Environmental Factors, Toxicants and Systemic Lupus Erythematosus



{kind=link}
Abstract
:1. Introduction
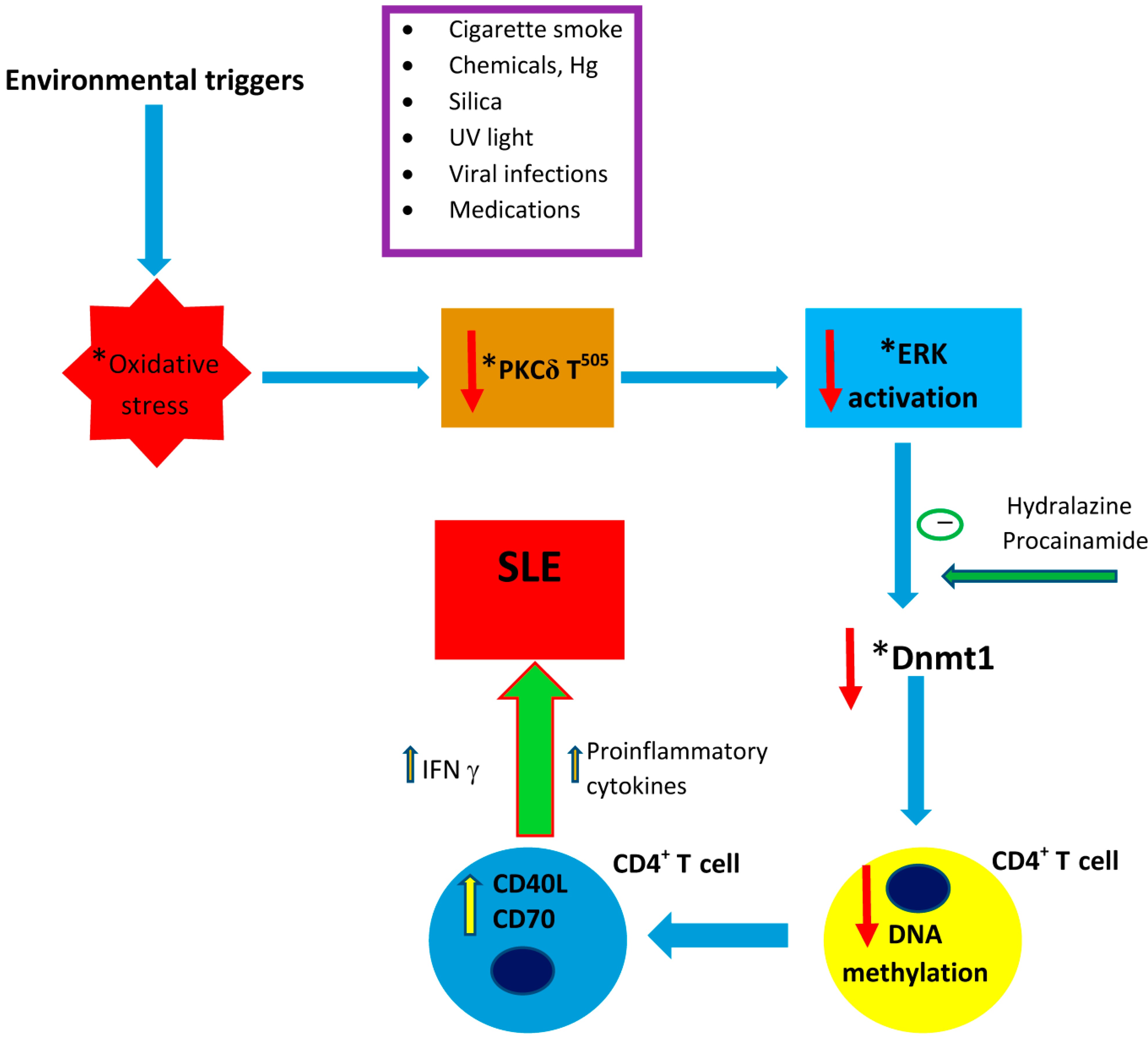
2. Potential Mechanisms of Induction of SLE-Interactions amongst Genetics, Epigenetics and Environmental Factors
2.1. Proposed Molecular Mechanisms
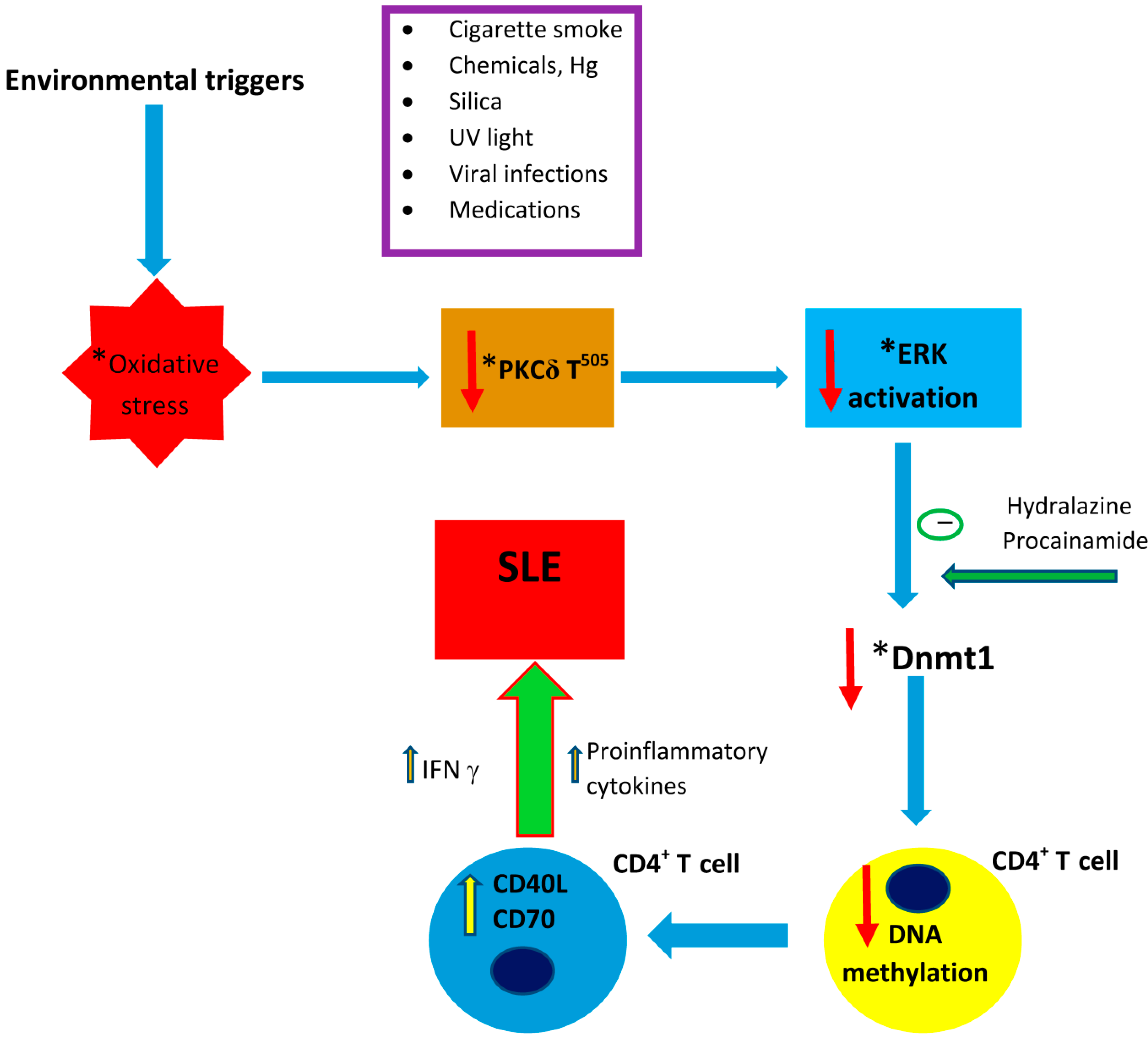
2.2. Evidence from Animal Models
3. Environmental Agents Data from Observational Studies
3.1. Ultraviolent Light
3.2. Vitamin D Deficiency
3.3. Smoking
3.4. Alcohol
3.5. Occupationally- and Non-Occupationally-Related Chemicals
4. Vaccinations
5. Medications
6. Conclusions and Future Directions
Author Contributions
Conflicts of Interest
References
- Wahren-Herlenius, M.; Dörner, T. Immunopathogenic mechanisms of systemic autoimmune disease. Lancet 2013, 382, 819–831. [Google Scholar] [CrossRef]
- Hewagama, A.; Richardson, B. The genetics and epigenetics of autoimmune diseases. J. Autoimmun. 2009, 33, 3–11. [Google Scholar] [CrossRef]
- Lisnevskaia, L.; Murphy, G.; Isenberg, D. Systemic lupus erythematosus. Lancet 2014. [Google Scholar] [CrossRef]
- Sawalha, A.H.; Jeffries, M.; Webb, R.; Lu, Q.; Gorelik, G.; Ray, D.; Osban, J.; Knowlton, N.; Johnson, K.; Richardson, B. Defective T-cell ERK signaling induces interferon-regulated gene expression and overexpression of methylation-sensitive genes similar to lupus patients. Genes Immun. 2008, 9, 368–378. [Google Scholar] [CrossRef]
- Wang, Z.; Patel, D.J. Combinatorial readout of dual histone modifications by paired chromatin-associated modules. J. Biol. Chem. 2011, 286, 18363–18368. [Google Scholar] [CrossRef]
- Heard, E. Recent advances in X-chromosome inactivation. Curr. Opin. Cell Biol. 2004, 16, 247–255. [Google Scholar] [CrossRef]
- Richardson, B. Primer: Epigenetics of autoimmunity. Nat. Clin. Pract. Rheumatol. 2007, 3, 521–527. [Google Scholar] [CrossRef]
- Basu, D.; Liu, Y.; Wu, A.; Yarlagadda, S.; Gorelik, G.J.; Kaplan, M.J.; Hewagama, A.; Hinderer, R.C.; Strickland, F.M.; Richardson, B.C. Stimulatory and inhibitory killer Ig-like receptor molecules are expressed and functional on lupus T cells. J. Immunol. 2009, 183, 3481–3487. [Google Scholar] [CrossRef]
- Wilson, C.B.; Rowell, E.; Sekimata, M. Epigenetic control of T-helper-cell differentiation. Nat. Rev. Immunol. 2009, 9, 91–105. [Google Scholar] [CrossRef]
- Lu, Q.; Wu, A.; Tesmer, L.; Ray, D.; Yousif, N.; Richardson, B. Demethylation of CD40LG on the inactive X in T cells from women with lupus. J. Immunol. 2007, 179, 6352–6358. [Google Scholar] [CrossRef]
- Mak, A.; Kow, N.Y. The pathology of T cells in systemic lupus erythematosus. J. Immunol. Res. 2014, 2014, 8. [Google Scholar]
- Deng, C.; Yang, J.; Scott, J.; Hanash, S.; Richardson, B.C. Role of the ras-MAPK signaling pathway in the DNA methyltransferase response to DNA hypomethylation. Biol. Chem. 1998, 379, 1113–1120. [Google Scholar]
- Deng, C.; Kaplan, M.J.; Yang, J.; Ray, D.; Zhang, Z.; McCune, W.J.; Hanash, S.M.; Richardson, B.C. Decreased Ras-mitogen-activated protein kinase signaling may cause DNA hypomethylation in T lymphocytes from lupus patients. Arthritis Rheumatol. 2001, 44, 397–407. [Google Scholar] [CrossRef]
- Gorelik, G.J.; Yarlagadda, S.; Patel, D.R.; Richardson, B.C. Protein kinase Cδ oxidation contributes to ERK inactivation in lupus T cells. Arthritis Rheumatol. 2012, 64, 2964–2974. [Google Scholar] [CrossRef]
- Somers, E.C.; Richardson, B.C. Environmental exposures, epigenetic changes and the risk of lupus. Lupus 2014, 23, 568–576. [Google Scholar] [CrossRef]
- Strickland, F.M.; Hewagama, A.; Lu, Q.; Wu, A.; Hinderer, R.; Webb, R.; Johnson, K.; Sawalha, A.H.; Delaney, C.; Yung, R.; et al. Environmental exposure, estrogen and two X chromosomes are required for disease development in an epigenetic model of lupus. J. Autoimmun. 2012, 38, J135–J143. [Google Scholar] [CrossRef]
- Leiss, H.; Niederreiter, B.; Bandur, T.; Schwarzecker, B.; Blüml, S.; Steiner, G.; Ulrich, W.; Smolen, J.S.; Stummvoll, G.H. Pristane-induced lupus as a model of human lupus arthritis: Evolvement of autoantibodies, internal organ and joint inflammation. Lupus 2013, 22, 778–792. [Google Scholar] [CrossRef]
- Reeves, W.H.; Lee, P.Y.; Weinstein, J.S.; Satoh, M.; Lu, L. Induction of autoimmunity by pristane and other naturally occurring hydrocarbons. Trends Immunol. 2009, 30, 455–464. [Google Scholar] [CrossRef]
- Satoh, M.; Richards, H.B.; Shaheen, V.M.; Yoshida, H.; Shaw, M.; Naim, J.O.; Wooley, P.H.; Reeves, W.H. Widespread susceptibility among inbred mouse strains to the induction of lupus autoantibodies by pristane. Clin. Exp. Immunol. 2000, 121, 399–405. [Google Scholar] [CrossRef]
- Bender, A.T.; Wu, Y.; Cao, Q.; Ding, Y.; Oestreicher, J.; Genest, M.; Akare, S.; Ishizaka, S.T.; Mackey, M.F. Assessment of the translational value of mouse lupus models using clinically relevant biomarkers. Transl. Res. 2014, 163, 515–532. [Google Scholar] [CrossRef]
- Bijl, M.; Kallenberg, C.G. Ultraviolet light and cutaneous lupus. Lupus 2006, 15, 724–727. [Google Scholar] [CrossRef]
- Caricchio, R.; McPhie, L.; Cohen, P.L.; Ultraviolet, B. radiation-induced cell death: Critical role of ultraviolet dose in inflammation and lupus autoantigen redistribution. J. Immunol. 2003, 171, 5778–5786. [Google Scholar] [CrossRef]
- Muñoz-Ortego, J.; Torrente-Segarra, V.; Prieto-Alhambra, D.; Salman-Monte, T.C.; Carbonell-Abello, J. Prevalence and predictors of vitamin D deficiency in non-supplemented women with systemic lupus erythematosus in the Mediterranean region: A cohort study. Scand. J. Rheumatol. 2012, 41, 472–475. [Google Scholar] [CrossRef]
- Chun, R.F.; Liu, P.T.; Modlin, R.L.; Adams, J.S.; Hewison, M. Impact of vitamin D on immune function: Lessons learned from genome-wide analysis. Front. Physiol. 2014. [Google Scholar] [CrossRef]
- Petri, M.; Bello, K.J.; Fang, H.; Magder, L.S. Vitamin D in systemic lupus erythematosus: Modest association with disease activity and the urine protein-to-creatinine ratio. Arthritis Rheumatol. 2013, 65, 1865–1871. [Google Scholar] [CrossRef]
- Ramagopalan, S.V.; Heger, A.; Berlanga, A.J.; Maugeri, N.J.; Lincoln, M.R.; Burrell, A.; Handunnetthi, L.; Handel, A.E.; Disanto, G.; Orton, S.M.; et al. ChIP-seq defined genome-wide map of vitamin D receptor binding: Associations with disease and evolution. Genome Res. 2010, 20, 1352–1360. [Google Scholar]
- Ben-Zvi, I.; Aranow, C.; Mackay, M.; Stanevsky, A.; Kamen, D.L.; Marinescu, L.M.; Collins, C.E.; Gilkeson, G.S.; Diamond, B.; Hardin, J.A. The impact of vitamin D on dendritic cell function in patients with systemic lupus erythematosus. PLoS One 2010, 5, e9193. [Google Scholar] [CrossRef]
- Ritterhouse, L.L.; Crowe, S.R.; Niewold, T.B.; Kamen, D.L.; Macwana, S.R.; Roberts, V.C.; Dedeke, A.B.; Harley, J.B.; Scofield, R.H.; Guthridge, J.M.; et al. Vitamin D deficiency is associated with an increased autoimmune response in healthy individuals and in patients with systemic lupus erythematosus. Ann. Rheum. Dis. 2011, 70, 1569–1574. [Google Scholar] [CrossRef]
- Linker-Israeli, M.; Elstner, E.; Klinenberg, J.R.; Wallace, D.J.; Koeffler, H.P. Vitamin D3 and its synthetic analogs inhibit the spontaneous in vitro immunoglobulin production by SLE-derived PBMC. Clin. Immunol. 2001, 99, 82–93. [Google Scholar] [CrossRef]
- Chen, S.; Sims, G.P.; Chen, X.X.; Gu, Y.Y.; Chen, S.; Lipsky, P.E. Modulatory effects of 1,25-dihydroxyvitamin D3 on human B cell differentiation. J. Immunol. 2007, 179, 1634–1647. [Google Scholar] [CrossRef]
- Linnik, M.D.; Hu, J.Z.; Heilbrunn, K.R.; Strand, V.; Hurley, F.L.; Joh, T. Relationship between anti-double-stranded DNA antibodies and exacerbation of renal disease in patients with systemic lupus erythematosus. Arthritis Rheumatol. 2005, 52, 1129–1137. [Google Scholar] [CrossRef]
- Robinson, A.B.; Thierry-Palmer, M.; Gibson, K.L.; Rabinovich, C.E. Disease activity, proteinuria, and vitamin D status in children with systemic lupus erythematosus and juvenile dermatomyositis. J. Pediatr. 2012, 160, 297–302. [Google Scholar] [CrossRef]
- Cohen, A.B.; Chenoweth, D.E.; Hugli, T.E. The release of elastase, myeloperoxidase, and lysozyme from human alveolar macrophages. Am. Rev. Respir. Dis. 1982, 126, 241–247. [Google Scholar]
- Costenbader, K.H.; Karlson, E.W. Cigarette smoking and systemic lupus erythematosus: A smoking gun? Autoimmunity 2005, 38, 541–547. [Google Scholar] [CrossRef]
- Costenbader, K.H.; Kim, D.J.; Peerzada, J.; Lockman, S.; Nobles-Knight, D.; Petri, M.; Karlson, E.W. Cigarette smoking and the risk of systemic lupus erythematosus: A meta-analysis. Arthritis Rheumatol. 2004, 50, 849–857. [Google Scholar] [CrossRef]
- Lim, S.S.; Drenkard, C. The epidemiology of lupus. In Dubois’Lupus Erythematosus and Related Syndromes, 8th ed.; Elsevier: Philadelphia, PA, USA, 2013. [Google Scholar]
- Kiyohara, C.; Washio, M.; Horiuchi, T.; Asami, T.; Ide, S.; Atsumi, T.; Kobashi, G.; Tada, Y.; Takahashi, H. Kyushu Sapporo SLE (KYSS) Study Group. Cigarette smoking, alcohol consumption, and risk of systemic lupus erythematosus: A case-control study in a Japanese population. J. Rheumatol. 2012, 39, 1363–1370. [Google Scholar] [CrossRef]
- Kiyohara, C.; Washio, M.; Horiuchi, T.; Tada, Y.; Asami, T.; Ide, S.; Atsumi, T.; Kobashi, G.; Takahashi, H. Kyushu Sapporo SLE (KYSS) Study Group. Cigarette smoking, STAT4 and TNFRSF1B polymorphisms, and systemic lupus erythematosus in a Japanese population. J. Rheumatol. 2009, 36, 2195–2203. [Google Scholar] [CrossRef]
- Washio, M.; Horiuchi, T.; Kiyohara, C.; Kodama, H.; Tada, Y.; Asami, T.; Takahashi, H.; Kobashi, G.; Abe, T.; Tanaka, H.; et al. Smoking, drinking, sleeping habits, and other lifestyle factors and the risk of systemic lupus erythematosus in Japanese females: Findings from the KYSS study. Mod. Rheumatol. 2006, 16, 143–150. [Google Scholar] [CrossRef]
- Ezra, N.; Jorizzo, J. Hydroxychloroquine and smoking in patients with cutaneous lupus erythematosus. Clin. Exp. Dermatol. 2012, 37, 327–334. [Google Scholar] [CrossRef]
- Takvorian, S.U.; Merola, J.F.; Costenbader, K.H. Cigarette smoking, alcohol consumption and risk of systemic lupus erythematosus. Lupus 2014, 23, 537–544. [Google Scholar] [CrossRef]
- Formica, M.K.; Palmer, J.R.; Rosenberg, L.; McAlindon, T.E. Smoking, alcohol consumption, and risk of systemic lupus erythematosus in the Black Women’s Health Study. J. Rheumatol. 2003, 30, 1222–1226. [Google Scholar]
- Wang, J.; Kay, A.B.; Fletcher, J.; Formica, M.K.; McAlindon, T.E. Alcohol consumption is not protective for systemic lupus erythematosus. Ann. Rheum. Dis. 2009, 68, 345–348. [Google Scholar] [CrossRef]
- Wang, J.; Pan, H.F.; Ye, D.Q.; Su, H.; Li, X.P. Moderate alcohol drinking might be protective for systemic lupus erythematosus: A systematic review and meta-analysis. Clin. Rheumatol. 2008, 27, 1557–1563. [Google Scholar] [CrossRef]
- Parks, C.G.; Cooper, G.S.; Nylander-French, L.A.; Sanderson, W.T.; Dement, J.M.; Cohen, P.L.; Dooley, M.A.; Treadwell, E.L.; St Clair, E.W.; Gilkeson, G.S.; et al. Occupational exposure to crystalline silica and risk of systemic lupus erythematosus: A population-based, case-control study in the southeastern United States. Arthritis Rheumatol. 2002, 46, 1840–1850. [Google Scholar] [CrossRef]
- Cooper, G.S.; Wither, J.; Bernatsky, S.; Claudio, J.O.; Clarke, A.; Rioux, J.D.; CaNIOS GenES Investigators; Fortin, P.R. Occupational and environmental exposures and risk of systemic lupus erythematosus: Silica, sunlight, solvents. Rheumatology 2010, 49, 2172–2180. [Google Scholar] [CrossRef]
- Finckh, A.; Cooper, G.S.; Chibnik, L.B.; Costenbader, K.H.; Watts, J.; Pankey, H.; Fraser, P.A.; Karlson, E.W. Occupational silica and solvent exposures and risk of systemic lupus erythematosus in urban women. Arthritis Rheumatol. 2006, 54, 3648–3654. [Google Scholar] [CrossRef]
- Makol, A.; Reilly, M.J.; Rosenman, K.D. Prevalence of connective tissue disease in silicosis (1985–2006)—A report from the state of Michigan surveillance system for silicosis. Am. J. Ind. Med. 2011, 54, 255–262. [Google Scholar] [CrossRef]
- Tsai, P.C.; Ko, Y.C.; Huang, W.; Liu, H.S.; Guo, Y.L. Increased liver and lupus mortalities in 24-year follow-up of the Taiwanese people highly exposed to polychlorinated biphenyls and dibenzofurans. Sci. Total Environ. 2007, 374, 216–222. [Google Scholar] [CrossRef]
- Parks, C.G.; de Roos, A.J. Pesticides, chemical and industrial exposures in relation to systemic lupus erythematosus. Lupus 2014, 23, 527–536. [Google Scholar] [CrossRef]
- Hultman, P.; Eneström, S. Mercury induced antinuclear antibodies in mice: Characterization and correlation with renal immune complex deposits. Clin. Exp. Immunol. 1988, 71, 269–274. [Google Scholar]
- Gardner, R.M.; Nyland, J.F.; Silva, I.A.; Ventura, A.M.; de Souza, J.M.; Silbergeld, E.K. Mercury exposure, serum antinuclear/antinucleolar antibodies, and serum cytokine levels in mining populations in Amazonian Brazil: A cross-sectional study. Environ. Res. 2010, 110, 345–354. [Google Scholar] [CrossRef]
- Shenker, B.J.; Mayro, J.S.; Rooney, C.; Vitale, L.; Shapiro, I.M. Immunotoxic effects of mercuric compounds on human lymphocytes and monocytes. IV. Alterations in cellular glutathione content. Immunopharmacol. Immunotoxicol. 1993, 15, 273–290. [Google Scholar] [CrossRef]
- Shenker, B.J.; Guo, T.L.; Shapiro, I.M. Low-Level methylmercury exposure causes human T-cells to undergo apoptosis: Evidence of mitochondrial dysfunction. Environ. Res. 1998, 77, 149–159. [Google Scholar] [CrossRef]
- Cooper, G.S.; Parks, C.G.; Treadwell, E.L.; St Clair, E.W.; Gilkeson, G.S.; Dooley, M.A. Occupational risk factors for the development of systemic lupus erythematosus. J. Rheumatol. 2004, 31, 1928–1933. [Google Scholar]
- Lim, S.Y.; Ghosh, S.K. Autoreactive responses to environmental factors: 3. Mouse strain-specific differences in induction and regulation of anti-DNA antibody responses due to phthalate-isomers. J. Autoimmun. 2005, 25, 33–45. [Google Scholar] [CrossRef]
- Wang, J.; Kay, A.B.; Fletcher, J.; Formica, M.K.; McAlindon, T.E. Is lipstick associated with the development of systemic lupus erythematosus (SLE)? Clin. Rheumatol. 2008, 27, 1183–1187. [Google Scholar] [CrossRef]
- Reidenberg, M.M. Aromatic amines and the pathogenesis of lupus erythematosus. Am. J. Med. 1983, 75, 1037–1042. [Google Scholar] [CrossRef]
- Cooper, G.; Dooley, M.A.; Treadwell, E.L.; St Clair, E.W.; Gilkeson, G.S. Smoking and use of hair treatments in relation to risk of developing systemic lupus erythematosus. J. Rheumatol. 2001, 28, 2653–2656. [Google Scholar]
- Jiménez-Alonso, J.1.; Sabio, J.M.; Pérez-Alvarez, F.; Reche, I.; Hidalgo, C.; Jáimez, L. Grupo Lupus Virgen de las Nieves. Hair dye treatment use and clinical course in patients with systemic lupus erythematosus and cutaneous lupus. Lupus 2002, 11, 430–434. [Google Scholar] [CrossRef]
- Colafrancesco, S.; Perricone, C.; Priori, R.; Valesini, G.; Shoenfeld, Y. Sjögren’s syndrome: Another facet of the autoimmune/inflammatory syndrome induced by adjuvants (ASIA). J. Autoimmun. 2014, 51, 10–16. [Google Scholar] [CrossRef]
- Shoenfeld, Y.; Aron-Maor, A. Vaccination and autoimmunity—Vaccinosis: A dangerous liaison? J. Autoimmun. 2000, 14, 1–10. [Google Scholar] [CrossRef]
- Kowal, C.; Weinstein, A.; Diamond, B. Molecular mimicry between bacterial and self antigen in a patient with systemic lupus erythematosus. Eur. J. Immunol. 1999, 29, 1901–1911. [Google Scholar] [CrossRef]
- Murdaca, G.; Orsi, A.; Spanò, F.; Puppo, F.; Durando, P.; Icardi, G.; Ansaldi, F. Influenza and pneumococcal vaccinations of patients with systemic lupus erythematosus: Current views upon safety and immunogenicity. Autoimmun. Rev. 2014, 13, 75–84. [Google Scholar] [CrossRef]
- Grimaldi-Bensouda, L.; le Guern, V.; Kone-Paut, I.; Aubrun, E.; Fain, O.; Ruel, M.; Machet, L.; Viallard, J.F.; Magy-Bertrand, N.; Daugas, E.; et al. The risk of systemic lupus erythematosus associated with vaccines: An international case-control study. Arthritis Rheumatol. 2014, 66, 1559–1567. [Google Scholar] [CrossRef]
- Mok, C.C.; Ho, L.Y.; Fong, L.S.; To, C.H. Immunogenicity and safety of a quadrivalent human papillomavirus vaccine in patients with systemic lupus erythematosus: A case-control study. Ann. Rheum. Dis. 2013, 72, 659–6564. [Google Scholar] [CrossRef]
- Scheinbart, L.S.; Johnson, M.A.; Gross, L.A.; Edelstein, S.R.; Richardson, B.C. Procainamide inhibits DNA methyltransferase in a human T cell line. J. Rheumatol. 1991, 18, 530–534. [Google Scholar]
- Lu, Q.; Wu, A.; Richardson, B.C. Demethylation of the same promoter sequence increases CD70 expression in lupus T cells and T cells treated with lupus-inducing drugs. J. Immunol. 2005, 174, 6212–6219. [Google Scholar] [CrossRef]
- Zhu, L.J.; Yang, X.; Yu, X.Q. Anti-TNFα therapies in systemic lupus erythematosus. J. Biomed. Biotechnol. 2010, 2010, 465898. [Google Scholar]
- Mok, C.C.; Lau, C.S. Pathogenesis of systemic lupus erythematosus. J. Clin. Pathol. 2003, 56, 481–490. [Google Scholar]
- Verthelyi, D. Sex hormones as immunomodulators in health and disease. Int. Immunopharmacol. 2001, 1, 983–993. [Google Scholar] [CrossRef]
- Bynoté, K.K.; Hackenberg, J.M.; Korach, K.S.; Lubahn, D.B.; Lane, P.H.; Gould, K.A. Estrogen receptor-α deficiency attenuates autoimmune disease in (NZB × NZW)F1 mice. Genes Immun. 2008, 9, 137–152. [Google Scholar] [CrossRef]
- Gilbert, E.L.; Mathis, K.W.; Ryan, M.J. 17β-Estradiol protects against the progression of hypertension during adulthood in a mouse model of systemic lupus erythematosus. Hypertension 2014, 63, 616–623. [Google Scholar] [CrossRef]
- Rider, V.; Jones, S.; Evans, M.; Bassiri, H.; Afsar, Z.; Abdou, N.I. Estrogen increases CD40 ligand expression in T cells from women with systemic lupus erythematosus. J. Rheumatol. 2001, 28, 2644–2649. [Google Scholar]
- McMurray, R.W.; Ndebele, K.; Hardy, K.J.; Jenkins, J.K. 17β-Estradiol suppresses IL-2 and IL-2 receptor. Cytokine 2001, 14, 324–333. [Google Scholar] [CrossRef]
- Petri, M.; Kim, M.Y.; Kalunian, K.C.; Grossman, J.; Hahn, B.H.; Sammaritano, L.R.; Lockshin, M.; Merrill, J.T.; Belmont, H.M.; Askanase, A.D.; et al. Combined oral contraceptives in women with systemic lupus erythematosus. N. Engl. J. Med. 2005, 353, 2550–2558. [Google Scholar] [CrossRef]
- Buyon, J.P.; Petri, M.A.; Kim, M.Y.; Kalunian, K.C.; Grossman, J.; Hahn, B.H.; Merrill, J.T.; Sammaritano, L.; Lockshin, M.; Alarcón, G.S.; et al. The effect of combined estrogen and progesterone hormone replacement therapy on disease activity in systemic lupus erythematosus: A randomized trial. Ann. Intern. Med. 2005, 142, 953–962. [Google Scholar] [CrossRef]
- Mak, A.; Cheung, M.W.; Ho, R.C.; Cheak, A.A.; Lau, C.S. Bisphosphonates and atrial fibrillation: Bayesian meta-analyses of randomized controlled trials and observational studies. BMC Musculoskelet. Disord. 2009, 10, 113. [Google Scholar] [CrossRef]
- Mak, A.; Almsherqi, Z.A.; Lai, Y.W.; Cheak, A.A.; Deng, Y. Intracellular tubulo-reticular structures of peripheral blood mononuclear cells as an ultra-structural marker of disease activity in systemic lupus erythematosus: A pilot study. Int. J. Rheum. Dis. 2013, 16, 692–697. [Google Scholar] [CrossRef]
- Helder, A.W.; Feltkamp-Vroom, T.M. Tubuloreticular structures and antinuclear antibodies in autoimmune and non-autoimmune diseases. J. Pathol. 1976, 119, 49–56. [Google Scholar] [CrossRef]
- Sinniah, R.; Feng, P.H. Lupus nephritis: Correlation between light, electron microscopic and immunofluorescent findings and renal function. Clin. Nephrol. 1976, 6, 340–351. [Google Scholar]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Mak, A.; Tay, S.H. Environmental Factors, Toxicants and Systemic Lupus Erythematosus. Int. J. Mol. Sci. 2014, 15, 16043-16056. https://doi.org/10.3390/ijms150916043
Mak A, Tay SH. Environmental Factors, Toxicants and Systemic Lupus Erythematosus. International Journal of Molecular Sciences. 2014; 15(9):16043-16056. https://doi.org/10.3390/ijms150916043
Chicago/Turabian StyleMak, Anselm, and Sen Hee Tay. 2014. "Environmental Factors, Toxicants and Systemic Lupus Erythematosus" International Journal of Molecular Sciences 15, no. 9: 16043-16056. https://doi.org/10.3390/ijms150916043
APA StyleMak, A., & Tay, S. H. (2014). Environmental Factors, Toxicants and Systemic Lupus Erythematosus. International Journal of Molecular Sciences, 15(9), 16043-16056. https://doi.org/10.3390/ijms150916043