
Elucidating Mechanisms of Toxicity Using Phenotypic Data from Primary Human Cell Systems—A Chemical Biology Approach for Thrombosis-Related Side Effects

Abstract
:
1. Introduction
2. Results and Discussion
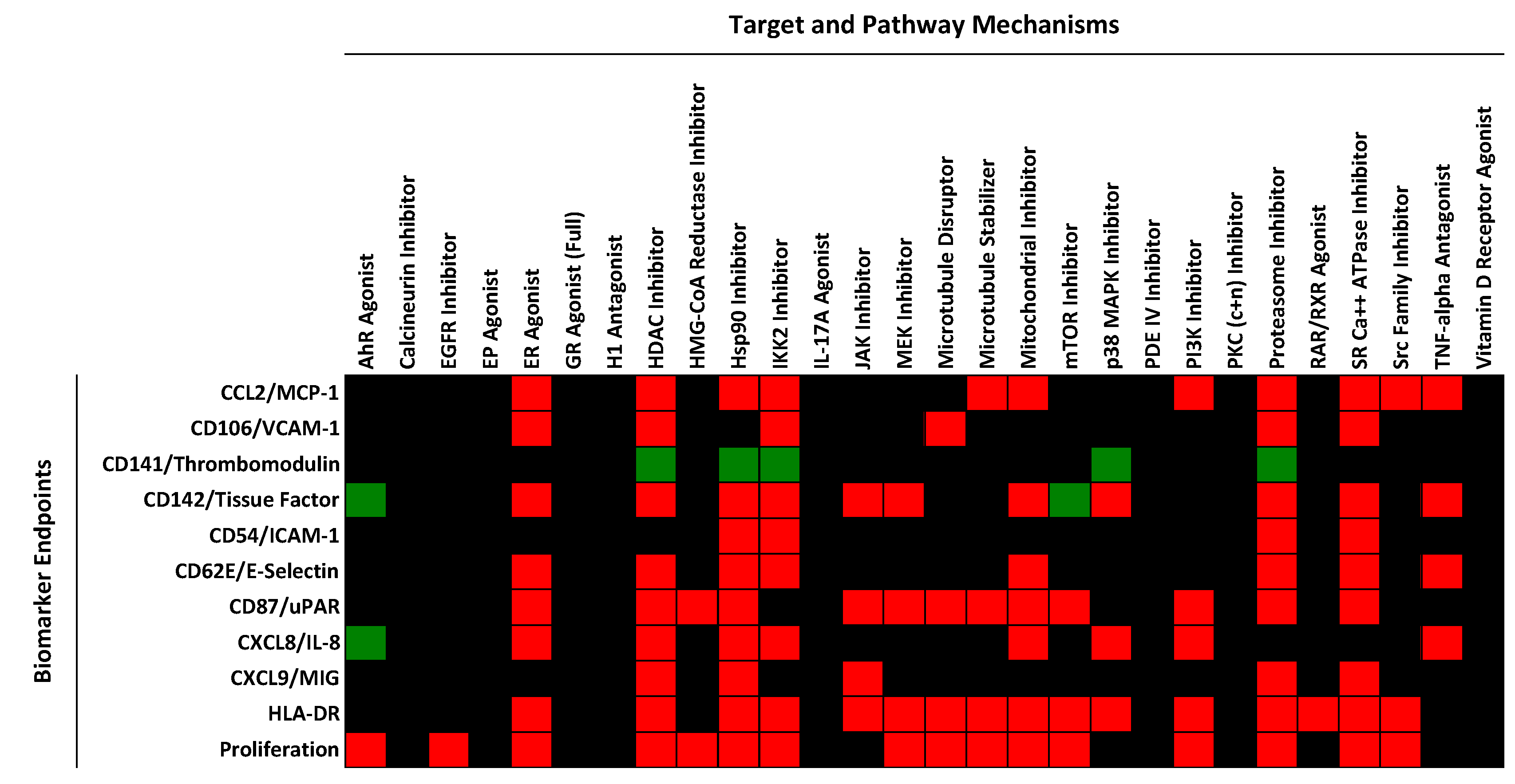
2.1. Regulation of Protein Biomarker Levels in a Human Primary Endothelial Cell Vascular Inflammation Model
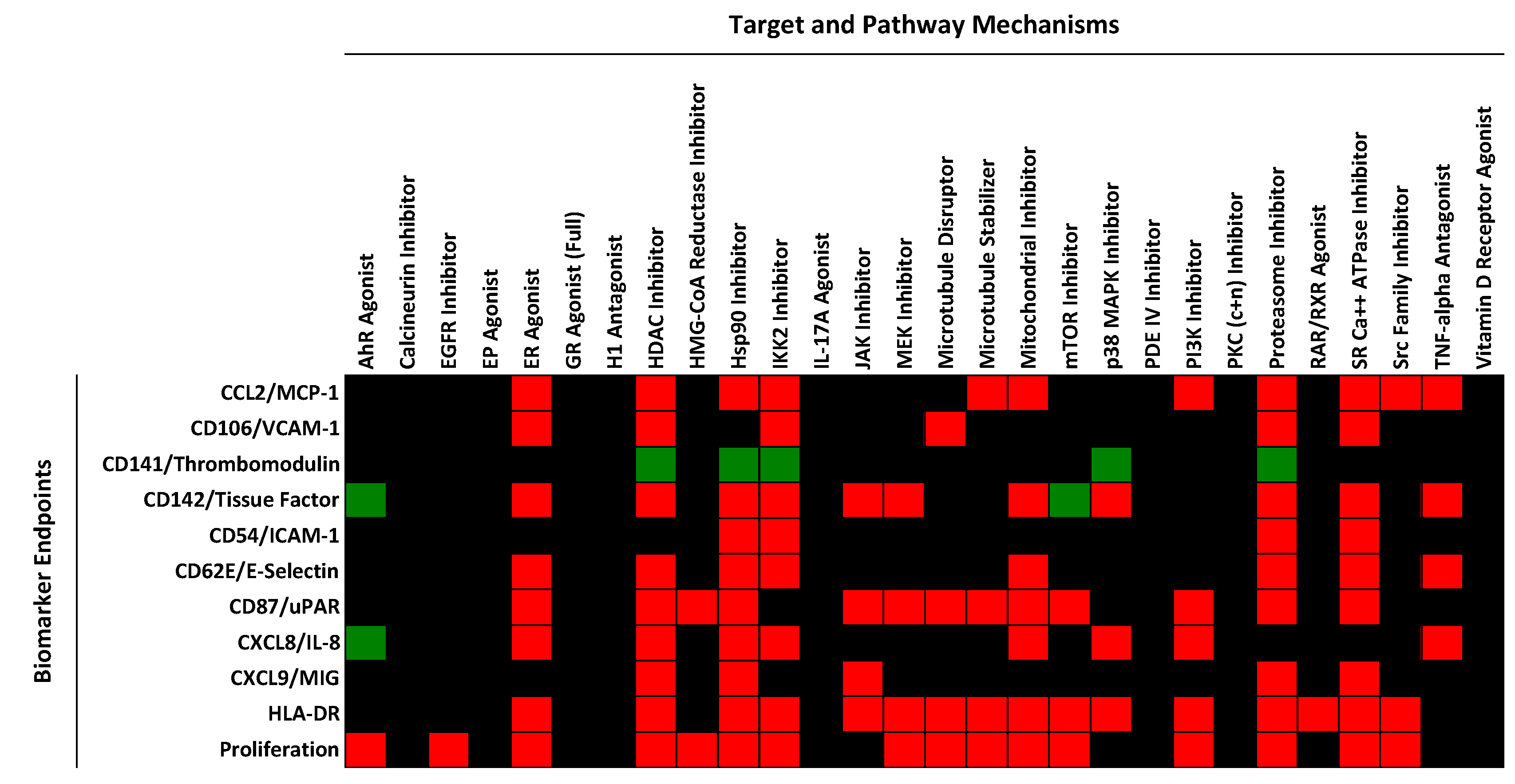
2.2. Identification of Mechanisms Controlling Increased Levels of TF in the BioMAP 3C System

{kind=link}
{kind=link}
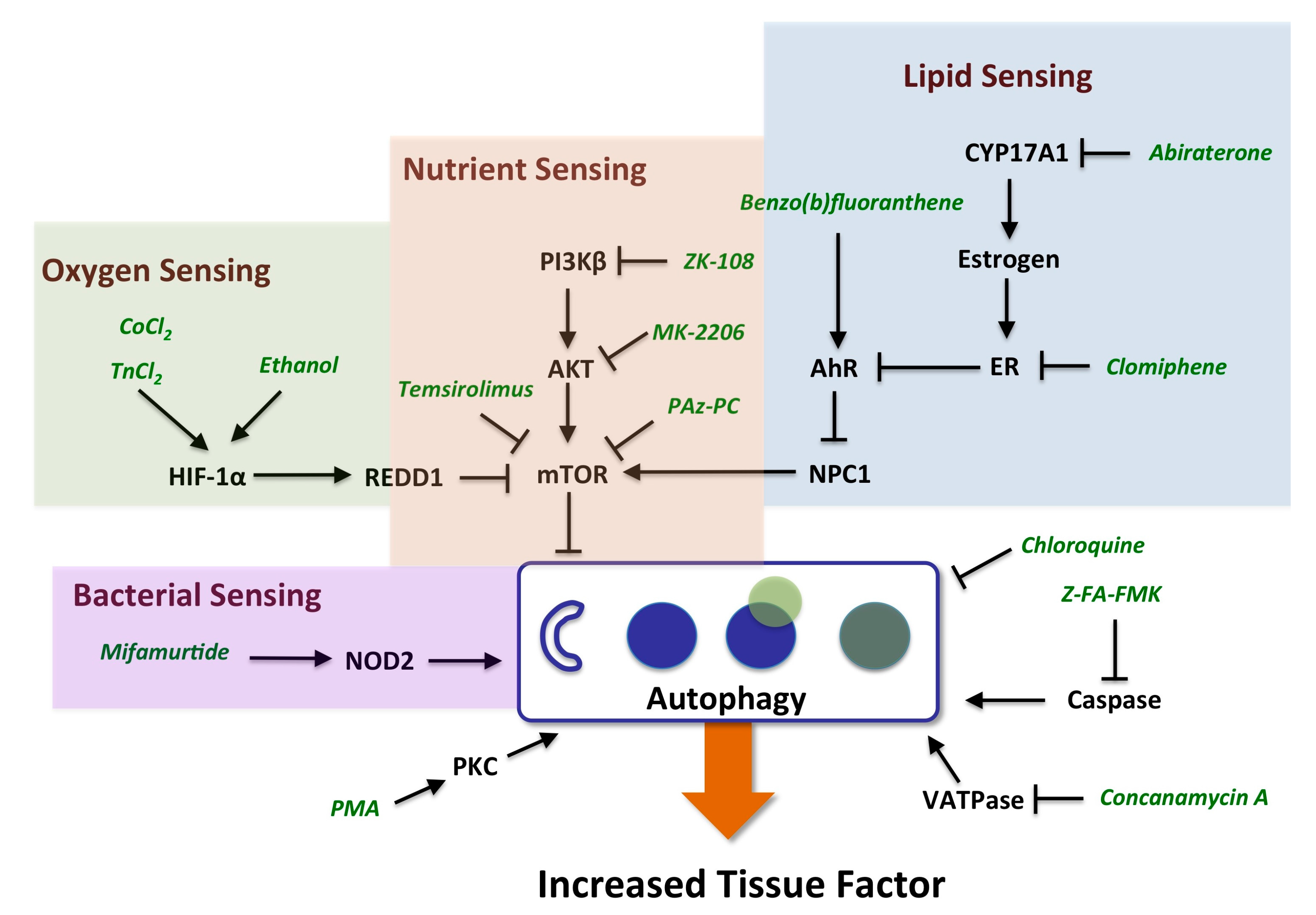{kind=link}
| Compound Name | Mechanism | Likelihood of Mechanism Involvement | Concentrations Active (nM) | Data Source |
|---|---|---|---|---|
| 2-Mercaptobenzothiazole | AhR agonist | Probable | 13,000, 40,000 | Reference [1] |
| 3-Hydroxyfluorene | AhR agonist | Probable | 4400, 13,000, 40,000 | Reference [1] |
| Benzo(b)fluoranthene | AhR agonist | Probable | 4400, 13,000, 40,000 | Reference [1] |
| C.I Solvent yellow 14 | AhR agonist | Probable | 13,000, 40,000 | Reference [1] |
| FICZ | AhR agonist | Probable | 0.15, 0.46, 1.4, 4.1, 12, 37, 111, 333, 1000 | This study |
| Abiraterone | CYP17A Inhibitor | Probable | 1100, 3300, 10,000 | This study |
| Ketoconazole | CYP17A Inhibitor | Probable | 3300, 10,000 | This study |
| Clomiphene citrate | Estrogen R Antagonist | Probable | 2100, 4200 | Reference [1] |
| Histamine | H1R agonist | Probable | 370, 1100, 3300, 10,000, 30,000, 90,000 | This study |
| Histamine Phosphate | H1R agonist | Probable | 1111, 3300, 10,000, 30,000 | This study |
| Cobalt(II) Chloride Hexahydrate | HIF-1α Inducer | Probable | 30,000, 100,000 | This study |
| Tin(II) Chloride | HIF-1α Inducer | Probable | 10,000, 30,000 | This study |
| Chloroquine Phosphate | Lysosome Inhibitor | Probable | 10,000, 30,000 | This study |
| Primaquine Diphosphate | Lysosome Inhibitor | Probable | 11,000, 33,000 | This study |
| Temsirolimus | mTOR Inhibitor | Probable | 0.51, 1.5, 4.6, 123 | This study |
| Torin-1 | mTOR Inhibitor | Probable | 0.46, 1.4, 4.1, 12, 37 | This study |
| Torin-2 | mTOR Inhibitor | Probable | 0.46, 1.4, 4.1, 12 | This study |
| Bryolog | PKC activator | Probable | 4, 12, 37, 110, 330, 1000 | This study |
| Bryostatin 1 | PKC activator | Probable | 12, 37, 110, 330, 1000 | This study |
| Bryostatin 2 | PKC activator | Probable | 0.15, 0.46, 1.4, 4.1, 12, 37, 111 | This study |
| Phorbol 12-myristate 13-acetate | PKC activator | Probable | 37, 41, 111, 120, 330, 370, 1000, 1100, 3300 | This study |
| Phorbol 12,13-didecanoate | PKC activator | Probable | 1.5, 4.6, 14, 41, 123, 370, 1100, 3300 | This study |
| Picolog | PKC activator | Probable | 3.7, 11, 33, 100 | This study |
| Z-FA-FMK | Cysteine protease Inhibitor | Possible | 1100, 3300, 10,000, 30,000 | This study |
| Mifamurtide | NOD2 agonist | Possible | 1111, 3300, 10,000, 30,000 | This study |
| Ethanol | Organic Solvent | Possible | 0.3%, 0.9% | This study |
| Oncostatin M | OSM R agonist | Possible | 0.037, 0.11, 0.33, 3 | This study |
| PAz-PC | Oxidized phospholipid | Possible | 10,000, 30,000 | This study |
| 3,5,3-Triiodothyronine | Thyroid H R agonist | Possible | 16,000, 32,000 | Reference [1] |
| Concanamycin A | Vacuolar ATPase Inhibitor | Possible | 0.14, 0.41, 1.2, 3.7, 11, 33, 100 | This study |
| MK-2206 | AKT Inhibitor | Unknown | 1111, 3300, 10,000 | This study |
| Crizotinib | ALK, c-met Inhibitor | Unknown | 1111, 3333 | This study |
| N-Ethylmaleimide | Alkylating agent | Unknown | 37, 110 | This study |
| Terconazole | Anti-fungal | Unknown | 9300, 9400 | This study |
| GDC-0879 | B-Raf Inhibitor | Unknown | 370, 1100 | This study |
| KN93 | CAMKII Inhibitor | Unknown | 1100, 3300 | This study |
| 8-Hydroxyquinoline | Chelating agent | Unknown | 18, 55 | Reference [1] |
| Linoleic Acid Ethyl Ester | Fatty Acid | Unknown | 10,000, 30,000, 90,000, 270,000 | This study |
| Tris(1,3-dichloro-2-propyl) phosphate | Flame retardant | Unknown | 13,000, 40,000 | Reference [1] |
| Fenaminosulf | Fungicide | Unknown | 13,000, 40,000 | Reference [1] |
| Mancozeb | Fungicide | Unknown | 20,000, 40,000 | Reference [1] |
| Primidone | GABA R agonist | Unknown | 1500, 4400 | This study |
| Mometasone furoate | GR agonist | Unknown | 1111, 3300 | This study |
| Desloratadine | H1R antagonist | Unknown | 10,000, 30,000 | This study |
| A 205804 | ICAM, E-selectin inhibitor | Unknown | 41, 123 | This study |
| Dodecylbenzene | Industrial chemical | Unknown | 1200, 2500 | Reference [1] |
| UO126 | MEK Inhibitor | Unknown | 4.6, 14, 41 | This study |
| Imatinib | PDGFR, c-Kit, Bcr-Abl Inhibitor | Unknown | 3300, 10,000 | This study |
| ZK-108 | PI-3K Inhibitor (βγ-selective) | Unknown | 1100, 3300, 10,000 | This study |
| GW9662 | PPARγ agonist | Unknown | 7400, 22,000 | This study |
| PP3 | SRC Kinase Inhibitor | Unknown | 3300, 10,000, 30,000 | This study |
| TX006146 | Unknown | Unknown | 13,000, 40,000 | Reference [1] |
| TX006237 | Unknown | Unknown | 1500, 4400 | Reference [1] |
| TX011661 | Unknown | Unknown | 5000, 10,000, 20,000 | Reference [1] |
| U-73343 | Unknown | Unknown | 560, 1700 | This study |
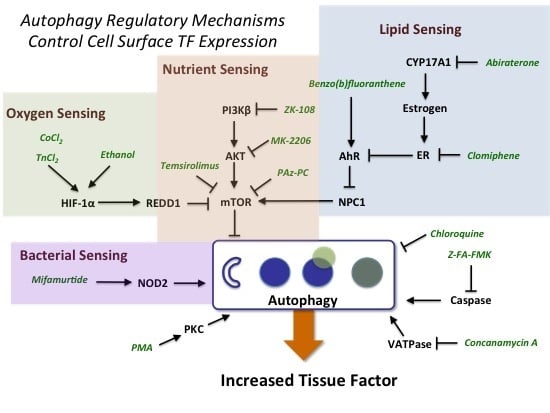
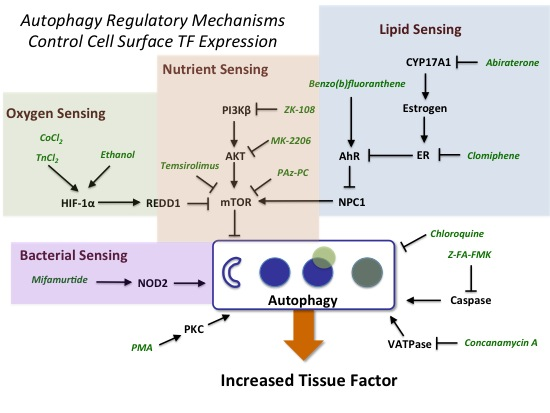
2.3. Evidence for the Role of Autophagy in Controlling Cell Surface TF Levels
| Target Class | Action | Compound | TF Level | Reference |
|---|---|---|---|---|
| AhR | Agonist | 2-Mercaptobenzothiazole | Increased | This study |
| AhR | Agonist | 3-Hydroxyfluorene | Increased | This study |
| AhR | Agonist | Benzo(b)fluoranthene | Increased | This study |
| AhR | Agonist | C.I Solvent yellow 14 | Increased | This study |
| AhR | Agonist | FICZ | Increased | Reference [21] |
| AhR | Antagonist | CH223191 | Decreased | Unpublished, 2014 |
| ER | Antagonist | Clomiphene citrate | Increased | This study |
| ER | Agonist | 17α-Ethynylestradiol | Decreased | Reference [21] |
| ER | Agonist | 17β-Estradiol | Decreased | Reference [21] |
| ER | Antagonist | Tamoxifen | Increased | Reference [1] |
| ER | Antagonist | Tamoxifen Citrate | Increased | Reference [1] |
| ER | Antagonist | Fulvestrant | Increased | Reference [1] |
| ER | Antagonist | Raloxifene hydrochloride | Increased | Reference [1] |
| H1R | Antagonist | Astemizole | Decreased | Reference [21] |
| H1R | Antagonist | Ketotifen Fumarate | Decreased | Reference [21] |
| H1R | Antagonist | trans-Triprolidine | Decreased | Reference [21] |
| H1R | Agonist | Histamine | Increased | This study |
| H1R | Agonist | Histamine Phosphate | Increased | This study |
| PKC | Activator | Bryolog | Increased | This study |
| PKC | Activator | Bryostatin | Increased | This study |
| PKC | Activator | Bryostatin 1 | Increased | This study |
| PKC | Activator | Phorbol 12-myristate 13-acetate | Increased | This study |
| PKC | Activator | Phorbol 12,13-didecanoate | Increased | This study |
| PKC | Activator | Picolog | Increased | This study |
| PKC | Inhibitor | GF 109203X | Decreased | Reference [21] |
| PKC | Inhibitor | Go 6983 | Decreased | Reference [21] |
| PKC | Inhibitor | Ro-32-0432 | Decreased | Reference [21] |
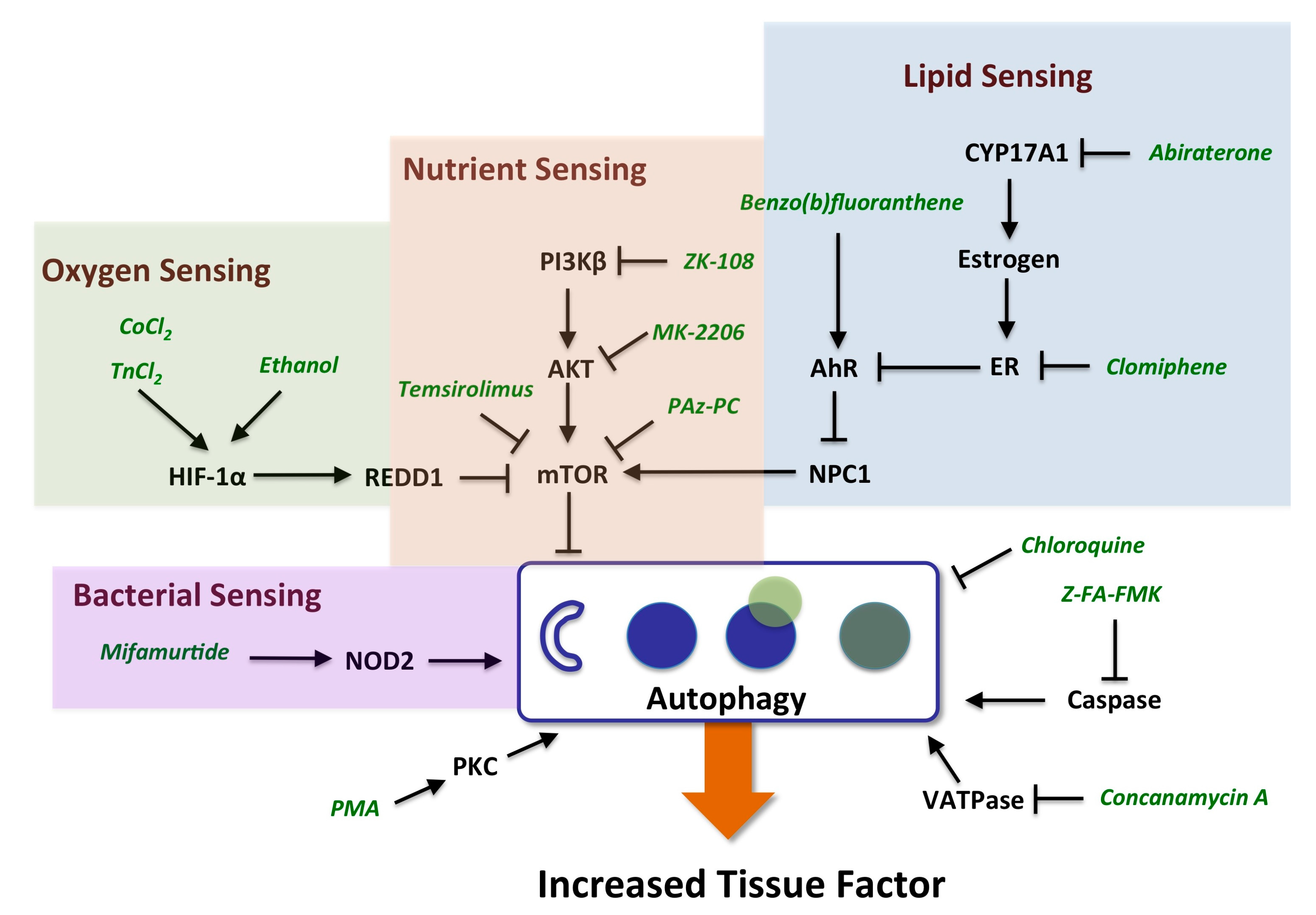
2.3.1. mTOR and HIF-1α Play a Key Role in Autophagy
2.3.2. AhR, Lipid Metabolism and Sterol Sensing in Autophagy
2.3.3. Autophagy-Dependent Bacterial Sensing Mechanisms Are Involved in the Regulation of TF
2.3.4. Other Mechanisms of Interest
2.4. Advantages and Limitations of Chemical Biology Approaches
2.5. Relationship between Tissue Factor Expression and Function
2.6. Applications for Risk Assessment and Adverse Outcome Pathways
3. Experimental Section
3.1. Materials and Reagents
3.2. Cell Culture Methods
3.3. Preparation of Test Agents
3.4. Endpoint Measurements
3.5. Statistical Analysis
3.6. Data Sources
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Kleinstreuer, N.C.; Yang, J.; Berg, E.L.; Knudsen, T.B.; Richard, A.M.; Martin, M.T.; Reif, D.M.; Judson, R.S.; Polokoff, M.; Dix, D.J.; et al. Phenotypic screening of the toxcast chemical library to classify toxic and therapeutic mechanisms. Nat. Biotechnol. 2014, 32, 583–591. [Google Scholar]
- Owens, A.P.; Mackman, N. Tissue factor and thrombosis: The clot starts here. Thromb. Haemost. 2010, 104, 432–439. [Google Scholar]
- Mackman, N. The many faces of tissue factor. J. Thromb. Haemost. 2009, 7, 136–139. [Google Scholar]
- Williams, J.C.; Mackman, N. Tissue factor in health and disease. Front. Biosci. 2012, 4, 358–372. [Google Scholar]
- McElroy, A.K.; Erickson, B.R.; Flietstra, T.D.; Rollin, P.E.; Nichol, S.T.; Towner, J.S.; Spiropoulou, C.F. Ebola hemorrhagic fever: Novel biomarker correlates of clinical outcome. J. Infect. Dis. 2014, 210, 558–566. [Google Scholar]
- Barua, R.S.; Ambrose, J.A. Mechanisms of coronary thrombosis in cigarette smoke exposure. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 1460–1467. [Google Scholar]
- Lipton, A.; Harvey, H.A.; Hamilton, R.W. Venous thrombosis as a side effect of tamoxifen treatment. Cancer Treat. Rep. 1984, 68, 887–889. [Google Scholar]
- Cummings, S.R.; Eckert, S.; Krueger, K.A.; Grady, D.; Powles, T.J.; Cauley, J.A.; Norton, L.; Nickelsen, T.; Bjarnason, N.H.; Morrow, M.; et al. The effect of raloxifene on risk of breast cancer in postmenopausal women: Results from the more randomized trial. Multiple outcomes of raloxifene evaluation. J. Am. Med. Assoc. 1999, 281, 2189–2197. [Google Scholar]
- Owens, A.P., 3rd; Mackman, N. Role of tissue factor in atherothrombosis. Curr. Atheroscler. Rep. 2012, 14, 394–401. [Google Scholar]
- Camici, G.G.; Steffel, J.; Amanovic, I.; Breitenstein, A.; Baldinger, J.; Keller, S.; Luscher, T.F.; Tanner, F.C. Rapamycin promotes arterial thrombosis in vivo: Implications for everolimus and zotarolimus eluting stents. Eur. Heart J. 2010, 31, 236–242. [Google Scholar]
- Judson, R.; Kavlock, R.; Martin, M.; Reif, D.; Houck, K.; Knudsen, T.; Richard, A.; Tice, R.R.; Whelan, M.; Xia, M.; et al. Perspectives on validation of high-throughput assays supporting 21st century toxicity testing. Altex 2013, 30, 51–56. [Google Scholar]
- Lounkine, E.; Keiser, M.J.; Whitebread, S.; Mikhailov, D.; Hamon, J.; Jenkins, J.L.; Lavan, P.; Weber, E.; Doak, A.K.; Cote, S.; et al. Large-scale prediction and testing of drug activity on side-effect targets. Nature 2012, 486, 361–367. [Google Scholar]
- Ankley, G.T.; Bennett, R.S.; Erickson, R.J.; Hoff, D.J.; Hornung, M.W.; Johnson, R.D.; Mount, D.R.; Nichols, J.W.; Russom, C.L.; Schmieder, P.K.; et al. Adverse outcome pathways: A conceptual framework to support ecotoxicology research and risk assessment. Environ. Toxicol. Chem. 2010, 29, 730–741. [Google Scholar]
- Landesmann, B.; Mennecozzi, M.; Berggren, E.; Whelan, M. Adverse outcome pathway-based screening strategies for an animal-free safety assessment of chemicals. Altern. Lab. Anim. ATLA 2013, 41, 461–471. [Google Scholar]
- Vinken, M. The adverse outcome pathway concept: A pragmatic tool in toxicology. Toxicology 2013, 312, 158–165. [Google Scholar]
- Berg, E.L.; Kunkel, E.J.; Hytopoulos, E.; Plavec, I. Characterization of compound mechanisms and secondary activities by biomap analysis. J. Pharmacol. Toxicol. Methods 2006, 53, 67–74. [Google Scholar]
- Berg, E.L.; Yang, J.; Melrose, J.; Nguyen, D.; Privat, S.; Rosler, E.; Kunkel, E.J.; Ekins, S. Chemical target and pathway toxicity mechanisms defined in primary human cell systems. J. Pharmacol. Toxicol. Methods 2010, 61, 3–15. [Google Scholar]
- Kunkel, E.J.; Dea, M.; Ebens, A.; Hytopoulos, E.; Melrose, J.; Nguyen, D.; Ota, K.S.; Plavec, I.; Wang, Y.; Watson, S.R.; et al. An integrative biology approach for analysis of drug action in models of human vascular inflammation. FASEB J. 2004, 18, 1279–1281. [Google Scholar]
- Kunkel, E.J.; Plavec, I.; Nguyen, D.; Melrose, J.; Rosler, E.S.; Kao, L.T.; Wang, Y.; Hytopoulos, E.; Bishop, A.C.; Bateman, R.; et al. Rapid structure-activity and selectivity analysis of kinase inhibitors by biomap analysis in complex human primary cell-based models. Assay Drug Dev. Technol. 2004, 2, 431–441. [Google Scholar]
- Houck, K.A.; Dix, D.J.; Judson, R.S.; Kavlock, R.J.; Yang, J.; Berg, E.L. Profiling bioactivity of the toxcast chemical library using biomap primary human cell systems. J. Biomol. Screen. 2009, 14, 1054–1066. [Google Scholar]
- Berg, E.L.; Yang, J.; Polokoff, M.A. Building predictive models for mechanism-of-action classification from phenotypic assay data sets. J. Biomol. Screen. 2013, 18, 1260–1269. [Google Scholar]
- Garrison, J.L.; Kunkel, E.J.; Hegde, R.S.; Taunton, J. A substrate-specific inhibitor of protein translocation into the endoplasmic reticulum. Nature 2005, 436, 285–289. [Google Scholar]
- Williams, O.; Houseman, B.T.; Kunkel, E.J.; Aizenstein, B.; Hoffman, R.; Knight, Z.A.; Shokat, K.M. Discovery of dual inhibitors of the immune cell PI3Ks p110δ and p110γ: A prototype for new anti-inflammatory drugs. Chem. Biol. 2010, 17, 123–134. [Google Scholar]
- Bergamini, G.; Bell, K.; Shimamura, S.; Werner, T.; Cansfield, A.; Muller, K.; Perrin, J.; Rau, C.; Ellard, K.; Hopf, C.; et al. A selective inhibitor reveals PI3Kγ dependence of TH17 cell differentiation. Nat. Chem. Biol. 2012, 8, 576–582. [Google Scholar]
- Xu, D.; Kim, Y.; Postelnek, J.; Vu, M.D.; Hu, D.Q.; Liao, C.; Bradshaw, M.; Hsu, J.; Zhang, J.; Pashine, A.; et al. Rn486, a selective brutonʼs tyrosine kinase inhibitor, abrogates immune hypersensitivity responses and arthritis in rodents. J. Pharmacol. Exp. Ther. 2012, 341, 90–103. [Google Scholar]
- Parry, G.C.; Mackman, N. Transcriptional regulation of tissue factor expression in human endothelial cells. Arterioscler. Thromb. Vasc. Biol. 1995, 15, 612–621. [Google Scholar]
- Fuste, B.; Serradell, M.; Escolar, G.; Cases, A.; Mazzara, R.; Castillo, R.; Ordinas, A.; Diaz-Ricart, M. Erythropoietin triggers a signaling pathway in endothelial cells and increases the thrombogenicity of their extracellular matrices in vitro. Thromb. Haemost. 2002, 88, 678–685. [Google Scholar]
- Bode, M.; Mackman, N. Regulation of tissue factor gene expression in monocytes and endothelial cells: Thromboxane a as a new player. Vasc. Pharmacol. 2014, 62, 57–62. [Google Scholar]
- Gondouin, B.; Cerini, C.; Dou, L.; Sallee, M.; Duval-Sabatier, A.; Pletinck, A.; Calaf, R.; Lacroix, R.; Jourde-Chiche, N.; Poitevin, S.; et al. Indolic uremic solutes increase tissue factor production in endothelial cells by the aryl hydrocarbon receptor pathway. Kidney Int. 2013, 84, 733–744. [Google Scholar]
- Nishibe, T.; Parry, G.; Ishida, A.; Aziz, S.; Murray, J.; Patel, Y.; Rahman, S.; Strand, K.; Saito, K.; Saito, Y.; et al. Oncostatin M promotes biphasic tissue factor expression in smooth muscle cells: Evidence for Erk-1/2 activation. Blood 2001, 97, 692–699. [Google Scholar]
- Ryter, S.W.; Choi, A.M. Autophagy: An integral component of the mammalian stress response. J. Biochem. Pharmacol. Res. 2013, 1, 176–188. [Google Scholar]
- Ni, H.M.; Williams, J.A.; Yang, H.; Shi, Y.H.; Fan, J.; Ding, W.X. Targeting autophagy for the treatment of liver diseases. Pharmacol. Res. 2012, 66, 463–474. [Google Scholar]
- Kambas, K.; Mitroulis, I.; Apostolidou, E.; Girod, A.; Chrysanthopoulou, A.; Pneumatikos, I.; Skendros, P.; Kourtzelis, I.; Koffa, M.; Kotsianidis, I.; et al. Autophagy mediates the delivery of thrombogenic tissue factor to neutrophil extracellular traps in human sepsis. PLoS One 2012, 7, e45427. [Google Scholar]
- Dunlop, E.A.; Tee, A.R. mTOR and autophagy: A dynamic relationship governed by nutrients and energy. Semin. Cell Dev. Biol. 2014, 36, 121–129. [Google Scholar]
- Yang, Z.J.; Chee, C.E.; Huang, S.; Sinicrope, F.A. The role of autophagy in cancer: Therapeutic implications. Mol. Cancer Ther. 2011, 10, 1533–1541. [Google Scholar]
- Jiang, P.; Lan, Y.; Luo, J.; Ren, Y.L.; Liu, D.G.; Pang, J.X.; Liu, J.; Li, J.; Wang, C.; Cai, J.P. Rapamycin promoted thrombosis and platelet adhesion to endothelial cells by inducing membrane remodeling. BMC Cell Biol. 2014, 15. [Google Scholar] [CrossRef]
- Chen, G.; Zhang, W.; Li, Y.P.; Ren, J.G.; Xu, N.; Liu, H.; Wang, F.Q.; Sun, Z.J.; Jia, J.; Zhao, Y.F. Hypoxia-induced autophagy in endothelial cells: A double-edged sword in the progression of infantile haemangioma? Cardiovasc. Res. 2013, 98, 437–448. [Google Scholar]
- Gallo, S.; Gatti, S.; Sala, V.; Albano, R.; Costelli, P.; Casanova, E.; Comoglio, P.M.; Crepaldi, T. Agonist antibodies activating the met receptor protect cardiomyoblasts from cobalt chloride-induced apoptosis and autophagy. Cell Death Dis. 2014, 5. [Google Scholar] [CrossRef]
- Katiyar, S.; Liu, E.; Knutzen, C.A.; Lang, E.S.; Lombardo, C.R.; Sankar, S.; Toth, J.I.; Petroski, M.D.; Ronai, Z.; Chiang, G.G. Redd1, an inhibitor of mtor signalling, is regulated by the cul4a-ddb1 ubiquitin ligase. EMBO Rep. 2009, 10, 866–872. [Google Scholar]
- Podechard, N.; le Ferrec, E.; Rebillard, A.; Fardel, O.; Lecureur, V. Npc1 repression contributes to lipid accumulation in human macrophages exposed to environmental aryl hydrocarbons. Cardiovasc. Res. 2009, 82, 361–370. [Google Scholar]
- Xu, J.; Dang, Y.; Ren, Y.R.; Liu, J.O. Cholesterol trafficking is required for mtor activation in endothelial cells. Proc. Natl. Acad. Sci. USA 2010, 107, 4764–4769. [Google Scholar]
- Sarkar, S.; Carroll, B.; Buganim, Y.; Maetzel, D.; Ng, A.H.; Cassady, J.P.; Cohen, M.A.; Chakraborty, S.; Wang, H.; Spooner, E.; et al. Impaired autophagy in the lipid-storage disorder niemann-pick type c1 disease. Cell Rep. 2013, 5, 1302–1315. [Google Scholar]
- Welch, C.L.; Sun, Y.; Arey, B.J.; Lemaitre, V.; Sharma, N.; Ishibashi, M.; Sayers, S.; Li, R.; Gorelik, A.; Pleskac, N.; et al. Spontaneous atherothrombosis and medial degradation in Apoe-/-, Npc1-/- mice. Circulation 2007, 116, 2444–2452. [Google Scholar]
- Travassos, L.H.; Carneiro, L.A.; Ramjeet, M.; Hussey, S.; Kim, Y.G.; Magalhaes, J.G.; Yuan, L.; Soares, F.; Chea, E.; le Bourhis, L.; et al. Nod1 and Nod2 direct autophagy by recruiting ATG16L1 to the plasma membrane at the site of bacterial entry. Nat. Immunol. 2010, 11, 55–62. [Google Scholar]
- Madaro, L.; Marrocco, V.; Carnio, S.; Sandri, M.; Bouche, M. Intracellular signaling in Er stress-induced autophagy in skeletal muscle cells. FASEB J. 2013, 27, 1990–2000. [Google Scholar]
- Page, E.L.; Robitaille, G.A.; Pouyssegur, J.; Richard, D.E. Induction of hypoxia-inducible factor-1α by transcriptional and translational mechanisms. J. Biol. Chem. 2002, 277, 48403–48409. [Google Scholar]
- Steffel, J.; Akhmedov, A.; Greutert, H.; Luscher, T.F.; Tanner, F.C. Histamine induces tissue factor expression: Implications for acute coronary syndromes. Circulation 2005, 112, 341–349. [Google Scholar]
- Stobbe-Maicherski, N.; Wolff, S.; Wolff, C.; Abel, J.; Sydlik, U.; Frauenstein, K.; Haarmann-Stemmann, T. The interleukin-6-type cytokine oncostatin m induces aryl hydrocarbon receptor expression in a STAT3-dependent manner in human HEPG2 hepatoma cells. FEBS J. 2013, 280, 6681–6690. [Google Scholar]
- Vollmer, S.; Kappler, V.; Kaczor, J.; Flugel, D.; Rolvering, C.; Kato, N.; Kietzmann, T.; Behrmann, I.; Haan, C. Hypoxia-inducible factor 1α is up-regulated by oncostatin m and participates in oncostatin m signaling. Hepatology 2009, 50, 253–260. [Google Scholar]
- Sinha, R.A.; You, S.H.; Zhou, J.; Siddique, M.M.; Bay, B.H.; Zhu, X.; Privalsky, M.L.; Cheng, S.Y.; Stevens, R.D.; Summers, S.A.; et al. Thyroid hormone stimulates hepatic lipid catabolism via activation of autophagy. J. Clin. Investig. 2012, 122, 2428–2438. [Google Scholar]
- Debeij, J.; Dekkers, O.M.; Asvold, B.O.; Christiansen, S.C.; Naess, I.A.; Hammerstrom, J.; Rosendaal, F.R.; Cannegieter, S.C. Increased levels of free thyroxine and risk of venous thrombosis in a large population-based prospective study. J. Thromb. Haemost. 2012, 10, 1539–1546. [Google Scholar]
- Podrez, E.A.; Poliakov, E.; Shen, Z.; Zhang, R.; Deng, Y.; Sun, M.; Finton, P.J.; Shan, L.; Febbraio, M.; Hajjar, D.P.; et al. A novel family of atherogenic oxidized phospholipids promotes macrophage foam cell formation via the scavenger receptor cd36 and is enriched in atherosclerotic lesions. J. Biol. Chem. 2002, 277, 38517–38523. [Google Scholar]
- Hutter, R.; Speidl, W.S.; Valdiviezo, C.; Sauter, B.; Corti, R.; Fuster, V.; Badimon, J.J. Macrophages transmit potent proangiogenic effects of oxldl in vitro and in vivo involving HIF-1α activation: A novel aspect of angiogenesis in atherosclerosis. J. Cardiovasc. Transl. Res. 2013, 6, 558–569. [Google Scholar]
- Zhang, Y.L.; Cao, Y.J.; Zhang, X.; Liu, H.H.; Tong, T.; Xiao, G.D.; Yang, Y.P.; Liu, C.F. The autophagy-lysosome pathway: A novel mechanism involved in the processing of oxidized LDL in human vascular endothelial cells. Biochem. Biophys. Res. Commun. 2010, 394, 377–382. [Google Scholar]
- Ding, W.X.; Manley, S.; Ni, H.M. The emerging role of autophagy in alcoholic liver disease. Exp. Biol. Med. 2011, 236, 546–556. [Google Scholar]
- Thomes, P.G.; Ehlers, R.A.; Trambly, C.S.; Clemens, D.L.; Fox, H.S.; Tuma, D.J.; Donohue, T.M. Multilevel regulation of autophagosome content by ethanol oxidation in HEPG2 cells. Autophagy 2013, 9, 63–73. [Google Scholar]
- Mestas, J.; Hughes, C.C.W. Of mice and not men: Differences between mouse and human immunology. J. Immunol. 2004, 172, 2731–2738. [Google Scholar]
- Berg, E.L.; Kunkel, E.J.; Hytopoulos, E. Biological complexity and drug discovery: A practical systems biology approach. Syst. Biol. 2005, 152, 201–206. [Google Scholar]
- Aberg, M.; Siegbahn, A. Tissue factor non-coagulant signaling—Molecular mechanisms and biological consequences with a focus on cell migration and apoptosis. J. Thromb. Haemost. 2013, 11, 817–825. [Google Scholar]
- Schaffner, F.; Ruf, W. Tissue factor and PAR2 signaling in the tumor microenvironment. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 1999–2004. [Google Scholar]
- Lupu, C.; Westmuckett, A.D.; Peer, G.; Ivanciu, L.; Zhu, H.; Taylor, F.B., Jr.; Lupu, F. Tissue factor-dependent coagulation is preferentially up-regulated within arterial branching areas in a baboon model of escherichia coli sepsis. Am. J. Pathol. 2005, 167, 1161–1172. [Google Scholar]
- Song, D.; Ye, X.; Xu, H.; Liu, S.F. Activation of endothelial intrinsic NF-κb pathway impairs protein C anticoagulation mechanism and promotes coagulation in endotoxemic mice. Blood 2009, 114, 2521–2529. [Google Scholar]
- Ahamed, J.; Ruf, W. Protease-activated receptor 2-dependent phosphorylation of the tissue factor cytoplasmic domain. J. Biol. Chem. 2004, 279, 23038–23044. [Google Scholar]
- Rao, L.V.; Pendurthi, U.R. Regulation of tissue factor coagulant activity on cell surfaces. J. Thromb. Haemost. 2012, 10, 2242–2253. [Google Scholar]
- Spronk, H.M.; ten Cate, H.; van der Meijden, P.E. Differential roles of tissue factor and phosphatidylserine in activation of coagulation. Thromb. Res. 2014, 133, S54–S56. [Google Scholar]
- Attene-Ramos, M.S.; Miller, N.; Huang, R.; Michael, S.; Itkin, M.; Kavlock, R.J.; Austin, C.P.; Shinn, P.; Simeonov, A.; Tice, R.R.; et al. The Tox21 robotic platform for the assessment of environmental chemicals—from vision to reality. Drug Discov. Today 2013, 18, 716–723. [Google Scholar]
- Tice, R.R.; Austin, C.P.; Kavlock, R.J.; Bucher, J.R. Improving the human hazard characterization of chemicals: A Tox21 update. Environ. Health Perspect. 2013, 121, 756–765. [Google Scholar]
- Rotroff, D.M.; Martin, M.T.; Dix, D.J.; Filer, D.L.; Houck, K.A.; Knudsen, T.B.; Sipes, N.S.; Reif, D.M.; Xia, M.; Huang, R.; et al. Predictive endocrine testing in the 21st century using in vitro assays of estrogen receptor signaling responses. Environ. Sci. Technol. 2014, 48, 8706–8716. [Google Scholar]
- Thomas, R.S.; Philbert, M.A.; Auerbach, S.S.; Wetmore, B.A.; Devito, M.J.; Cote, I.; Rowlands, J.C.; Whelan, M.P.; Hays, S.M.; Andersen, M.E.; et al. Incorporating new technologies into toxicity testing and risk assessment: Moving from 21st century vision to a data-driven framework. Toxicol. Sci. 2013, 136, 4–18. [Google Scholar]
- Papazisis, K.T.; Geromichalos, G.D.; Dimitriadis, K.A.; Kortsaris, A.H. Optimization of the sulforhodamine b colorimetric assay. J. Immunol. Methods 1997, 208, 151–158. [Google Scholar]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Berg, E.L.; Polokoff, M.A.; O'Mahony, A.; Nguyen, D.; Li, X. Elucidating Mechanisms of Toxicity Using Phenotypic Data from Primary Human Cell Systems—A Chemical Biology Approach for Thrombosis-Related Side Effects. Int. J. Mol. Sci. 2015, 16, 1008-1029. https://doi.org/10.3390/ijms16011008
Berg EL, Polokoff MA, O'Mahony A, Nguyen D, Li X. Elucidating Mechanisms of Toxicity Using Phenotypic Data from Primary Human Cell Systems—A Chemical Biology Approach for Thrombosis-Related Side Effects. International Journal of Molecular Sciences. 2015; 16(1):1008-1029. https://doi.org/10.3390/ijms16011008
Chicago/Turabian StyleBerg, Ellen L., Mark A. Polokoff, Alison O'Mahony, Dat Nguyen, and Xitong Li. 2015. "Elucidating Mechanisms of Toxicity Using Phenotypic Data from Primary Human Cell Systems—A Chemical Biology Approach for Thrombosis-Related Side Effects" International Journal of Molecular Sciences 16, no. 1: 1008-1029. https://doi.org/10.3390/ijms16011008
APA StyleBerg, E. L., Polokoff, M. A., O'Mahony, A., Nguyen, D., & Li, X. (2015). Elucidating Mechanisms of Toxicity Using Phenotypic Data from Primary Human Cell Systems—A Chemical Biology Approach for Thrombosis-Related Side Effects. International Journal of Molecular Sciences, 16(1), 1008-1029. https://doi.org/10.3390/ijms16011008