticles for rheumatoid arthritis. Mol. Ther. 2014, 22, 397–408. [Google Scholar] [CrossRef]
- Komano, Y.; Yagi, N.; Onoue, I.; Kaneko, K.; Miyasaka, N.; Nanki, T. Arthritic joint-targeting small interfering RNA-encapsulated liposome: Implication for treatment strategy for rheumatoid arthritis. J. Pharmacol. Exp. Ther. 2012, 340, 109–113. [Google Scholar] [CrossRef] [PubMed]
- Scheinman, R.I.; Trivedi, R.; Vermillion, S.; Kompella, U.B. Functionalized STAT1 siRNA nanoparticles regress rheumatoid arthritis in a mouse model. Nanomedicine 2011, 6, 1669–1682. [Google Scholar] [CrossRef] [PubMed]
- Xue, Y.; Yang, Y.; Su, Z.; Barnie, P.A.; Zheng, D.; Zhang, Y.; Xu, Y.; Wang, S.; Xu, H. Local delivery of T-bet shRNA reduces inflammation in collagen II-induced arthritis via downregulation of IFN-γ and IL-17. Mol. Med. Rep. 2014, 9, 899–903. [Google Scholar] [PubMed]
- Fedorov, Y.; Anderson, E.M.; Birmingham, A.; Reynolds, A.; Karpilow, J.; Robinson, K.; Leake, D.; Marshall, W.S.; Khvorova, A. Off-target effects by sirna can induce toxic phenotype. RNA 2006, 12, 1188–1196. [Google Scholar] [CrossRef] [PubMed]
- Kleinman, M.E.; Yamada, K.; Takeda, A.; Chandrasekaran, V.; Nozaki, M.; Baffi, J.Z.; Albuquerque, R.J.; Yamasaki, S.; Itaya, M.; Pan, Y.; et al. Sequence- and target-independent angiogenesis suppression by sirna via TLR3. Nature 2008, 452, 591–597. [Google Scholar] [CrossRef] [PubMed]
- Satpute, S.R.; Durai, M.; Moudgil, K.D. Antigen-specific tolerogenic and immunomodulatory strategies for the treatment of autoimmune arthritis. Semin. Arthritis Rheum. 2008, 38, 195–207. [Google Scholar] [CrossRef] [PubMed]
- Satpute, S.R.; Soukhareva, N.; Scott, D.W.; Moudgil, K.D. Mycobacterial Hsp65-IgG-expressing tolerogenic B cells confer protection against adjuvant-induced arthritis in Lewis rats. Arthritis Rheumatol. 2007, 56, 1490–1496. [Google Scholar] [CrossRef]
- Thomas, C.E.; Ehrhardt, A.; Kay, M.A. Progress and problems with the use of viral vectors for gene therapy. Nat. Rev. Genet. 2003, 4, 346–358. [Google Scholar] [CrossRef] [PubMed]
- Williams, D.A.; Thrasher, A.J. Concise review: Lessons learned from clinical trials of gene therapy in monogenic immunodeficiency diseases. Stem Cells Transl. Med. 2014, 3, 636–642. [Google Scholar] [CrossRef]
- Sade, R.M.; Khushf, G. Gene therapy: Ethical and social issues. J. South Carol. Med. Assoc. 1998, 94, 406–410. [Google Scholar]
- Wivel, N.A.; Walters, L. Germ-line gene modification and disease prevention: Some medical and ethical perspectives. Science 1993, 262, 533–538. [Google Scholar] [CrossRef] [PubMed]
- Kim, S. Interleukin-32 in inflammatory autoimmune diseases. Immune Netw. 2014, 14, 123–127. [Google Scholar] [CrossRef] [PubMed]
- Joosten, L.A.; Netea, M.G.; Kim, S.H.; Yoon, D.Y.; Oppers-Walgreen, B.; Radstake, T.R.; Barrera, P.; van de Loo, F.A.; Dinarello, C.A.; van den Berg, W.B. Il-32, a proinflammatory cytokine in rheumatoid arthritis. Proc. Natl. Acad. Sci. USA 2006, 103, 3298–3303. [Google Scholar] [CrossRef] [PubMed]
- Shoda, H.; Fujio, K.; Yamaguchi, Y.; Okamoto, A.; Sawada, T.; Kochi, Y.; Yamamoto, K. Interactions between IL-32 and tumor necrosis factor alpha contribute to the exacerbation of immune-inflammatory diseases. Arthritis Res. Ther. 2006, 8, R166. [Google Scholar] [CrossRef] [PubMed]
- Alsaleh, G.; Sparsa, L.; Chatelus, E.; Ehlinger, M.; Gottenberg, J.E.; Wachsmann, D.; Sibilia, J. Innate immunity triggers IL-32 expression by fibroblast-like synoviocytes in rheumatoid arthritis. Arthritis Res. Ther. 2010, 12, R135. [Google Scholar] [CrossRef] [PubMed]
- Jeong, H.J.; Nam, S.Y.; Oh, H.A.; Han, N.R.; Kim, Y.S.; Moon, P.D.; Shin, S.Y.; Kim, M.H.; Kim, H.M. Interleukin-32-induced thymic stromal lymphopoietin plays a critical role in macrophage differentiation through the activation of caspase-1 in vitro. Arthritis Res. Ther. 2012, 14, R259. [Google Scholar] [CrossRef] [PubMed]
- Son, M.H.; Jung, M.Y.; Choi, S.; Cho, D.; Kim, T.S. IL-32γ induces chemotaxis of activated t cells via dendritic cell-derived ccl5. Biochem. Biophys. Res. Commun. 2014, 450, 30–35. [Google Scholar] [CrossRef] [PubMed]
- Moon, Y.M.; Yoon, B.Y.; Her, Y.M.; Oh, H.J.; Lee, J.S.; Kim, K.W.; Lee, S.Y.; Woo, Y.J.; Park, K.S.; Park, S.H.; et al. IL-32 and IL-17 interact and have the potential to aggravate osteoclastogenesis in rheumatoid arthritis. Arthritis Res. Ther. 2012, 14, R246. [Google Scholar] [CrossRef] [PubMed]
- Dinarello, C.A.; Kim, S.H. IL-32, a novel cytokine with a possible role in disease. Ann. Rheum. Dis. 2006, 65 (Suppl. S3), iii61–iii64. [Google Scholar] [CrossRef]
- Bostrom, E.A.; Lundberg, P. The newly discovered cytokine IL-34 is expressed in gingival fibroblasts, shows enhanced expression by pro-inflammatory cytokines, and stimulates osteoclast differentiation. PLoS One 2013, 8, e81665. [Google Scholar] [CrossRef] [PubMed]
- Hwang, S.J.; Choi, B.; Kang, S.S.; Chang, J.H.; Kim, Y.G.; Chung, Y.H.; Sohn, D.H.; So, M.W.; Lee, C.K.; Robinson, W.H.; et al. Interleukin-34 produced by human fibroblast-like synovial cells in rheumatoid arthritis supports osteoclastogenesis. Arthritis Res. Ther. 2012, 14, R14. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Buki, K.; Vaaraniemi, J.; Gu, G.; Vaananen, H.K. The critical role of IL-34 in osteoclastogenesis. PLoS One 2011, 6, e18689. [Google Scholar] [CrossRef] [PubMed]
- Baud'huin, M.; Renault, R.; Charrier, C.; Riet, A.; Moreau, A.; Brion, R.; Gouin, F.; Duplomb, L.; Heymann, D. Interleukin-34 is expressed by giant cell tumours of bone and plays a key role in rankl-induced osteoclastogenesis. J. Pathol. 2010, 221, 77–86. [Google Scholar] [CrossRef] [PubMed]
- Chang, S.H.; Choi, B.Y.; Choi, J.; Yoo, J.J.; Ha, Y.J.; Cho, H.J.; Kang, E.H.; Song, Y.W.; Lee, Y.J. Baseline serum interleukin-34 levels independently predict radiographic progression in patients with rheumatoid arthritis. Rheumatol. Int. 2014. [Google Scholar] [CrossRef]
- Chemel, M.; Le Goff, B.; Brion, R.; Cozic, C.; Berreur, M.; Amiaud, J.; Bougras, G.; Touchais, S.; Blanchard, F.; Heymann, M.F.; et al. Interleukin 34 expression is associated with synovitis severity in rheumatoid arthritis patients. Ann. Rheum. Dis. 2012, 71, 150–154. [Google Scholar] [CrossRef] [PubMed]
- Tian, Y.; Shen, H.; Xia, L.; Lu, J. Elevated serum and synovial fluid levels of interleukin-34 in rheumatoid arthritis: Possible association with disease progression via interleukin-17 production. J. Interferon Cytokine Res. 2013, 33, 398–401. [Google Scholar] [CrossRef] [PubMed]
- Moon, S.J.; Hong, Y.S.; Ju, J.H.; Kwok, S.K.; Park, S.H.; Min, J.K. Increased levels of interleukin 34 in serum and synovial fluid are associated with rheumatoid factor and anticyclic citrullinated peptide antibody titers in patients with rheumatoid arthritis. J. Rheumatol. 2013, 40, 1842–1849. [Google Scholar] [CrossRef] [PubMed]
- Collison, L.W.; Chaturvedi, V.; Henderson, A.L.; Giacomin, P.R.; Guy, C.; Bankoti, J.; Finkelstein, D.; Forbes, K.; Workman, C.J.; Brown, S.A.; et al. IL-35-mediated induction of a potent regulatory t cell population. Nat. Immunol. 2010, 11, 1093–1101. [Google Scholar] [CrossRef] [PubMed]
- Collison, L.W.; Delgoffe, G.M.; Guy, C.S.; Vignali, K.M.; Chaturvedi, V.; Fairweather, D.; Satoskar, A.R.; Garcia, K.C.; Hunter, C.A.; Drake, C.G.; et al. The composition and signaling of the IL-35 receptor are unconventional. Nat. Immunol. 2012, 13, 290–299. [Google Scholar] [CrossRef] [PubMed]
- Niedbala, W.; Wei, X.Q.; Cai, B.; Hueber, A.J.; Leung, B.P.; McInnes, I.B.; Liew, F.Y. IL-35 is a novel cytokine with therapeutic effects against collagen-induced arthritis through the expansion of regulatory T cells and suppression of th17 cells. Eur. J. Immunol. 2007, 37, 3021–3029. [Google Scholar] [CrossRef] [PubMed]
- Kochetkova, I.; Golden, S.; Holderness, K.; Callis, G.; Pascual, D.W. IL-35 stimulation of CD39+ regulatory T cells confers protection against collagen II-induced arthritis via the production of IL-10. J. Immunol. 2010, 184, 7144–7153. [Google Scholar] [CrossRef] [PubMed]
- Bettini, M.; Castellaw, A.H.; Lennon, G.P.; Burton, A.R.; Vignali, D.A. Prevention of autoimmune diabetes by ectopic pancreatic beta-cell expression of interleukin-35. Diabetes 2012, 61, 1519–1526. [Google Scholar] [CrossRef] [PubMed]
- Wirtz, S.; Billmeier, U.; McHedlidze, T.; Blumberg, R.S.; Neurath, M.F. Interleukin-35 mediates mucosal immune responses that protect against T-cell-dependent colitis. Gastroenterology 2011, 141, 1875–1886. [Google Scholar] [CrossRef] [PubMed]
- Thiolat, A.; Denys, A.; Petit, M.; Biton, J.; Lemeiter, D.; Herve, R.; Lutomski, D.; Boissier, M.C.; Bessis, N. Interleukin-35 gene therapy exacerbates experimental rheumatoid arthritis in mice. Cytokine 2014, 69, 87–93. [Google Scholar] [CrossRef] [PubMed]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Venkatesha, S.H.; Dudics, S.; Acharya, B.; Moudgil, K.D. Cytokine-Modulating Strategies and Newer Cytokine Targets for Arthritis Therapy. Int. J. Mol. Sci. 2015, 16, 887-906. https://doi.org/10.3390/ijms16010887
Venkatesha SH, Dudics S, Acharya B, Moudgil KD. Cytokine-Modulating Strategies and Newer Cytokine Targets for Arthritis Therapy. International Journal of Molecular Sciences. 2015; 16(1):887-906. https://doi.org/10.3390/ijms16010887
Chicago/Turabian StyleVenkatesha, Shivaprasad H., Steven Dudics, Bodhraj Acharya, and Kamal D. Moudgil. 2015. "Cytokine-Modulating Strategies and Newer Cytokine Targets for Arthritis Therapy" International Journal of Molecular Sciences 16, no. 1: 887-906. https://doi.org/10.3390/ijms16010887
APA StyleVenkatesha, S. H., Dudics, S., Acharya, B., & Moudgil, K. D. (2015). Cytokine-Modulating Strategies and Newer Cytokine Targets for Arthritis Therapy. International Journal of Molecular Sciences, 16(1), 887-906. https://doi.org/10.3390/ijms16010887