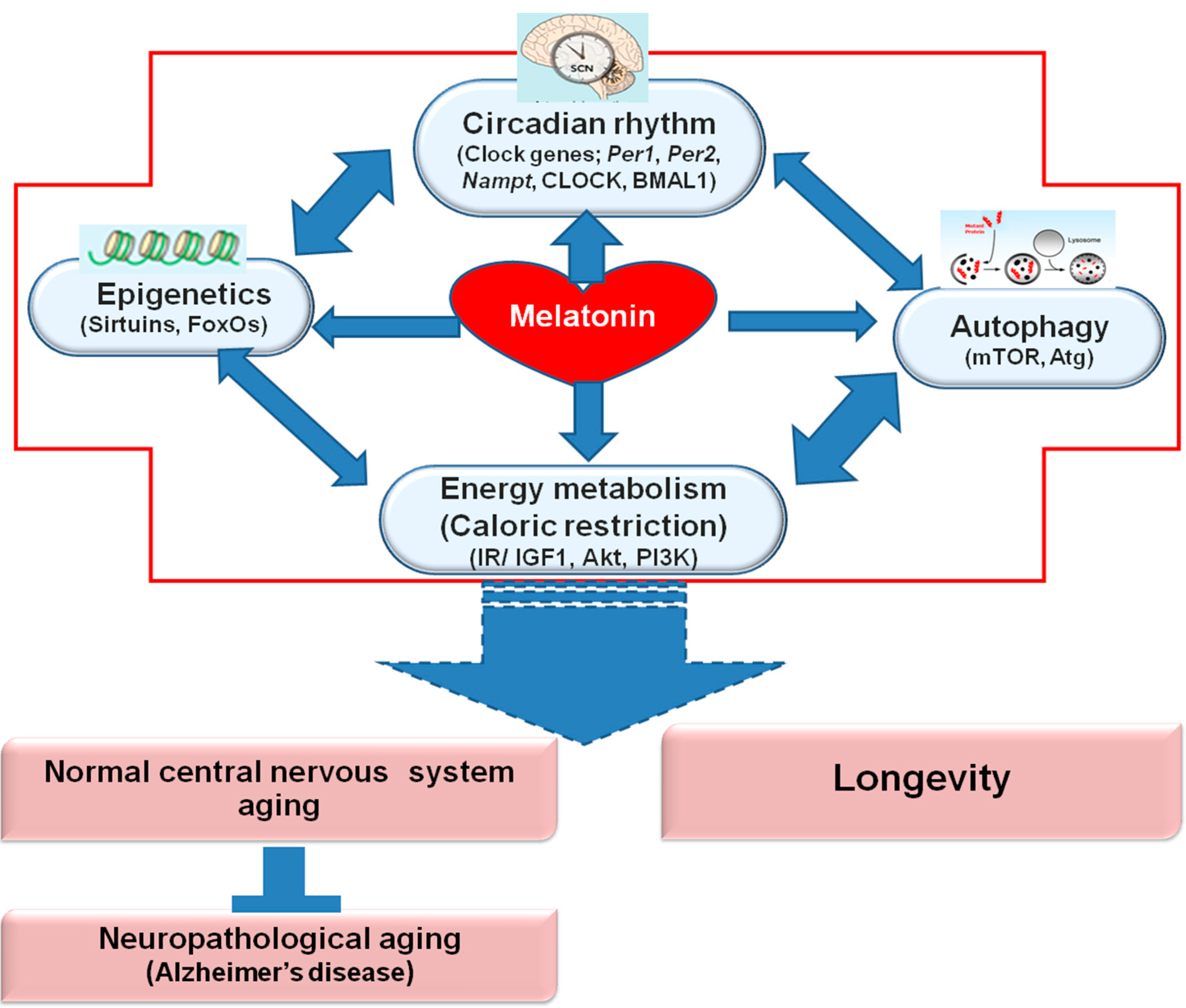
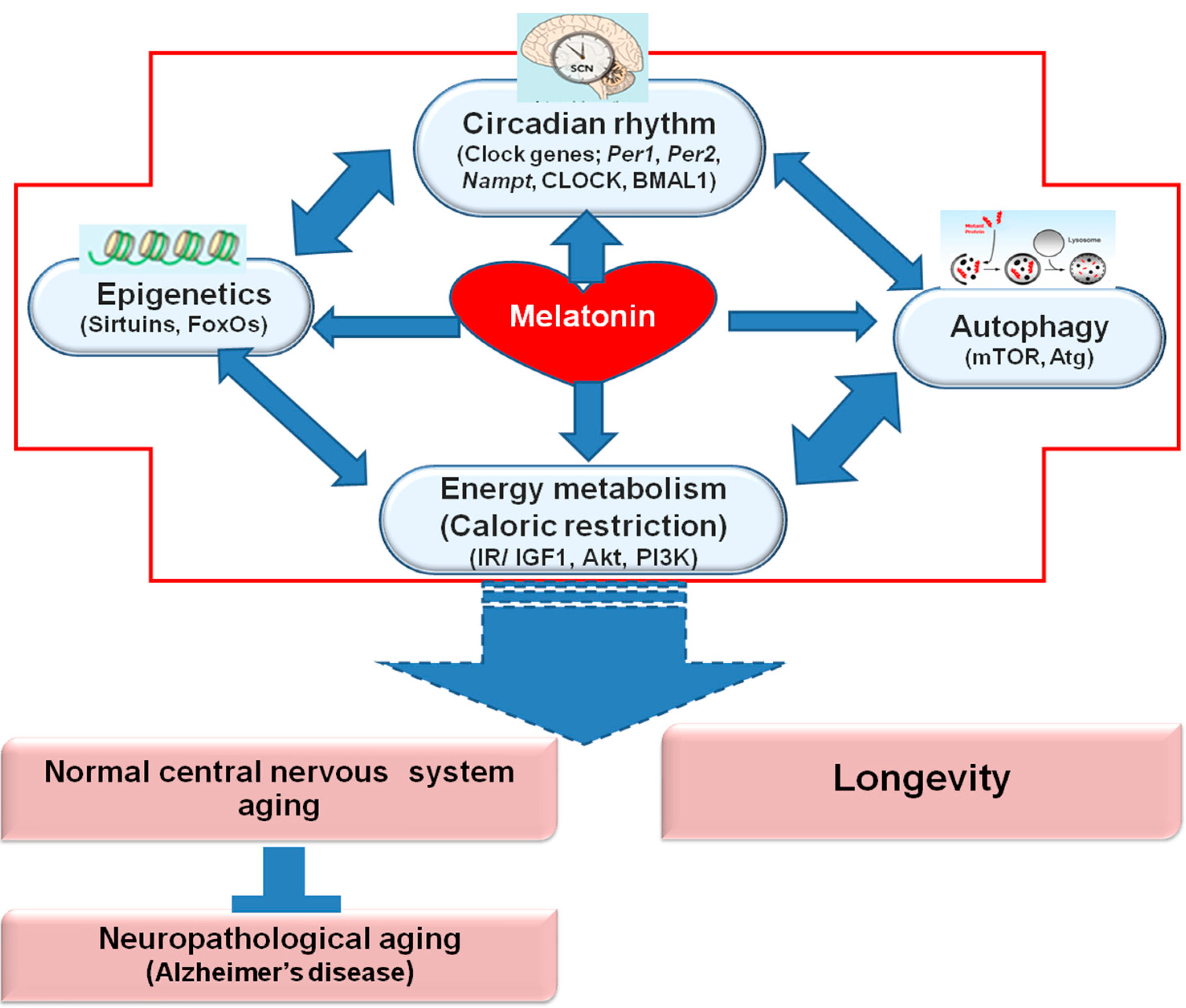
Melatonin Regulates Aging and Neurodegeneration through Energy Metabolism, Epigenetics, Autophagy and Circadian Rhythm Pathways

{kind=link}
Abstract
:1. Introduction
2. Brain Energy Metabolism
2.1. Insulin/IGF-1 (Insulin-Like Growth Factor 1) Signaling Pathways and Brain Energy
2.2. Melatonin and Metabolic Pathways
2.3. Epigenetics and Aging
2.4. Sirtuins
2.5. Forkhead Box O (FoxO)
2.6. Melatonin and Epigenetics
2.7. Melatonin and Sirtuin System
3. Autophagy
3.1. Autophagy and the Aging Process
3.2. Autophagy and Sirtuin Pathways
3.3. Autophagy and Caloric Restriction (CR)
3.4. Autophagy and Neuroinflammation
3.5. Melatonin and Autophagy
4. The Circadian System
4.1. Melatonin and the Regulation of Clock Genes
4.2. Epigenetic Regulation of Clock Genes
4.3. Connection among the Circadian Clock, Epigenetic Variation and Metabolism
4.4. Melatonin, Circadian Clock and Aging
4.5. Circadian Regulation and Autophagy
4.6. Role of Melatonin and SIRT1 as Circadian Modulators in Memory Processing
5. Conclusions
Acknowledgments
Conflicts of Interest
References
- Hardeland, R. Melatonin, hormone of darkness and more: Occurrence, control mechanisms, actions and bioactive metabolites. Cell. Mol. Life Sci. 2008, 65, 2001–2018. [Google Scholar] [CrossRef] [PubMed]
- Reiter, R.J. Melatonin: The chemical expression of darkness. Mol. Cell Endocrinol. 1991, 79, C153–C158. [Google Scholar] [CrossRef] [PubMed]
- Ostrowska, Z.; Kos-Kudla, B.; Swietochowska, E.; Marek, B.; Kajdaniuk, D.; Ciesielska-Kopacz, N. Influence of pinealectomy and long-term melatonin administration on GH-IGF-I axis function in male rats. Neuro. Endocrinol. Lett. 2001, 22, 255–262. [Google Scholar] [PubMed]
- Vriend, J.; Sheppard, M.S.; Borer, K.T. Melatonin increases serum growth hormone and insulin-like growth factor I (IGF-I) levels in male Syrian hamsters via hypothalamic neurotransmitters. Growth Dev. Aging 1990, 54, 165–171. [Google Scholar] [PubMed]
- Gutierrez-Cuesta, J.; Tajes, M.; Jimenez, A.; Coto-Montes, A.; Camins, A.; Pallas, M. Evaluation of potential pro-survival pathways regulated by melatonin in a murine senescence model. J. Pineal Res. 2008, 45, 497–505. [Google Scholar] [CrossRef] [PubMed]
- Tajes, M.; Gutierrez-Cuesta, J.; Ortuno-Sahagun, D.; Camins, A.; Pallas, M. Anti-aging properties of melatonin in an in vitro murine senescence model: Involvement of the sirtuin 1 pathway. J. Pineal Res. 2009, 47, 228–237. [Google Scholar] [CrossRef] [PubMed]
- Cristofol, R.; Porquet, D.; Corpas, R.; Coto-Montes, A.; Serret, J.; Camins, A.; Pallas, M.; Sanfeliu, C. Neurons from senescence-accelerated SAMP8 mice are protected against frailty by the sirtuin 1 promoting agents melatonin and resveratrol. J. Pineal Res. 2012, 52, 271–281. [Google Scholar] [CrossRef] [PubMed]
- Spielman, L.J.; Little, J.P.; Klegeris, A. Inflammation and insulin/IGF-1 resistance as the possible link between obesity and neurodegeneration. J. Neuroimmunol. 2014, 273, 8–21. [Google Scholar] [CrossRef] [PubMed]
- Long, Y.C.; Tan, T.M.; Takao, I.; Tang, B.L. The biochemistry and cell biology of aging: Metabolic regulation through mitochondrial signaling. Am. J. Physiol. Endocrinol. MeTab. 2014, 306, E581–E591. [Google Scholar] [CrossRef] [PubMed]
- Yin, F.; Jiang, T.; Cadenas, E. Metabolic triad in brain aging: Mitochondria, insulin/IGF-1 signalling and JNK signalling. Biochem. Soc. Trans. 2013, 41, 101–105. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Tindall, D.J. Dynamic FoxO transcription factors. J. Cell Sci. 2007, 120, 2479–2487. [Google Scholar] [CrossRef] [PubMed]
- Aberg, N.D.; Brywe, K.G.; Isgaard, J. Aspects of growth hormone and insulin-like growth factor-I related to neuroprotection, regeneration, and functional plasticity in the adult brain. Sci. World J. 2006, 6, 53–80. [Google Scholar] [CrossRef]
- Wada, A.; Yokoo, H.; Yanagita, T.; Kobayashi, H. New twist on neuronal insulin receptor signaling in health, disease, and therapeutics. J. Pharmacol. Sci. 2005, 99, 128–143. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.Y.; Al-Regaiey, K.; Masternak, M.M.; Wang, J.; Bartke, A. Local expression of GH and IGF-1 in the hippocampus of GH-deficient long-lived mice. Neurobiol. Aging 2005, 26, 929–937. [Google Scholar] [CrossRef] [PubMed]
- Bartke, A. Impact of reduced insulin-like growth factor-1/insulin signaling on aging in mammals: Novel findings. Aging Cell 2008, 7, 285–290. [Google Scholar] [CrossRef] [PubMed]
- Simpson, J.E.; Ince, P.G.; Shaw, P.J.; Heath, P.R.; Raman, R.; Garwood, C.J.; Gelsthorpe, C.; Baxter, L.; Forster, G.; Matthews, F.E.; et al. Microarray analysis of the astrocyte transcriptome in the aging brain: Relationship to Alzheimer’s pathology and APOE genotype. Neurobiol. Aging 2011, 32, 1795–1807. [Google Scholar] [CrossRef] [PubMed]
- O’Neill, C.; Kiely, A.P.; Coakley, M.F.; Manning, S.; Long-Smith, C.M. Insulin and IGF-1 signalling: Longevity, protein homoeostasis and Alzheimer's disease. Biochem. Soc. Trans. 2012, 40, 721–727. [Google Scholar] [CrossRef] [PubMed]
- Westwood, A.J.; Beiser, A.; Decarli, C.; Harris, T.B.; Chen, T.C.; He, X.M.; Roubenoff, R.; Pikula, A.; Au, R.; Braverman, L.E.; et al. Insulin-like growth factor-1 and risk of Alzheimer dementia and brain atrophy. Neurology 2014, 82, 1613–1619. [Google Scholar] [CrossRef] [PubMed]
- Trueba-Saiz, A.; Cavada, C.; Fernandez, A.M.; Leon, T.; Gonzalez, D.A.; Fortea Ormaechea, J.; Lleo, A.; del Ser, T.; Nunez, A.; Torres-Aleman, I. Loss of serum IGF-I input to the brain as an early biomarker of disease onset in Alzheimer mice. Transl. Psychiatry 2013, 3. [Google Scholar] [CrossRef]
- De Bruijn, R.F.; Janssen, J.A.; Brugts, M.P.; van Duijn, C.M.; Hofman, A.; Koudstaal, P.J.; Ikram, M.A. Insulin-like growth factor-I receptor stimulating activity is associated with dementia. J. Alzheimers Dis. 2014, 42, 137–142. [Google Scholar]
- Zemva, J.; Schubert, M. The role of neuronal insulin/insulin-like growth factor-1 signaling for the pathogenesis of Alzheimer’s disease: Possible therapeutic implications. CNS Neurol. Disord. Drug Targets 2014, 13, 322–337. [Google Scholar] [CrossRef] [PubMed]
- Desai, G.; Zheng, C.; Geetha, T.; Mathews, S.T.; White, B.D.; Huggins, K.W.; Zizza, C.A.; Broderick, T.L.; Babu, J.R. The pancreas-brain axis: Insight into disrupted mechanisms associating type 2 diabetes and Alzheimer’s disease. J. Alzheimers Dis. 2014, 42, 347–356. [Google Scholar]
- Kim, J.H.; Choi, J.S.; Lee, B.H. PI3K/Akt and MAPK pathways evoke activation of FoxO transcription factor to undergo neuronal apoptosis in brain of the silkworm Bombyx mori (Lepidoptera: Bombycidae). Cell Mol. Biol. 2012, 58 (Suppl.), OL1780–OL1785. [Google Scholar]
- Cheng, Z.; White, M.F. Targeting Forkhead box O1 from the concept to metabolic diseases: Lessons from mouse models. Antioxid. Redox Signal. 2011, 14, 649–661. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Kihara, T.; Akaike, A.; Niidome, T.; Sugimoto, H. PI3K/Akt/mTOR signaling regulates glutamate transporter 1 in astrocytes. Biochem. Biophys. Res. Commun. 2010, 393, 514–518. [Google Scholar] [CrossRef] [PubMed]
- Erren, T.C.; Reiter, R.J. A generalized theory of carcinogenesis due to chronodisruption. Neuro Endocrinol. Lett. 2008, 29, 815–821. [Google Scholar] [PubMed]
- Jung, B.; Ahmad, N. Melatonin in cancer management: Progress and promise. Cancer Res. 2006, 66, 9789–9793. [Google Scholar] [CrossRef] [PubMed]
- Peyrot, F.; Ducrocq, C. Potential role of tryptophan derivatives in stress responses characterized by the generation of reactive oxygen and nitrogen species. J. Pineal Res. 2008, 45, 235–246. [Google Scholar] [CrossRef] [PubMed]
- Tan, D.X.; Manchester, L.C.; Terron, M.P.; Flores, L.J.; Reiter, R.J. One molecule, many derivatives: A never-ending interaction of melatonin with reactive oxygen and nitrogen species? J. Pineal Res. 2007, 42, 28–42. [Google Scholar] [CrossRef]
- Puchalski, S.S.; Green, J.N.; Rasmussen, D.D. Melatonin effect on rat body weight regulation in response to high-fat diet at middle age. Endocrine 2003, 21, 163–167. [Google Scholar] [CrossRef] [PubMed]
- Wolden-Hanson, T.; Mitton, D.R.; McCants, R.L.; Yellon, S.M.; Wilkinson, C.W.; Matsumoto, A.M.; Rasmussen, D.D. Daily melatonin administration to middle-aged male rats suppresses body weight, intraabdominal adiposity, and plasma leptin and insulin independent of food intake and total body fat. Endocrinology 2000, 141, 487–497. [Google Scholar] [PubMed]
- She, M.; Deng, X.; Guo, Z.; Laudon, M.; Hu, Z.; Liao, D.; Hu, X.; Luo, Y.; Shen, Q.; Su, Z.; et al. NEU-P11, a novel melatonin agonist, inhibits weight gain and improves insulin sensitivity in high-fat/high-sucrose-fed rats. Pharmacol. Res. 2009, 59, 248–253. [Google Scholar] [CrossRef] [PubMed]
- Zanquetta, M.M.; Seraphim, P.M.; Sumida, D.H.; Cipolla-Neto, J.; Machado, U.F. Calorie restriction reduces pinealectomy-induced insulin resistance by improving GLUT4 gene expression and its translocation to the plasma membrane. J. Pineal Res. 2003, 35, 141–148. [Google Scholar] [CrossRef] [PubMed]
- Peschke, E. Melatonin, endocrine pancreas and diabetes. J. Pineal Res. 2008, 44, 26–40. [Google Scholar] [PubMed]
- Mantele, S.; Otway, D.T.; Middleton, B.; Bretschneider, S.; Wright, J.; Robertson, M.D.; Skene, D.J.; Johnston, J.D. Daily rhythms of plasma melatonin, but not plasma leptin or leptin mRNA, vary between lean, obese and type 2 diabetic men. PLoS One 2012, 7, e37123. [Google Scholar] [CrossRef] [PubMed]
- Srinivasan, V.; Ohta, Y.; Espino, J.; Pariente, J.A.; Rodriguez, A.B.; Mohamed, M.; Zakaria, R. Metabolic syndrome, its pathophysiology and the role of melatonin. Recent Pat. Endocr. Metab. Immune Drug Discov. 2013, 7, 11–25. [Google Scholar] [CrossRef] [PubMed]
- Piccinetti, C.C.; Migliarini, B.; Olivotto, I.; Simoniello, M.P.; Giorgini, E.; Carnevali, O. Melatonin and peripheral circuitries: Insights on appetite and metabolism in Danio rerio. Zebrafish 2013, 10, 275–282. [Google Scholar] [CrossRef] [PubMed]
- Marques, F.Z.; Markus, M.A.; Morris, B.J. The molecular basis of longevity, and clinical implications. Maturitas 2010, 65, 87–91. [Google Scholar] [CrossRef] [PubMed]
- Vaquero, A.; Reinberg, D. Calorie restriction and the exercise of chromatin. Genes Dev. 2009, 23, 1849–1869. [Google Scholar] [CrossRef] [PubMed]
- Berdichevsky, A.; Viswanathan, M.; Horvitz, H.R.; Guarente, L. C. elegans SIR-2.1 interacts with 14-3-3 proteins to activate DAF-16 and extend life span. Cell 2006, 125, 1165–1177. [Google Scholar]
- Berryman, D.E.; Christiansen, J.S.; Johannsson, G.; Thorner, M.O.; Kopchick, J.J. Role of the GH/IGF-1 axis in lifespan and healthspan: Lessons from animal models. Growth Horm. IGF Res. 2008, 18, 455–471. [Google Scholar] [CrossRef] [PubMed]
- Hekimi, S.; Guarente, L. Genetics and the specificity of the aging process. Science 2003, 299, 1351–1354. [Google Scholar] [CrossRef] [PubMed]
- Weeks, K.R.; Dwyer, D.S.; Aamodt, E.J. Antipsychotic drugs activate the C. elegans Akt pathway via the DAF-2 insulin/IGF-1 receptor. ACS Chem. Neurosci. 2010; 1, 463–473. [Google Scholar]
- Liang, R.; Khanna, A.; Muthusamy, S.; Li, N.; Sarojini, H.; Kopchick, J.J.; Masternak, M.M.; Bartke, A.; Wang, E. Post-transcriptional regulation of IGF1R by key microRNAs in long-lived mutant mice. Aging Cell 2011, 10, 1080–1088. [Google Scholar] [CrossRef] [PubMed]
- Swindell, W.R. Gene expression profiling of long-lived dwarf mice: Longevity-associated genes and relationships with diet, gender and aging. BMC Genomics 2007, 8. [Google Scholar] [CrossRef] [PubMed]
- Shimokawa, I.; Trindade, L.S. Dietary restriction and aging in rodents: A current view on its molecular mechanisms. Aging Dis. 2010, 1, 89–107. [Google Scholar]
- Nishida, S.; Sato, R.; Murai, I.; Nakagawa, S. Effect of pinealectomy on plasma levels of insulin and leptin and on hepatic lipids in type 2 diabetic rats. J. Pineal Res. 2003, 35, 251–256. [Google Scholar] [CrossRef] [PubMed]
- Nduhirabandi, F.; du Toit, E.F.; Lochner, A. Melatonin and the metabolic syndrome: A tool for effective therapy in obesity-associated abnormalities? Acta Physiol. 2012, 205, 209–223. [Google Scholar] [CrossRef]
- Ge, D.; Dauchy, R.T.; Liu, S.; Zhang, Q.; Mao, L.; Dauchy, E.M.; Blask, D.E.; Hill, S.M.; Rowan, B.G.; Brainard, G.C.; et al. Insulin and IGF1 enhance IL-17-induced chemokine expression through a GSK3B-dependent mechanism: A new target for melatonin’s anti-inflammatory action. J. Pineal Res. 2013, 55, 377–387. [Google Scholar] [PubMed]
- Nassar, E.; Mulligan, C.; Taylor, L.; Kerksick, C.; Galbreath, M.; Greenwood, M.; Kreider, R.; Willoughby, D.S. Effects of a single dose of N-Acetyl-5-methoxytryptamine (Melatonin) and resistance exercise on the growth hormone/IGF-1 axis in young males and females. J. Int. Soc. Sports Nutr. 2007, 4. [Google Scholar] [CrossRef] [PubMed]
- Koh, P.O. Melatonin prevents the injury-induced decline of Akt/forkhead transcription factors phosphorylation. J. Pineal Res. 2008, 45, 199–203. [Google Scholar] [PubMed]
- Lee, S.H.; Chun, W.; Kong, P.J.; Han, J.A.; Cho, B.P.; Kwon, O.Y.; Lee, H.J.; Kim, S.S. Sustained activation of Akt by melatonin contributes to the protection against kainic acid-induced neuronal death in hippocampus. J. Pineal Res. 2006, 40, 79–85. [Google Scholar] [CrossRef] [PubMed]
- Tajes Orduna, M.; Pelegri Gabalda, C.; Vilaplana Hortensi, J.; Pallas Lliberia, M.; Camins Espuny, A. An evaluation of the neuroprotective effects of melatonin in an in vitro experimental model of age-induced neuronal apoptosis. J. Pineal Res. 2009, 46, 262–267. [Google Scholar]
- Calvanese, V.; Lara, E.; Kahn, A.; Fraga, M.F. The role of epigenetics in aging and age-related diseases. Ageing Res. Rev. 2009, 8, 268–276. [Google Scholar] [CrossRef] [PubMed]
- Bandyopadhyay, D.; Medrano, E.E. The emerging role of epigenetics in cellular and organismal aging. Exp. Gerontol. 2003, 38, 1299–1307. [Google Scholar] [CrossRef] [PubMed]
- Munoz-Najar, U.; Sedivy, J.M. Epigenetic control of aging. Antioxid. Redox Signal. 2011, 14, 241–259. [Google Scholar] [CrossRef] [PubMed]
- Coppieters, N.; Dieriks, B.V.; Lill, C.; Faull, R.L.; Curtis, M.A.; Dragunow, M. Global changes in DNA methylation and hydroxymethylation in Alzheimer’s disease human brain. Neurobiol. Aging 2014, 35, 1334–1344. [Google Scholar] [CrossRef] [PubMed]
- Dagnas, M.; Mons, N. Region- and age-specific patterns of histone acetylation related to spatial and cued learning in the water maze. Hippocampus 2013, 23, 581–591. [Google Scholar] [PubMed]
- Dos Santos Sant’ Anna, G.; Rostirola Elsner, V.; Moyses, F.; Reck Cechinel, L.; Agustini Lovatel, G.; Rodrigues Siqueira, I. Histone deacetylase activity is altered in brain areas from aged rats. Neurosci. Lett. 2013, 556, 152–154. [Google Scholar]
- Sen, N. Epigenetic regulation of memory by acetylation and methylation of chromatin: Implications in neurological disorders, aging, and addiction. Neuromolecul. Med. 2014. [Google Scholar] [CrossRef]
- Wang, C.M.; Tsai, S.N.; Yew, T.W.; Kwan, Y.W.; Ngai, S.M. Identification of histone methylation multiplicities patterns in the brain of senescence-accelerated prone mouse 8. Biogerontology 2010, 11, 87–102. [Google Scholar] [CrossRef] [PubMed]
- Cosin-Tomas, M.; Alvarez-Lopez, M.J.; Sanchez-Roige, S.; Lalanza, J.F.; Bayod, S.; Sanfeliu, C.; Pallas, M.; Escorihuela, R.M.; Kaliman, P. Epigenetic alterations in hippocampus of SAMP8 senescent mice and modulation by voluntary physical exercise. Front. Aging Neurosci. 2014, 6. [Google Scholar] [CrossRef] [PubMed]
- Dagnas, M.; Guillou, J.L.; Prevot, T.; Mons, N. HDAC inhibition facilitates the switch between memory systems in young but not aged mice. J. Neurosci. 2013, 33, 1954–1963. [Google Scholar] [CrossRef] [PubMed]
- Castellano, J.F.; Fletcher, B.R.; Patzke, H.; Long, J.M.; Sewal, A.; Kim, D.H.; Kelley-Bell, B.; Rapp, P.R. Reassessing the effects of histone deacetylase inhibitors on hippocampal memory and cognitive aging. Hippocampus 2014, 24, 1006–1016. [Google Scholar] [CrossRef] [PubMed]
- Tomas Pereira, I.; Coletta, C.E.; Perez, E.V.; Kim, D.H.; Gallagher, M.; Goldberg, I.G.; Rapp, P.R. CREB-binding protein levels in the rat hippocampus fail to predict chronological or cognitive aging. Neurobiol. Aging 2013, 34, 832–844. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Xiao, H.; Isobe, K. Histone acetyltransferase activities of cAMP-regulated enhancer-binding protein and p300 in tissues of fetal, young, and old mice. J. Gerontol. A Biol. Sci. Med. Sci. 2002, 57, B93–B98. [Google Scholar] [CrossRef] [PubMed]
- Bousiges, O.; Vasconcelos, A.P.; Neidl, R.; Cosquer, B.; Herbeaux, K.; Panteleeva, I.; Loeffler, J.P.; Cassel, J.C.; Boutillier, A.L. Spatial memory consolidation is associated with induction of several lysine-acetyltransferase (histone acetyltransferase) expression levels and H2B/H4 acetylation-dependent transcriptional events in the rat hippocampus. Neuropsychopharmacology 2010, 35, 2521–2537. [Google Scholar] [CrossRef] [PubMed]
- Brown, R.; Imran, S.A.; Ur, E.; Wilkinson, M. Valproic acid and CEBPα-mediated regulation of adipokine gene expression in hypothalamic neurons and 3T3-L1 adipocytes. Neuroendocrinology 2008, 88, 25–34. [Google Scholar] [CrossRef] [PubMed]
- Hodgson, N.; Trivedi, M.; Muratore, C.; Li, S.; Deth, R. Soluble oligomers of amyloid-β cause changes in redox state, DNA methylation, and gene transcription by inhibiting EAAT3 mediated cysteine uptake. J. Alzheimers Dis. 2013, 36, 197–209. [Google Scholar] [PubMed]
- Bardai, F.H.; Price, V.; Zaayman, M.; Wang, L.; D’Mello, S.R. Histone deacetylase-1 (HDAC1) is a molecular switch between neuronal survival and death. J. Biol. Chem. 2012, 287, 35444–35453. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, T.; Guarente, L. Sirtuins at a glance. J. Cell Sci. 2011, 124, 833–838. [Google Scholar] [CrossRef] [PubMed]
- Braidy, N.; Guillemin, G.J.; Mansour, H.; Chan-Ling, T.; Poljak, A.; Grant, R. Age related changes in NAD+ metabolism oxidative stress and Sirt1 activity in wistar rats. PLoS One 2011, 6, e19194. [Google Scholar] [PubMed]
- Imai, S. Dissecting systemic control of metabolism and aging in the NAD World: The importance of SIRT1 and NAMPT-mediated NAD biosynthesis. FEBS Lett. 2011, 585, 1657–1662. [Google Scholar] [CrossRef] [PubMed]
- Zakhary, S.M.; Ayubcha, D.; Dileo, J.N.; Jose, R.; Leheste, J.R.; Horowitz, J.M.; Torres, G. Distribution analysis of deacetylase SIRT1 in rodent and human nervous systems. Anat. Rec. Hoboken 2010, 293, 1024–1032. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, T.; Kitamura, T. Roles of FoxO1 and Sirt1 in the central regulation of food intake. Endocr. J. 2010, 57, 939–946. [Google Scholar] [CrossRef] [PubMed]
- Cakir, I.; Perello, M.; Lansari, O.; Messier, N.J.; Vaslet, C.A.; Nillni, E.A. Hypothalamic Sirt1 regulates food intake in a rodent model system. PLoS One 2009, 4, e8322. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, T.; Kim, H.J.; Kobayashi, M.; Kitamura, Y.I.; Yokota-Hashimoto, H.; Shiuchi, T.; Minokoshi, Y.; Kitamura, T. Induction of hypothalamic Sirt1 leads to cessation of feeding via agouti-related peptide. Endocrinology 2010, 151, 2556–2566. [Google Scholar] [CrossRef] [PubMed]
- Greco, S.J.; Hamzelou, A.; Johnston, J.M.; Smith, M.A.; Ashford, J.W.; Tezapsidis, N. Leptin boosts cellular metabolism by activating AMPK and the sirtuins to reduce tau phosphorylation and β-amyloid in neurons. Biochem. Biophys. Res. Commun. 2011, 414, 170–174. [Google Scholar] [CrossRef]
- Dietrich, M.O.; Antunes, C.; Geliang, G.; Liu, Z.W.; Borok, E.; Nie, Y.; Xu, A.W.; Souza, D.O.; Gao, Q.; Diano, S.; et al. Agrp neurons mediate Sirt1’s action on the melanocortin system and energy balance: Roles for Sirt1 in neuronal firing and synaptic plasticity. J. Neurosci. 2010, 30, 11815–11825. [Google Scholar] [CrossRef] [PubMed]
- Parmentier, F.; Lejeune, F.X.; Neri, C. Pathways to decoding the clinical potential of stress response FOXO-interaction networks for Huntington’s disease: Of gene prioritization and context dependence. Front. Aging Neurosci. 2013, 5. [Google Scholar] [CrossRef] [PubMed]
- Ro, S.H.; Liu, D.; Yeo, H.; Paik, J.H. FoxOs in neural stem cell fate decision. Arch. Biochem. Biophys. 2013, 534, 55–63. [Google Scholar] [CrossRef] [PubMed]
- Shimokawa, I.; Higami, Y. Leptin signaling and aging: Insight from caloric restriction. Mech. Ageing Dev. 2001, 122, 1511–1519. [Google Scholar] [CrossRef]
- Kim, D.H.; Zhang, T.; Lee, S.; Dong, H.H. FoxO6 in glucose metabolism (FoxO6). J. Diabetes 2013, 5, 233–240. [Google Scholar] [CrossRef] [PubMed]
- Chaves, I.; van der Horst, G.T.; Schellevis, R.; Nijman, R.M.; Koerkamp, M.G.; Holstege, F.C.; Smidt, M.P.; Hoekman, M.F. Insulin-FOXO3 signaling modulates circadian rhythms via regulation of clock transcription. Curr. Biol. 2014, 24, 1248–1255. [Google Scholar] [CrossRef] [PubMed]
- Moll, Lorna; Schubert, M. The role of insulin and insulin-like growth factor-1/FoxO-mediated transcription for the pathogenesis of obesity-associated dementia. Curr. Gerontol. Geriatr. Res. 2012, 2012, 1–13. [Google Scholar]
- Salih, D.A.; Rashid, A.J.; Colas, D.; de la Torre-Ubieta, L.; Zhu, R.P.; Morgan, A.A.; Santo, E.E.; Ucar, D.; Devarajan, K.; Cole, C.J.; et al. FoxO6 regulates memory consolidation and synaptic function. Genes Dev. 2012, 26, 2780–2801. [Google Scholar] [CrossRef]
- Exil, V.; Ping, L.; Yu, Y.; Chakraborty, S.; Caito, S.W.; Wells, K.S.; Karki, P.; Lee, E.; Aschner, M. Activation of MAPK and FoxO by manganese (Mn) in rat neonatal primary astrocyte cultures. PLoS One 2014, 9, e94753. [Google Scholar] [CrossRef] [PubMed]
- Akhter, R.; Sanphui, P.; Biswas, S.C. The essential role of p53-up-regulated modulator of apoptosis (Puma) and its regulation by FoxO3a transcription factor in β-amyloid-induced neuron death. J. Biol. Chem. 2014, 289, 10812–10822. [Google Scholar] [CrossRef] [PubMed]
- Pino, E.; Amamoto, R.; Zheng, L.; Cacquevel, M.; Sarria, J.C.; Knott, G.W.; Schneider, B.L. FOXO3 determines the accumulation of α-synuclein and controls the fate of dopaminergic neurons in the substantia nigra. Hum. Mol. Genet. 2014, 23, 1435–1452. [Google Scholar] [CrossRef] [PubMed]
- Mediavilla, M.D.; Sanchez-Barcelo, E.J.; Tan, D.X.; Manchester, L.; Reiter, R.J. Basic mechanisms involved in the anti-cancer effects of melatonin. Curr. Med. Chem. 2010, 17, 4462–4481. [Google Scholar] [PubMed]
- Korkmaz, A.; Sanchez-Barcelo, E.J.; Tan, D.X.; Reiter, R.J. Role of melatonin in the epigenetic regulation of breast cancer. Breast Cancer Res. Treat. 2009, 115, 13–27. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.E.; Kim, S.J.; Yoon, H.J.; Yu, S.Y.; Yang, H.; Jeong, S.I.; Hwang, S.Y.; Park, C.S.; Park, Y.S. Genome-wide profiling in melatonin-exposed human breast cancer cell lines identifies differentially methylated genes involved in the anticancer effect of melatonin. J. Pineal Res. 2013, 54, 80–88. [Google Scholar]
- Nakamura, E.; Kozaki, K.; Tsuda, H.; Suzuki, E.; Pimkhaokham, A.; Yamamoto, G.; Irie, T.; Tachikawa, T.; Amagasa, T.; Inazawa, J.; et al. Frequent silencing of a putative tumor suppressor gene melatonin receptor 1 A (MTNR1A) in oral squamous-cell carcinoma. Cancer Sci. 2008, 99, 1390–1400. [Google Scholar] [PubMed]
- Tiao, M.M.; Huang, L.T.; Chen, C.J.; Sheen, J.M.; Tain, Y.L.; Chen, C.C.; Kuo, H.C.; Huang, Y.H.; Tang, K.S.; Chu, E.W.; et al. Melatonin in the Regulation of Liver Steatosis following Prenatal Glucocorticoid Exposure. Biomed. Res. Int. 2014, 2014. [Google Scholar] [CrossRef]
- Tain, Y.L.; Chen, C.C.; Sheen, J.M.; Yu, H.R.; Tiao, M.M.; Kuo, H.C.; Huang, L.T. Melatonin attenuates prenatal dexamethasone-induced blood pressure increase in a rat model. J. Am. Soc. Hypertens. 2014, 8, 216–226. [Google Scholar]
- Sharma, R.; Ottenhof, T.; Rzeczkowska, P.A.; Niles, L.P. Epigenetic targets for melatonin: Induction of histone H3 hyperacetylation and gene expression in C17.2 neural stem cells. J. Pineal Res. 2008, 45, 277–284. [Google Scholar] [CrossRef]
- Kim, B.; Rincon Castro, L.M.; Jawed, S.; Niles, L.P. Clinically relevant concentrations of valproic acid modulate melatonin MT(1) receptor, HDAC and MeCP2 mRNA expression in C6 glioma cells. Eur. J. Pharmacol. 2008, 589, 45–48. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Xiao, X.; Zhang, Y.; Shi, D.; Chen, W.; Fu, L.; Liu, L.; Xie, F.; Kang, T.; Huang, W.; et al. Simultaneous modulation of COX-2, p300, Akt, and Apaf-1 signaling by melatonin to inhibit proliferation and induce apoptosis in breast cancer cells. J. Pineal Res. 2012, 53, 77–90. [Google Scholar] [CrossRef] [PubMed]
- Shi, D.; Xiao, X.; Wang, J.; Liu, L.; Chen, W.; Fu, L.; Xie, F.; Huang, W.; Deng, W. Melatonin suppresses proinflammatory mediators in lipopolysaccharide-stimulated CRL1999 cells via targeting MAPK, NF-κB, c/EBPβ, and p300 signaling. J. Pineal Res. 2012, 53, 154–165. [Google Scholar] [CrossRef] [PubMed]
- Permpoonputtana, K.; Govitrapong, P. The anti-inflammatory effect of melatonin on methamphetamine-induced proinflammatory mediators in human neuroblastoma dopamine SH-SY5Y cell lines. Neurotox. Res. 2013, 23, 189–199. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Ma, C.; Meng, C.J.; Zhu, G.Q.; Sun, X.B.; Huo, L.; Zhang, J.; Liu, H.X.; He, W.C.; Shen, X.M.; et al. Melatonin activates the Nrf2-ARE pathway when it protects against early brain injury in a subarachnoid hemorrhage model. J. Pineal Res. 2012, 53, 129–137. [Google Scholar] [CrossRef] [PubMed]
- Negi, G.; Kumar, A.; Sharma, S.S. Melatonin modulates neuroinflammation and oxidative stress in experimental diabetic neuropathy: Effects on NF-κB and Nrf2 cascades. J. Pineal Res. 2011, 50, 124–131. [Google Scholar] [PubMed]
- Sun, Z.; Chin, Y.E.; Zhang, D.D. Acetylation of Nrf2 by p300/CBP augments promoter-specific DNA binding of Nrf2 during the antioxidant response. Mol. Cell Biol. 2009, 29, 2658–2672. [Google Scholar] [CrossRef] [PubMed]
- Korkmaz, A.; Rosales-Corral, S.; Reiter, R.J. Gene regulation by melatonin linked to epigenetic phenomena. Gene 2012, 503, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Chang, H.M.; Wu, U.I.; Lan, C.T. Melatonin preserves longevity protein (sirtuin 1) expression in the hippocampus of total sleep-deprived rats. J. Pineal Res. 2009, 47, 211–220. [Google Scholar] [CrossRef] [PubMed]
- Jung-Hynes, B.; Schmit, T.L.; Reagan-Shaw, S.R.; Siddiqui, I.A.; Mukhtar, H.; Ahmad, N. Melatonin, a novel Sirt1 inhibitor, imparts antiproliferative effects against prostate cancer in vitro in culture and in vivo in TRAMP model. J. Pineal Res. 2011, 50, 140–149. [Google Scholar] [PubMed]
- Jung-Hynes, B.; Reiter, R.J.; Ahmad, N. Sirtuins, melatonin and circadian rhythms: Building a bridge between aging and cancer. J. Pineal Res. 2010, 48, 9–19. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; Cai, L.; Jiang, P.; Wang, J.; Gao, C.; Feng, H.; Wang, C.; Pan, H.; Yang, Y. SIRT1 inhibition by melatonin exerts antitumor activity in human osteosarcoma cells. Eur. J. Pharmacol. 2013, 715, 219–229. [Google Scholar] [CrossRef] [PubMed]
- Tan, D.X.; Manchester, L.C.; Sainz, R.M.; Mayo, J.C.; Leon, J.; Hardeland, R.; Poeggeler, B.; Reiter, R.J. Interactions between melatonin and nicotinamide nucleotide: NADH preservation in cells and in cell-free systems by melatonin. J. Pineal Res. 2005, 39, 185–194. [Google Scholar] [CrossRef] [PubMed]
- Swerdlow, R.H. Brain aging, Alzheimer’s disease, and mitochondria. Biochim. Biophys. Acta 2011, 1812, 1630–1639. [Google Scholar] [CrossRef]
- Duan, W. Sirtuins: From metabolic regulation to brain aging. Front. Aging Neurosci. 2013, 5. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N.; Levine, B.; Cuervo, A.M.; Klionsky, D.J. Autophagy fights disease through cellular self-digestion. Nature 2008, 451, 1069–1075. [Google Scholar] [CrossRef] [PubMed]
- Inoki, K.; Corradetti, M.N.; Guan, K.L. Dysregulation of the TSC-mTOR pathway in human disease. Nat. Genet. 2005, 37, 19–24. [Google Scholar] [CrossRef] [PubMed]
- Todde, V.; Veenhuis, M.; van der Klei, I.J. Autophagy: Principles and significance in health and disease. Biochim. Biophys. Acta 2009, 1792, 3–13. [Google Scholar] [CrossRef] [PubMed]
- Shang, Y.C.; Chong, Z.Z.; Wang, S.; Maiese, K. Tuberous sclerosis protein 2 (TSC2) modulates CCN4 cytoprotection during apoptotic amyloid toxicity in microglia. Curr. Neurovasc. Res. 2013, 10, 29–38. [Google Scholar] [CrossRef] [PubMed]
- Vellai, T.; Takacs-Vellai, K.; Zhang, Y.; Kovacs, A.L.; Orosz, L.; Muller, F. Genetics: Influence of TOR kinase on lifespan in C. elegans. Nature 2003, 426. [Google Scholar] [CrossRef]
- Levine, B.; Klionsky, D.J. Development by self-digestion: Molecular mechanisms and biological functions of autophagy. Dev. Cell 2004, 6, 463–477. [Google Scholar] [CrossRef] [PubMed]
- Rajawat, Y.S.; Bossis, I. Autophagy in aging and in neurodegenerative disorders. Hormones 2008, 7, 46–61. [Google Scholar] [CrossRef] [PubMed]
- Jia, K.; Hart, A.C.; Levine, B. Autophagy genes protect against disease caused by polyglutamine expansion proteins in Caenorhabditis elegans. Autophagy 2007, 3, 21–25. [Google Scholar] [CrossRef] [PubMed]
- Melendez, A.; Talloczy, Z.; Seaman, M.; Eskelinen, E.L.; Hall, D.H.; Levine, B. Autophagy genes are essential for dauer development and life-span extension in C. elegans. elegans. Science 2003, 301, 1387–1391. [Google Scholar]
- Nixon, R.A. Autophagy in neurodegenerative disease: Friend, foe or turncoat? Trends Neurosci. 2006, 29, 528–535. [Google Scholar]
- Donati, A. The involvement of macroautophagy in aging and anti-aging interventions. Mol. Asp. Med. 2006, 27, 455–470. [Google Scholar] [CrossRef]
- Lionaki, E.; Markaki, M.; Tavernarakis, N. Autophagy and ageing: Insights from invertebrate model organisms. Ageing Res. Rev. 2013, 12, 413–428. [Google Scholar] [CrossRef]
- Low, P.; Varga, A.; Pircs, K.; Nagy, P.; Szatmari, Z.; Sass, M.; Juhasz, G. Impaired proteasomal degradation enhances autophagy via hypoxia signaling in Drosophila. BMC Cell Biol. 2013, 14. [Google Scholar] [CrossRef] [PubMed]
- Shibata, M.; Lu, T.; Furuya, T.; Degterev, A.; Mizushima, N.; Yoshimori, T.; MacDonald, M.; Yankner, B.; Yuan, J. Regulation of intracellular accumulation of mutant Huntingtin by Beclin 1. J. Biol. Chem. 2006, 281, 14474–14485. [Google Scholar] [CrossRef] [PubMed]
- Gupta, V.K.; Scheunemann, L.; Eisenberg, T.; Mertel, S.; Bhukel, A.; Koemans, T.S.; Kramer, J.M.; Liu, K.S.; Schroeder, S.; Stunnenberg, H.G.; et al. Restoring polyamines protects from age-induced memory impairment in an autophagy-dependent manner. Nat. Neurosci. 2013; 16, 1453–1460. [Google Scholar]
- Orr, M.E.; Oddo, S. Autophagic/lysosomal dysfunction in Alzheimer’s disease. Alzheimers Res. Ther. 2013, 5. [Google Scholar] [CrossRef]
- Hardy, J.; Selkoe, D.J. The amyloid hypothesis of Alzheimer’s disease: Progress and problems on the road to therapeutics. Science 2002, 297, 353–356. [Google Scholar] [CrossRef]
- Bokov, A.F.; Garg, N.; Ikeno, Y.; Thakur, S.; Musi, N.; DeFronzo, R.A.; Zhang, N.; Erickson, R.C.; Gelfond, J.; Hubbard, G.B.; et al. Does reduced IGF-1R signaling in Igf1r+/− mice alter aging? PLoS One 2011, 6, e26891. [Google Scholar] [CrossRef]
- Toth, M.L.; Sigmond, T.; Borsos, E.; Barna, J.; Erdelyi, P.; Takacs-Vellai, K.; Orosz, L.; Kovacs, A.L.; Csikos, G.; Sass, M.; et al. Longevity pathways converge on autophagy genes to regulate life span in Caenorhabditis elegans. Autophagy 2008, 4, 330–338. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, H.S.; McBurney, M.; Robbins, P.D. SIRT1 negatively regulates the mammalian target of rapamycin. PLoS One 2010, 5, e9199. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Li, X.; Zhu, J.X.; Xie, W.; Le, W.; Fan, Z.; Jankovic, J.; Pan, T. Resveratrol-activated AMPK/SIRT1/autophagy in cellular models of Parkinson's disease. Neurosignals 2011, 19, 163–174. [Google Scholar] [CrossRef]
- Canto, C.; Gerhart-Hines, Z.; Feige, J.N.; Lagouge, M.; Noriega, L.; Milne, J.C.; Elliott, P.J.; Puigserver, P.; Auwerx, J. AMPK regulates energy expenditure by modulating NAD+ metabolism and SIRT1 activity. Nature 2009, 458, 1056–1060. [Google Scholar] [CrossRef] [PubMed]
- Lee, I.H.; Cao, L.; Mostoslavsky, R.; Lombard, D.B.; Liu, J.; Bruns, N.E.; Tsokos, M.; Alt, F.W.; Finkel, T. A role for the NAD-dependent deacetylase Sirt1 in the regulation of autophagy. Proc. Natl. Acad. Sci. USA 2008, 105, 3374–3379. [Google Scholar] [CrossRef] [PubMed]
- Baur, J.A.; Pearson, K.J.; Price, N.L.; Jamieson, H.A.; Lerin, C.; Kalra, A.; Prabhu, V.V.; Allard, J.S.; Lopez-Lluch, G.; Lewis, K.; et al. Resveratrol improves health and survival of mice on a high-calorie diet. Nature 2006, 444, 337–342. [Google Scholar] [CrossRef] [PubMed]
- Jeong, H.; Then, F.; Melia, T.J., Jr.; Mazzulli, J.R.; Cui, L.; Savas, J.N.; Voisine, C.; Paganetti, P.; Tanese, N.; Hart, A.C.; et al. Acetylation targets mutant huntingtin to autophagosomes for degradation. Cell 2009, 137, 60–72. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.Y.; Li, T.Y.; Liu, Q.; Zhang, C.; Li, X.; Chen, Y.; Zhang, S.M.; Lian, G.; Ruan, K.; Wang, Z.; et al. Protein phosphorylation-acetylation cascade connects growth factor deprivation to autophagy. Autophagy 2012, 8, 1385–1386. [Google Scholar] [PubMed]
- Lin, S.Y.; Li, T.Y.; Liu, Q.; Zhang, C.; Li, X.; Chen, Y.; Zhang, S.M.; Lian, G.; Ruan, K.; Wang, Z.; et al. GSK3-TIP60-ULK1 signaling pathway links growth factor deprivation to autophagy. Science 2012, 336, 477–481. [Google Scholar] [CrossRef] [PubMed]
- Lee, I.H.; Finkel, T. Regulation of autophagy by the p300 acetyltransferase. J. Biol. Chem. 2009, 284, 6322–6328. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, M.; Bartlett, J.D. Sirtuin1 and autophagy protect cells from fluoride-induced cell stress. Biochim. Biophys. Acta 2014, 1842, 245–255. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Zhou, Y.; Mueller-Steiner, S.; Chen, L.F.; Kwon, H.; Yi, S.; Mucke, L.; Gan, L. SIRT1 protects against microglia-dependent amyloid-β toxicity through inhibiting NF-κB signaling. J. Biol. Chem. 2005, 280, 40364–40374. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, S.; Ray, D.; Lekli, I.; Bak, I.; Tosaki, A.; Das, D.K. Effects of Longevinex (modified resveratrol) on cardioprotection and its mechanisms of action. Can. J. Physiol. Pharmacol. 2010, 88, 1017–1025. [Google Scholar] [CrossRef] [PubMed]
- Medema, R.H.; Jaattela, M. Cytosolic FoxO1: Alive and killing. Nat. Cell Biol. 2010, 12, 642–643. [Google Scholar] [PubMed]
- Ng, F.; Tang, B.L. Sirtuins’ modulation of autophagy. J. Cell Physiol. 2013, 228, 2262–2270. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Kanasaki, M.; He, J.; Kitada, M.; Nagao, K.; Jinzu, H.; Noguchi, Y.; Maegawa, H.; Kanasaki, K.; Koya, D. Ketogenic essential amino acids replacement diet ameliorated hepatosteatosis with altering autophagy-associated molecules. Biochim. Biophys. Acta 2013, 1832, 1605–1612. [Google Scholar] [CrossRef] [PubMed]
- Hariharan, N.; Maejima, Y.; Nakae, J.; Paik, J.; Depinho, R.A.; Sadoshima, J. Deacetylation of FoxO by Sirt1 plays an essential role in mediating starvation-induced autophagy in cardiac myocytes. Circ. Res. 2010, 107, 1470–1482. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Lee, H.Y.; Gustafsson, A.B. Regulation of autophagy by metabolic and stress signaling pathways in the heart. J. Cardiovasc. Pharmacol. 2012, 60, 118–124. [Google Scholar] [CrossRef] [PubMed]
- Luo, L.; Lu, A.M.; Wang, Y.; Hong, A.; Chen, Y.; Hu, J.; Li, X.; Qin, Z.H. Chronic resistance training activates autophagy and reduces apoptosis of muscle cells by modulating IGF-1 and its receptors, Akt/mTOR and Akt/FOXO3a signaling in aged rats. Exp. Gerontol. 2013, 48, 427–436. [Google Scholar] [CrossRef] [PubMed]
- Sengupta, A.; Molkentin, J.D.; Yutzey, K.E. FoxO transcription factors promote autophagy in cardiomyocytes. J. Biol. Chem. 2009, 284, 28319–28331. [Google Scholar] [CrossRef] [PubMed]
- Park, K.J.; Lee, S.H.; Lee, C.H.; Jang, J.Y.; Chung, J.; Kwon, M.H.; Kim, Y.S. Upregulation of Beclin-1 expression and phosphorylation of Bcl-2 and p53 are involved in the JNK-mediated autophagic cell death. Biochem. Biophys. Res. Commun. 2009, 382, 726–729. [Google Scholar] [CrossRef] [PubMed]
- Samara, C.; Syntichaki, P.; Tavernarakis, N. Autophagy is required for necrotic cell death in Caenorhabditis elegans. Cell Death Differ. 2008, 15, 105–112. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Chong, Z.Z.; Shang, Y.C.; Maiese, K. WISP1 neuroprotection requires FoxO3a post-translational modulation with autoregulatory control of SIRT1. Curr. Neurovasc. Res. 2013, 10, 54–69. [Google Scholar] [CrossRef]
- Fontana, L.; Partridge, L.; Longo, V.D. Extending healthy life span—From yeast to humans. Science 2010, 328, 321–326. [Google Scholar] [CrossRef] [PubMed]
- Speakman, J.R.; Mitchell, S.E. Caloric restriction. Mol. Asp. Med. 2011, 32, 159–221. [Google Scholar] [CrossRef]
- Shi, L.; Adams, M.M.; Linville, M.C.; Newton, I.G.; Forbes, M.E.; Long, A.B.; Riddle, D.R.; Brunso-Bechtold, J.K. Caloric restriction eliminates the aging-related decline in NMDA and AMPA receptor subunits in the rat hippocampus and induces homeostasis. Exp. Neurol. 2007, 206, 70–79. [Google Scholar] [CrossRef] [PubMed]
- Fontan-Lozano, A.; Saez-Cassanelli, J.L.; Inda, M.C.; de los Santos-Arteaga, M.; Sierra-Dominguez, S.A.; Lopez-Lluch, G.; Delgado-Garcia, J.M.; Carrion, A.M. Caloric restriction increases learning consolidation and facilitates synaptic plasticity through mechanisms dependent on NR2B subunits of the NMDA receptor. J. Neurosci. 2007, 27, 10185–10195. [Google Scholar] [CrossRef] [PubMed]
- Komatsu, T.; Chiba, T.; Yamaza, H.; Yamashita, K.; Shimada, A.; Hoshiyama, Y.; Henmi, T.; Ohtani, H.; Higami, Y.; de Cabo, R.; et al. Manipulation of caloric content but not diet composition, attenuates the deficit in learning and memory of senescence-accelerated mouse strain P8. Exp. Gerontol. 2008, 43, 339–346. [Google Scholar] [CrossRef] [PubMed]
- Halagappa, V.K.; Guo, Z.; Pearson, M.; Matsuoka, Y.; Cutler, R.G.; Laferla, F.M.; Mattson, M.P. Intermittent fasting and caloric restriction ameliorate age-related behavioral deficits in the triple-transgenic mouse model of Alzheimer’s disease. Neurobiol. Dis. 2007, 26, 212–220. [Google Scholar] [CrossRef] [PubMed]
- Wu, P.; Shen, Q.; Dong, S.; Xu, Z.; Tsien, J.Z.; Hu, Y. Calorie restriction ameliorates neurodegenerative phenotypes in forebrain-specific presenilin-1 and presenilin-2 double knockout mice. Neurobiol. Aging 2008, 29, 1502–1511. [Google Scholar] [CrossRef] [PubMed]
- Rotermund, C.; Truckenmuller, F.M.; Schell, H.; Kahle, P.J. Diet-induced obesity accelerates the onset of terminal phenotypes in α-synuclein transgenic mice. J. Neurochem. 2014. [Google Scholar] [CrossRef]
- Morris, J.K.; Bomhoff, G.L.; Stanford, J.A.; Geiger, P.C. Neurodegeneration in an animal model of Parkinson’s disease is exacerbated by a high-fat diet. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2010, 299, R1082–R1090. [Google Scholar] [CrossRef] [PubMed]
- Hars, E.S.; Qi, H.; Ryazanov, A.G.; Jin, S.; Cai, L.; Hu, C.; Liu, L.F. Autophagy regulates ageing in C. elegans. Autophagy 2007, 3, 93–95. [Google Scholar] [CrossRef]
- Pattingre, S.; Espert, L.; Biard-Piechaczyk, M.; Codogno, P. Regulation of macroautophagy by mTOR and Beclin 1 complexes. Biochimie 2008, 90, 313–323. [Google Scholar] [CrossRef] [PubMed]
- Kaeberlein, M.; Powers, R.W., 3rd; Steffen, K.K.; Westman, E.A.; Hu, D.; Dang, N.; Kerr, E.O.; Kirkland, K.T.; Fields, S.; Kennedy, B.K. Regulation of yeast replicative life span by TOR and Sch9 in response to nutrients. Science 2005, 310, 1193–1196. [Google Scholar]
- Sekito, T.; Kawamata, T.; Ichikawa, R.; Suzuki, K.; Ohsumi, Y. Atg17 recruits Atg9 to organize the pre-autophagosomal structure. Genes Cells 2009, 14, 525–538. [Google Scholar] [CrossRef] [PubMed]
- Morselli, E.; Maiuri, M.C.; Markaki, M.; Megalou, E.; Pasparaki, A.; Palikaras, K.; Criollo, A.; Galluzzi, L.; Malik, S.A.; Vitale, I.; et al. Caloric restriction and resveratrol promote longevity through the Sirtuin-1-dependent induction of autophagy. Cell Death Dis. 2010, 1. [Google Scholar] [CrossRef]
- Caballero, B.; Coto-Montes, A. An insight into the role of autophagy in cell responses in the aging and neurodegenerative brain. Histol. Histopathol. 2012, 27, 263–275. [Google Scholar] [PubMed]
- Levine, B.; Mizushima, N.; Virgin, H.W. Autophagy in immunity and inflammation. Nature 2011, 469, 323–335. [Google Scholar] [CrossRef] [PubMed]
- Virgin, H.W.; Levine, B. Autophagy genes in immunity. Nat. Immunol. 2009, 10, 461–470. [Google Scholar] [CrossRef] [PubMed]
- Djavaheri-Mergny, M.; Amelotti, M.; Mathieu, J.; Besancon, F.; Bauvy, C.; Souquere, S.; Pierron, G.; Codogno, P. NF-κB activation represses tumor necrosis factor-α-induced autophagy. J. Biol. Chem. 2006, 281, 30373–30382. [Google Scholar] [CrossRef] [PubMed]
- Schlottmann, S.; Buback, F.; Stahl, B.; Meierhenrich, R.; Walter, P.; Georgieff, M.; Senftleben, U. Prolonged classical NF-κB activation prevents autophagy upon E. coli stimulation in vitro: A potential resolving mechanism of inflammation. Mediators Inflamm. 2008; 2008. [Google Scholar] [CrossRef]
- Salminen, A.; Hyttinen, J.M.; Kauppinen, A.; Kaarniranta, K. Context-dependent regulation of autophagy by IKK-NF-κB signaling: Impact on the aging process. Int. J. Cell Biol. 2012, 2012. [Google Scholar] [CrossRef]
- Salminen, A.; Kaarniranta, K.; Kauppinen, A. Beclin 1 interactome controls the crosstalk between apoptosis, autophagy and inflammasome activation: Impact on the aging process. Ageing Res. Rev. 2013, 12, 520–534. [Google Scholar] [CrossRef] [PubMed]
- Dan, H.C.; Cooper, M.J.; Cogswell, P.C.; Duncan, J.A.; Ting, J.P.; Baldwin, A.S. Akt-dependent regulation of NF-κB is controlled by mTOR and Raptor in association with IKK. Genes Dev. 2008, 22, 1490–1500. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.F.; Kuo, H.P.; Chen, C.T.; Wei, Y.; Chou, C.K.; Hung, J.Y.; Yen, C.J.; Hung, M.C. IKKβ suppression of TSC1 function links the mTOR pathway with insulin resistance. Int. J. Mol. Med. 2008, 22, 633–638. [Google Scholar] [PubMed]
- Lee, D.F.; Kuo, H.P.; Chen, C.T.; Hsu, J.M.; Chou, C.K.; Wei, Y.; Sun, H.L.; Li, L.Y.; Ping, B.; Huang, W.C.; et al. IKKβ suppression of TSC1 links inflammation and tumor angiogenesis via the mTOR pathway. Cell 2007, 130, 440–455. [Google Scholar] [CrossRef] [PubMed]
- Nivon, M.; Richet, E.; Codogno, P.; Arrigo, A.P.; Kretz-Remy, C. Autophagy activation by NF-κB is essential for cell survival after heat shock. Autophagy 2009, 5, 766–783. [Google Scholar] [CrossRef] [PubMed]
- Herranz, D.; Munoz-Martin, M.; Canamero, M.; Mulero, F.; Martinez-Pastor, B.; Fernandez-Capetillo, O.; Serrano, M. Sirt1 improves healthy ageing and protects from metabolic syndrome-associated cancer. Nat. Commun. 2010, 1. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Zhang, W.; Pan, H.; Feldser, H.G.; Lainez, E.; Miller, C.; Leung, S.; Zhong, Z.; Zhao, H.; Sweitzer, S.; et al. SIRT1 activators suppress inflammatory responses through promotion of p65 deacetylation and inhibition of NF-κB activity. PLoS One 2012, 7, e46364. [Google Scholar] [CrossRef] [PubMed]
- Polyakova, O.; Borman, S.; Grimley, R.; Vamathevan, J.; Hayes, B.; Solari, R. Identification of novel interacting partners of Sirtuin6. PLoS One 2012, 7, e51555. [Google Scholar] [CrossRef] [PubMed]
- Yeung, F.; Hoberg, J.E.; Ramsey, C.S.; Keller, M.D.; Jones, D.R.; Frye, R.A.; Mayo, M.W. Modulation of NF-κB-dependent transcription and cell survival by the SIRT1 deacetylase. EMBO J. 2004, 23, 2369–2380. [Google Scholar] [CrossRef] [PubMed]
- Rothgiesser, K.M.; Erener, S.; Waibel, S.; Luscher, B.; Hottiger, M.O. SIRT2 regulates NF-κB dependent gene expression through deacetylation of p65 Lys310. J. Cell Sci. 2010, 123, 4251–4258. [Google Scholar] [CrossRef] [PubMed]
- Kawahara, T.L.; Michishita, E.; Adler, A.S.; Damian, M.; Berber, E.; Lin, M.; McCord, R.A.; Ongaigui, K.C.; Boxer, L.D.; Chang, H.Y.; et al. SIRT6 links histone H3 lysine 9 deacetylation to NF-κB-dependent gene expression and organismal life span. Cell 2009, 136, 62–74. [Google Scholar] [CrossRef] [PubMed]
- Klongpanichapak, S.; Phansuwan-Pujito, P.; Ebadi, M.; Govitrapong, P. Melatonin protects SK-N-SH neuroblastoma cells from amphetamine-induced neurotoxicity. J. Pineal Res. 2007, 43, 65–73. [Google Scholar] [CrossRef] [PubMed]
- Klongpanichapak, S.; Phansuwan-Pujito, P.; Ebadi, M.; Govitrapong, P. Melatonin inhibits amphetamine-induced increase in α-synuclein and decrease in phosphorylated tyrosine hydroxylase in SK-N-SH cells. Neurosci. Lett. 2008, 436, 309–313. [Google Scholar] [CrossRef] [PubMed]
- Kongsuphol, P.; Mukda, S.; Nopparat, C.; Villarroel, A.; Govitrapong, P. Melatonin attenuates methamphetamine-induced deactivation of the mammalian target of rapamycin signaling to induce autophagy in SK-N-SH cells. J. Pineal Res. 2009, 46, 199–206. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, S.; Kanaseki, T.; Mizushima, N.; Mizuta, T.; Arakawa-Kobayashi, S.; Thompson, C.B.; Tsujimoto, Y. Role of Bcl-2 family proteins in a non-apoptotic programmed cell death dependent on autophagy genes. Nat. Cell Biol. 2004, 6, 1121–1128. [Google Scholar]
- Khapre, R.V.; Samsa, W.E.; Kondratov, R.V. Circadian regulation of cell cycle: Molecular connections between aging and the circadian clock. Ann. Med. 2010, 42, 404–415. [Google Scholar] [CrossRef] [PubMed]
- Pattingre, S.; Tassa, A.; Qu, X.; Garuti, R.; Liang, X.H.; Mizushima, N.; Packer, M.; Schneider, M.D.; Levine, B. Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell 2005, 122, 927–939. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Pattingre, S.; Sinha, S.; Bassik, M.; Levine, B. JNK1-mediated phosphorylation of Bcl-2 regulates starvation-induced autophagy. Mol. Cell. 2008, 30, 678–688. [Google Scholar] [CrossRef] [PubMed]
- Nopparat, C.; Porter, J.E.; Ebadi, M.; Govitrapong, P. The mechanism for the neuroprotective effect of melatonin against methamphetamine-induced autophagy. J. Pineal Res. 2010, 49, 382–389. [Google Scholar] [CrossRef] [PubMed]
- Jeong, J.K.; Moon, M.H.; Lee, Y.J.; Seol, J.W.; Park, S.Y. Melatonin-induced autophagy protects against human prion protein-mediated neurotoxicity. J. Pineal Res. 2012, 53, 138–146. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.F.; Huang, H.J.; Lee, H.C.; Hung, K.C.; Wu, R.T.; Lin, A.M. Melatonin attenuates kainic acid-induced neurotoxicity in mouse hippocampus via inhibition of autophagy and α-synuclein aggregation. J. Pineal Res. 2012, 52, 312–321. [Google Scholar] [CrossRef] [PubMed]
- Weinert, D. Age-dependent changes of the circadian system. Chronobiol. Int. 2000, 17, 261–283. [Google Scholar] [CrossRef] [PubMed]
- Reppert, S.M.; Weaver, D.R. Coordination of circadian timing in mammals. Nature 2002, 418, 935–941. [Google Scholar] [CrossRef] [PubMed]
- Vitaterna, M.H.; King, D.P.; Chang, A.M.; Kornhauser, J.M.; Lowrey, P.L.; McDonald, J.D.; Dove, W.F.; Pinto, L.H.; Turek, F.W.; Takahashi, J.S. Mutagenesis and mapping of a mouse gene, Clock, essential for circadian behavior. Science 1994, 264, 719–725. [Google Scholar] [CrossRef] [PubMed]
- Asher, G.; Schibler, U. A CLOCK-less clock. Trends Cell Biol. 2006, 16, 547–549. [Google Scholar] [CrossRef] [PubMed]
- Debruyne, J.P.; Noton, E.; Lambert, C.M.; Maywood, E.S.; Weaver, D.R.; Reppert, S.M. A clock shock: Mouse CLOCK is not required for circadian oscillator function. Neuron 2006, 50, 465–477. [Google Scholar] [CrossRef] [PubMed]
- Froy, O.; Chang, D.C.; Reppert, S.M. Redox potential: Differential roles in dCRY and mCRY1 functions. Curr. Biol. 2002, 12, 147–152. [Google Scholar] [CrossRef] [PubMed]
- Dunlap, J.C. Molecular bases for circadian clocks. Cell 1999, 96, 271–290. [Google Scholar] [CrossRef] [PubMed]
- Preitner, N.; Damiola, F.; Lopez-Molina, L.; Zakany, J.; Duboule, D.; Albrecht, U.; Schibler, U. The orphan nuclear receptor REV-ERBα controls circadian transcription within the positive limb of the mammalian circadian oscillator. Cell 2002, 110, 251–260. [Google Scholar] [CrossRef] [PubMed]
- Sato, T.K.; Panda, S.; Miraglia, L.J.; Reyes, T.M.; Rudic, R.D.; McNamara, P.; Naik, K.A.; FitzGerald, G.A.; Kay, S.A.; Hogenesch, J.B. A functional genomics strategy reveals Rora as a component of the mammalian circadian clock. Neuron 2004, 43, 527–537. [Google Scholar] [CrossRef] [PubMed]
- Ueda, H.R.; Hayashi, S.; Chen, W.; Sano, M.; Machida, M.; Shigeyoshi, Y.; Iino, M.; Hashimoto, S. System-level identification of transcriptional circuits underlying mammalian circadian clocks. Nat. Genet. 2005, 37, 187–192. [Google Scholar] [CrossRef] [PubMed]
- Harms, E.; Kivimae, S.; Young, M.W.; Saez, L. Posttranscriptional and posttranslational regulation of clock genes. J. Biol. Rhythm. 2004, 19, 361–373. [Google Scholar] [CrossRef]
- Jasser, S.A.; Blask, D.E.; Brainard, G.C. Light during darkness and cancer: Relationships in circadian photoreception and tumor biology. Cancer Causes Control 2006, 17, 515–523. [Google Scholar] [CrossRef] [PubMed]
- Van Gelder, R.N. Non-visual photoreception: Sensing light without sight. Curr. Biol. 2008, 18, R38–R39. [Google Scholar] [CrossRef] [PubMed]
- Pevet, P.; Challet, E. Melatonin: Both master clock output and internal time-giver in the circadian clocks network. J. Physiol. Paris 2011, 105, 170–182. [Google Scholar] [CrossRef] [PubMed]
- Schroeder, A.M.; Colwell, C.S. How to fix a broken clock. Trends Pharmacol. Sci. 2013, 34, 605–619. [Google Scholar] [CrossRef] [PubMed]
- Torres-Farfan, C.; Seron-Ferre, M.; Dinet, V.; Korf, H.W. Immunocytochemical demonstration of day/night changes of clock gene protein levels in the murine adrenal gland: Differences between melatonin-proficient (C3H) and melatonin-deficient (C57BL) mice. J. Pineal Res. 2006, 40, 64–70. [Google Scholar] [CrossRef] [PubMed]
- Uz, T.; Akhisaroglu, M.; Ahmed, R.; Manev, H. The pineal gland is critical for circadian Period1 expression in the striatum and for circadian cocaine sensitization in mice. Neuropsychopharmacology 2003, 28, 2117–2123. [Google Scholar] [PubMed]
- Imbesi, M.; Arslan, A.D.; Yildiz, S.; Sharma, R.; Gavin, D.; Tun, N.; Manev, H.; Uz, T. The melatonin receptor MT1 is required for the differential regulatory actions of melatonin on neuronal “clock” gene expression in striatal neurons in vitro. J. Pineal Res. 2009, 46, 87–94. [Google Scholar] [CrossRef] [PubMed]
- Zeman, M.; Szantoova, K.; Stebelova, K.; Mravec, B.; Herichova, I. Effect of rhythmic melatonin administration on clock gene expression in the suprachiasmatic nucleus and the heart of hypertensive TGR(mRen2)27 rats. J. Hypertens. Suppl. 2009, 27, S21–S26. [Google Scholar] [PubMed]
- Crosio, C.; Cermakian, N.; Allis, C.D.; Sassone-Corsi, P. Light induces chromatin modification in cells of the mammalian circadian clock. Nat. Neurosci. 2000, 3, 1241–1247. [Google Scholar] [CrossRef] [PubMed]
- Etchegaray, J.P.; Lee, C.; Wade, P.A.; Reppert, S.M. Rhythmic histone acetylation underlies transcription in the mammalian circadian clock. Nature 2003, 421, 177–182. [Google Scholar] [CrossRef] [PubMed]
- Masri, S.; Sassone-Corsi, P. Plasticity and specificity of the circadian epigenome. Nat. Neurosci. 2010, 13, 1324–1329. [Google Scholar] [CrossRef] [PubMed]
- Ripperger, J.A.; Schibler, U. Rhythmic CLOCK-BMAL1 binding to multiple E-box motifs drives circadian Dbp transcription and chromatin transitions. Nat. Genet. 2006, 38, 369–374. [Google Scholar] [CrossRef] [PubMed]
- Welberg, L. Circadian rhythms: Methylation mediates clock plasticity. Nat. Rev. Neurosci. 2014, 15. [Google Scholar] [CrossRef]
- Azzi, A.; Dallmann, R.; Casserly, A.; Rehrauer, H.; Patrignani, A.; Maier, B.; Kramer, A.; Brown, S.A. Circadian behavior is light-reprogrammed by plastic DNA methylation. Nat. Neurosci. 2014, 17, 377–382. [Google Scholar] [CrossRef] [PubMed]
- Katada, S.; Sassone-Corsi, P. The histone methyltransferase MLL1 permits the oscillation of circadian gene expression. Nat. Struct. Mol. Biol. 2010, 17, 1414–1421. [Google Scholar] [CrossRef] [PubMed]
- Doi, M.; Hirayama, J.; Sassone-Corsi, P. Circadian regulator CLOCK is a histone acetyltransferase. Cell 2006, 125, 497–508. [Google Scholar] [CrossRef] [PubMed]
- Hirayama, J.; Sahar, S.; Grimaldi, B.; Tamaru, T.; Takamatsu, K.; Nakahata, Y.; Sassone-Corsi, P. CLOCK-mediated acetylation of BMAL1 controls circadian function. Nature 2007, 450, 1086–1090. [Google Scholar] [CrossRef] [PubMed]
- Hochberg, I.; Hochberg, Z. Expanding the definition of hypothalamic obesity. Obes. Rev. 2010, 11, 709–721. [Google Scholar] [CrossRef] [PubMed]
- Herichova, I. Changes of physiological functions induced by shift work. Endocr. Regul. 2013, 47, 159–170. [Google Scholar] [CrossRef] [PubMed]
- Sookoian, S.; Pirola, C.J. Epigenetics of insulin resistance: An emerging field in translational medicine. Curr. Diab. Rep. 2013, 13, 229–237. [Google Scholar] [CrossRef] [PubMed]
- Nakabayashi, H.; Ohta, Y.; Yamamoto, M.; Susuki, Y.; Taguchi, A.; Tanabe, K.; Kondo, M.; Hatanaka, M.; Nagao, Y.; Tanizawa, Y. Clock-controlled output gene Dbp is a regulator of Arnt/Hif-1β gene expression in pancreatic islet β-cells. Biochem. Biophys. Res. Commun. 2013, 434, 370–375. [Google Scholar] [CrossRef] [PubMed]
- Sahar, S.; Sassone-Corsi, P. Circadian rhythms and memory formation: Regulation by chromatin remodeling. Front. Mol. Neurosci. 2012, 5. [Google Scholar] [CrossRef] [PubMed]
- Bishop, N.A.; Guarente, L. Genetic links between diet and lifespan: Shared mechanisms from yeast to humans. Nat. Rev. Genet. 2007, 8, 835–844. [Google Scholar] [CrossRef] [PubMed]
- Asher, G.; Gatfield, D.; Stratmann, M.; Reinke, H.; Dibner, C.; Kreppel, F.; Mostoslavsky, R.; Alt, F.W.; Schibler, U. SIRT1 regulates circadian clock gene expression through PER2 deacetylation. Cell 2008, 134, 317–328. [Google Scholar] [CrossRef] [PubMed]
- Grimaldi, B.; Nakahata, Y.; Kaluzova, M.; Masubuchi, S.; Sassone-Corsi, P. Chromatin remodeling, metabolism and circadian clocks: The interplay of CLOCK and SIRT1. Int. J. Biochem. Cell Biol. 2009, 41, 81–86. [Google Scholar] [CrossRef] [PubMed]
- Nakahata, Y.; Sahar, S.; Astarita, G.; Kaluzova, M.; Sassone-Corsi, P. Circadian control of the NAD+ salvage pathway by CLOCK-SIRT1. Science 2009, 324, 654–657. [Google Scholar] [CrossRef]
- Nakahata, Y.; Kaluzova, M.; Grimaldi, B.; Sahar, S.; Hirayama, J.; Chen, D.; Guarente, L.P.; Sassone-Corsi, P. The NAD+-dependent deacetylase SIRT1 modulates CLOCK-mediated chromatin remodeling and circadian control. Cell 2008, 134, 329–340. [Google Scholar] [CrossRef] [PubMed]
- Ramsey, K.M.; Yoshino, J.; Brace, C.S.; Abrassart, D.; Kobayashi, Y.; Marcheva, B.; Hong, H.K.; Chong, J.L.; Buhr, E.D.; Lee, C.; et al. Circadian clock feedback cycle through NAMPT-mediated NAD+ biosynthesis. Science 2009, 324, 651–654. [Google Scholar] [CrossRef] [PubMed]
- Garaulet, M.; Esteban Tardido, A.; Lee, Y.C.; Smith, C.E.; Parnell, L.D.; Ordovas, J.M. SIRT1 and CLOCK 3111T>C combined genotype is associated with evening preference and weight loss resistance in a behavioral therapy treatment for obesity. Int. J. Obes. 2012, 36, 1436–1441. [Google Scholar] [CrossRef]
- Kondratov, R.V.; Kondratova, A.A.; Gorbacheva, V.Y.; Vykhovanets, O.V.; Antoch, M.P. Early aging and age-related pathologies in mice deficient in BMAL1, the core componentof the circadian clock. Genes Dev. 2006, 20, 1868–1873. [Google Scholar] [CrossRef] [PubMed]
- Dubrovsky, Y.V.; Samsa, W.E.; Kondratov, R.V. Deficiency of circadian protein CLOCK reduces lifespan and increases age-related cataract development in mice. Aging 2010, 2, 936–944. [Google Scholar] [PubMed]
- Antoch, M.P.; Gorbacheva, V.Y.; Vykhovanets, O.; Toshkov, I.A.; Kondratov, R.V.; Kondratova, A.A.; Lee, C.; Nikitin, A.Y. Disruption of the circadian clock due to the Clock mutation has discrete effects on aging and carcinogenesis. Cell Cycle 2008, 7, 1197–1204. [Google Scholar] [CrossRef] [PubMed]
- Fu, L.; Pelicano, H.; Liu, J.; Huang, P.; Lee, C. The circadian gene Period2 plays an important role in tumor suppression and DNA damage response in vivo. Cell 2002, 111, 41–50. [Google Scholar] [CrossRef] [PubMed]
- Kondratova, A.A.; Kondratov, R.V. The circadian clock and pathology of the ageing brain. Nat. Rev. Neurosci. 2012, 13, 325–335. [Google Scholar] [PubMed]
- Nakamura, T.J.; Nakamura, W.; Yamazaki, S.; Kudo, T.; Cutler, T.; Colwell, C.S.; Block, G.D. Age-related decline in circadian output. J. Neurosci. 2011, 31, 10201–10205. [Google Scholar] [CrossRef] [PubMed]
- Weinert, D. Circadian temperature variation and ageing. Ageing Res. Rev. 2010, 9, 51–60. [Google Scholar] [CrossRef] [PubMed]
- Jagota, A.; Kalyani, D. Effect of melatonin on age induced changes in daily serotonin rhythms in suprachiasmatic nucleus of male Wistar rat. Biogerontology 2010, 11, 299–308. [Google Scholar] [CrossRef] [PubMed]
- Palomba, M.; Nygard, M.; Florenzano, F.; Bertini, G.; Kristensson, K.; Bentivoglio, M. Decline of the presynaptic network, including GABAergic terminals, in the aging suprachiasmatic nucleus of the mouse. J. Biol. Rhythm. 2008, 23, 220–231. [Google Scholar] [CrossRef]
- Manikonda, P.K.; Jagota, A. Melatonin administration differentially affects age-induced alterations in daily rhythms of lipid peroxidation and antioxidant enzymes in male rat liver. Biogerontology 2012, 13, 511–524. [Google Scholar] [CrossRef] [PubMed]
- von Gall, C.; Weaver, D.R. Loss of responsiveness to melatonin in the aging mouse suprachiasmatic nucleus. Neurobiol. Aging 2008, 29, 464–470. [Google Scholar] [CrossRef] [PubMed]
- Karasek, M. Melatonin, human aging, and age-related diseases. Exp. Gerontol. 2004, 39, 1723–1729. [Google Scholar] [CrossRef] [PubMed]
- Poeggeler, B. Melatonin, aging, and age-related diseases: Perspectives for prevention, intervention, and therapy. Endocrine 2005, 27, 201–212. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.H.; Swaab, D.F. The human pineal gland and melatonin in aging and Alzheimer’s disease. J. Pineal Res. 2005, 38, 145–152. [Google Scholar] [CrossRef] [PubMed]
- Anisimov, V.N.; Vinogradova, I.A.; Panchenko, A.V.; Popovich, I.G.; Zabezhinski, M.A. Light-at-night-induced circadian disruption, cancer and aging. Curr. Aging Sci. 2012, 5, 170–177. [Google Scholar] [CrossRef] [PubMed]
- Vinogradova, I.A.; Anisimov, V.N.; Bukalev, A.V.; Semenchenko, A.V.; Zabezhinski, M.A. Circadian disruption induced by light-at-night accelerates aging and promotes tumorigenesis in rats. Aging 2009, 1, 855–865. [Google Scholar] [PubMed]
- Kondratov, R.V.; Vykhovanets, O.; Kondratova, A.A.; Antoch, M.P. Antioxidant N-acetyl-l-cysteine ameliorates symptoms of premature aging associated with the deficiency of the circadian protein BMAL1. Aging 2009, 1, 979–987. [Google Scholar] [PubMed]
- Mattam, U.; Jagota, A. Differential role of melatonin in restoration of age-induced alterations in daily rhythms of expression of various clock genes in suprachiasmatic nucleus of male Wistar rats. Biogerontology 2014, 15, 257–268. [Google Scholar] [CrossRef] [PubMed]
- Pfeifer, U. Inverted diurnal rhythm of cellular autophagy in liver cells of rats fed a single daily meal. Virchows Arch. B Cell Pathol. 1972, 10, 1–3. [Google Scholar] [PubMed]
- Pfeifer, U. Cellular autophagy and cell atrophy in the rat liver during long-term starvation. A quantitative morphological study with regard to diurnal variations. Virchows Arch. B Cell Pathol. 1973, 12, 195–211. [Google Scholar]
- Pfeifer, U.; Scheller, H. A morphometric study of cellular autophagy including diurnal variations in kidney tubules of normal rats. J. Cell Biol. 1975, 64, 608–621. [Google Scholar] [CrossRef] [PubMed]
- Reme, C.E.; Sulser, M. Diurnal variation of autophagy in rod visual cells in the rat. Albrecht Von Graefes Arch. Klin. Exp. Ophthalmol. 1977, 203, 261–270. [Google Scholar] [CrossRef] [PubMed]
- Pfeifer, U.; Scheller, H.; Ormanns, W. Diurnal rhythm of lysosomal organelle decomposition in liver, kidney and pancreas. Acta. Histochem. Suppl. 1976, 16, 205–210. [Google Scholar] [PubMed]
- Reme, C.; Wirz-Justice, A.; Rhyner, A.; Hofmann, S. Circadian rhythm in the light response of rat retinal disk-shedding and autophagy. Brain Res. 1986, 369, 356–360. [Google Scholar] [CrossRef] [PubMed]
- Pfeifer, U.; Bertling, J. A morphometric study of the inhibition of autophagic degradation during restorative growth of liver cells in rats re-fed after starvation. Virchows Arch. B Cell Pathol. 1977, 24, 109–120. [Google Scholar] [PubMed]
- Sachdeva, U.M.; Thompson, C.B. Diurnal rhythms of autophagy: Implications for cell biology and human disease. Autophagy 2008, 4, 581–589. [Google Scholar] [CrossRef] [PubMed]
- Ma, D.; Lin, J.D. Circadian regulation of autophagy rhythm through transcription factor C/EBPβ. Autophagy 2012, 8, 124–125. [Google Scholar] [CrossRef] [PubMed]
- Ma, D.; Li, S.; Molusky, M.M.; Lin, J.D. Circadian autophagy rhythm: A link between clock and metabolism? Trends Endocrinol. MeTab. 2012, 23, 319–325. [Google Scholar]
- Cao, R.; Anderson, F.E.; Jung, Y.J.; Dziema, H.; Obrietan, K. Circadian regulation of mammalian target of rapamycin signaling in the mouse suprachiasmatic nucleus. Neuroscience 2011, 181, 79–88. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Sehgal, A. AKT and TOR signaling set the pace of the circadian pacemaker. Curr. Biol. 2010, 20, 1203–1208. [Google Scholar] [CrossRef] [PubMed]
- Cao, R.; Li, A.; Cho, H.Y.; Lee, B.; Obrietan, K. Mammalian target of rapamycin signaling modulates photic entrainment of the suprachiasmatic circadian clock. J. Neurosci. 2010, 30, 6302–6314. [Google Scholar] [CrossRef] [PubMed]
- Khapre, R.V.; Kondratova, A.A.; Patel, S.; Dubrovsky, Y.; Wrobel, M.; Antoch, M.P.; Kondratov, R.V. BMAL1-dependent regulation of the mTOR signaling pathway delays aging. Aging 2014, 6, 48–57. [Google Scholar] [PubMed]
- Gerstner, J.R.; Yin, J.C. Circadian rhythms and memory formation. Nat. Rev. Neurosci. 2010, 11, 577–588. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.M.; Dragich, J.M.; Kudo, T.; Odom, I.H.; Welsh, D.K.; O'Dell, T.J.; Colwell, C.S. Expression of the circadian clock gene Period2 in the hippocampus: Possible implications for synaptic plasticity and learned behaviour. ASN Neuro 2009, 1. [Google Scholar] [CrossRef]
- Chaudhury, D.; Wang, L.M.; Colwell, C.S. Circadian regulation of hippocampal long-term potentiation. J. Biol. Rhythms. 2005, 20, 225–236. [Google Scholar] [CrossRef] [PubMed]
- Eckel-Mahan, K.L.; Phan, T.; Han, S.; Wang, H.; Chan, G.C.; Scheiner, Z.S.; Storm, D.R. Circadian oscillation of hippocampal MAPK activity and cAmp: Implications for memory persistence. Nat. Neurosci. 2008, 11, 1074–1082. [Google Scholar] [CrossRef]
- Bass, J.; Takahashi, J.S. Circadian integration of metabolism and energetics. Science 2010, 330, 1349–1354. [Google Scholar] [CrossRef] [PubMed]
- Gery, S.; Koeffler, H.P. Circadian rhythms and cancer. Cell Cycle 2010, 9, 1097–1103. [Google Scholar] [CrossRef] [PubMed]
- Kyriacou, C.P.; Hastings, M.H. Circadian clocks: Genes, sleep, and cognition. Trends Cogn. Sci. 2010, 14, 259–267. [Google Scholar] [CrossRef] [PubMed]
- Young, M.E. The circadian clock within the heart: Potential influence on myocardial gene expression, metabolism, and function. Am. J. Physiol. Heart Circ. Physiol. 2006, 290, H1–H16. [Google Scholar] [CrossRef] [PubMed]
- Dardente, H.; Cermakian, N. Molecular circadian rhythms in central and peripheral clocks in mammals. Chronobiol. Int. 2007, 24, 195–213. [Google Scholar] [CrossRef] [PubMed]
- Gan, L.; Mucke, L. Paths of convergence: Sirtuins in aging and neurodegeneration. Neuron 2008, 58, 10–14. [Google Scholar] [CrossRef] [PubMed]
- Donmez, G.; Wang, D.; Cohen, D.E.; Guarente, L. SIRT1 suppresses β-amyloid production by activating the α-secretase gene ADAM10. Cell 2010, 142, 320–332. [Google Scholar] [CrossRef] [PubMed]
- Min, S.W.; Cho, S.H.; Zhou, Y.; Schroeder, S.; Haroutunian, V.; Seeley, W.W.; Huang, E.J.; Shen, Y.; Masliah, E.; Mukherjee, C.; et al. Acetylation of tau inhibits its degradation and contributes to tauopathy. Neuron 2010, 67, 953–966. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Nguyen, M.D.; Dobbin, M.M.; Fischer, A.; Sananbenesi, F.; Rodgers, J.T.; Delalle, I.; Baur, J.A.; Sui, G.; Armour, S.M.; et al. SIRT1 deacetylase protects against neurodegeneration in models for Alzheimer’s disease and amyotrophic lateral sclerosis. EMBO J. 2007, 26, 3169–3179. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Wang, W.Y.; Mao, Y.W.; Graff, J.; Guan, J.S.; Pan, L.; Mak, G.; Kim, D.; Su, S.C.; Tsai, L.H. A novel pathway regulates memory and plasticity via SIRT1 and miR-134. Nature 2010, 466, 1105–1109. [Google Scholar] [CrossRef] [PubMed]
- Michan, S.; Li, Y.; Chou, M.M.; Parrella, E.; Ge, H.; Long, J.M.; Allard, J.S.; Lewis, K.; Miller, M.; Xu, W.; et al. SIRT1 is essential for normal cognitive function and synaptic plasticity. J. Neurosci. 2010, 30, 9695–9707. [Google Scholar] [CrossRef] [PubMed]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Jenwitheesuk, A.; Nopparat, C.; Mukda, S.; Wongchitrat, P.; Govitrapong, P. Melatonin Regulates Aging and Neurodegeneration through Energy Metabolism, Epigenetics, Autophagy and Circadian Rhythm Pathways. Int. J. Mol. Sci. 2014, 15, 16848-16884. https://doi.org/10.3390/ijms150916848
Jenwitheesuk A, Nopparat C, Mukda S, Wongchitrat P, Govitrapong P. Melatonin Regulates Aging and Neurodegeneration through Energy Metabolism, Epigenetics, Autophagy and Circadian Rhythm Pathways. International Journal of Molecular Sciences. 2014; 15(9):16848-16884. https://doi.org/10.3390/ijms150916848
Chicago/Turabian StyleJenwitheesuk, Anorut, Chutikorn Nopparat, Sujira Mukda, Prapimpun Wongchitrat, and Piyarat Govitrapong. 2014. "Melatonin Regulates Aging and Neurodegeneration through Energy Metabolism, Epigenetics, Autophagy and Circadian Rhythm Pathways" International Journal of Molecular Sciences 15, no. 9: 16848-16884. https://doi.org/10.3390/ijms150916848
APA StyleJenwitheesuk, A., Nopparat, C., Mukda, S., Wongchitrat, P., & Govitrapong, P. (2014). Melatonin Regulates Aging and Neurodegeneration through Energy Metabolism, Epigenetics, Autophagy and Circadian Rhythm Pathways. International Journal of Molecular Sciences, 15(9), 16848-16884. https://doi.org/10.3390/ijms150916848