Data Mining of Atherosclerotic Plaque Transcriptomes Predicts STAT1-Dependent Inflammatory Signal Integration in Vascular Disease

Abstract
:1. Introduction
2. Results
2.1. STAT1 Target Genes Are Profoundly Present in Coronary Plaques

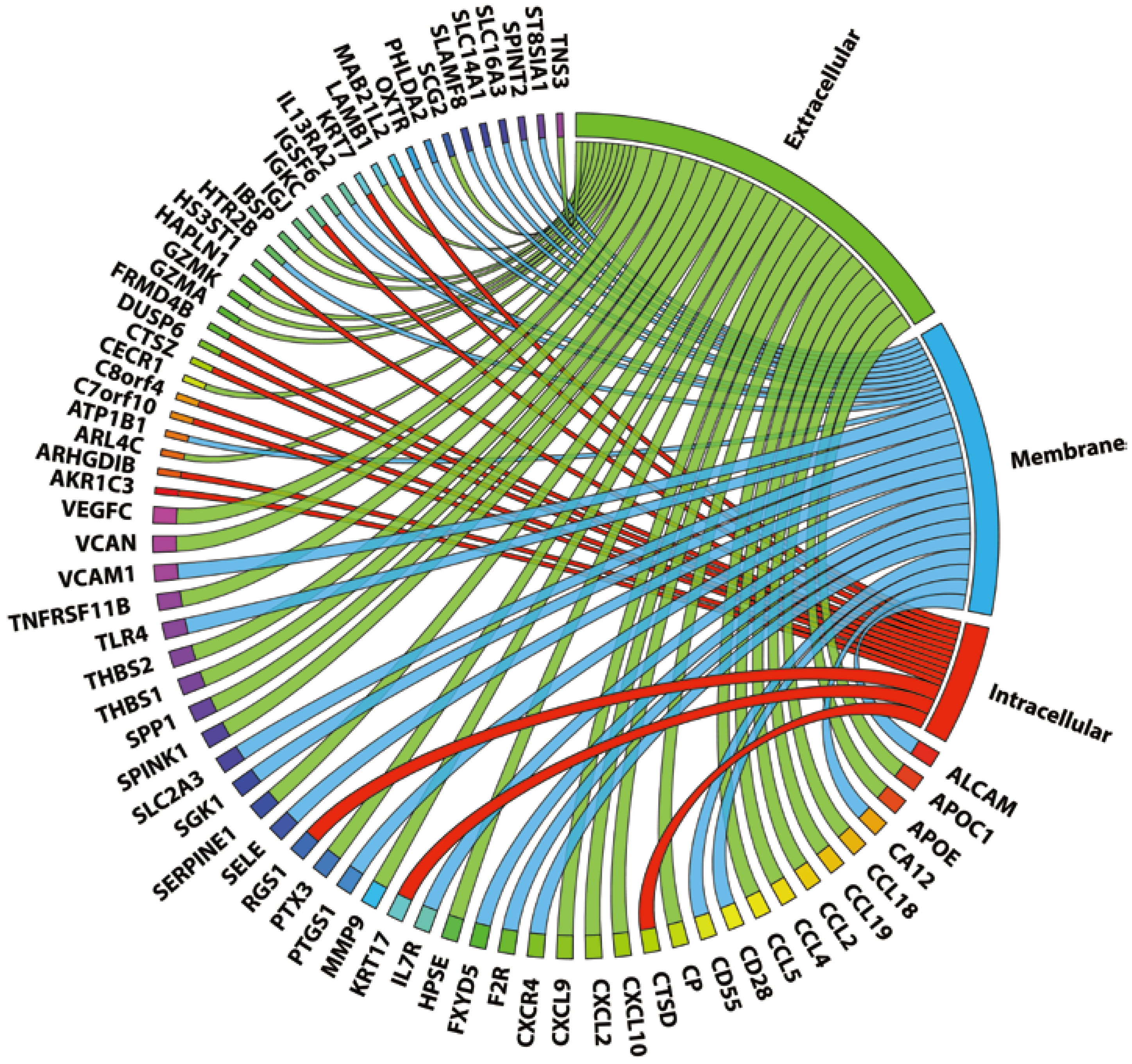{kind=link}
{kind=link}
| GO Term | GO ID | log10 p-Value |
|---|---|---|
| response to external stimulus | GO:0009605 | −12.1409 |
| biological adhesion | GO:0022610 | −11.2716 |
| single-multicellular organism process | GO:0044707 | −11.2716 |
| locomotion | GO:0040011 | −11.0788 |
| multicellular organismal process | GO:0032501 | −10.5817 |
| single-organism process | GO:0044699 | −10.0362 |
| developmental process | GO:0032502 | −9.0752 |
| anatomical structure development | GO:0048856 | −8.7645 |
| single-organism cellular process | GO:0044763 | −8.4034 |
| response to stimulus | GO:0050896 | −8.2916 |
| signalling | GO:0023052 | −5.8633 |
| single organism signalling | GO:0044700 | −5.8633 |
| localization of cell | GO:0051674 | −5.4306 |
| response to chemical stimulus | GO:0042221 | −4.3116 |
| immune system process | GO:0002376 | −3.6253 |
| regulation of biological quality | GO:0065008 | −3.4145 |
| behaviour | GO:0007610 | −3.2549 |
| response to abiotic stimulus | GO:0009628 | −2.6289 |
| rhythmic process | GO:0048511 | −2.3675 |
| STAT1 | NFκB | IRF | ||
|---|---|---|---|---|
| GAS | ISRE | |||
| coronary | 5.14 | 3.63 | 5.05 | 3.66 |
| carotid | 4.92 | 6.39 | 5.23 | 6.29 |
| common | 3.24 | 2.06 | 2.87 | 4.86 |
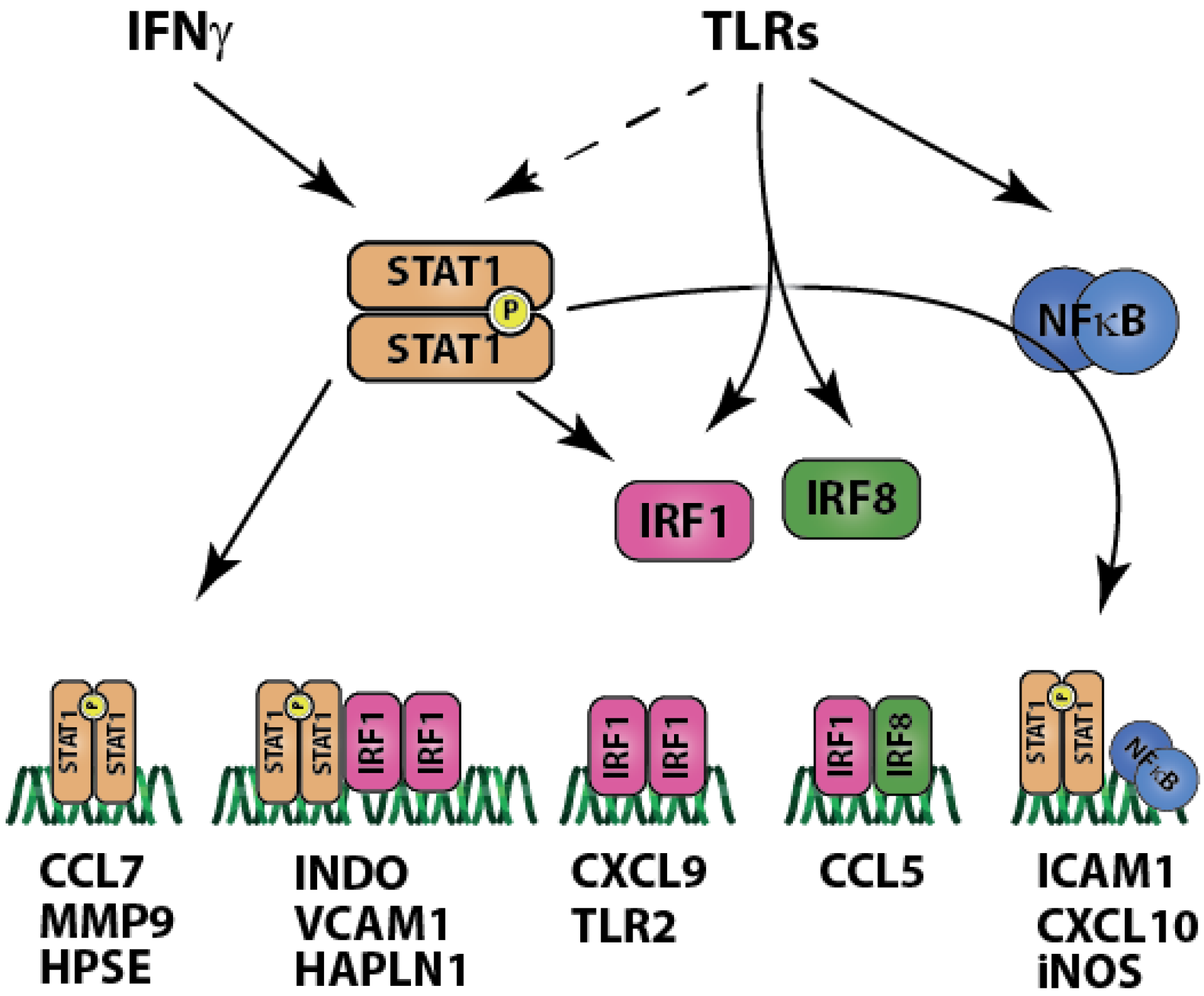
2.2. Carotid Plaques Share with Coronary Plaques Similar Pro-Inflammatory Gene Expression Patterns Governed by STAT1
| GO Term | GO ID | log10 p-Value |
|---|---|---|
| immune system process | GO:0002376 | −133.4342 |
| immune response | GO:0006955 | −120.0516 |
| response to stimulus | GO:0050896 | −77.3635 |
| response to biotic stimulus | GO:0009607 | −55.3516 |
| response to stress | GO:0006950 | −54.1255 |
| response to chemical stimulus | GO:0042221 | −53.7212 |
| multi-organism process | GO:0051704 | −47.5129 |
| signalling | GO:0023052 | −28.9431 |
| single organism signalling | GO:0044700 | −28.9431 |
| single-organism cellular process | GO:0044763 | −25.752 |
| single-organism process | GO:0044699 | −25.5622 |
| biological adhesion | GO:0022610 | −23.9788 |
| regulation of biological process | GO:0050789 | −23.9706 |
| immune effector process | GO:0002252 | −23.7825 |
| biological regulation | GO:0065007 | −22.2381 |
| interspecies interaction between organisms | GO:0044419 | −22.1319 |
| locomotion | GO:0040011 | −22.0991 |
| regulation of molecular function | GO:0065009 | −20.7825 |
| regulation of biological quality | GO:0065008 | −18.4841 |
2.3. 72 Genes Are Expressed in both Coronary and Carotid Plaques and Form “Plaque Signature”
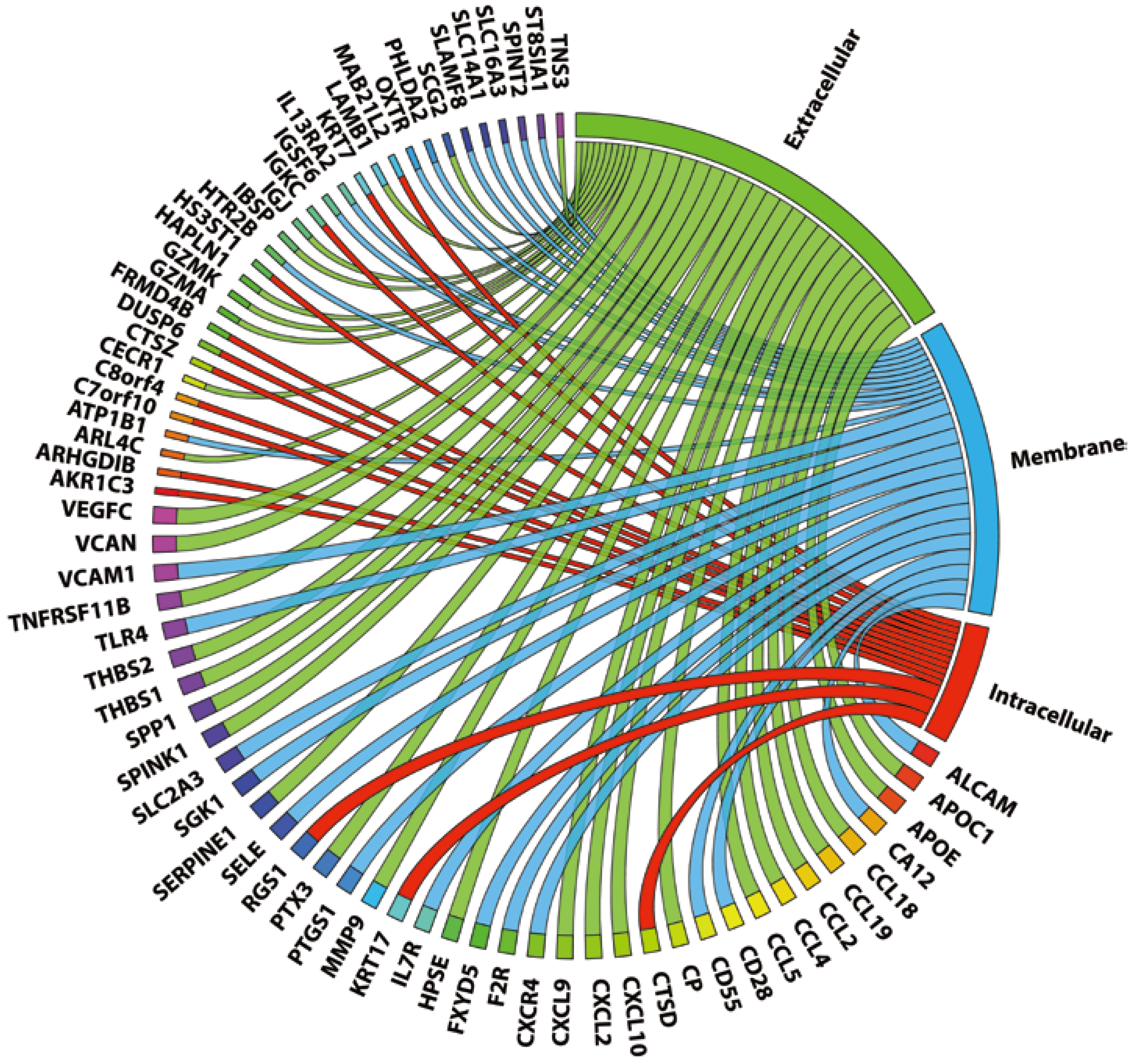
| Gene Symbol | Known STAT1 Regulation | ISRE | STAT1-NFκB | STAT1-IRF | |
|---|---|---|---|---|---|
| APOC1 | no | + | − | − | |
| APOE | no | + | − | − | |
| CCL18 | no | + | + | + | |
| CCL19 | no | + | + | + | |
| CCL2 | yes [30,31] | + | + | + | |
| CCL4 | yes [32] | + | + | + | |
| CCL5 | yes [33] | + | + | + | |
| CP | no | + | − | + | |
| CXCL10 | yes [34] | + | + | − | |
| CXCL2 | no | − | + | − | |
| CXCL9 | yes [34] | + | − | − | |
| GZMA | no | + | − | + | |
| GZMK | no | + | − | + | |
| HAPLN1 | no | + | + | + | |
| HPSE | no | − | − | − | |
| IBSP | no | + | − | − | |
| IGJ | no | − | + | + | |
| LAMB1 | no | + | + | + | |
| MMP9 | yes [35] | − | + | + | |
| PTX3 | no | + | − | + | |
| SCG2 | no | + | − | + | |
| SERPINE1 | yes [36] | + | + | + | |
| SPINK1 | no | + | − | + | |
| SPP1 | no | + | + | + | |
| THBS1 | no | + | − | + | |
| THBS2 | no | + | − | − | |
| TNFRSF11B | no | + | + | + | |
| TNS3 | no | + | − | − | |
| VCAN | no | + | + | + | |
| VEGFC | yes [37] | + | − | + | |
2.4. Genes Common for Carotid and Coronary Plaques Are Strongly Involved in Plaque Formation Processes and Could Be Regulated by STAT1
| GO Term | GO ID | log10 p-Value |
|---|---|---|
| biological adhesion | GO:0022610 | −7.8861 |
| immune system process | GO:0002376 | −6.2765 |
| locomotion | GO:0040011 | −6.2765 |
| single-organism process | GO:0044699 | −5.5575 |
| single-organism cellular process | GO:0044763 | −5.2832 |
| immune response | GO:0006955 | −4.8041 |
| response to stress | GO:0006950 | −4.6402 |
| localization of cell | GO:0051674 | −4.5607 |
| response to external stimulus | GO:0009605 | −4.1475 |
| response to chemical stimulus | GO:0042221 | −4.0329 |
| multi-organism process | GO:0051704 | −3.2677 |
| multi-multicellular organism process | GO:0044706 | −2.7118 |
| regulation of biological quality | GO:0065008 | −2.7082 |
| response to biotic stimulus | GO:0009607 | −2.6917 |
| localization | GO:0051179 | −2.5934 |
| response to stimulus | GO:0050896 | −2.121 |
| response to abiotic stimulus | GO:0009628 | −2.1199 |
| single-organism developmental process | GO:0044767 | −2.0234 |
3. Discussion
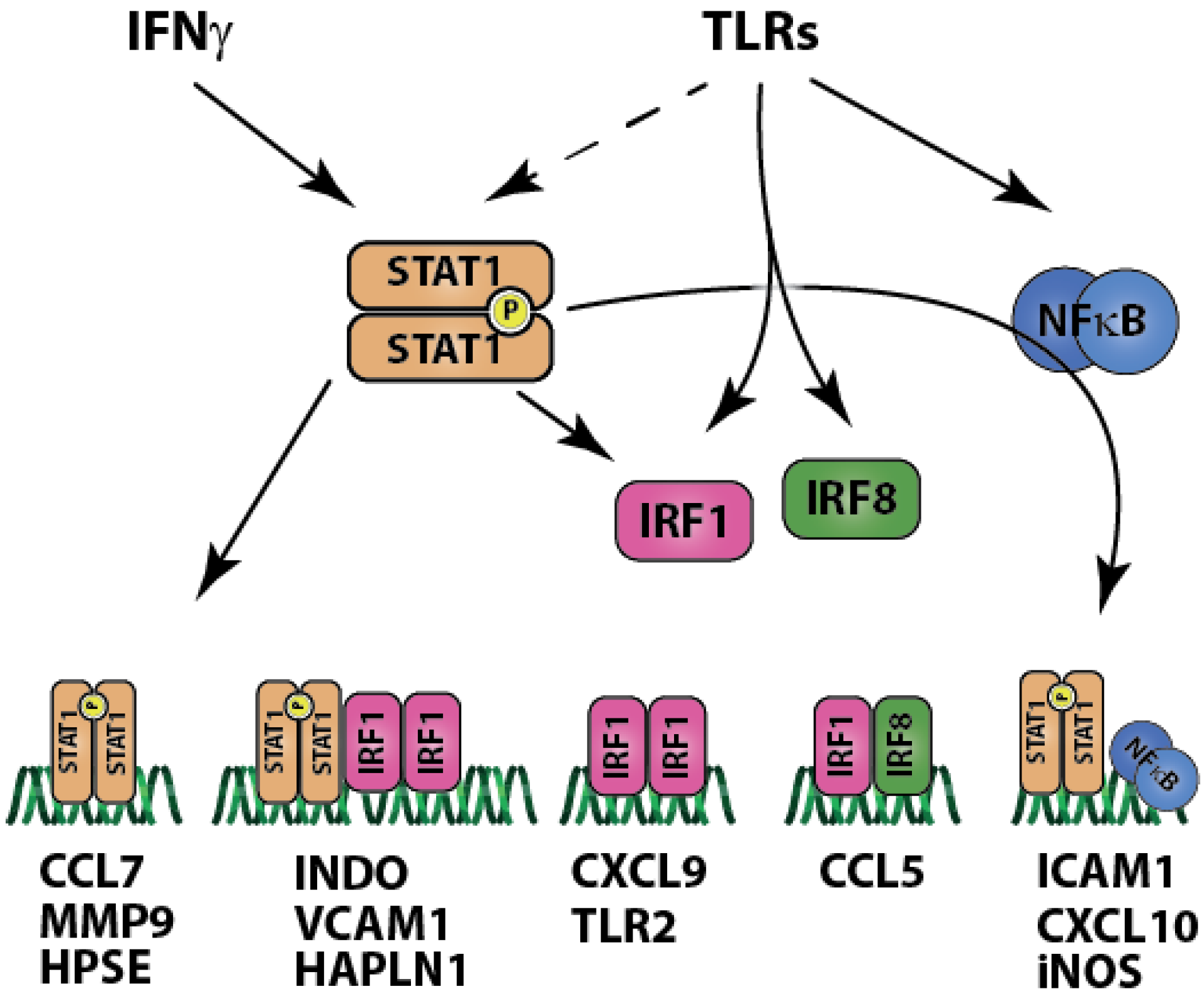
4. Materials and Methods
4.1. Microarray Data Normalization and Analysis
4.2. Gene Ontology Enrichment Studies
4.3. Pathway Analysis and Literature Mining
4.4. Promoter Analysis
4.5. Supplementary Data
5. Conclusions
Supplementary Files
Supplementary File 1Acknowledgments
Author Contributions
Conflicts of Interest
References
- Raines, E.W. The extracellular matrix can regulate vascular cell migration, proliferation, and survival: Relationships to vascular disease. Int. J. Exp. Pathol. 2000, 81, 173–182. [Google Scholar] [CrossRef]
- Ameriso, S.F.; Fridman, E.A.; Leiguarda, R.C.; Sevlever, G.E. Detection of helicobacter pylori in human carotid atherosclerotic plaques. Stroke J. Cereb. Circ. 2001, 32, 385–391. [Google Scholar]
- Sitia, S.; Tomasoni, L.; Atzeni, F.; Ambrosio, G.; Cordiano, C.; Catapano, A.; Tramontana, S.; Perticone, F.; Naccarato, P.; Camici, P.; et al. From endothelial dysfunction to atherosclerosis. Autoimmun. Rev. 2010, 9, 830–834. [Google Scholar] [CrossRef]
- Dauphinee, S.M.; Karsan, A. Lipopolysaccharide signaling in endothelial cells. Lab. Investig. J. Tech. Methods Pathol. 2006, 86, 9–22. [Google Scholar]
- Robertson, A.K.; Hansson, G.K. T cells in atherogenesis: For better or for worse? Arterioscler. Thromb. Vasc. Biol. 2006, 26, 2421–2432. [Google Scholar] [CrossRef]
- Hansson, G.K.; Robertson, A.K.; Soderberg-Naucler, C. Inflammation and atherosclerosis. Annu. Rev. Pathol. 2006, 1, 297–329. [Google Scholar]
- Hidalgo, L.G.; Halloran, P.F. Role of ifn-gamma in allograft rejection. Crit. Rev. Immunol. 2002, 22, 317–349. [Google Scholar]
- Tellides, G.; Tereb, D.A.; Kirkiles-Smith, N.C.; Kim, R.W.; Wilson, J.H.; Schechner, J.S.; Lorber, M.I.; Pober, J.S. Interferon-gamma elicits arteriosclerosis in the absence of leukocytes. Nature 2000, 403, 207–211. [Google Scholar]
- Tamura, T.; Yanai, H.; Savitsky, D.; Taniguchi, T. The irf family transcription factors in immunity and oncogenesis. Annu. Rev. Immunol. 2008, 26, 535–584. [Google Scholar]
- Liu, J.; Guan, X.; Ma, X. Interferon regulatory factor 1 is an essential and direct transcriptional activator for interferon {gamma}-induced rantes/ccl5 expression in macrophages. J. Biol. Chem. 2005, 280, 24347–24355. [Google Scholar]
- Ohmori, Y.; Hamilton, T.A. The interferon-stimulated response element and a kappa b site mediate synergistic induction of murine ip-10 gene transcription by ifn-gamma and tnf-alpha. J. Immunol. 1995, 154, 5235–5244. [Google Scholar]
- Methe, H.; Kim, J.O.; Kofler, S.; Weis, M.; Nabauer, M.; Koglin, J. Expansion of circulating toll-like receptor 4-positive monocytes in patients with acute coronary syndrome. Circulation 2005, 111, 2654–2661. [Google Scholar]
- Michelsen, K.S.; Wong, M.H.; Shah, P.K.; Zhang, W.; Yano, J.; Doherty, T.M.; Akira, S.; Rajavashisth, T.B.; Arditi, M. Lack of toll-like receptor 4 or myeloid differentiation factor 88 reduces atherosclerosis and alters plaque phenotype in mice deficient in apolipoprotein e. Proc. Natl. Acad. Sci. USA 2004, 101, 10679–10684. [Google Scholar]
- Sakaguchi, S.; Negishi, H.; Asagiri, M.; Nakajima, C.; Mizutani, T.; Takaoka, A.; Honda, K.; Taniguchi, T. Essential role of irf-3 in lipopolysaccharide-induced interferon-beta gene expression and endotoxin shock. Biochem. Biophys. Res. Commun. 2003, 306, 860–866. [Google Scholar]
- Sikorski, K.; Chmielewski, S.; Olejnik, A.; Wesoly, J.; Heemann, U.; Baumann, M.; Bluyssen, H.A. Stat1 as a central mediator of ifnγ and tlr4 signal integration in vascular dysfunction. JAK-STAT 2012, 1, 241–249. [Google Scholar] [CrossRef]
- Ohmori, Y.; Schreiber, R.D.; Hamilton, T.A. Synergy between interferon-gamma and tumor necrosis factor-alpha in transcriptional activation is mediated by cooperation between signal transducer and activator of transcription 1 and nuclear factor kappab. J. Biol. Chem. 1997, 272, 14899–14907. [Google Scholar]
- Sikorski, K.; Chmielewski, S.; Przybyl, L.; Heemann, U.; Wesoly, J.; Baumann, M.; Bluyssen, H.A. Stat1-mediated signal integration between ifnγ and lps leads to increased ec and smc activation and monocyte adhesion. Am. J. Physiol. Cell Physiol. 2011, 300, C1337–C1344. [Google Scholar]
- Hiroi, M.; Ohmori, Y. The transcriptional coactivator creb-binding protein cooperates with stat1 and nf-kappa b for synergistic transcriptional activation of the cxc ligand 9/monokine induced by interferon-gamma gene. J. Biol. Chem. 2003, 278, 651–660. [Google Scholar]
- Collins, T.; Read, M.A.; Neish, A.S.; Whitley, M.Z.; Thanos, D.; Maniatis, T. Transcriptional regulation of endothelial cell adhesion molecules: Nf-kappa b and cytokine-inducible enhancers. FASEB J. 1995, 9, 899–909. [Google Scholar]
- Ganster, R.W.; Guo, Z.; Shao, L.; Geller, D.A. Differential effects of tnf-alpha and ifn-gamma on gene transcription mediated by nf-kappab-stat1 interactions. J. Interferon Cytokine Res. 2005, 25, 707–719. [Google Scholar]
- Chon, S.Y.; Hassanain, H.H.; Gupta, S.L. Cooperative role of interferon regulatory factor 1 and p91 (stat1) response elements in interferon-gamma-inducible expression of human indoleamine 2,3-dioxygenase gene. J. Biol. Chem. 1996, 271, 17247–17252. [Google Scholar]
- Brucet, M.; Marques, L.; Sebastian, C.; Lloberas, J.; Celada, A. Regulation of murine tap1 and lmp2 genes in macrophages by interferon gamma is mediated by stat1 and irf-1. Genes Immun. 2004, 5, 26–35. [Google Scholar]
- Pietila, T.E.; Veckman, V.; Lehtonen, A.; Lin, R.; Hiscott, J.; Julkunen, I. Multiple nf-kappab and ifn regulatory factor family transcription factors regulate ccl19 gene expression in human monocyte-derived dendritic cells. J. Immunol. 2007, 178, 253–261. [Google Scholar]
- Tamassia, N.; Calzetti, F.; Ear, T.; Cloutier, A.; Gasperini, S.; Bazzoni, F.; McDonald, P.P.; Cassatella, M.A. Molecular mechanisms underlying the synergistic induction of cxcl10 by lps and ifn-gamma in human neutrophils. Eur. J. Immunol. 2007, 37, 2627–2634. [Google Scholar]
- Hagg, S.; Skogsberg, J.; Lundstrom, J.; Noori, P.; Nilsson, R.; Zhong, H.; Maleki, S.; Shang, M.M.; Brinne, B.; Bradshaw, M.; et al. Multi-organ expression profiling uncovers a gene module in coronary artery disease involving transendothelial migration of leukocytes and lim domain binding 2: The stockholm atherosclerosis gene expression (stage) study. PLoS Genet. 2009, 5, e1000754. [Google Scholar] [CrossRef]
- Folkersen, L.; Persson, J.; Ekstrand, J.; Agardh, H.E.; Hansson, G.K.; Gabrielsen, A.; Hedin, U.; Paulsson-Berne, G. Prediction of ischemic events on the basis of transcriptomic and genomic profiling in patients undergoing carotid endarterectomy. Mol. Med. 2012, 18, 669–675. [Google Scholar]
- Canfield, A.E.; Farrington, C.; Dziobon, M.D.; Boot-Handford, R.P.; Heagerty, A.M.; Kumar, S.N.; Roberts, I.S. The involvement of matrix glycoproteins in vascular calcification and fibrosis: An immunohistochemical study. J. Pathol. 2002, 196, 228–234. [Google Scholar]
- Cantor, H.; Shinohara, M.L. Regulation of t-helper-cell lineage development by osteopontin: The inside story. Nat. Rev. Immunol. 2009, 9, 137–141. [Google Scholar]
- Chen, T.; Yan, H.; Li, Z.; Jing, T.; Zhu, W.; Ge, J.; Zheng, X.; Pan, X.; Zhu, J. Microrna-155 regulates lipid uptake, adhesion/chemokine marker secretion and scg2 expression in oxldl-stimulated dendritic cells/macrophages. Int. J. Cardiol. 2011, 147, 446–447. [Google Scholar]
- Scharl, M.; Hruz, P.; McCole, D.F. Protein tyrosine phosphatase non-receptor type 2 regulates ifn-gamma-induced cytokine signaling in thp-1 monocytes. Inflamm. Bowel Dis. 2010, 16, 2055–2064. [Google Scholar]
- Kok, S.H.; Hong, C.Y.; Kuo, M.Y.; Wang, C.C.; Hou, K.L.; Lin, Y.T.; Galson, D.L.; Lin, S.K. Oncostatin m-induced ccl2 transcription in osteoblastic cells is mediated by multiple levels of stat-1 and stat-3 signaling: An implication for the pathogenesis of arthritis. Arthritis Rheumatol. 2009, 60, 1451–1462. [Google Scholar]
- Dai, X.; Sayama, K.; Tohyama, M.; Shirakata, Y.; Yang, L.; Hirakawa, S.; Tokumaru, S.; Hashimoto, K. The nf-kappab, p38 mapk and stat1 pathways differentially regulate the dsrna-mediated innate immune responses of epidermal keratinocytes. Int. Immunol. 2008, 20, 901–909. [Google Scholar]
- Fleetwood, A.J.; Dinh, H.; Cook, A.D.; Hertzog, P.J.; Hamilton, J.A. Gm-csf- and m-csf-dependent macrophage phenotypes display differential dependence on type i interferon signaling. J. Leukoc. Biol. 2009, 86, 411–421. [Google Scholar]
- Hardison, S.E.; Herrera, G.; Young, M.L.; Hole, C.R.; Wozniak, K.L.; Wormley, F.L., Jr. Protective immunity against pulmonary cryptococcosis is associated with stat1-mediated classical macrophage activation. J. Immunol. 2012, 189, 4060–4068. [Google Scholar]
- Tyagi, A.; Agarwal, C.; Dwyer-Nield, L.D.; Singh, R.P.; Malkinson, A.M.; Agarwal, R. Silibinin modulates tnf-alpha and ifn-gamma mediated signaling to regulate cox2 and inos expression in tumorigenic mouse lung epithelial lm2 cells. Mol. Carcinog. 2012, 51, 832–842. [Google Scholar]
- Kosaka, H.; Yoshimoto, T.; Fujimoto, J.; Nakanishi, K. Interferon-gamma is a therapeutic target molecule for prevention of postoperative adhesion formation. Nat. Med. 2008, 14, 437–441. [Google Scholar]
- Eastabrook, G.D.; Hu, Y.; Tan, R.; Dutz, J.P.; Maccalman, C.D.; von Dadelszen, P. Decidual nk cell-derived conditioned medium (dnk-cm) mediates vegf-c secretion in extravillous cytotrophoblasts. Am. J. Reprod. Immunol. 2012, 67, 101–111. [Google Scholar]
- Hu, X.; Ivashkiv, L.B. Cross-regulation of signaling pathways by interferon-gamma: Implications for immune responses and autoimmune diseases. Immunity 2009, 31, 539–550. [Google Scholar]
- Schroder, K.; Sweet, M.J.; Hume, D.A. Signal integration between ifngamma and tlr signalling pathways in macrophages. Immunobiology 2006, 211, 511–524. [Google Scholar]
- Cagnin, S.; Biscuola, M.; Patuzzo, C.; Trabetti, E.; Pasquali, A.; Laveder, P.; Faggian, G.; Iafrancesco, M.; Mazzucco, A.; Pignatti, P.F.; et al. Reconstruction and functional analysis of altered molecular pathways in human atherosclerotic arteries. BMC Genomics 2009, 10, 13. [Google Scholar]
- Hansson, G.K. Immune mechanisms in atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2001, 21, 1876–1890. [Google Scholar]
- Poston, R.N.; Davies, D.F. Immunity and inflammation in the pathogenesis of atherosclerosis. A review. Atherosclerosis 1974, 19, 353–367. [Google Scholar] [CrossRef]
- Toshchakov, V.; Jones, B.W.; Perera, P.Y.; Thomas, K.; Cody, M.J.; Zhang, S.; Williams, B.R.; Major, J.; Hamilton, T.A.; Fenton, M.J.; et al. Tlr4, but not tlr2, mediates ifn-beta-induced stat1alpha/beta-dependent gene expression in macrophages. Nat. Immunol. 2002, 3, 392–398. [Google Scholar] [CrossRef]
- Clarke, D.L.; Clifford, R.L.; Jindarat, S.; Proud, D.; Pang, L.; Belvisi, M.; Knox, A.J. Tnfalpha and ifngamma synergistically enhance transcriptional activation of cxcl10 in human airway smooth muscle cells via stat-1, nf-kappab, and the transcriptional coactivator creb-binding protein. J. Biol. Chem. 2010, 285, 29101–29110. [Google Scholar]
- Qi, X.F.; Kim, D.H.; Yoon, Y.S.; Jin, D.; Huang, X.Z.; Li, J.H.; Deung, Y.K.; Lee, K.J. Essential involvement of cross-talk between ifn-gamma and tnf-alpha in cxcl10 production in human thp-1 monocytes. J. Cell. Physiol. 2009, 220, 690–697. [Google Scholar]
- Gao, J.; Morrison, D.C.; Parmely, T.J.; Russell, S.W.; Murphy, W.J. An interferon-gamma-activated site (gas) is necessary for full expression of the mouse inos gene in response to interferon-gamma and lipopolysaccharide. J. Biol. Chem. 1997, 272, 1226–1230. [Google Scholar]
- Butcher, M.J.; Galkina, E.V. Phenotypic and functional heterogeneity of macrophages and dendritic cell subsets in the healthy and atherosclerosis-prone aorta. Front. Physiol. 2012, 3, 44. [Google Scholar]
- Rohde, L.E.; Lee, R.T. Pathophysiology of atherosclerotic plaque development and rupture: An overview. Semin. Vasc. Med. 2003, 3, 347–354. [Google Scholar]
- Jashari, F.; Ibrahimi, P.; Nicoll, R.; Bajraktari, G.; Wester, P.; Henein, M.Y. Coronary and carotid atherosclerosis: Similarities and differences. Atherosclerosis 2013, 227, 193–200. [Google Scholar]
- Prasad, A.; Zhu, J.; Halcox, J.P.; Waclawiw, M.A.; Epstein, S.E.; Quyyumi, A.A. Predisposition to atherosclerosis by infections: Role of endothelial dysfunction. Circulation 2002, 106, 184–190. [Google Scholar]
- Xu, Q. Role of heat shock proteins in atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2002, 22, 1547–1559. [Google Scholar]
- Frantz, S.; Ertl, G.; Bauersachs, J. Mechanisms of disease: Toll-like receptors in cardiovascular disease. Nat. Clin. Pract. Cardiovasc. Med. 2007, 4, 444–454. [Google Scholar]
- Rauch, S.J.; Rosenkranz, A.C.; Bohm, A.; Meyer-Kirchrath, J.; Hohlfeld, T.; Schror, K.; Rauch, B.H. Cholesterol induces apoptosis-associated loss of the activated leukocyte cell adhesion molecule (alcam) in human monocytes. Vasc. Pharmacol. 2011, 54, 93–99. [Google Scholar]
- Kurata, H.; Matsumoto, A.; Fujiwara, Y.; Kondo, K.; Itakura, H.; Mitchell, A.; Fidge, N. A candidate high density lipoprotein (hdl) receptor, hb2, with possible multiple functions shows sequence homology with adhesion molecules. J. Atheroscler. Thromb. 1998, 4, 112–117. [Google Scholar] [CrossRef]
- Barks, J.L.; McQuillan, J.J.; Iademarco, M.F. Tnf-alpha and il-4 synergistically increase vascular cell adhesion molecule-1 expression in cultured vascular smooth muscle cells. J. Immunol. 1997, 159, 4532–4538. [Google Scholar]
- Murooka, T.T.; Rahbar, R.; Platanias, L.C.; Fish, E.N. Ccl5-mediated t-cell chemotaxis involves the initiation of mrna translation through mtor/4e-bp1. Blood 2008, 111, 4892–4901. [Google Scholar]
- Yurchenko, E.; Tritt, M.; Hay, V.; Shevach, E.M.; Belkaid, Y.; Piccirillo, C.A. Ccr5-dependent homing of naturally occurring cd4+ regulatory t cells to sites of leishmania major infection favors pathogen persistence. J. Exp. Med. 2006, 203, 2451–2460. [Google Scholar]
- Ottoson, N.C.; Pribila, J.T.; Chan, A.S.; Shimizu, Y. Cutting edge: T cell migration regulated by cxcr4 chemokine receptor signaling to zap-70 tyrosine kinase. J. Immunol. 2001, 167, 1857–1861. [Google Scholar]
- Cangemi, C.; Skov, V.; Poulsen, M.K.; Funder, J.; Twal, W.O.; Gall, M.A.; Hjortdal, V.; Jespersen, M.L.; Kruse, T.A.; Aagard, J.; et al. Fibulin-1 is a marker for arterial extracellular matrix alterations in type 2 diabetes. Clin. Chem. 2011, 57, 1556–1565. [Google Scholar]
- Kallio, M.A.; Tuimala, J.T.; Hupponen, T.; Klemela, P.; Gentile, M.; Scheinin, I.; Koski, M.; Kaki, J.; Korpelainen, E.I. Chipster: User-friendly analysis software for microarray and other high-throughput data. BMC Genomics 2011, 12, 507. [Google Scholar]
- Johnson, W.E.; Li, C.; Rabinovic, A. Adjusting batch effects in microarray expression data using empirical bayes methods. Biostatistics 2007, 8, 118–127. [Google Scholar]
- Zheng, Q.; Wang, X.J. Goeast: A web-based software toolkit for gene ontology enrichment analysis. Nucleic Acids Res. 2008, 36, W358–W363. [Google Scholar]
- Data Analysis Suite GENOMATIX. Available online: www.genomatix.de (accessed on 14 August 2014).
- Kanehisa, M.; Goto, S. Kegg: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar]
- BioCarta. Available online: http://www.biocarta.com (accessed on 20 February 2014).
- Croft, D.; O’Kelly, G.; Wu, G.; Haw, R.; Gillespie, M.; Matthews, L.; Caudy, M.; Garapati, P.; Gopinath, G.; Jassal, B.; et al. Reactome: A database of reactions, pathways and biological processes. Nucleic Acids Res. 2011, 39, D691–D697. [Google Scholar]
- Krzywinski, M.; Schein, J.; Birol, I.; Connors, J.; Gascoyne, R.; Horsman, D.; Jones, S.J.; Marra, M.A. Circos: An information aesthetic for comparative genomics. Genome Res. 2009, 19, 1639–1645. [Google Scholar]
- Genomatix Regionminer Manual. Available online: http://www.genomatix.de/online_help/help_regionminer/overrepresented_TFs.html (accessed on 20 February 2014).
- Sikorski, K.; Czerwoniec, A.; Bujnicki, J.M.; Wesoly, J.; Bluyssen, H.A. Stat1 as a novel therapeutical target in pro-atherogenic signal integration of ifngamma, tlr4 and il-6 in vascular disease. Cytokine Growth Factor Rev. 2011, 22, 211–219. [Google Scholar]
- Kuvin, J.T.; Karas, R.H. Clinical utility of endothelial function testing: Ready for prime time? Circulation 2003, 107, 3243–3247. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Sikorski, K.; Wesoly, J.; Bluyssen, H.A.R. Data Mining of Atherosclerotic Plaque Transcriptomes Predicts STAT1-Dependent Inflammatory Signal Integration in Vascular Disease. Int. J. Mol. Sci. 2014, 15, 14313-14331. https://doi.org/10.3390/ijms150814313
Sikorski K, Wesoly J, Bluyssen HAR. Data Mining of Atherosclerotic Plaque Transcriptomes Predicts STAT1-Dependent Inflammatory Signal Integration in Vascular Disease. International Journal of Molecular Sciences. 2014; 15(8):14313-14331. https://doi.org/10.3390/ijms150814313
Chicago/Turabian StyleSikorski, Krzysztof, Joanna Wesoly, and Hans A. R. Bluyssen. 2014. "Data Mining of Atherosclerotic Plaque Transcriptomes Predicts STAT1-Dependent Inflammatory Signal Integration in Vascular Disease" International Journal of Molecular Sciences 15, no. 8: 14313-14331. https://doi.org/10.3390/ijms150814313
APA StyleSikorski, K., Wesoly, J., & Bluyssen, H. A. R. (2014). Data Mining of Atherosclerotic Plaque Transcriptomes Predicts STAT1-Dependent Inflammatory Signal Integration in Vascular Disease. International Journal of Molecular Sciences, 15(8), 14313-14331. https://doi.org/10.3390/ijms150814313