Apoptotic Death of Cancer Stem Cells for Cancer Therapy


Abstract
:1. Introduction
2. Cancer Stem Cells (CSCs)
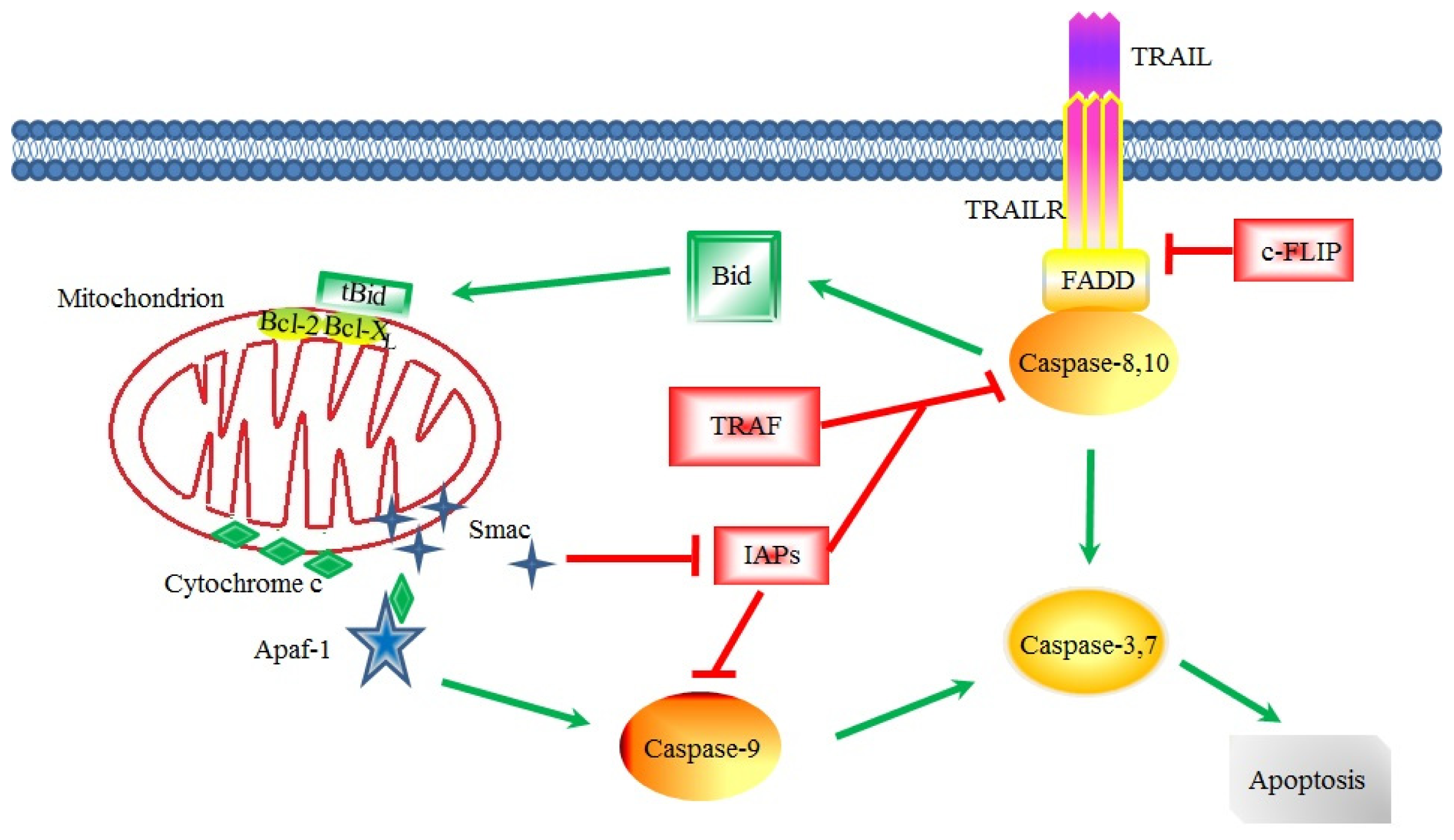
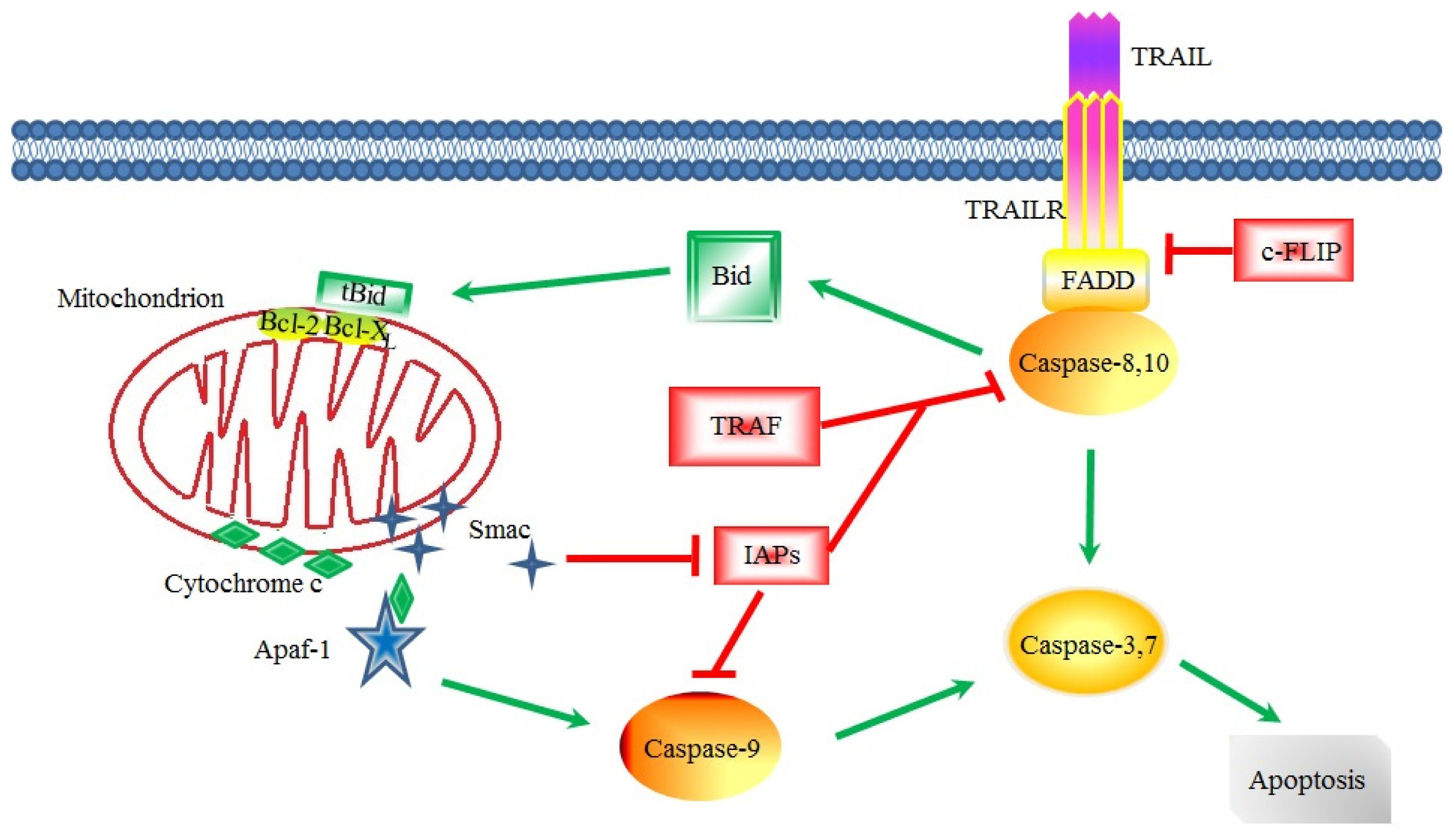
3. Apoptotic Signaling in CSCs
4. Apoptotic Inducers of CSCs
4.1. Natural Compounds
4.1.1. Natural Compounds from Traditional Chinese Medicines
4.1.2. Antibiotics
4.2. Synthetic Compounds
4.3. Antibodies and Recombinant Proteins
4.4. Oligonucleotides
4.5. Combined Application of Apoptotic Inducers
5. Prospects
Acknowledgments
Conflicts of Interest
- Author ContributionsY.-C.H. wrote the draft, F.-L.Z. helped with literature search, Y.S. helped with revision, and D.-F.L. and D.C. revised and proofed of the manuscript.
References
- Julius Cohnheim (1839–1884) experimental pathologist. J. Am. Med. Assoc 1968, 206, 1561–1562.
- Lapidot, T.; Sirard, C.; Vormoor, J.; Murdoch, B.; Hoang, T.; Caceres-Cortes, J.; Minden, M.; Paterson, B.; Caligiuri, M.; Dick, J. A cell initiating human acute myeloid leukaemia after transplantation into SCID mice. Nature 1994, 367, 645–648. [Google Scholar]
- Bonnet, D.; Dick, J. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat. Med 1997, 3, 730–737. [Google Scholar]
- Al-Hajj, M.; Wicha, M.S.; Benito-Hernandez, A.; Morrison, S.J.; Clarke, M.F. Prospective identification of tumorigenic breast cancer cells. Proc. Natl. Acad. Sci. USA 2003, 100, 3983–3988. [Google Scholar]
- Clarke, M.F.; Dick, J.E.; Dirks, P.B.; Eaves, C.J.; Jamieson, C.H.M.; Jones, D.L.; Visvader, J.; Weissman, I.L.; Wahl, G.M. Cancer stem cells—Perspectives on current status and future directions: AACR workshop on cancer stem cells. Cancer Res 2006, 66, 9339–9344. [Google Scholar]
- Bruce, W.R.; van der Gaag, H. A quantitative assay for the number of murine lymphoma cells capable of proliferation in vivo. Nature 1963, 199, 79–80. [Google Scholar]
- Bergsagel, D.E.; Valeriote, F.A. Growth characteristics of a mouse plasma cell tumor. Cancer Res 1968, 28, 2187–2196. [Google Scholar]
- Hamburger, A.; Salmon, S. Primary bioassay of human tumor stem cells. Science 1977, 197, 461–463. [Google Scholar]
- Chaffer, C.L.; Weinberg, R.A. A perspective on cancer cell metastasis. Science 2011, 331, 1559–1564. [Google Scholar]
- Zhang, P.; Zhang, Y.; Mao, L.; Zhang, Z.; Chen, W. Side population in oral squamous cell carcinoma possesses tumor stem cell phenotypes. Cancer Lett 2009, 277, 227–234. [Google Scholar]
- Anderson, K.; Lutz, C.; van Delft, F.W.; Bateman, C.M.; Guo, Y.; Colman, S.M.; Kempski, H.; Moorman, A.V.; Titley, I.; Swansbury, J.; et al. Genetic variegation of clonal architecture and propagating cells in leukaemia. Nature 2011, 469, 356–361. [Google Scholar]
- Zhang, Y.; Young, E.D.; Bill, K.; Belousov, R.; Peng, T.; Lazar, A.J.; Pollock, R.E.; Simmons, P.J.; Lev, D.; Kolonin, M.G. Heterogeneity and immunophenotypic plasticity of malignant cells in human liposarcomas. Stem Cell Res 2013, 11, 772–781. [Google Scholar]
- Goardon, N.; Marchi, E.; Atzberger, A.; Quek, L.; Schuh, A.; Soneji, S.; Woll, P.; Mead, A.; Alford, K.A.; Rout, R.; et al. Coexistence of LMPP-like and GMP-like leukemia stem cells in acute myeloid leukemia. Cancer Cell 2011, 19, 138–152. [Google Scholar]
- Meyer, M.J.; Fleming, J.M.; Lin, A.F.; Hussnain, S.A.; Ginsburg, E.; Vonderhaar, B.K. CD44posCD49fhiCD133/2hi defines xenograft-initiating cells in estrogen receptor-negative breast cancer. Cancer Res 2010, 70, 4624–4633. [Google Scholar]
- Schober, M.; Fuchs, E. Tumor-initiating stem cells of squamous cell carcinomas and their control by TGF-beta and integrin/focal adhesion kinase (FAK) signaling. Proc. Natl. Acad. Sci. USA 2011, 108, 10544–10549. [Google Scholar]
- Stewart, J.M.; Shaw, P.A.; Gedye, C.; Bernardini, M.Q.; Neel, B.G.; Ailles, L.E. Phenotypic heterogeneity and instability of human ovarian tumor-initiating cells. Proc. Natl. Acad. Sci. USA 2011, 108, 6468–6473. [Google Scholar]
- Dieter, S.M.; Ball, C.R.; Hoffmann, C.M.; Nowrouzi, A.; Herbst, F.; Zavidij, O.; Abel, U.; Arens, A.; Weichert, W.; Brand, K.; et al. Distinct types of tumor-initiating cells form human colon cancer tumors and metastases. Cell Stem Cell 2011, 9, 357–365. [Google Scholar]
- Chen, R.; Nishimura, M.; Bumbaca, S.; Kharbanda, S.; Forrest, W.; Kasman, I.; Greve, J.; Soriano, R.; Gilmour, L.; Rivers, C.; et al. A hierarchy of self-renewing tumor-initiating cell types in glioblastoma. Cancer Cell 2010, 17, 362–375. [Google Scholar]
- Akunuru, S.; James Zhai, Q.; Zheng, Y. Non-small cell lung cancer stem/progenitor cells are enriched in multiple distinct phenotypic subpopulations and exhibit plasticity. Cell Death Dis 2012, 3, e352. [Google Scholar]
- Zhang, H.; Wu, H.; Zheng, J.; Yu, P.; Xu, L.; Jiang, P.; Gao, J.; Wang, H.; Zhang, Y. Transforming growth factor beta1 signal is crucial for dedifferentiation of cancer cells to cancer stem cells in osteosarcoma. Stem Cells 2013, 31, 433–446. [Google Scholar]
- Chaffer, C.L.; Brueckmann, I.; Scheel, C.; Kaestli, A.J.; Wiggins, P.A.; Rodrigues, L.O.; Brooks, M.; Reinhardt, F.; Su, Y.; Polyak, K.; et al. Normal and neoplastic nonstem cells can spontaneously convert to a stem-like state. Proc. Natl. Acad. Sci. USA 2011, 108, 7950–7955. [Google Scholar]
- Dey-Guha, I.; Wolfer, A.; Yeh, A.C.; Albeck, J.G.; Darp, R.; Leon, E.; Wulfkuhle, J.; Petricoin, E.F., III; Wittner, B.S.; Ramaswamy, S. Asymmetric cancer cell division regulated by AKT. Proc. Natl. Acad. Sci. USA 2011, 108, 12845–12850. [Google Scholar]
- Vermeulen, L.; de Sousa, E.; Melo, F.; van der Heijden, M.; Cameron, K.; de Jong, J.; Borovski, T.; Tuynman, J.; Todaro, M.; Merz, C.; et al. Wnt activity defines colon cancer stem cells and is regulated by the microenvironment. Nat. Cell Biol 2010, 12, 468–476. [Google Scholar]
- Heddleston, J.; Li, Z.; McLendon, R.; Hjelmeland, A.; Rich, J. The hypoxic microenvironment maintains glioblastoma stem cells and promotes reprogramming towards a cancer stem cell phenotype. Cell Cycle 2009, 8, 3274–3284. [Google Scholar]
- Li, Z.; Bao, S.; Wu, Q.; Wang, H.; Eyler, C.; Sathornsumetee, S.; Shi, Q.; Cao, Y.; Lathia, J.; McLendon, R.; et al. Hypoxia-inducible factors regulate tumorigenic capacity of glioma stem cells. Cancer Cell 2009, 15, 501–513. [Google Scholar]
- Mao, X.; Yan, M.; Xue, X.; Zhang, X.; Ren, H.; Guo, G.; Wang, P.; Zhang, W.; Huo, J. Overexpression of ZNF217 in glioblastoma contributes to the maintenance of glioma stem cells regulated by hypoxia-inducible factors. Lab. Investig 2011, 91, 1068–1078. [Google Scholar]
- Kerr, J.F.; Wyllie, A.H.; Currie, A.R. Apoptosis: A basic biological phenomenon with wide-ranging implications in tissue kinetics. Br. J. Cancer 1972, 26, 239–257. [Google Scholar]
- Taylor, R.C.; Cullen, S.P.; Martin, S.J. Apoptosis: Controlled demolition at the cellular level. Nat. Rev. Mol. Cell Biol 2008, 9, 231–241. [Google Scholar]
- Ashkenazi, A. Targeting the extrinsic apoptosis pathway in cancer. Cytokine Growth Factor Rev 2008, 19, 325–331. [Google Scholar]
- Scaffidi, C.; Fulda, S.; Srinivasan, A.; Friesen, C.; Li, F.; Tomaselli, K.J.; Debatin, K.M.; Krammer, P.H.; Peter, M.E. Two CD95 (APO-1/Fas) signaling pathways. EMBO J 1998, 17, 1675–1687. [Google Scholar]
- Kischkel, F.C.; Lawrence, D.A.; Chuntharapai, A.; Schow, P.; Kim, K.J.; Ashkenazi, A. Apo2L/TRAIL-dependent recruitment of endogenous FADD and caspase-8 to death receptors 4 and 5. Immunity 2000, 12, 611–620. [Google Scholar]
- Kischkel, F.C.; Lawrence, D.A.; Tinel, A.; LeBlanc, H.; Virmani, A.; Schow, P.; Gazdar, A.; Blenis, J.; Arnott, D.; Ashkenazi, A. Death receptor recruitment of endogenous caspase-10 and apoptosis initiation in the absence of caspase-8. J. Biol. Chem 2001, 276, 46639–46646. [Google Scholar]
- Falschlehner, C.; Emmerich, C.H.; Gerlach, B.; Walczak, H. TRAIL signalling: Decisions between life and death. Int. J. Biochem. Cell Biol 2007, 39, 1462–1475. [Google Scholar]
- Lavrik, I.; Golks, A.; Krammer, P.H. Death receptor signaling. J. Cell Sci 2005, 118, 265–267. [Google Scholar]
- Song, X.; Kim, S.Y.; Lee, Y.J. Evidence for two modes of synergistic induction of apoptosis by mapatumumab and oxaliplatin in combination with hyperthermia in human colon cancer cells. PLoS One 2013, 8, e73654. [Google Scholar]
- Fulda, S.; Galluzzi, L.; Kroemer, G. Targeting mitochondria for cancer therapy. Nat. Rev. Drug Discov 2010, 9, 447–464. [Google Scholar]
- Ma, X.; Zhou, J.; Zhang, C.X.; Li, X.Y.; Li, N.; Ju, R.J.; Shi, J.F.; Sun, M.G.; Zhao, W.Y.; Mu, L.M.; et al. Modulation of drug-resistant membrane and apoptosis proteins of breast cancer stem cells by targeting berberine liposomes. Biomaterials 2013, 34, 4452–4465. [Google Scholar]
- Eckelman, B.P.; Salvesen, G.S.; Scott, F.L. Human inhibitor of apoptosis proteins: Why XIAP is the black sheep of the family. EMBO Rep 2006, 7, 988–994. [Google Scholar]
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar]
- Lowe, S.W.; Cepero, E.; Evan, G. Intrinsic tumour suppression. Nature 2004, 432, 307–315. [Google Scholar]
- Signore, M.; Ricci-Vitiani, L.; de Maria, R. Targeting apoptosis pathways in cancer stem cells. Cancer Lett 2013, 332, 374–382. [Google Scholar]
- Sussman, R.T.; Ricci, M.S.; Hart, L.S.; Sun, S.Y.; El-Deiry, W.S. Chemotherapy-resistant side-population of colon cancer cells has a higher sensitivity to TRAIL than the non-SP, a higher expression of c-Myc and TRAIL-receptor DR4. Cancer Biol. Ther 2007, 6, 1490–1495. [Google Scholar]
- Kataoka, T. The caspase-8 modulator c-FLIP. Crit. Rev. Immunol 2005, 25, 31–58. [Google Scholar]
- Micheau, O. Cellular FLICE-inhibitory protein: An update. In Apoptosis and Cancer Therapy; Debatin, K., Pulda, S., Eds.; Wiley-VCH: Heidelberg, Germany, 2006; pp. 120–157. [Google Scholar]
- Liu, G.; Yuan, X.; Zeng, Z.; Tunici, P.; Ng, H.; Abdulkadir, I.R.; Lu, L.; Irvin, D.; Black, K.L.; Yu, J.S. Analysis of gene expression and chemoresistance of CD133+ cancer stem cells in glioblastoma. Mol. Cancer 2006, 5, 67. [Google Scholar]
- Zobalova, R.; McDermott, L.; Stantic, M.; Prokopova, K.; Dong, L.F.; Neuzil, J. CD133-positive cells are resistant to TRAIL due to up-regulation of FLIP. Biochem. Biophys. Res. Commun 2008, 373, 567–571. [Google Scholar]
- Zobalova, R.; Stantic, M.; Prokopova, K.; Dong, L.F.; Neuzil, J. Cancer cells with high expression of CD133 exert FLIP upregulation and resistance to TRAIL-induced apoptosis. Biofactors 2008, 34, 231–235. [Google Scholar]
- Piggott, L.; Omidvar, N.; Marti Perez, S.; Eberl, M.; Clarkson, R.W. Suppression of apoptosis inhibitor c-FLIP selectively eliminates breast cancer stem cell activity in response to the anti-cancer agent, TRAIL. Breast Cancer Res 2011, 13, R88. [Google Scholar]
- Ding, L.; Yuan, C.; Wei, F.; Wang, G.; Zhang, J.; Bellail, A.C.; Zhang, Z.; Olson, J.J.; Hao, C. Cisplatin restores TRAIL apoptotic pathway in glioblastoma-derived stem cells through up-regulation of DR5 and down-regulation of c-FLIP. Cancer Investig 2011, 29, 511–520. [Google Scholar]
- Fukuda, S.; Foster, R.G.; Porter, S.B.; Pelus, L.M. The antiapoptosis protein survivin is associated with cell cycle entry of normal cord blood CD34(+) cells and modulates cell cycle and proliferation of mouse hematopoietic progenitor cells. Blood 2002, 100, 2463–2471. [Google Scholar]
- Leung, C.G.; Xu, Y.; Mularski, B.; Liu, H.; Gurbuxani, S.; Crispino, J.D. Requirements for survivin in terminal differentiation of erythroid cells and maintenance of hematopoietic stem and progenitor cells. J. Exp. Med 2007, 204, 1603–1611. [Google Scholar]
- Pennartz, S.; Belvindrah, R.; Tomiuk, S.; Zimmer, C.; Hofmann, K.; Conradt, M.; Bosio, A.; Cremer, H. Purification of neuronal precursors from the adult mouse brain: Comprehensive gene expression analysis provides new insights into the control of cell migration, differentiation, and homeostasis. Mol. Cell. Neurosci 2004, 25, 692–706. [Google Scholar]
- Carter, B.Z.; Qiu, Y.; Huang, X.; Diao, L.; Zhang, N.; Coombes, K.R.; Mak, D.H.; Konopleva, M.; Cortes, J.; Kantarjian, H.M.; et al. Survivin is highly expressed in CD34(+)38(−) leukemic stem/progenitor cells and predicts poor clinical outcomes in AML. Blood 2012, 120, 173–180. [Google Scholar]
- Jin, F.; Zhao, L.; Zhao, H.Y.; Guo, S.G.; Feng, J.; Jiang, X.B.; Zhang, S.L.; Wei, Y.J.; Fu, R.; Zhao, J.S. Comparison between cells and cancer stem-like cells isolated from glioblastoma and astrocytoma on expression of anti-apoptotic and multidrug resistance-associated protein genes. Neuroscience 2008, 154, 541–550. [Google Scholar]
- Kelly, P.N.; Puthalakath, H.; Adams, J.M.; Strasser, A. Endogenous bcl-2 is not required for the development of Emu-myc-induced B-cell lymphoma. Blood 2007, 109, 4907–4913. [Google Scholar]
- Madjd, Z.; Mehrjerdi, A.Z.; Sharifi, A.M.; Molanaei, S.; Shahzadi, S.Z.; Asadi-Lari, M. CD44+ cancer cells express higher levels of the anti-apoptotic protein Bcl-2 in breast tumours. Cancer Immun 2009, 9, 4. [Google Scholar]
- Tagscherer, K.E.; Fassl, A.; Campos, B.; Farhadi, M.; Kraemer, A.; Bock, B.C.; Macher-Goeppinger, S.; Radlwimmer, B.; Wiestler, O.D.; Herold-Mende, C.; et al. Apoptosis-based treatment of glioblastomas with ABT-737, a novel small molecule inhibitor of Bcl-2 family proteins. Oncogene 2008, 27, 6646–6656. [Google Scholar]
- Wu, S.; Wang, X.; Chen, J.; Chen, Y. Autophagy of cancer stem cells is involved with chemoresistance of colon cancer cells. Biochem. Biophys. Res. Commun 2013, 434, 898–903. [Google Scholar]
- Wang, L.; Guo, H.; Yang, L.; Dong, L.; Lin, C.; Zhang, J.; Lin, P.; Wang, X. Morusin inhibits human cervical cancer stem cell growth and migration through attenuation of NF-kappaB activity and apoptosis induction. Mol. Cell. Biochem 2013, 379, 7–18. [Google Scholar]
- Vazquez, A.; Bond, E.E.; Levine, A.J.; Bond, G.L. The genetics of the p53 pathway, apoptosis and cancer therapy. Nat. Rev. Drug Discov 2008, 7, 979–987. [Google Scholar]
- Bao, S.; Wu, Q.; McLendon, R.E.; Hao, Y.; Shi, Q.; Hjelmeland, A.B.; Dewhirst, M.W.; Bigner, D.D.; Rich, J.N. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature 2006, 444, 756–760. [Google Scholar]
- Guzman, M.L.; Neering, S.J.; Upchurch, D.; Grimes, B.; Howard, D.S.; Rizzieri, D.A.; Luger, S.M.; Jordan, C.T. Nuclear factor-kappaB is constitutively activated in primitive human acute myelogenous leukemia cells. Blood 2001, 98, 2301–2307. [Google Scholar]
- Warner, J.K.; Wang, J.C.; Hope, K.J.; Jin, L.; Dick, J.E. Concepts of human leukemic development. Oncogene 2004, 23, 7164–7177. [Google Scholar]
- Zhou, J.; Zhang, H.; Gu, P.; Bai, J.; Margolick, J.B.; Zhang, Y. NF-kappaB pathway inhibitors preferentially inhibit breast cancer stem-like cells. Breast Cancer Res. Treat 2008, 111, 419–427. [Google Scholar]
- Birnie, R.; Bryce, S.D.; Roome, C.; Dussupt, V.; Droop, A.; Lang, S.H.; Berry, P.A.; Hyde, C.F.; Lewis, J.L.; Stower, M.J.; et al. Gene expression profiling of human prostate cancer stem cells reveals a pro-inflammatory phenotype and the importance of extracellular matrix interactions. Genome Biol 2008, 9, R83. [Google Scholar]
- Ju, J.H.; Jang, K.; Lee, K.M.; Kim, M.; Kim, J.; Yi, J.Y.; Noh, D.Y.; Shin, I. CD24 enhances DNA damage-induced apoptosis by modulating NF-kappaB signaling in CD44-expressing breast cancer cells. Carcinogenesis 2011, 32, 1474–1483. [Google Scholar]
- Dalmay, T.; Edwards, D.R. MicroRNAs and the hallmarks of cancer. Oncogene 2006, 25, 6170–6175. [Google Scholar]
- Ruan, K.; Fang, X.; Ouyang, G. MicroRNAs: Novel regulators in the hallmarks of human cancer. Cancer Lett 2009, 285, 116–126. [Google Scholar]
- Tu, H.F.; Lin, S.C.; Chang, K.W. MicroRNA aberrances in head and neck cancer: Pathogenetic and clinical significance. Curr. Opin. Otolaryngol. Head Neck Surg 2013, 21, 104–111. [Google Scholar]
- Wong, Q.W.; Lung, R.W.; Law, P.T.; Lai, P.B.; Chan, K.Y.; To, K.F.; Wong, N. MicroRNA-223 is commonly repressed in hepatocellular carcinoma and potentiates expression of Stathmin1. Gastroenterology 2008, 135, 257–269. [Google Scholar]
- Coulouarn, C.; Factor, V.M.; Andersen, J.B.; Durkin, M.E.; Thorgeirsson, S.S. Loss of miR-122 expression in liver cancer correlates with suppression of the hepatic phenotype and gain of metastatic properties. Oncogene 2009, 28, 3526–3536. [Google Scholar]
- Pineau, P.; Volinia, S.; McJunkin, K.; Marchio, A.; Battiston, C.; Terris, B.; Mazzaferro, V.; Lowe, S.W.; Croce, C.M.; Dejean, A. MiR-221 overexpression contributes to liver tumorigenesis. Proc. Natl. Acad. Sci. USA 2010, 107, 264–269. [Google Scholar]
- Ma, S.; Tang, K.H.; Chan, Y.P.; Lee, T.K.; Kwan, P.S.; Castilho, A.; Ng, I.; Man, K.; Wong, N.; To, K.F.; et al. MiR-130b Promotes CD133(+) liver tumor-initiating cell growth and self-renewal via tumor protein 53-induced nuclear protein 1. Cell Stem Cell 2010, 7, 694–707. [Google Scholar]
- Wong, Q.W.; Ching, A.K.; Chan, A.W.; Choy, K.W.; To, K.F.; Lai, P.B.; Wong, N. MiR-222 overexpression confers cell migratory advantages in hepatocellular carcinoma through enhancing AKT signaling. Clin. Cancer Res 2010, 16, 867–875. [Google Scholar]
- Bao, B.; Li, Y.; Ahmad, A.; Azmi, A.S.; Bao, G.; Ali, S.; Banerjee, S.; Kong, D.; Sarkar, F.H. Targeting CSC-related miRNAs for cancer therapy by natural agents. Curr. Drug Targets 2012, 13, 1858–1868. [Google Scholar]
- Ji, Q.; Hao, X.; Zhang, M.; Tang, W.; Yang, M.; Li, L.; Xiang, D.; Desano, J.T.; Bommer, G.T.; Fan, D.; et al. MicroRNA miR-34 inhibits human pancreatic cancer tumor-initiating cells. PLoS One 2009, 4, e6816. [Google Scholar]
- Guessous, F.; Zhang, Y.; Kofman, A.; Catania, A.; Li, Y.; Schiff, D.; Purow, B.; Abounader, R. MicroRNA-34a is tumor suppressive in brain tumors and glioma stem cells. Cell Cycle 2010, 9, 1031–1036. [Google Scholar]
- Golestaneh, A.F.; Atashi, A.; Langroudi, L.; Shafiee, A.; Ghaemi, N.; Soleimani, M. MiRNAs expressed differently in cancer stem cells and cancer cells of human gastric cancer cell line MKN-45. Cell Biochem. Funct 2012, 30, 411–418. [Google Scholar]
- Pannuti, A.; Foreman, K.; Rizzo, P.; Osipo, C.; Golde, T.; Osborne, B.; Miele, L. Targeting Notch to target cancer stem cells. Clin. Cancer Res 2010, 16, 3141–3152. [Google Scholar]
- Frank, N.Y.; Schatton, T.; Frank, M.H. The therapeutic promise of the cancer stem cell concept. J. Clin. Investig 2010, 120, 41–50. [Google Scholar]
- Caba, O.; Diaz-Gavilan, M.; Rodriguez-Serrano, F.; Boulaiz, H.; Aranega, A.; Gallo, M.A.; Marchal, J.A.; Campos, J.M. Anticancer activity and cDNA microarray studies of a (RS)-1,2,3,5-tetrahydro-4,1-benzoxazepine-3-yl]-6-chloro-9H-purine, and an acyclic (RS)-O,N-acetalic 6-chloro-7H-purine. Eur. J. Med. Chem 2011, 46, 3802–3809. [Google Scholar]
- Caba, O.; Rodriguez-Serrano, F.; Diaz-Gavilan, M.; Conejo-Garcia, A.; Ortiz, R.; Martinez-Amat, A.; Alvarez, P.; Gallo, M.A.; Campos, J.M.; Marchal, J.A.; et al. The selective cytotoxic activity in breast cancer cells by an anthranilic alcohol-derived acyclic 5-fluorouracil O,N-acetal is mediated by endoplasmic reticulum stress-induced apoptosis. Eur. J. Med. Chem 2012, 50, 376–382. [Google Scholar]
- Wang, Z.; Zhang, Y.; Li, Y.; Banerjee, S.; Liao, J.; Sarkar, F.H. Down-regulation of Notch-1 contributes to cell growth inhibition and apoptosis in pancreatic cancer cells. Mol. Cancer Ther 2006, 5, 483–493. [Google Scholar]
- Montales, M.T.; Rahal, O.M.; Kang, J.; Rogers, T.J.; Prior, R.L.; Wu, X.; Simmen, R.C. Repression of mammosphere formation of human breast cancer cells by soy isoflavone genistein and blueberry polyphenolic acids suggests diet-mediated targeting of cancer stem-like/progenitor cells. Carcinogenesis 2012, 33, 652–660. [Google Scholar]
- Montales, M.T.; Rahal, O.M.; Nakatani, H.; Matsuda, T.; Simmen, R.C. Repression of mammary adipogenesis by genistein limits mammosphere formation of human MCF-7 cells. J. Endocrinol 2013, 218, 135–149. [Google Scholar]
- Han, B.; Jiang, Y.; Zhang, C.; Bremer, E.; van Dam, G.; Kroesen, B.J.; de Leij, L.; Helfrich, W. Effect of 20(S)-ginsenoside Rg3 on cell proliferation and apoptosis of colon CSCs. Chin. J. Gerontol 2012, 32, 4431–4433. [Google Scholar]
- Alvero, A.B.; Montagna, M.K.; Holmberg, J.C.; Craveiro, V.; Brown, D.; Mor, G. Targeting the mitochondria activates two independent cell death pathways in ovarian cancer stem cells. Mol. Cancer Ther 2011, 10, 1385–1393. [Google Scholar]
- Guo, M.; Wang, M.; Zhang, X.; Deng, H.; Wang, Z.Y. Broussoflavonol B restricts growth of ER-negative breast cancer stem-like cells. Anticancer Res 2013, 33, 1873–1879. [Google Scholar]
- Zhang, F.L.; Wang, P.; Liu, Y.H.; Liu, L.B.; Liu, X.B.; Li, Z.; Xue, Y.X. Topoisomerase I inhibitors, shikonin and topotecan, inhibit growth and induce apoptosis of glioma cells and glioma stem cells. PLoS One 2013, 8, e81815. [Google Scholar]
- Wang, X.; QIU, J.; Yang, G.; Altman, A.R.; Reisman, D.S.; Higginson, J.S.; Davis, I.S. The radiosensitizing effect of curcumin on CD133+ rectal cancer cells. Chin. J. Gen. Surg 2013, 28, 134–137. [Google Scholar]
- Kakarala, M.; Brenner, D.E.; Korkaya, H.; Cheng, C.; Tazi, K.; Ginestier, C.; Liu, S.; Dontu, G.; Wicha, M.S. Targeting breast stem cells with the cancer preventive compounds curcumin and piperine. Breast Cancer Res. Treat 2010, 122, 777–785. [Google Scholar]
- Pandey, P.R.; Okuda, H.; Watabe, M.; Pai, S.K.; Liu, W.; Kobayashi, A.; Xing, F.; Fukuda, K.; Hirota, S.; Sugai, T.; et al. Resveratrol suppresses growth of cancer stem-like cells by inhibiting fatty acid synthase. Breast Cancer Res. Treat 2011, 130, 387–398. [Google Scholar]
- Gupta, P.B.; Onder, T.T.; Jiang, G.; Tao, K.; Kuperwasser, C.; Weinberg, R.A.; Lander, E.S. Identification of selective inhibitors of cancer stem cells by high-throughput screening. Cell 2009, 138, 645–659. [Google Scholar]
- Parajuli, B.; Shin, S.J.; Kwon, S.H.; Cha, S.D.; Chung, R.; Park, W.J.; Lee, H.G.; Cho, C.H. Salinomycin induces apoptosis via death receptor-5 up-regulation in cisplatin-resistant ovarian cancer cells. Anticancer Res 2013, 33, 1457–1462. [Google Scholar]
- Zhang, H.; Mi, J.Q.; Fang, H.; Wang, Z.; Wang, C.; Wu, L.; Zhang, B.; Minden, M.; Yang, W.T.; Wang, H.W.; et al. Preferential eradication of acute myelogenous leukemia stem cells by fenretinide. Proc. Natl. Acad. Sci. USA 2013, 110, 5606–5611. [Google Scholar]
- Sachlos, E.; Risueno, R.M.; Laronde, S.; Shapovalova, Z.; Lee, J.H.; Russell, J.; Malig, M.; McNicol, J.D.; Fiebig-Comyn, A.; Graham, M.; et al. Identification of drugs including a dopamine receptor antagonist that selectively target cancer stem cells. Cell 2012, 149, 1284–1297. [Google Scholar]
- Chen, X.; Liao, Y.; Zhao, K.; Yang, J.C.; Zhao, S.J.; Ma, Z.J.; Yin, R.L.; Luo, G.B.; Zhao, Z.H. The effect of aspirin on the expression of tumor stem cell marker Lgr 5 in human colorectal cancer cells. Acta Acad. Med. Zunyi 2012, 35, 287–290. [Google Scholar]
- Gomez-Cabrero, A.; Wrasidlo, W.; Reisfeld, R.A. IMD-0354 targets breast cancer stem cells: A novel approach for an adjuvant to chemotherapy to prevent multidrug resistance in a murine model. PLoS One 2013, 8, e73607. [Google Scholar]
- Nanta, R.; Kumar, D.; Meeker, D.; Rodova, M.; van Veldhuizen, P.J.; Shankar, S.; Srivastava, R.K. NVP-LDE-225 (Erismodegib) inhibits epithelial-mesenchymal transition and human prostate cancer stem cell growth in NOD/SCID IL2Rgamma null mice by regulating Bmi-1 and microRNA-128. Oncogenesis 2013, 2, e42. [Google Scholar]
- Zhang, C.C.; Yan, Z.; Zong, Q.; Fang, D.D.; Painter, C.; Zhang, Q.; Chen, E.; Lira, M.E.; John-Baptiste, A.; Christensen, J.G. Synergistic effect of the γ-secretase inhibitor PF-03084014 and docetaxel in breast cancer models. Stem Cells Transl. Med 2013, 2, 233–242. [Google Scholar]
- Galuppo, R.; Maynard, E.; Shah, M.; Daily, M.F.; Chen, C.; Spear, B.T.; Gedaly, R. Synergistic inhibition of HCC and liver cancer stem cell proliferation by targeting RAS/RAF/MAPK and WNT/beta-catenin pathways. Anticancer Res 2014, 34, 1709–1713. [Google Scholar]
- Ashizawa, T.; Miyata, H.; Iizuka, A.; Komiyama, M.; Oshita, C.; Kume, A.; Nogami, M.; Yagoto, M.; Ito, I.; Oishi, T.; et al. Effect of the STAT3 inhibitor STX-0119 on the proliferation of cancer stem-like cells derived from recurrent glioblastoma. Int. J. Oncol 2013, 43, 219–227. [Google Scholar]
- Ashkenazi, A. Directing cancer cells to self-destruct with pro-apoptotic receptor agonists. Nat. Rev. Drug Discov 2008, 7, 1001–1012. [Google Scholar]
- Calabrese, C.; Poppleton, H.; Kocak, M.; Hogg, T.L.; Fuller, C.; Hamner, B.; Oh, E.Y.; Gaber, M.W.; Finklestein, D.; Allen, M.; et al. A perivascular niche for brain tumor stem cells. Cancer Cell 2007, 11, 69–82. [Google Scholar]
- Burkhardt, J.K.; Hofstetter, C.P.; Santillan, A.; Shin, B.J.; Foley, C.P.; Ballon, D.J.; Pierre Gobin, Y.; Boockvar, J.A. Orthotopic glioblastoma stem-like cell xenograft model in mice to evaluate intra-arterial delivery of bevacizumab: From bedside to bench. J. Clin. Neurosci. Off. J. Neurosurg. Soc. Australas 2012, 19, 1568–1572. [Google Scholar]
- Todaro, M.; Alea, M.P.; di Stefano, A.B.; Cammareri, P.; Vermeulen, L.; Iovino, F.; Tripodo, C.; Russo, A.; Gulotta, G.; Medema, J.P.; et al. Colon cancer stem cells dictate tumor growth and resist cell death by production of interleukin-4. Cell Stem Cell 2007, 1, 389–402. [Google Scholar]
- Chao, M.P.; Alizadeh, A.A.; Tang, C.; Jan, M.; Weissman-Tsukamoto, R.; Zhao, F.; Park, C.Y.; Weissman, I.L.; Majeti, R. Therapeutic antibody targeting of CD47 eliminates human acute lymphoblastic leukemia. Cancer Res 2011, 71, 1374–1384. [Google Scholar]
- Lee, Y.; Ahn, C.; Han, J.; Choi, H.; Kim, J.; Yim, J.; Lee, J.; Provost, P.; Radmark, O.; Kim, S.; et al. The nuclear RNase III Drosha initiates microRNA processing. Nature 2003, 425, 415–419. [Google Scholar]
- Bartel, D.P. MicroRNAs: Target recognition and regulatory functions. Cell 2009, 136, 215–233. [Google Scholar]
- Ambros, V. MicroRNA pathways in flies and worms: Growth, death, fat, stress, and timing. Cell 2003, 113, 673–676. [Google Scholar]
- Cimmino, A.; Calin, G.A.; Fabbri, M.; Iorio, M.V.; Ferracin, M.; Shimizu, M.; Wojcik, S.E.; Aqeilan, R.I.; Zupo, S.; Dono, M.; et al. MiR-15 and miR-16 induce apoptosis by targeting BCL2. Proc. Natl. Acad. Sci. USA 2005, 102, 13944–13949. [Google Scholar]
- Selcuklu, S.D.; Donoghue, M.T.; Spillane, C. MiR-21 as a key regulator of oncogenic processes. Biochem. Soc. Trans 2009, 37, 918–925. [Google Scholar]
- Hu, Y.; Cherton-Horvat, G.; Dragowska, V.; Baird, S.; Korneluk, R.G.; Durkin, J.P.; Mayer, L.D.; LaCasse, E.C. Antisense oligonucleotides targeting XIAP induce apoptosis and enhance chemotherapeutic activity against human lung cancer cells in vitro and in vivo. Clin. Cancer Res. 2003, 9, 2826–2836. [Google Scholar]
- LaCasse, E.C.; Mahoney, D.J.; Cheung, H.H.; Plenchette, S.; Baird, S.; Korneluk, R.G. IAP-targeted therapies for cancer. Oncogene 2008, 27, 6252–6275. [Google Scholar]
- Marian, C.O.; Cho, S.K.; McEllin, B.M.; Maher, E.A.; Hatanpaa, K.J.; Madden, C.J.; Mickey, B.E.; Wright, W.E.; Shay, J.W.; Bachoo, R.M. The telomerase antagonist, imetelstat, efficiently targets glioblastoma tumor-initiating cells leading to decreased proliferation and tumor growth. Clin. Cancer Res 2010, 16, 154–163. [Google Scholar]
- Li, X.; Lewis, M.T.; Huang, J.; Gutierrez, C.; Osborne, C.K.; Wu, M.F.; Hilsenbeck, S.G.; Pavlick, A.; Zhang, X.; Chamness, G.C.; et al. Intrinsic resistance of tumorigenic breast cancer cells to chemotherapy. J. Natl. Cancer Inst 2008, 100, 672–679. [Google Scholar]
- Khdair, A.; Chen, D.; Patil, Y.; Ma, L.; Dou, Q.P.; Shekhar, M.P.; Panyam, J. Nanoparticle-mediated combination chemotherapy and photodynamic therapy overcomes tumor drug resistance. J. Control. Release 2010, 141, 137–144. [Google Scholar]
- Liu, Y.; Lu, W.L.; Guo, J.; Du, J.; Li, T.; Wu, J.W.; Wang, G.L.; Wang, J.C.; Zhang, X.; Zhang, Q. A potential target associated with both cancer and cancer stem cells: A combination therapy for eradication of breast cancer using vinorelbine stealthy liposomes plus parthenolide stealthy liposomes. J. Control. Release 2008, 129, 18–25. [Google Scholar]
- Hirsch, H.A.; Iliopoulos, D.; Tsichlis, P.N.; Struhl, K. Metformin selectively targets cancer stem cells, and acts together with chemotherapy to block tumor growth and prolong remission. Cancer Res 2009, 69, 7507–7511. [Google Scholar]
- Guo, J.; Zhou, J.; Ying, X.; Men, Y.; Li, R.J.; Zhang, Y.; Du, J.; Tian, W.; Yao, H.J.; Wang, X.X.; et al. Effects of stealth liposomal daunorubicin plus tamoxifen on the breast cancer and cancer stem cells. J. Pharm. Pharm. Sci 2010, 13, 136–151. [Google Scholar]
- Zhang, G.N.; Liang, Y.; Zhou, L.J.; Chen, S.P.; Chen, G.; Zhang, T.P.; Kang, T.; Zhao, Y.P. Combination of salinomycin and gemcitabine eliminates pancreatic cancer cells. Cancer Lett 2011, 313, 137–144. [Google Scholar]
- Zhu, Y.; Yu, F.; Jiao, Y.; Feng, J.; Tang, W.; Yao, H.; Gong, C.; Chen, J.; Su, F.; Zhang, Y.; et al. Reduced miR-128 in breast tumor-initiating cells induces chemotherapeutic resistance via Bmi-1 and ABCC5. Clin. Cancer Res 2011, 17, 7105–7115. [Google Scholar]
- Yang, Y.P.; Chien, Y.; Chiou, G.Y.; Cherng, J.Y.; Wang, M.L.; Lo, W.L.; Chang, Y.L.; Huang, P.I.; Chen, Y.W.; Shih, Y.H.; et al. Inhibition of cancer stem cell-like properties and reduced chemoradioresistance of glioblastoma using microRNA145 with cationic polyurethane-short branch PEI. Biomaterials 2012, 33, 1462–1476. [Google Scholar]
- Tazzari, P.L.; Tabellini, G.; Ricci, F.; Papa, V.; Bortul, R.; Chiarini, F.; Evangelisti, C.; Martinelli, G.; Bontadini, A.; Cocco, L.; et al. Synergistic proapoptotic activity of recombinant TRAIL plus the Akt inhibitor Perifosine in acute myelogenous leukemia cells. Cancer Res 2008, 68, 9394–9403. [Google Scholar]

{kind=link}
| Natural compound | Sources | Tumor types | Ref. |
|---|---|---|---|
| Genistein | Soy | Pancreatic CSCs | [83] |
| Genistein | Soy | Breast CSCs | [84,85] |
| Blueberry Polyphenolic Acids | Blueberry | Breast CSCs | [84] |
| 20(S)-Ginsenoside Rg3 | Panax Ginsen | Colon CSCs | [86] |
| Nv-128 | Soy (Isoflavone Derivative) | Ovarian CSCs | [87] |
| Broussoflavonol B | Broussonetia Papyrifera | Breast CSCs | [88] |
| Shikonin | Arnebia Euchroma | Glioma CSCs | [89] |
| Curcumin | Curcuma Longa L. | Rectal CSCs, Breast CSCs | [90,91] |
| Piperine | Pepper | Breast CSCs | [91] |
| Resveratrol | Grape, Peanut, Polygonum Cuspidatum | Breast CSCs | [92] |
| Morusin | Morus Alba L. | Cervical CSCs | [59] |
© 2014 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
He, Y.-C.; Zhou, F.-L.; Shen, Y.; Liao, D.-F.; Cao, D. Apoptotic Death of Cancer Stem Cells for Cancer Therapy. Int. J. Mol. Sci. 2014, 15, 8335-8351. https://doi.org/10.3390/ijms15058335
He Y-C, Zhou F-L, Shen Y, Liao D-F, Cao D. Apoptotic Death of Cancer Stem Cells for Cancer Therapy. International Journal of Molecular Sciences. 2014; 15(5):8335-8351. https://doi.org/10.3390/ijms15058335
Chicago/Turabian StyleHe, Ying-Chun, Fang-Liang Zhou, Yi Shen, Duan-Fang Liao, and Deliang Cao. 2014. "Apoptotic Death of Cancer Stem Cells for Cancer Therapy" International Journal of Molecular Sciences 15, no. 5: 8335-8351. https://doi.org/10.3390/ijms15058335
APA StyleHe, Y.-C., Zhou, F.-L., Shen, Y., Liao, D.-F., & Cao, D. (2014). Apoptotic Death of Cancer Stem Cells for Cancer Therapy. International Journal of Molecular Sciences, 15(5), 8335-8351. https://doi.org/10.3390/ijms15058335