
Designed Surface Residue Substitutions in [NiFe] Hydrogenase that Improve Electron Transfer Characteristics

Abstract
:1. Introduction
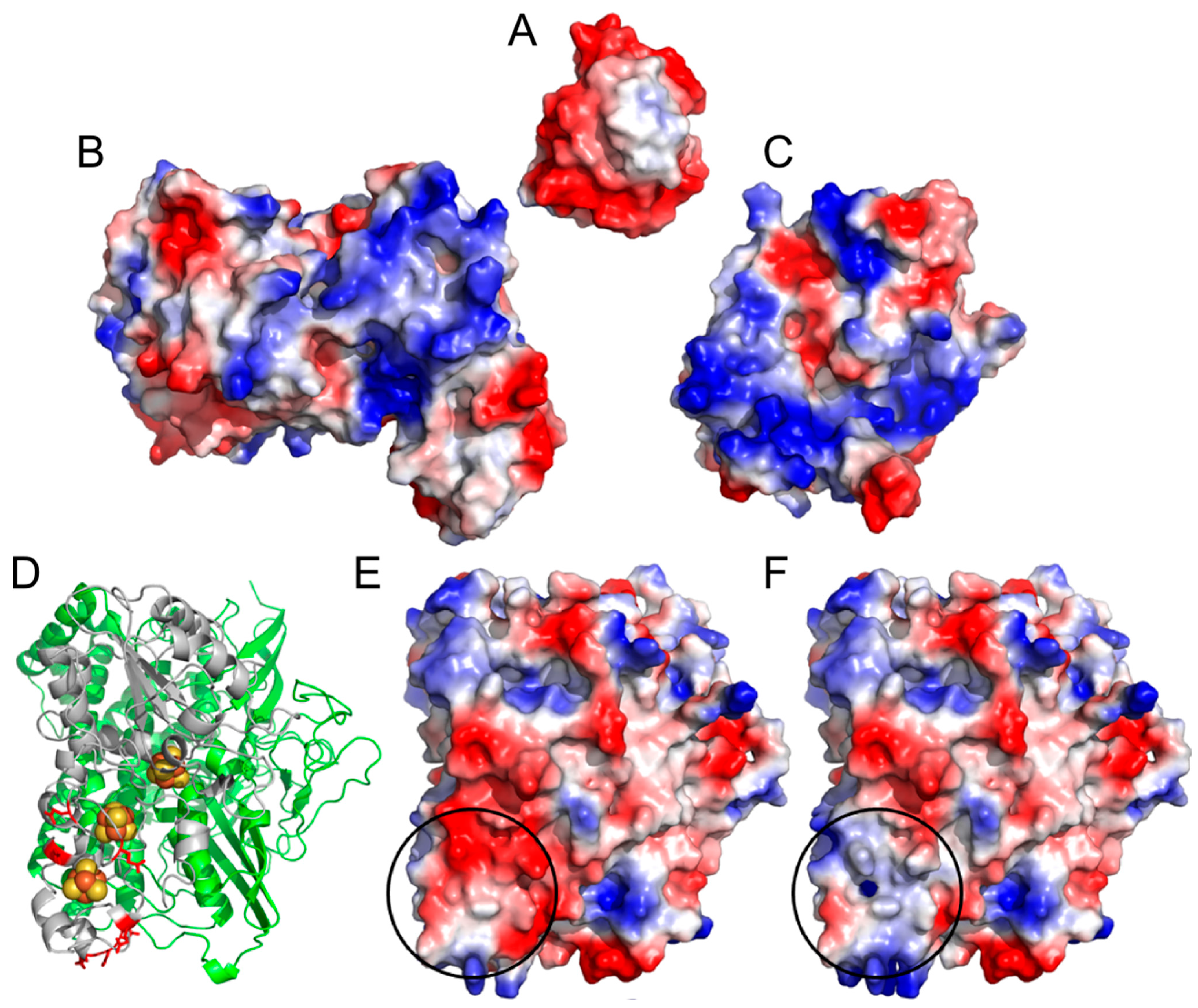
2. Results and Discussion
2.1. Progressive Modification of Surface Residues
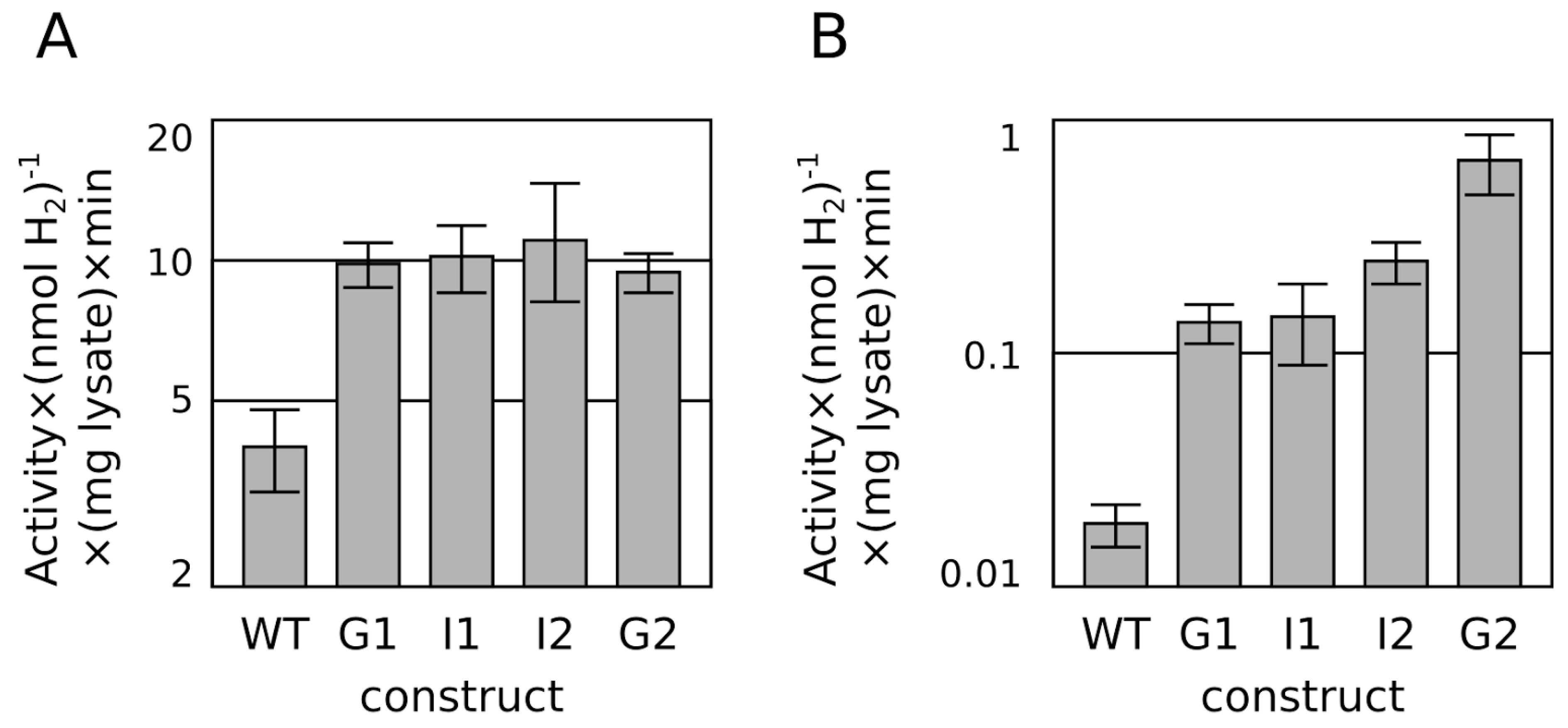

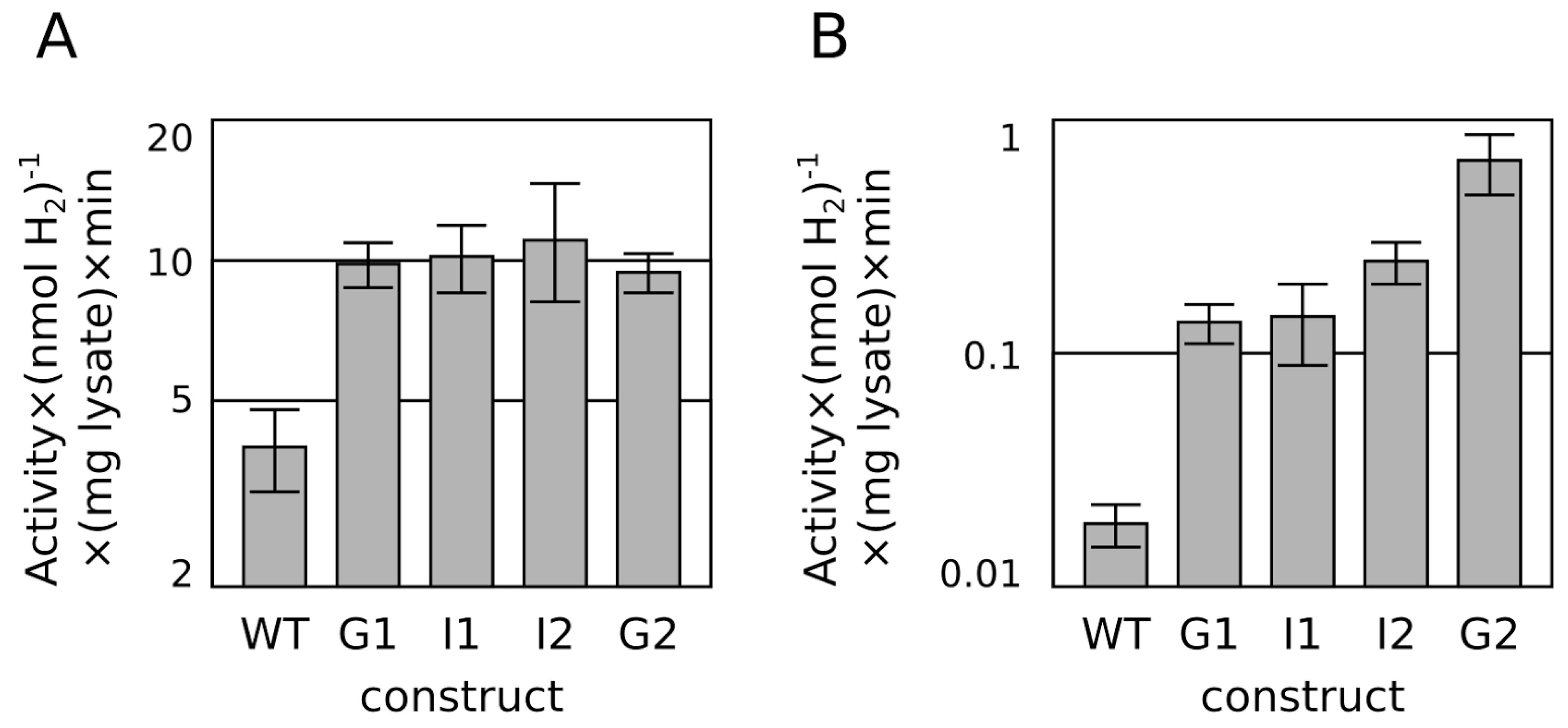
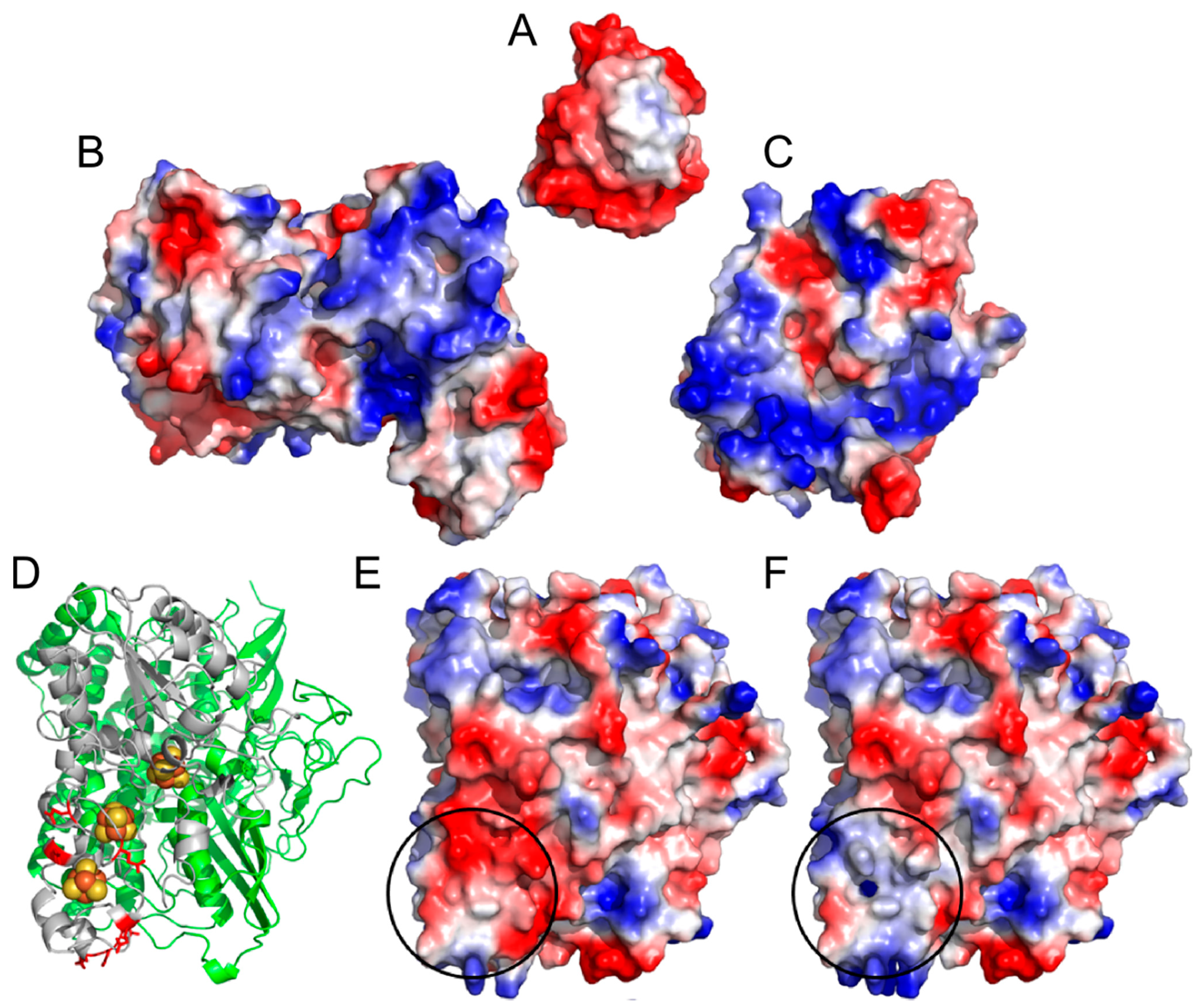{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Taxon | Sequence | |||||||
|---|---|---|---|---|---|---|---|---|
| Species | ||||||||
| Accession | 225 | 230 | 240 | 250 | 260 | 270 | 280 | Name |
| Gammaproteobacteria Alteromonas macleodii AEA96483 | FGESI | HDRCYR RPFF | EQRKFA KSFD | DEGAKNG WCL | FE-LGC KGPET | FNACAT VKWN | QGTSF PIE | “WT” |
| Engineered proteins Alteromonas macleodii AEA96483 derivatives | FGESI | DRCYR RPFF | EQRKFA KSFD | DEGAKNG WCL | FE-LGC KGPET | FNACAT VKWN | QGTSF IE | |
| FGESI | DRCYR RPFF | EQRKFA KSFD | DEGAKNG WCL | FLGC KGPT | FNACAT VKWN | QGTSF IE | “I1” | |
| FGSI | DRCYR RPFF | EQRKFA KSFD | DEGAKNG WCL | FLGC KGPT | FNACAT VKWN | QGTSF IE | “I2” | |
| FGSI | RCYR RPFF | EQRKFA KSFD | DEGAKNG WCL | FLGC KGPT | FNACAT VKWN | QGTSF IE | “G2” | |
| FGSI | RCYR RPFF | QRKFA KSFD | DEGAKNG WCL | FLGC KGPT | FNACAT VKWN | QGTSF IE | ALL+ | |
| FGSI | RCYR RPFF | EQRKFA KSF | GAK NGWCL | F-LGC KGPT | FNACAT VKWN | QGTSF IE | ΔDDE249 | |
| Cyanobacteria Crocosphaera watsonii EHJ10291 | FRSFT | QTGCTR NMHF | SYKATT QDF | GQR TG-CL | FYDMGCRGP MT | HSSCNRI LWN | RVSS- KTR | |
| Actinobacteria Collinsella tanakaei ZP_08853311 | FNTV | HDNCPR RGHF | ENGEFV YQFG | SAEEAKG YCL | YP-LGC RGPT | FTVCPVT RWN | QSVSW VE | |
| Gammaproteobacteria Beggiatoa alba ZP_10114366 | FGTI | HDRCYR RPFY | DKGLFA DTFD | DEGAKQG WCL | Y-LGC KGPTT | YNACAT LKWN | DGVSF PIE | |
| Deltaproteobacteria Desulfovibrio africanus str. “Walvis Bay” YP_005053084 | YGTV | HEQCPR LKFF | EEDKFA PSFD | SEEARQG YCL | Y-LGC KGPYT | YNNCPT AKFN | Q-VNW PVQ | |
| Betaproteobacteria Azoarcus sp. BH72 YP_935309 | ADLV | HGCSR NEFY | EFKASAE KPS | DLGCM | AHADCN LRPW | NGSGS TS | ||
| Alphaproteobacteria Novosphingobium nitrogenifigens ZP_08207308 | ADHLV | HACPK NEFY | EYKASA RALS | EMG CM | MEHLGC IGT-Q | AVGDCNI RPW | NGQGS TR | |
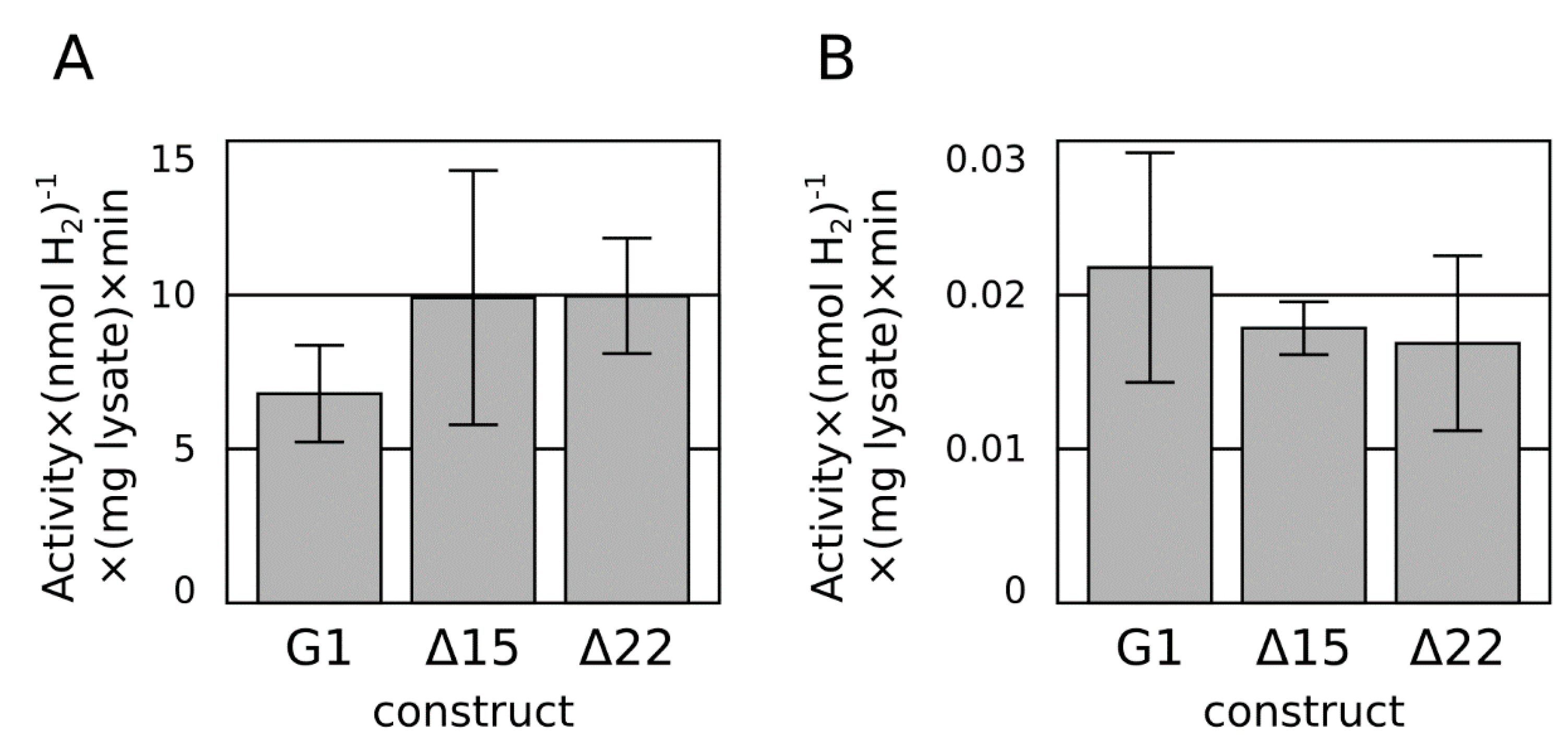
2.2. C-Terminal Truncation
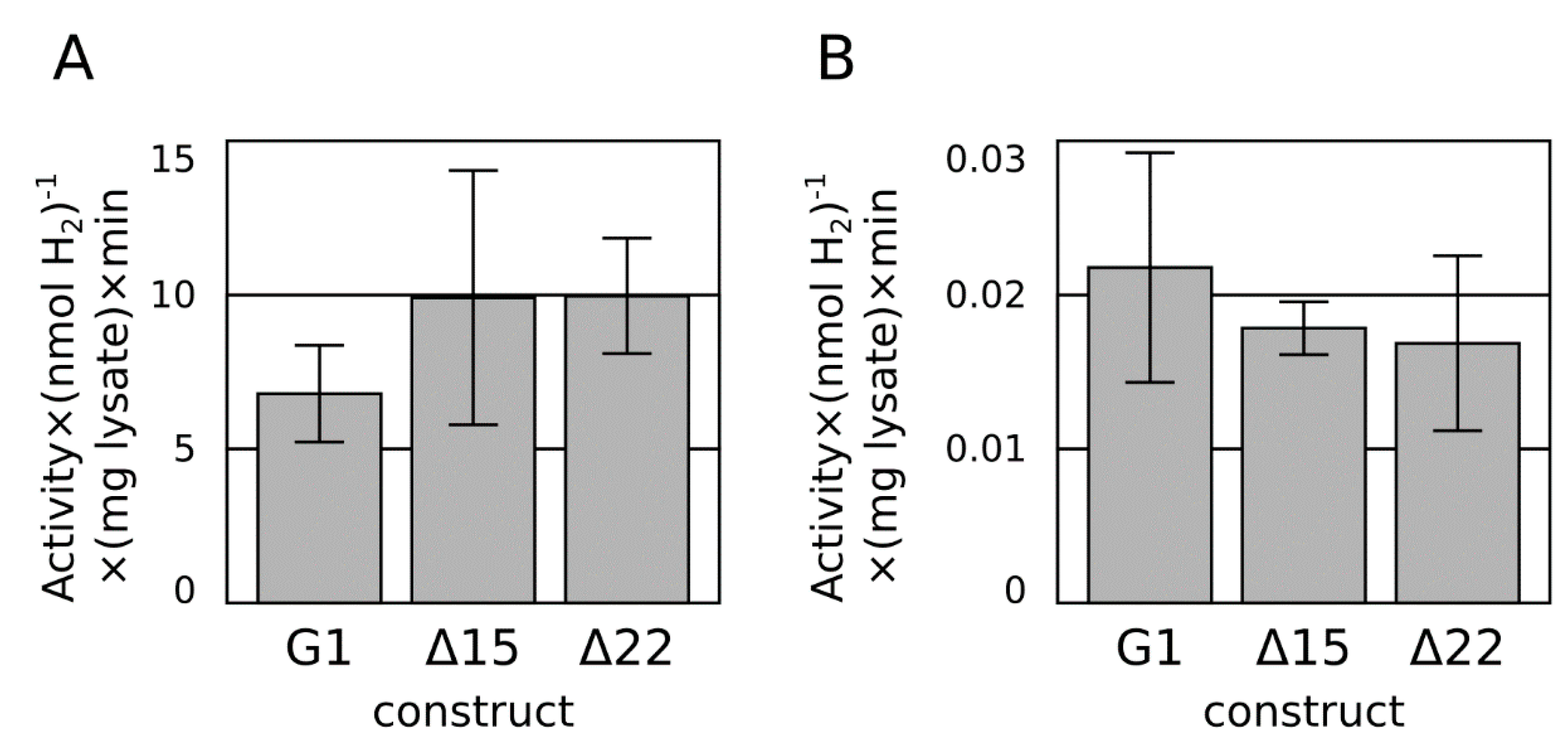
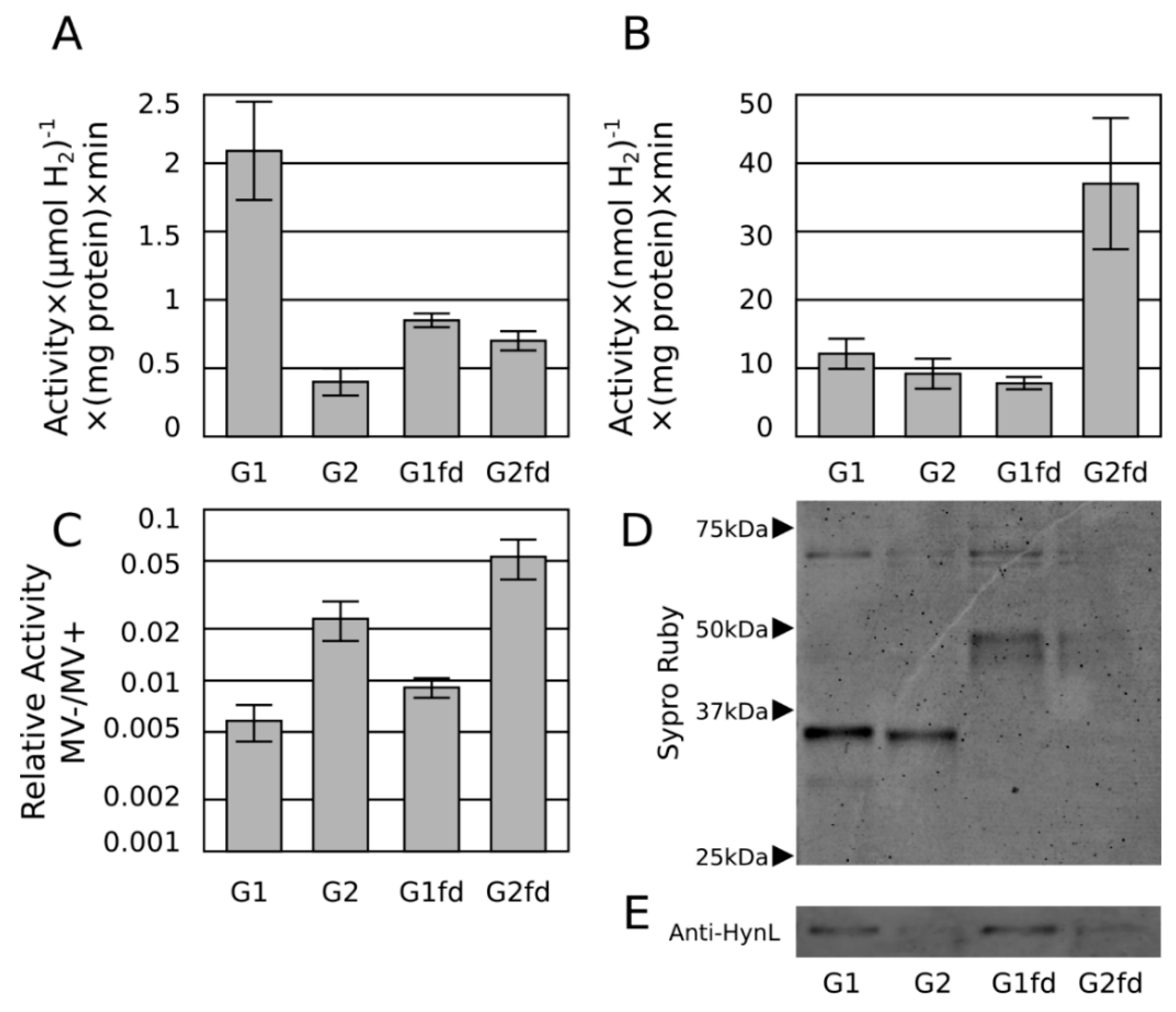
2.3. Ferredoxin Fusion to Hydrogenase
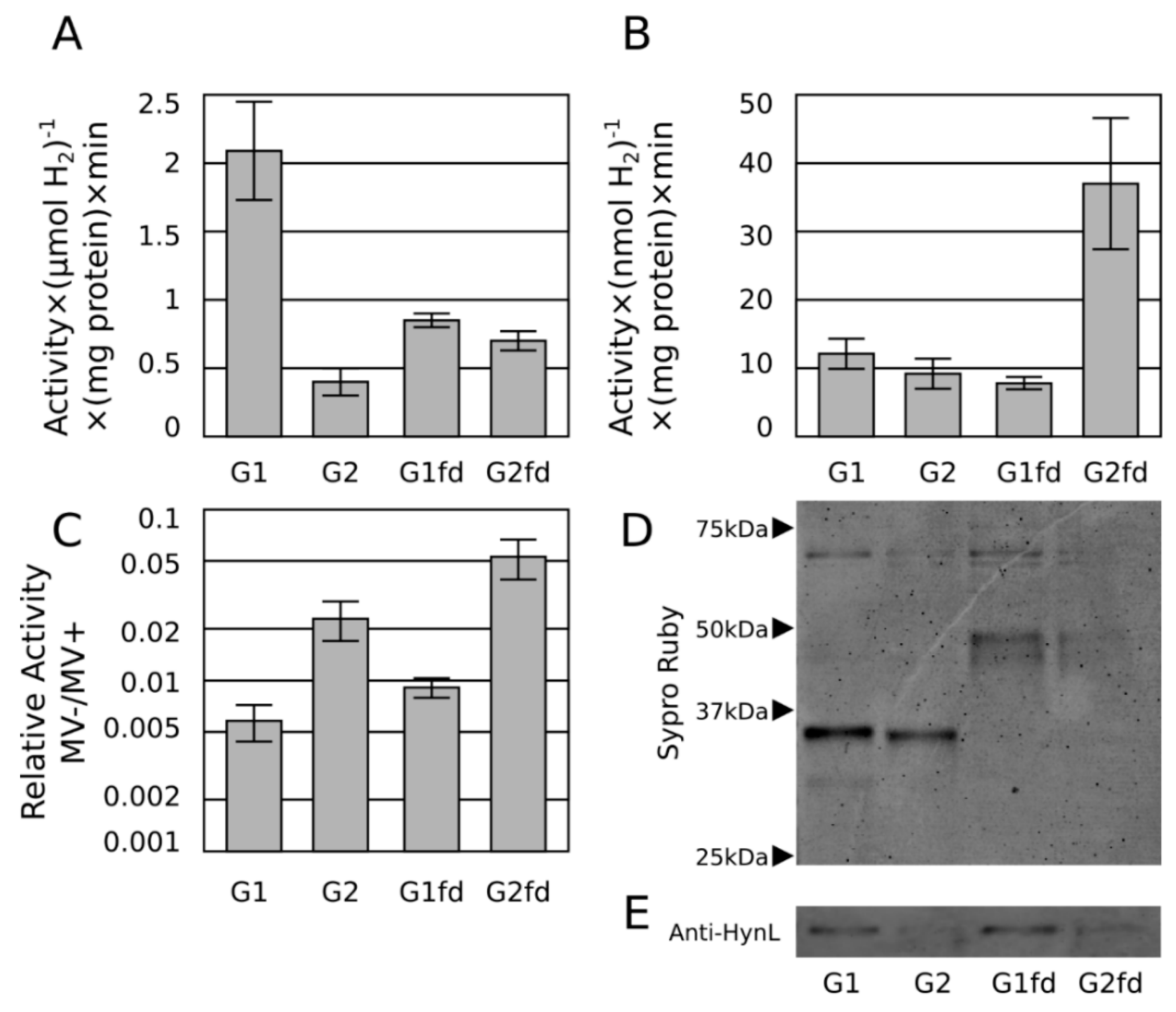
2.4. Discussion
3. Experimental Section
3.1. Molecular Biology
3.2. Hydrogenase Cleared Lysate Preparation
3.3. Tandem IMAC/Streptactin Hydrogenase Preparation
3.4. Hydrogenase Assay
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Schirmer, A.; Rude, M.A.; Li, X.; Popova, E.; del Cardayre, S.B. Microbial biosynthesis of alkanes. Science 2010, 329, 559–562. [Google Scholar] [CrossRef] [PubMed]
- Coates, R.C.; Podell, S.; Korobeynikov, A.; Lapidus, A.; Pevzner, P.; Sherman, D.H.; Allen, E.E.; Gerwick, L.; Gerwick, W.H. Characterization of cyanobacterial hydrocarbon composition and distribution of biosynthetic pathways. PLoS One 2014, 9, e85140. [Google Scholar] [CrossRef] [PubMed]
- Fischer, C.R.; Klein-Marcuschamer, D.; Stephanopoulos, G. Selection and optimization of microbial hosts for biofuels production. Metab. Eng. 2008, 10, 295–304. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Sawaya, M.R.; Eisenberg, D.S.; Liao, J.C. Expanding metabolism for biosynthesis of nonnatural alcohols. Proc. Natl. Acad. Sci. USA 2008, 105, 20653–20658. [Google Scholar] [CrossRef] [PubMed]
- Sage, R.F.; Sage, T.L.; Kocacinar, F. Photorespiration and the evolution of C4 photosynthesis. Annu. Rev. Plant Biol. 2012, 63, 19–47. [Google Scholar] [CrossRef] [PubMed]
- Beer, L.L.; Boyd, E.S.; Peters, J.W.; Posewitz, M.C. Engineering algae for biohydrogen and biofuel production. Curr. Opin. Biotechnol. 2009, 20, 264–271. [Google Scholar] [CrossRef] [PubMed]
- Ghirardi, M.L.; Posewitz, M.C.; Maness, P.C.; Dubini, A.; Yu, J.P.; Seibert, M. Hydrogenases and hydrogen photoproduction in oxygenic photosynthetic organisms. Annu. Rev. Plant Biol. 2007, 58, 71–91. [Google Scholar] [CrossRef] [PubMed]
- Tamagnini, P.; Axelsson, R.; Lindberg, P.; Oxelfelt, F.; Wunschiers, R.; Lindblad, P. Hydrogenases and hydrogen metabolism of cyanobacteria. Microbiol. Mol. Biol. Rev. 2002, 66, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Melis, A.; Zhang, L.P.; Forestier, M.; Ghirardi, M.L.; Seibert, M. Sustained photobiological hydrogen gas production upon reversible inactivation of oxygen evolution in the green alga Chlamydomonas reinhardtii. Plant Physiol. 2000, 122, 127–135. [Google Scholar] [CrossRef] [PubMed]
- Maroti, G.; Tong, Y.; Yooseph, S.; Baden-Tillson, H.; Smith, H.O.; Kovacs, K.L.; Frazier, M.; Venter, J.C.; Xu, Q. Discovery of a [NiFe]-hydrogenase in metagenomic sargasso sea DNA: Cloning and functional analysis in thiocapsa roseopersicina. Appl Envrion. Microbiol. 2009, 75, 5821–5830. [Google Scholar] [CrossRef]
- Vargas, W.A.; Weyman, P.D.; Tong, Y.; Smith, H.O.; Xu, Q. A [NiFe]-hydrogenase from Alteromonas macleodii with unusual stability in the presence of oxygen and high temperature. Appl. Environ. Microbiol. 2011, 77, 1990–1998. [Google Scholar] [CrossRef] [PubMed]
- Yonemoto, I.T.; Matteri, C.W.; Nguyen, T.A.; Smith, H.O.; Weyman, P.D. Dual organism design cycle reveals small subunit substitutions that improve [NiFe] hydrogenase hydrogen evolution. J. Biol. Eng. 2013, 7, 17. [Google Scholar] [CrossRef] [PubMed]
- Dementin, S.; Belle, V.; Bertrand, P.; Guigliarelli, B.; Adryanczyk-Perrier, G.; de Lacey, A.L.; Fernandez, V.M.; Rousset, M.; Leger, C. Changing the ligation of the distal [4Fe4S] cluster in NiFe hydrogenase impairs inter- and intramolecular electron transfers. J. Am. Chem. Soc. 2006, 128, 5209–5218. [Google Scholar] [CrossRef] [PubMed]
- Rousset, M.; Montet, Y.; Guigliarelli, B.; Forget, N.; Asso, M.; Bertrand, P.; Fontecilla-Camps, J.C.; Hatchikian, E.C. [3Fe-4S] to [4Fe-4S] cluster conversion in Desulfovibrio fructosovorans [NiFe] hydrogenase by site-directed mutagenesis. Proc. Natl. Acad. Sci. USA 1998, 95, 11625–11630. [Google Scholar] [CrossRef] [PubMed]
- Yacoby, I.; Pochekailov, S.; Toporik, H.; Ghirardi, M.L.; King, P.W.; Zhang, S. Photosynthetic electron partitioning between [FeFe]-hydrogenase and ferredoxin: NADP+-oxidoreductase (FNR) enzymes in vitro. Proc. Natl. Acad. Sci. USA 2011, 108, 9396–9401. [Google Scholar] [CrossRef] [PubMed]
- Agapakis, C.M.; Ducat, D.C.; Boyle, P.M.; Wintermute, E.H.; Way, J.C.; Silver, P.A. Insulation of a synthetic hydrogen metabolism circuit in bacteria. J. Biol. Eng. 2010, 4, 3. [Google Scholar] [CrossRef] [PubMed]
- Abdullatypov, A.; Zorin, N.; Tsygankov, A. Interaction of HydSL hydrogenase from the purple sulfur bacterium thiocapsa roseopersicina BBS with methyl viologen and positively charged polypeptides. Biochem. Biokhimii͡a 2014, 79, 805–811. [Google Scholar] [CrossRef]
- Lawrence, M.S.; Phillips, K.J.; Liu, D.R. Supercharging proteins can impart unusual resilience. J. Am. Chem. Soc. 2007, 129, 10110–10112. [Google Scholar] [CrossRef] [PubMed]
- Ihara, M.; Nishihara, H.; Yoon, K.-S.; Lenz, O.; Friedrich, B.; Nakamoto, H.; Kojima, K.; Honma, D.; Kamachi, T.; Okura, I. Light-driven hydrogen production by a hybrid complex of a [NiFe]-hydrogenase and the cyanobacterial photosystem I. Photochem. Photobiol. 2006, 82, 676–682. [Google Scholar] [CrossRef] [PubMed]
- Volbeda, A.; Darnault, C.; Parkin, A.; Sargent, F.; Armstrong, F.A.; Fontecilla-Camps, J.C. Crystal structure of the O(2)-tolerant membrane-bound hydrogenase 1 from Escherichia coli in complex with its cognate cytochrome b. Structure 2013, 21, 184–190. [Google Scholar] [CrossRef] [PubMed]
- Yonemoto, I.T.; Clarkson, B.R.; Smith, H.O.; Weyman, P.D. A broad survey reveals substitution tolerance of residues ligating FeS clusters in [NiFe] hydrogenase. BMC Biochem. 2014, 15, 10. [Google Scholar] [CrossRef] [PubMed]
- Sybirna, K.; Ezanno, P.; Baffert, C.; Léger, C.; Bottin, H. Arginine171 of Chlamydomonas reinhardtii [Fe–Fe] hydrogenase HydA1 plays a crucial role in electron transfer to its catalytic center. Int. J. Hydrog. Energy 2013, 38, 2998–3002. [Google Scholar] [CrossRef]
- Kuchenreuther, J.M.; Grady-Smith, C.S.; Bingham, A.S.; George, S.J.; Cramer, S.P.; Swartz, J.R. High-yield expression of heterologous [FeFe] hydrogenases in Escherichia coli. PLoS One 2010, 5, e15491. [Google Scholar] [CrossRef] [PubMed]
- Dementin, S.; Burlat, B.; Fourmond, V.; Leroux, F.; Liebgott, P.-P.; Abou Hamdan, A.; Léger, C.; Rousset, M.; Guigliarelli, B.; Bertrand, P. Rates of intra- and intermolecular electron transfers in hydrogenase deduced from steady-state activity measurements. J. Am. Chem. Soc. 2011, 133, 10211–10221. [Google Scholar] [CrossRef] [PubMed]
- Fontecilla-Camps, J.C.; Volbeda, A.; Cavazza, C.; Nicolet, Y. Structure/function relationships of [NiFe]- and [FeFe]-hydrogenases. Chem. Rev. 2007, 107, 4273–4303. [Google Scholar] [CrossRef] [PubMed]
- Shi, L.; Belchik, S.M.; Plymale, A.E.; Heald, S.; Dohnalkova, A.C.; Sybirna, K.; Bottin, H.; Squier, T.C.; Zachara, J.M.; Fredrickson, J.K. Purification and characterization of the [NiFe]-hydrogenase of Shewanella oneidensis MR-1. Appl. Environ. Microbiol. 2011, 77, 5584–5890. [Google Scholar] [CrossRef] [PubMed]
- Tam, K.Y.; Wang, R.L.; Lee, C.W.; Compton, R.G. Applications of the channel flow cell for UV-visible spectroelectrochemical studies: The kinetics of dimerization of the methyl viologen radical cation. Electroanalysis 1997, 9, 219–224. [Google Scholar] [CrossRef]
- Gibson, D.G.; Young, L.; Chuang, R.-Y.; Venter, J.C.; Hutchison, C.A.; Smith, H.O. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat. Meth. 2009, 6, 343–345. [Google Scholar] [CrossRef]
- Weyman, P.D.; Vargas, W.A.; Chuang, R.-Y.; Chang, Y.; Smith, H.O.; Xu, Q. Heterologous expression of Alteromonas macleodii and Thiocapsa roseopersicina [NiFe] hydrogenases in Escherichia coli. Microbiology 2011, 157, 1363–1374. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Mori, T.; Johnson, C.H. Circadian clock-protein expression in cyanobacteria: Rhythms and phase setting. EMBO J. 2000, 19, 3349–3357. [Google Scholar] [CrossRef] [PubMed]
- Wells, M.A.; Mercer, J.; Mott, R.A.; Pereira-Medrano, A.G.; Burja, A.M.; Radianingtyas, H.; Wright, P.C. Engineering a non-native hydrogen production pathway into Escherichia coli via a cyanobacterial [NiFe] hydrogenase. Metab. Eng. 2011, 13, 445–453. [Google Scholar] [CrossRef] [PubMed]
- Studier, F. Protein production by auto-induction in high-density shaking cultures. Protein Expr. Purif. 2005, 41, 207–234. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yonemoto, I.T.; Smith, H.O.; Weyman, P.D. Designed Surface Residue Substitutions in [NiFe] Hydrogenase that Improve Electron Transfer Characteristics. Int. J. Mol. Sci. 2015, 16, 2020-2033. https://doi.org/10.3390/ijms16012020
Yonemoto IT, Smith HO, Weyman PD. Designed Surface Residue Substitutions in [NiFe] Hydrogenase that Improve Electron Transfer Characteristics. International Journal of Molecular Sciences. 2015; 16(1):2020-2033. https://doi.org/10.3390/ijms16012020
Chicago/Turabian StyleYonemoto, Isaac T., Hamilton O. Smith, and Philip D. Weyman. 2015. "Designed Surface Residue Substitutions in [NiFe] Hydrogenase that Improve Electron Transfer Characteristics" International Journal of Molecular Sciences 16, no. 1: 2020-2033. https://doi.org/10.3390/ijms16012020
APA StyleYonemoto, I. T., Smith, H. O., & Weyman, P. D. (2015). Designed Surface Residue Substitutions in [NiFe] Hydrogenase that Improve Electron Transfer Characteristics. International Journal of Molecular Sciences, 16(1), 2020-2033. https://doi.org/10.3390/ijms16012020