Large-Scale Domain Motions and Pyridoxal-5'-Phosphate Assisted Radical Catalysis in Coenzyme B12-Dependent Aminomutases †


Abstract
:1. Introduction
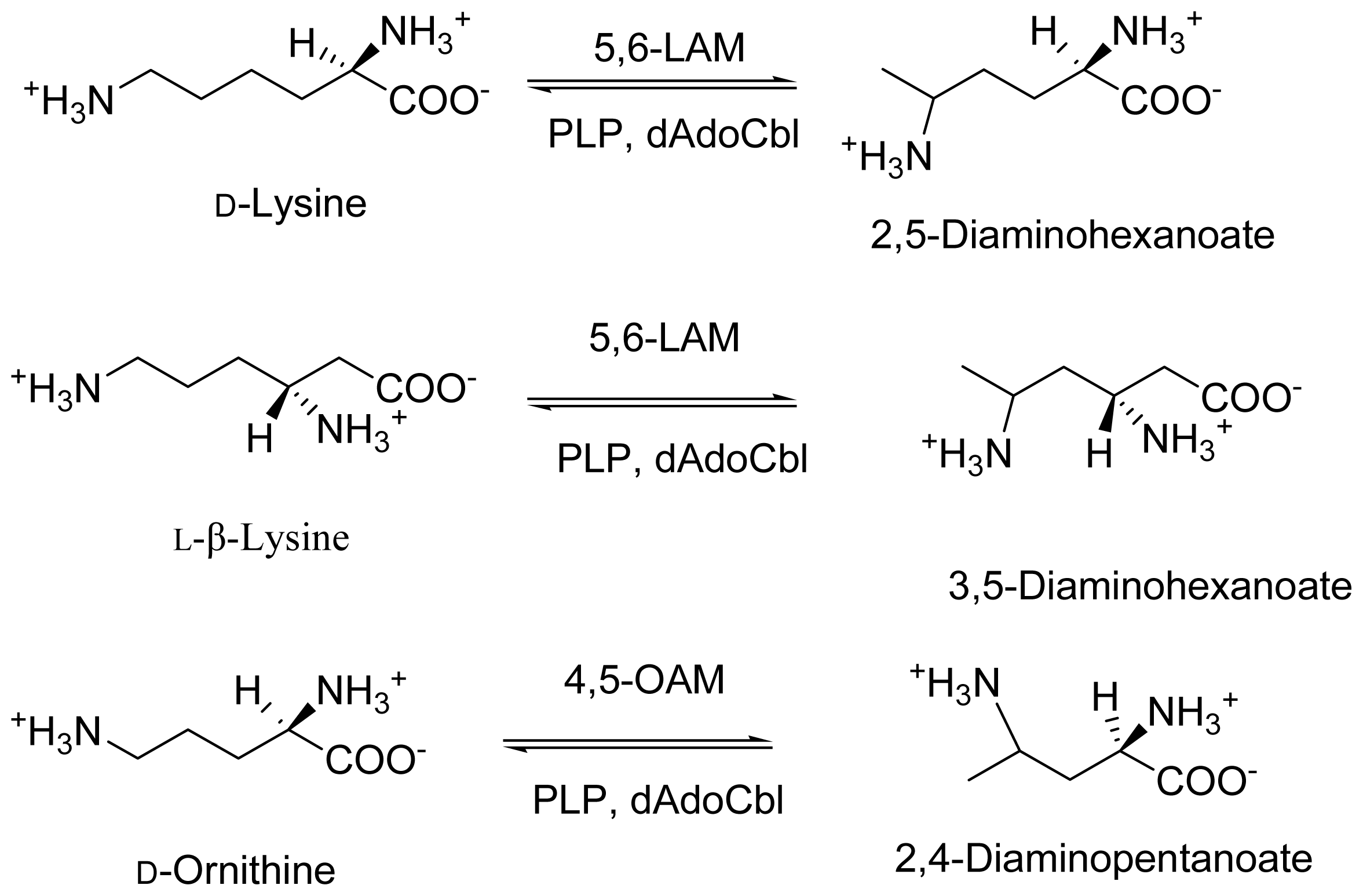
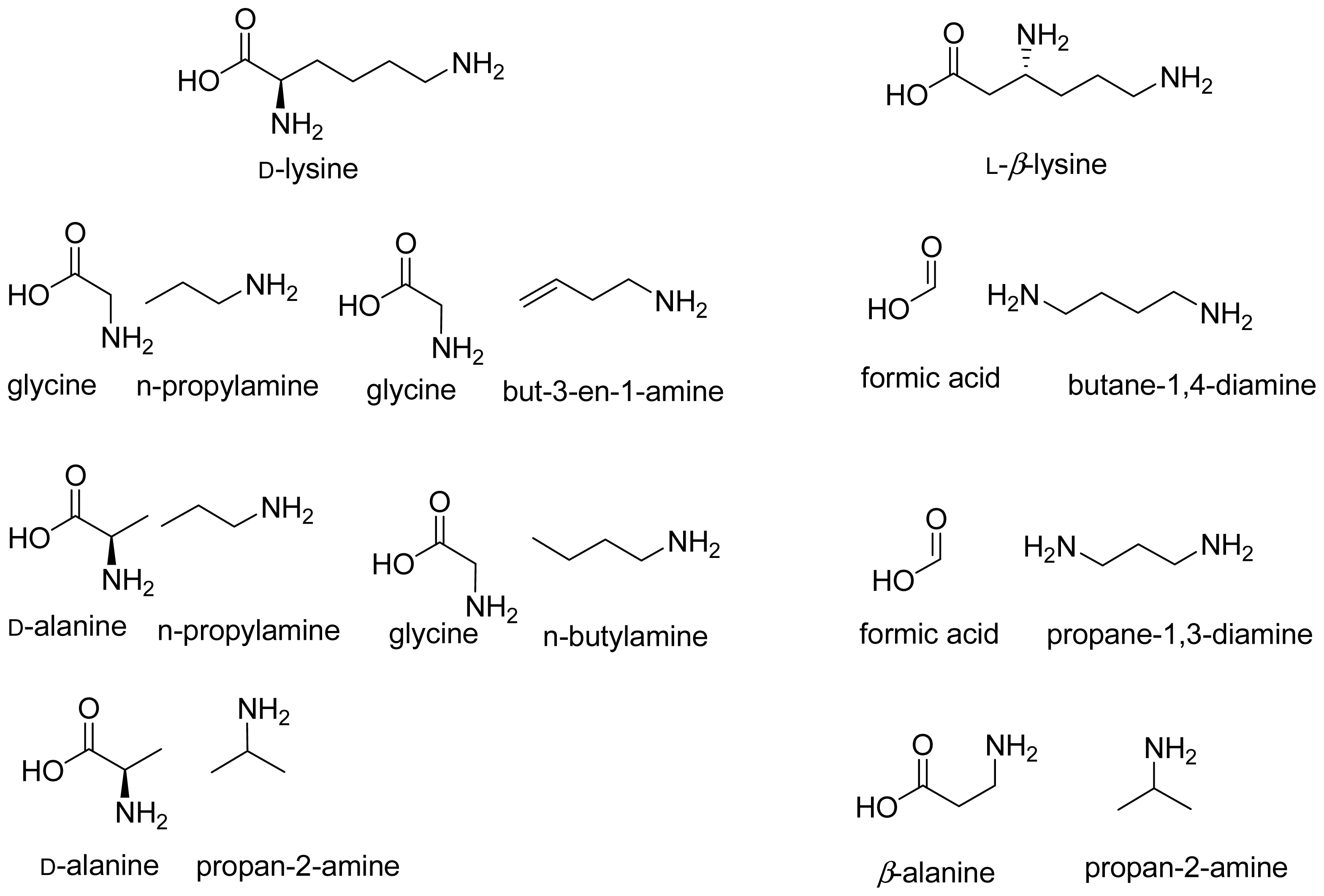
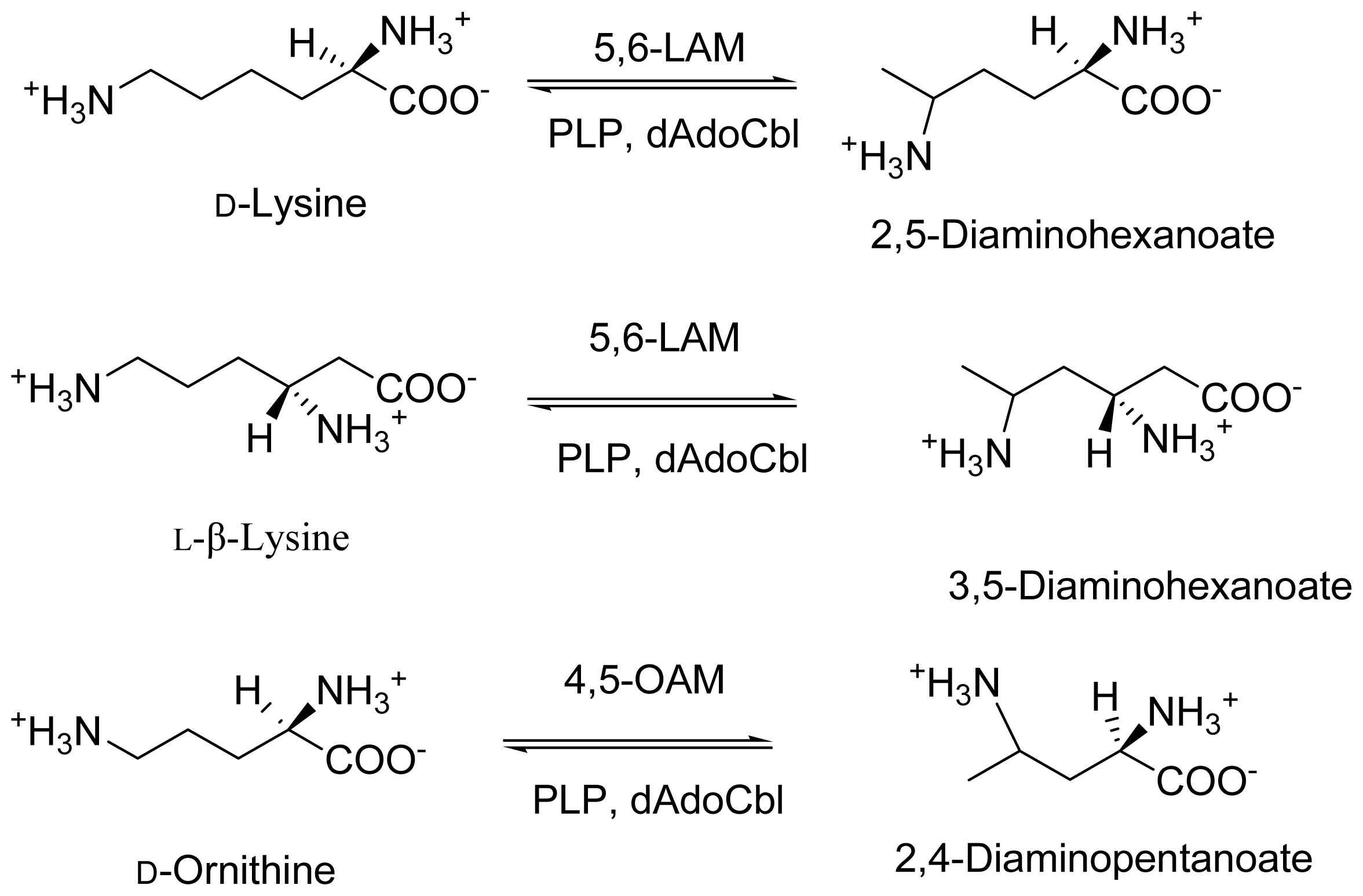
2. Discovery and Physiological Role
3. Cloning and Expression of Recombinant Aminomutases
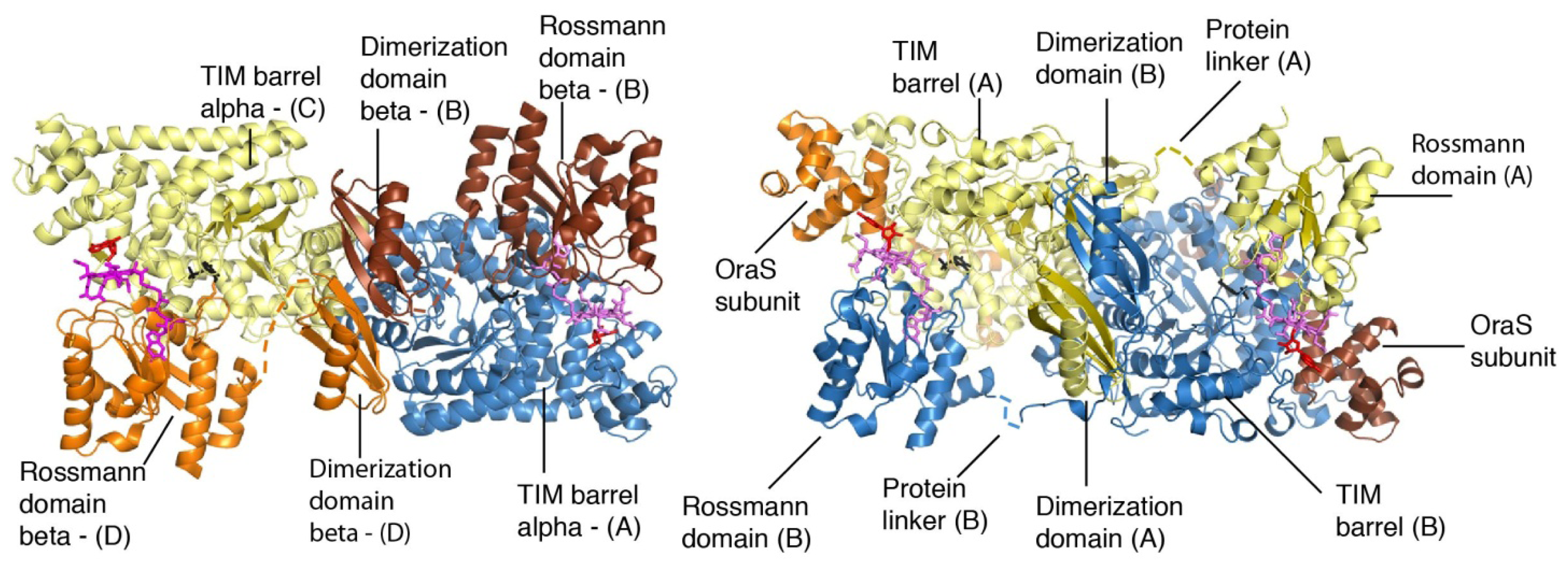
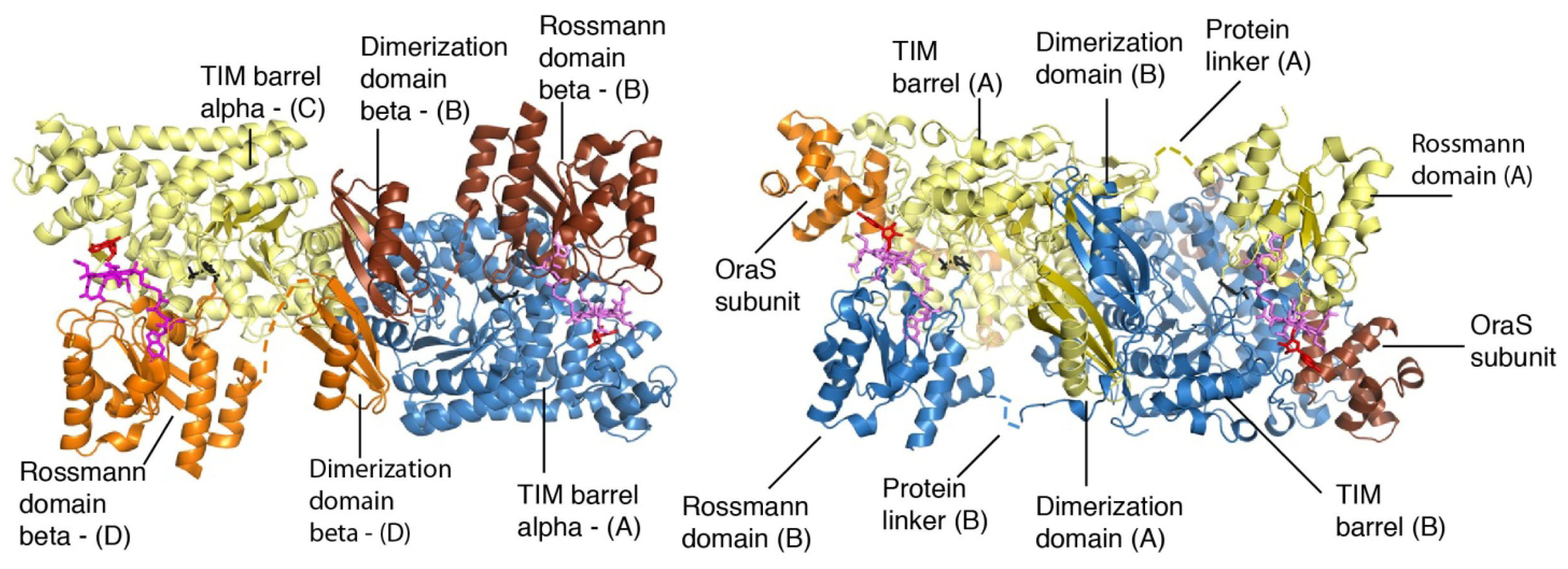
4. Structural Studies
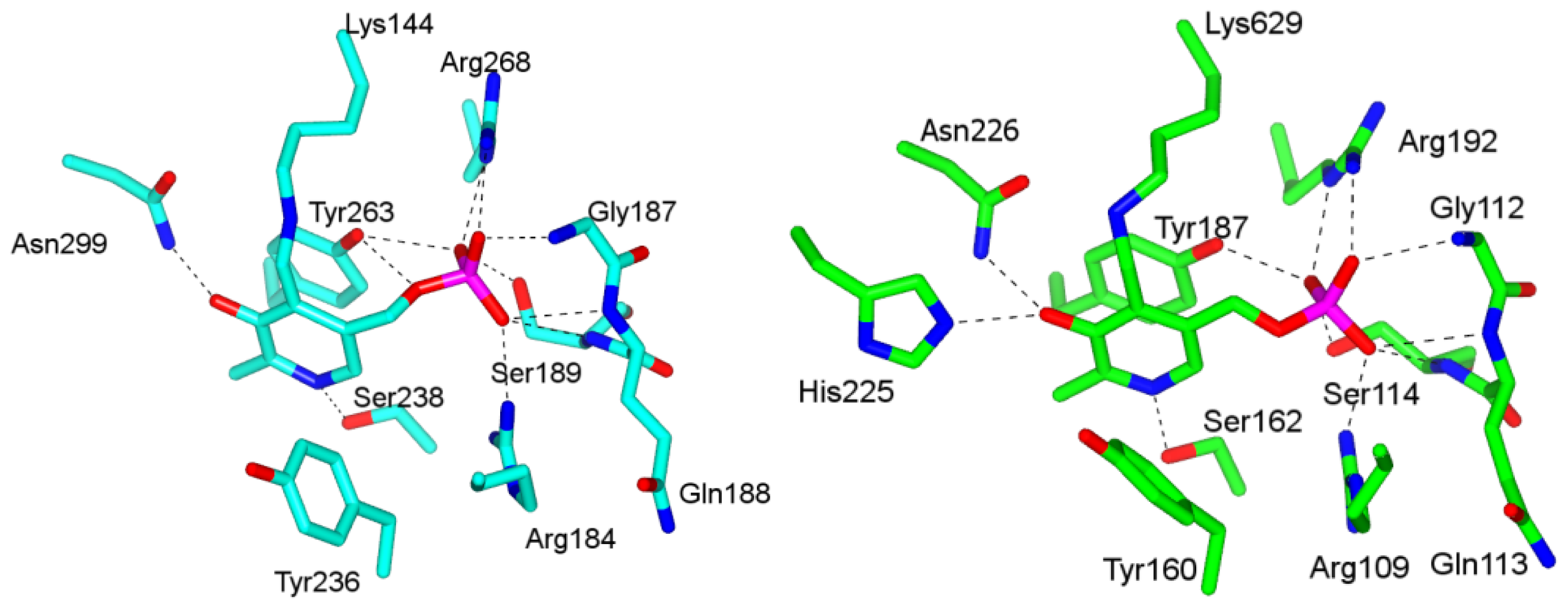
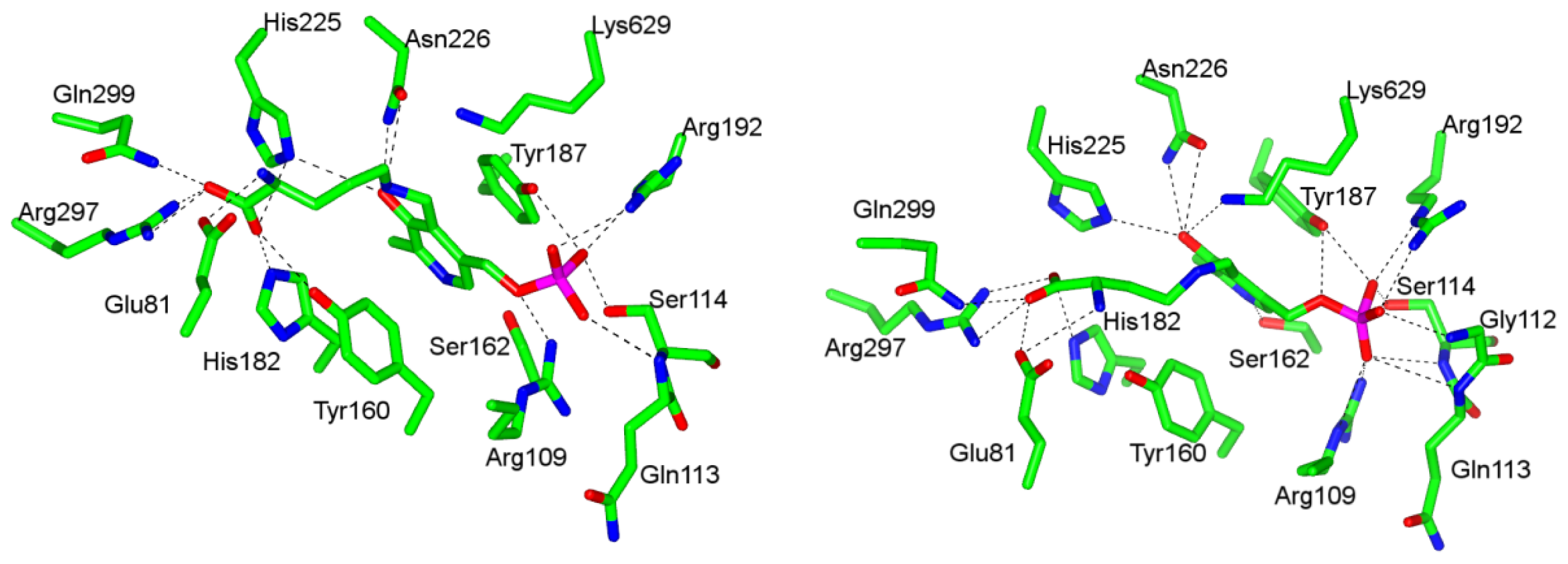
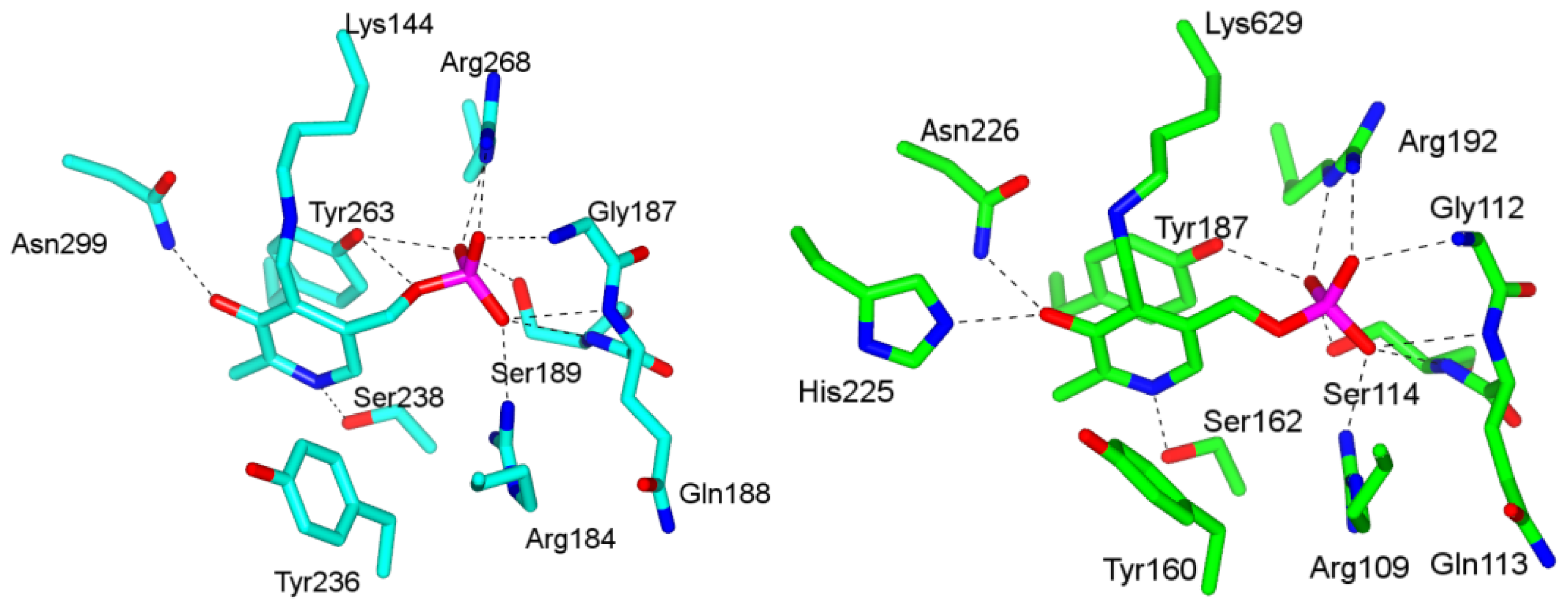
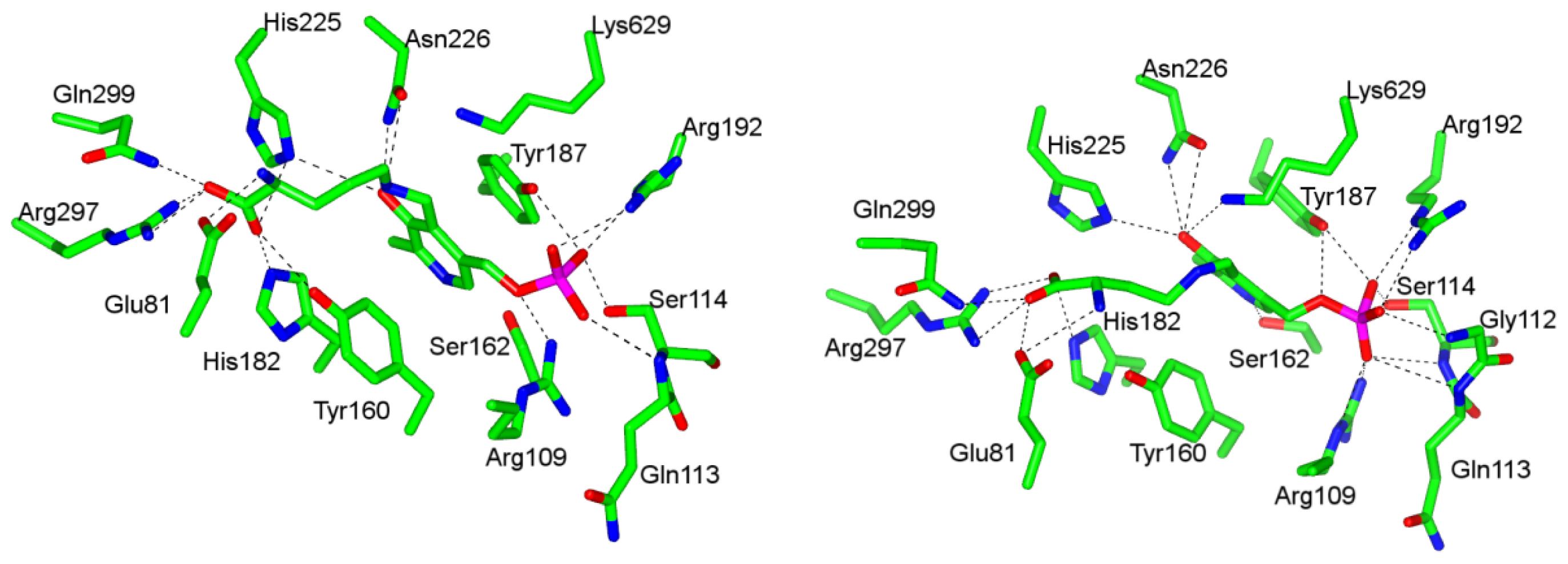
4.1. Pyridoxal-5′-Phosphate (PLP) Binding Site and Active Site Residues
4.2. “Base-off”/“His-on” Binding Mode of dAdoCbl
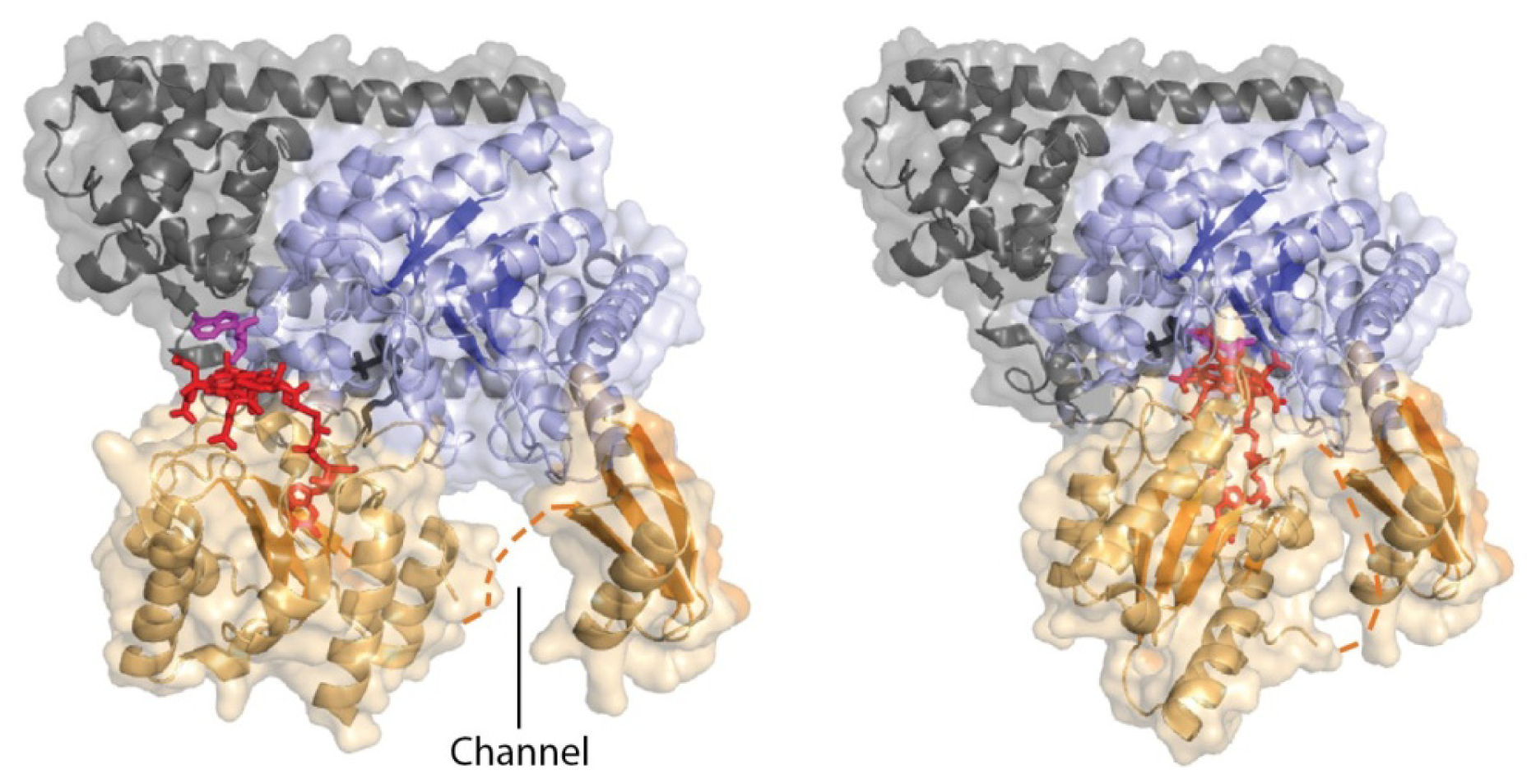
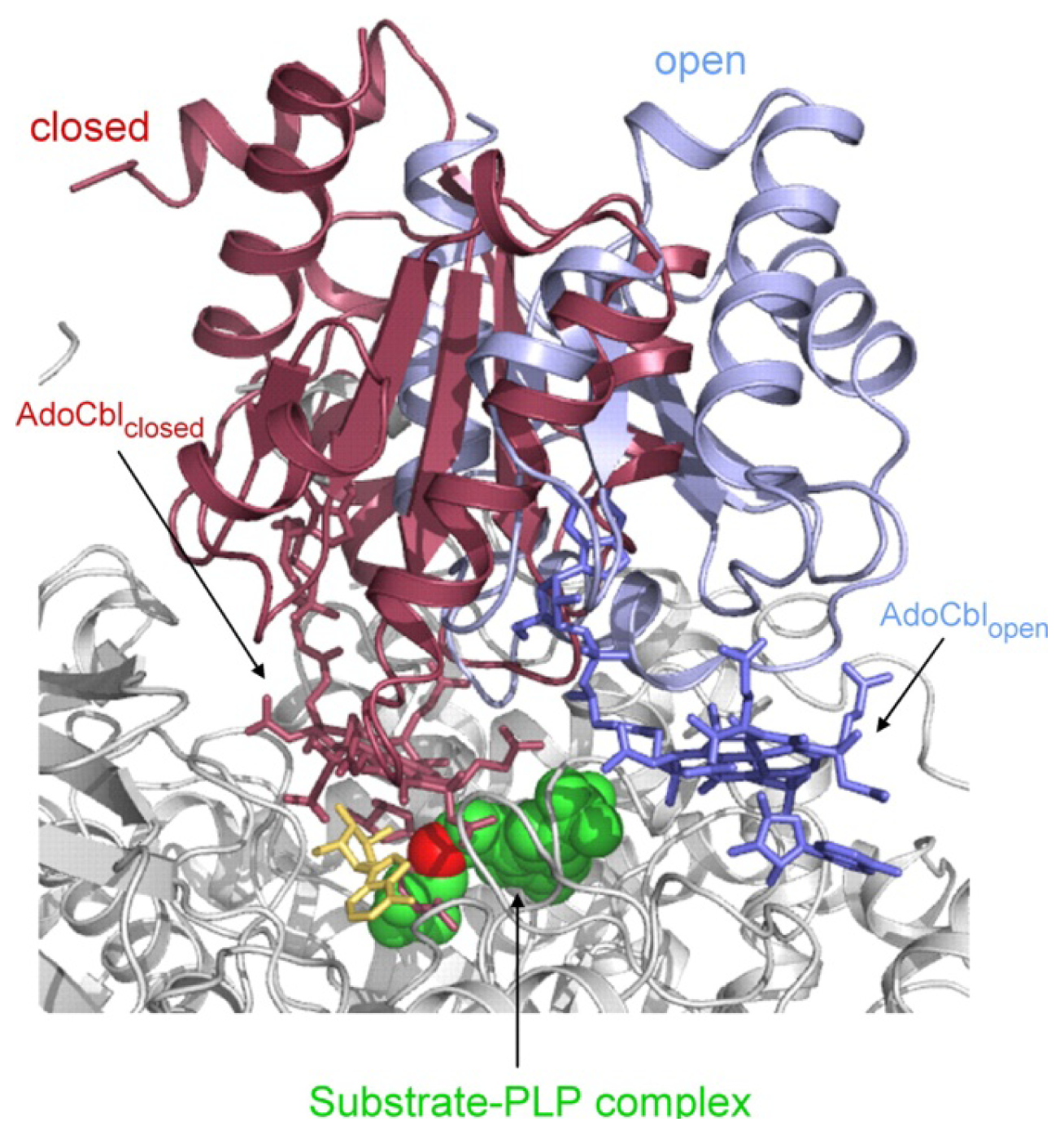
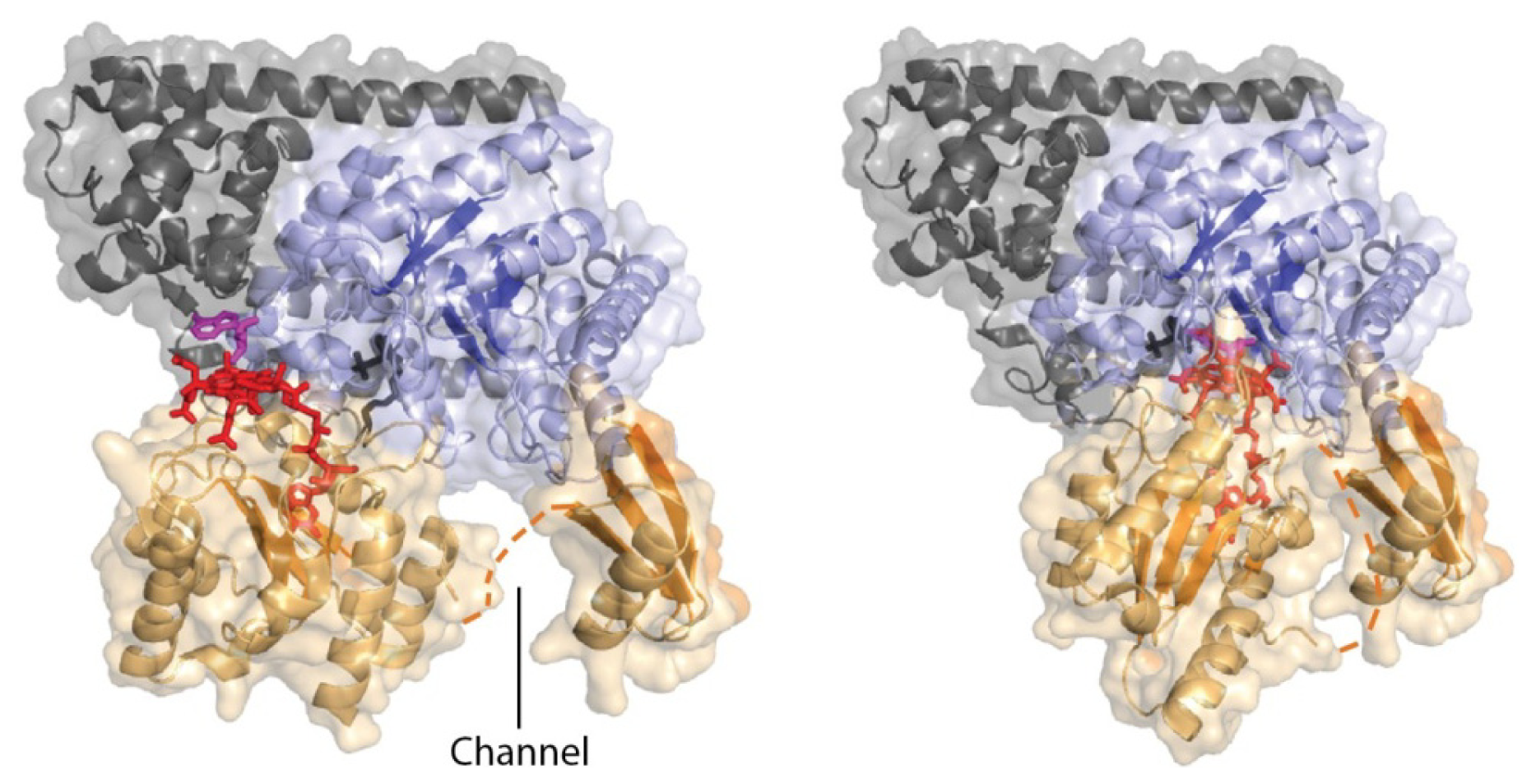
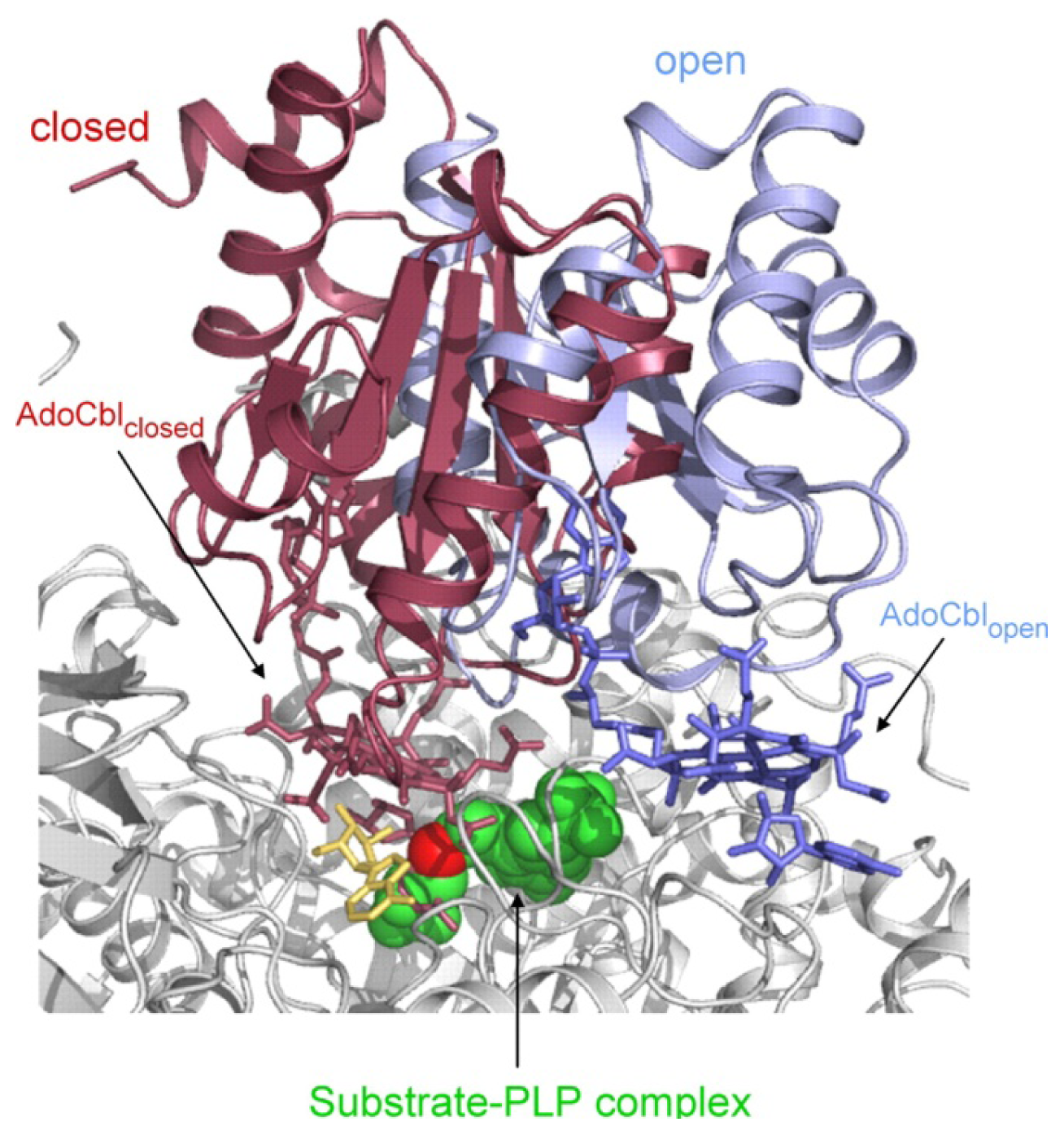
4.3. Models of Closed Conformation
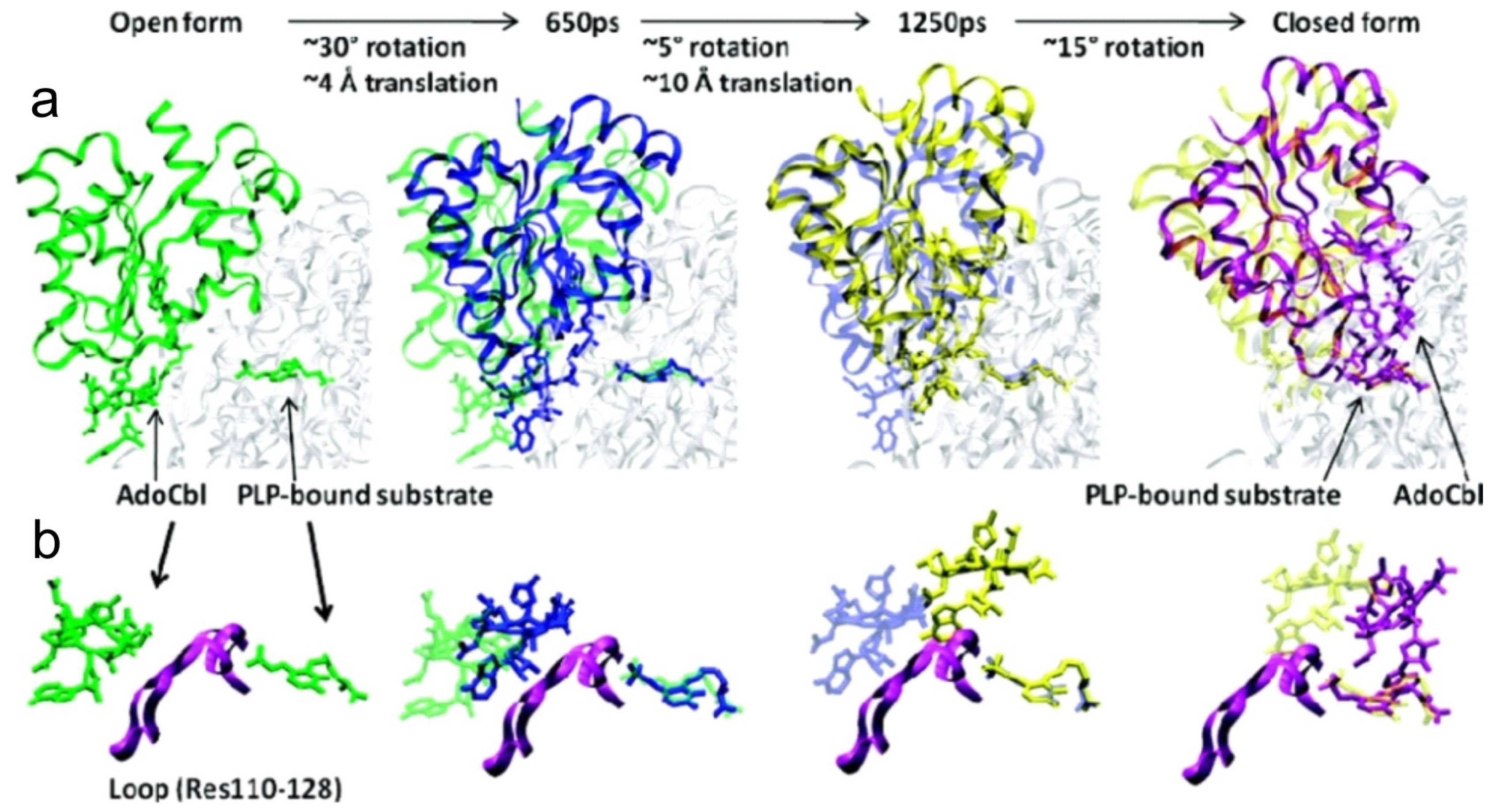
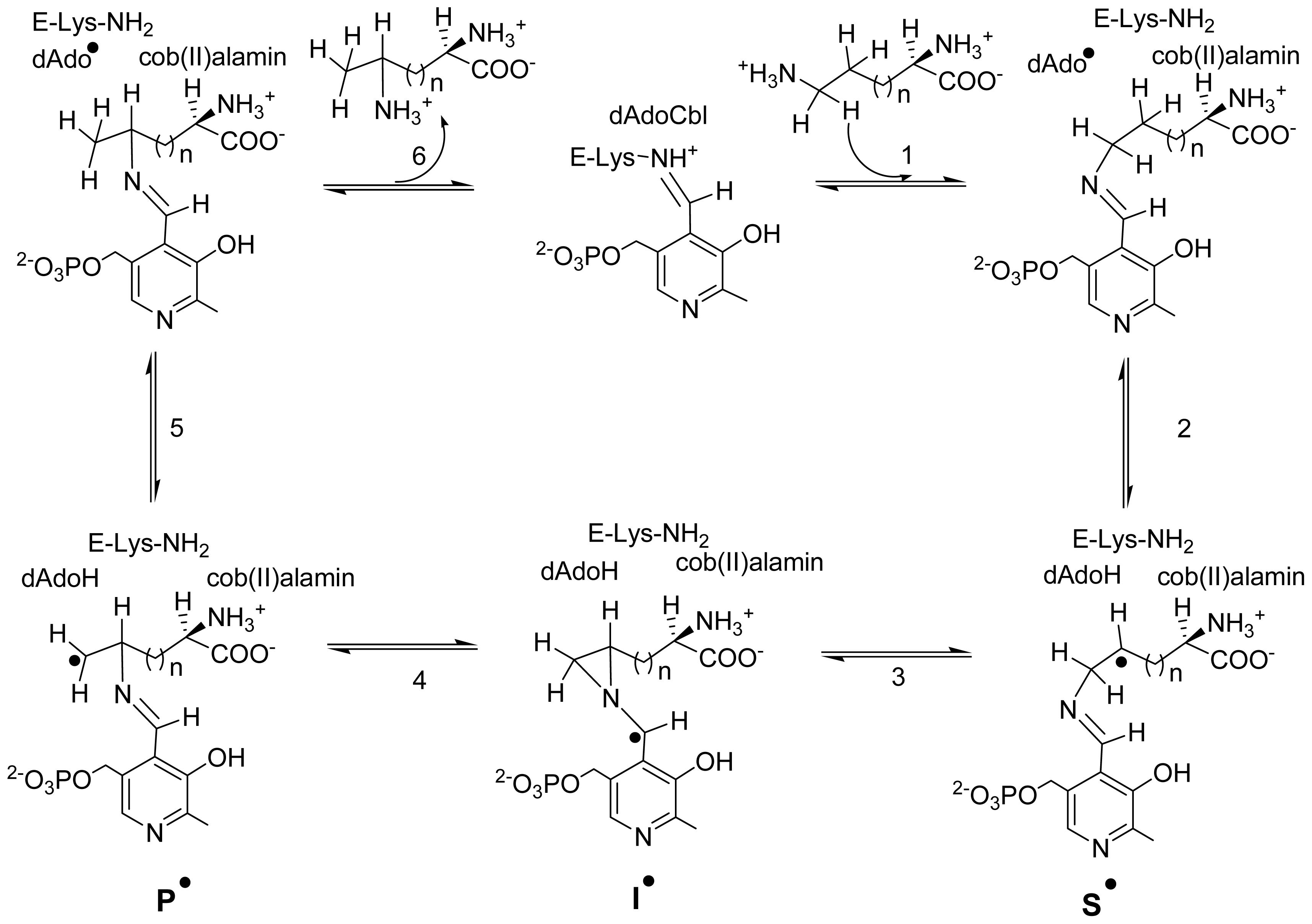
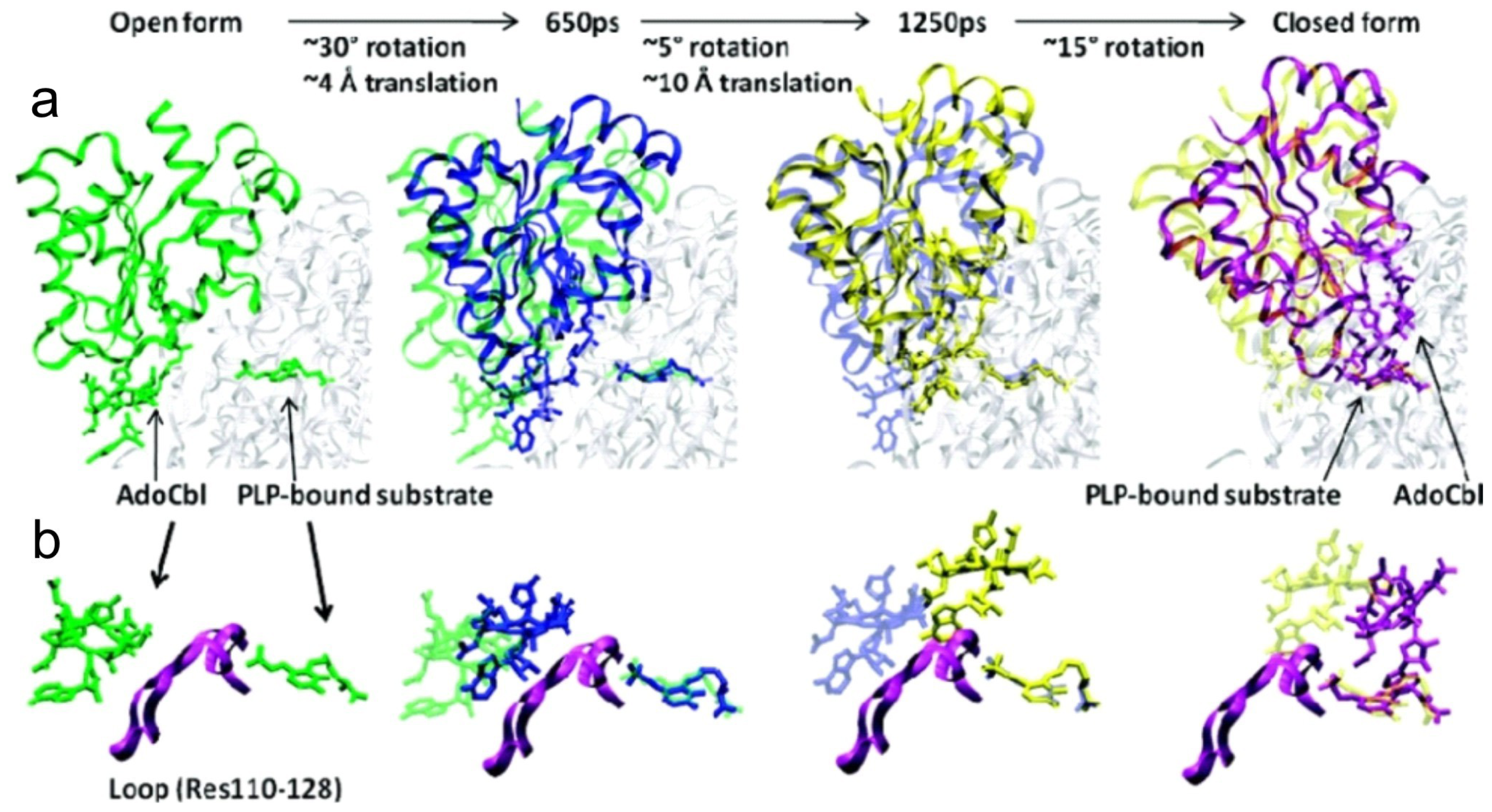
5. Domain Motions
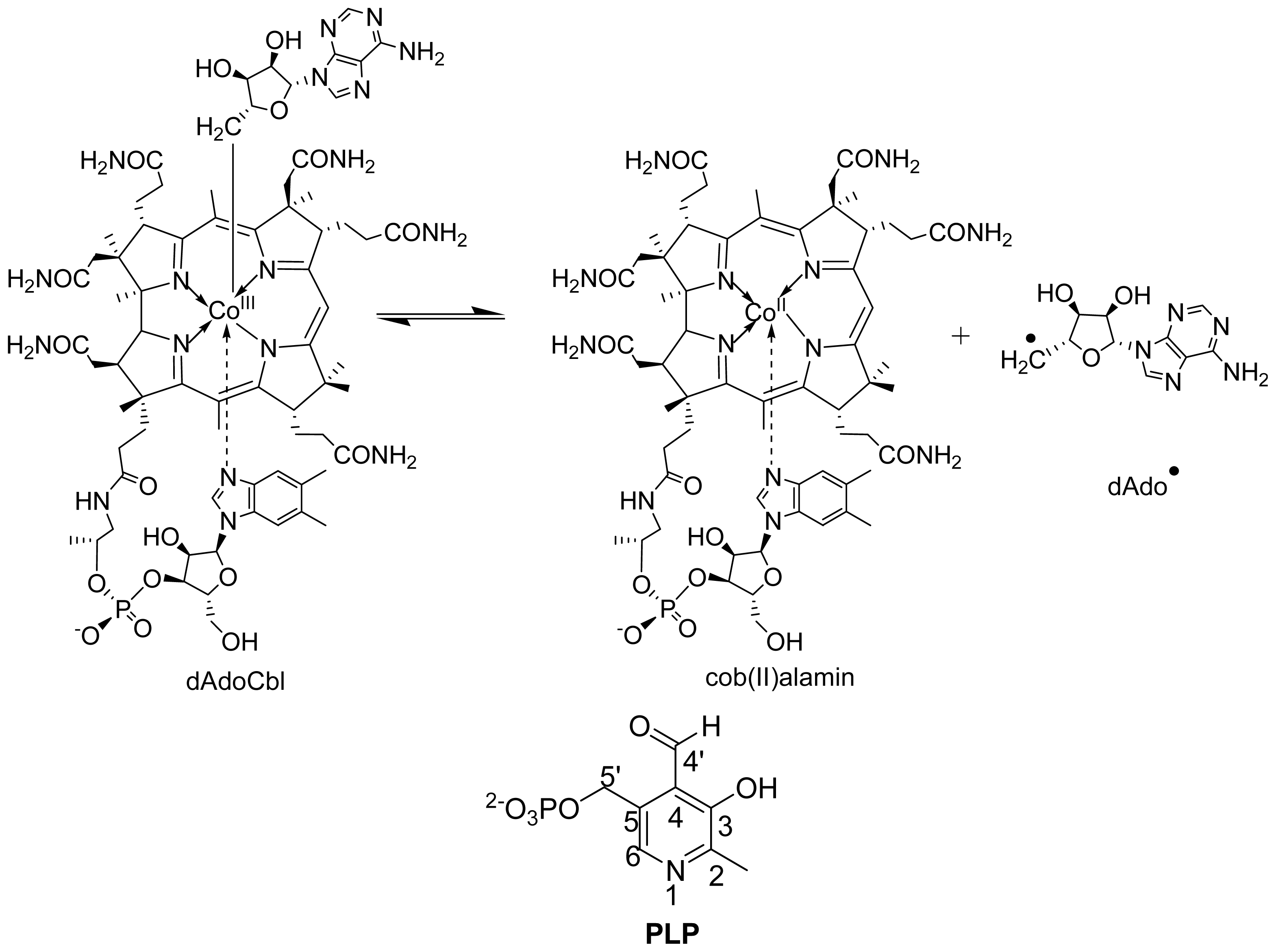
6. Mechanistic Studies
6.1. UV-Visible Spectroscopic Studies
6.1.1. Spectral Changes Originating from PLP
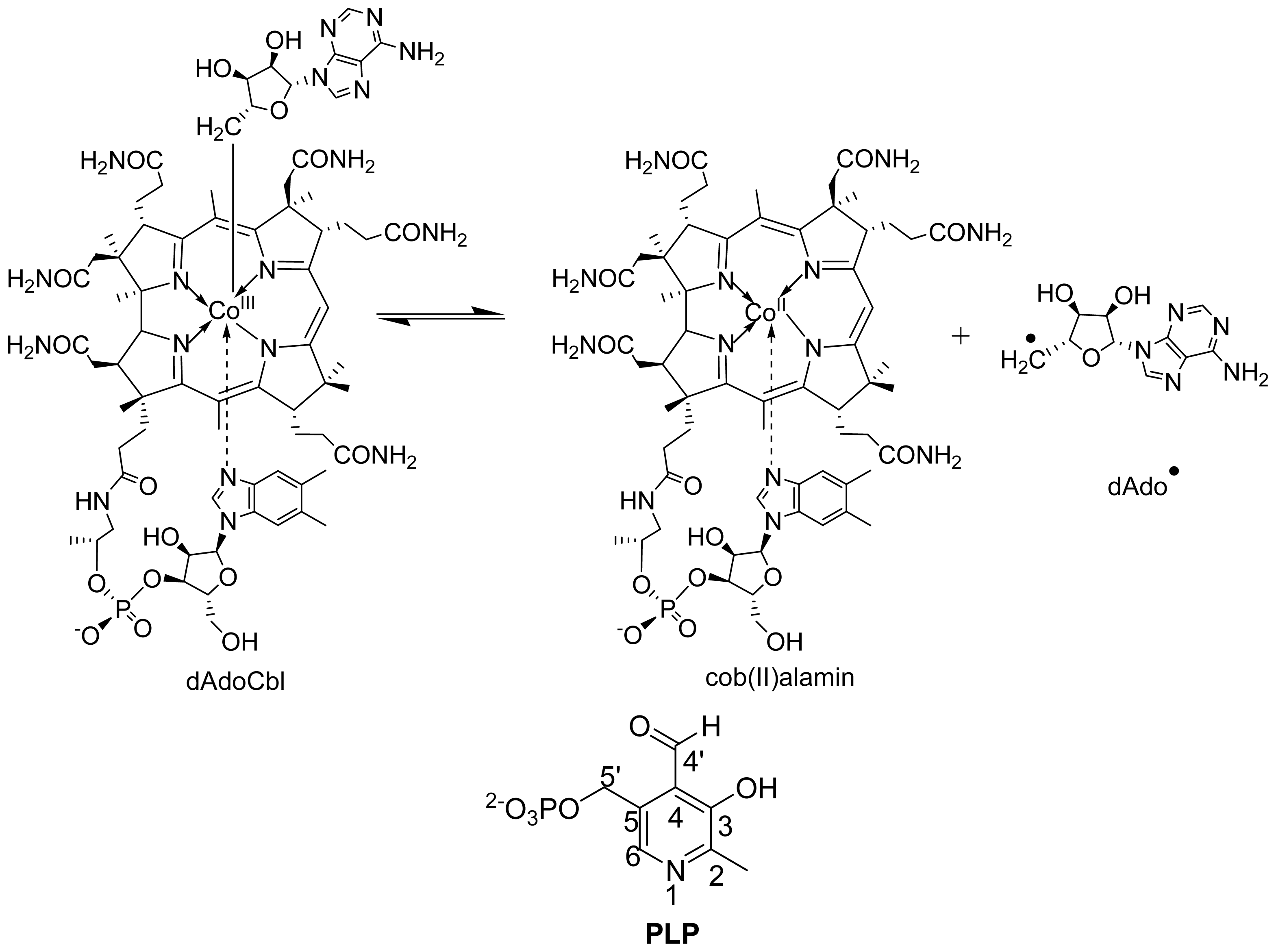
6.1.2. Spectral Changes Originating from dAdoCbl
6.2. Stopped-Flow Analysis
6.3. Electron Paramagnetic Resonance (EPR) and Electron Nuclear Double Resonance (ENDOR) Spectroscopy
6.3.1. Distance between PLP-Substrate/Analogue Radical and Cob (II) Alamin
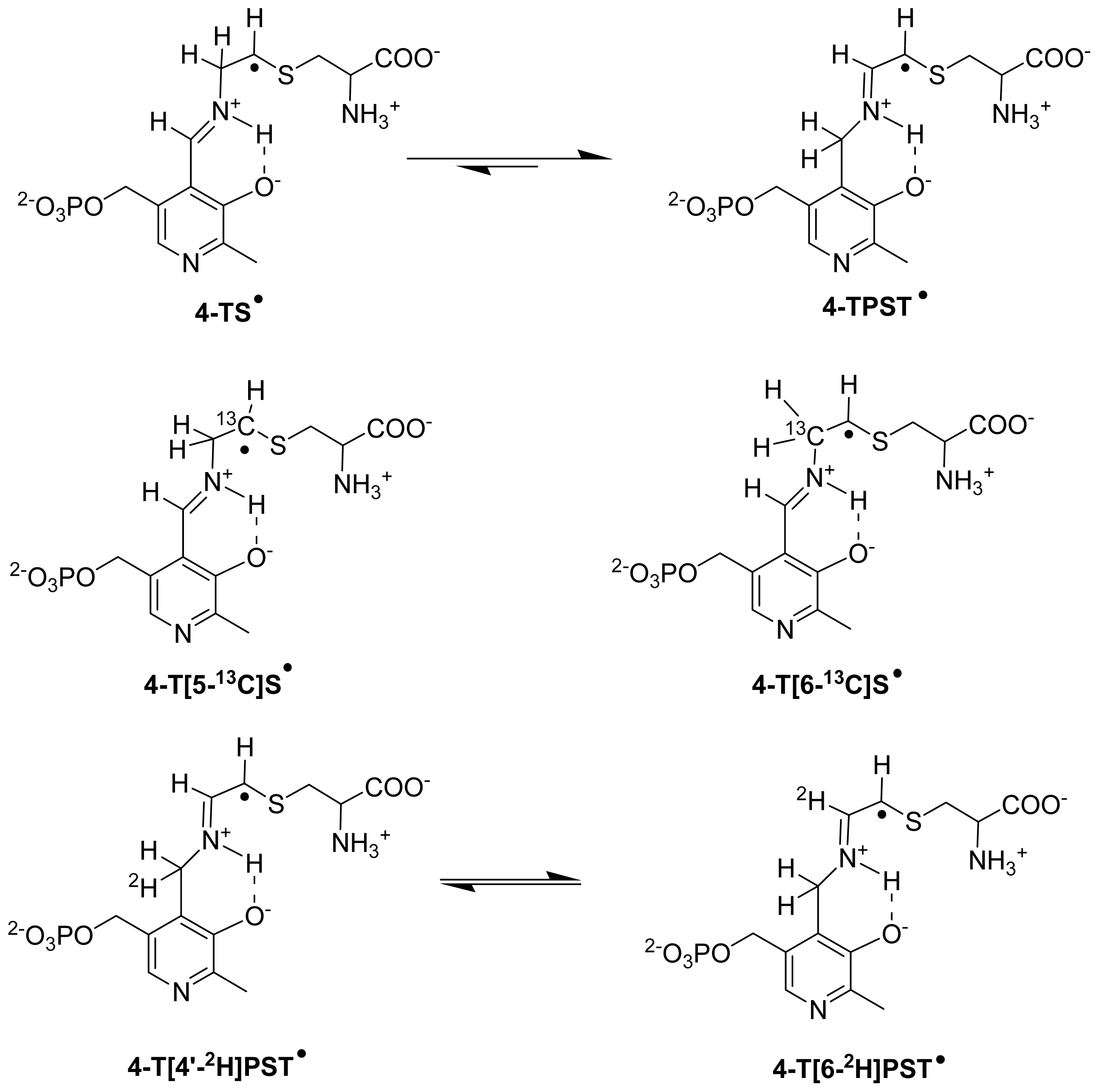
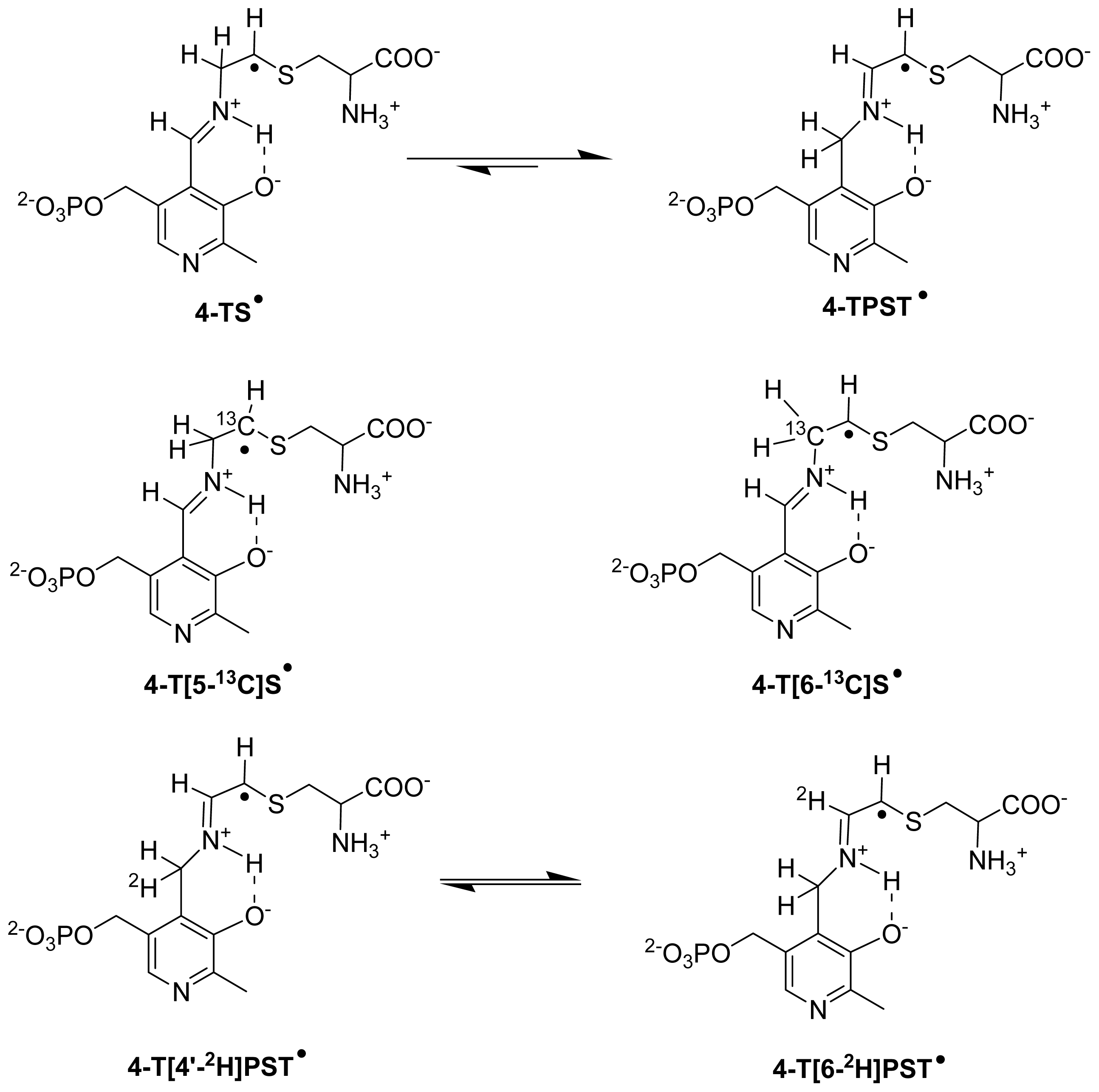
6.3.2. Isotope-Edited EPR and ENDOR Spectroscopy for Characterization of Radical Intermediates
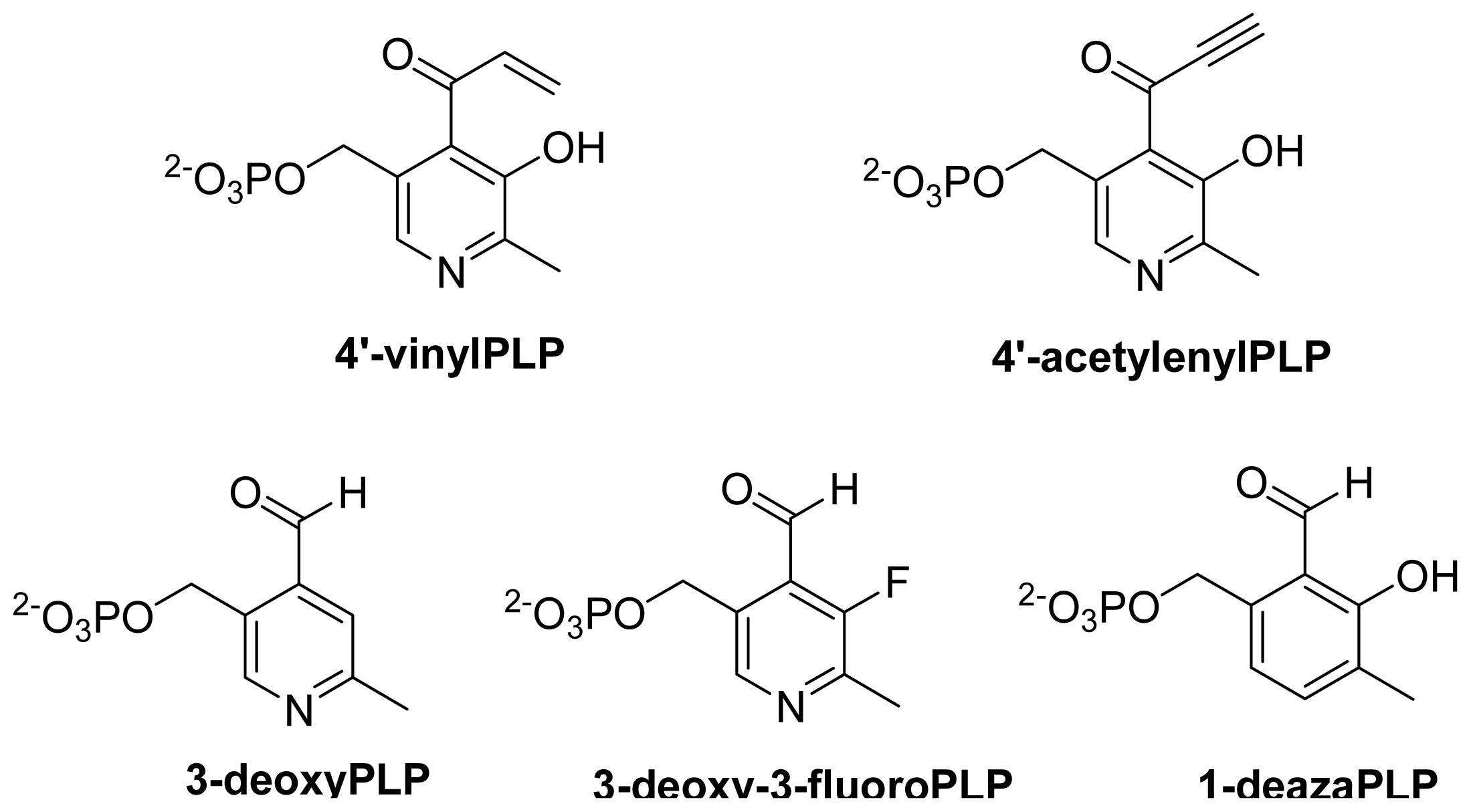
6.3.3. Role of Conserved Tyr Residue
6.3.4. Transition between Open and Closed State
6.4. Computational Studies
7. Outlook and Perspectives
Acknowledgments
Conflicts of Interest
- †This paper is dedicated to Emeritus Professor Perry A. Frey (Biochemistry Department, University of Wisconsin at Madison, Madison, WI, USA) on the occasion of his 78th birthday.
References
- Stryer, L. Biochemistry, 4th Ed. ed; W.H. Freeman and Company: New York, NY, USA, 1995. [Google Scholar]
- Dolphin, D. B12; John Wiley & Sons: New York, NY, USA, 1982; Volume 1. [Google Scholar]
- Dolphin, D. B12; John Wiley & Sons: New York, NY, USA, 1982; Volume 2. [Google Scholar]
- Banerjee, R. Chemistry and Biochemistry of B12; Wiley-Interscience: New York, NY, USA, 1999. [Google Scholar]
- Mooney, S.; Leuendorf, J.E.; Hendrickson, C.; Hellmann, H. Vitamin B6: A long known compound of surprising complexity. Molecules 2009, 14, 329–351. [Google Scholar]
- Kräutler, B.; Arigoni, D.; Golding, B.T. Vitamin B12 and B12-Proteins; Wiley-VCH: Weinheim, Germany, 1998. [Google Scholar]
- Zhang, X.H.; Ma, J.; Smith-Warner, S.A.; Lee, J.E.; Giovannucci, E. Vitamin B6 and colorectal cancer: Current evidence and future directions. World J. Gastroenterol 2013, 19, 1005–1010. [Google Scholar]
- Frey, P.A.; Hegeman, A.D.; Reed, G.H. Free radical mechanisms in enzymology. Chem. Rev 2006, 106, 3302–3316. [Google Scholar]
- Brown, K.L. Chemistry and enzymology of vitamin B12. Chem. Rev 2005, 105, 2075–2149. [Google Scholar]
- Banerjee, R. Radical carbon skeleton rearrangements: Catalysis by coenzyme B12-dependent mutases. Chem. Rev 2003, 103, 2083–2094. [Google Scholar]
- Toraya, T. Radical catalysis in coenzyme B12-dependent isomerization (eliminating) reactions. Chem. Rev 2003, 103, 2095–2127. [Google Scholar]
- Pyridoxal Phosphate Enzymology. In Biochimica et Biophysica Acta; Toney, M.D. (Ed.) Elsevier: Miamisburg, OH, USA, 2011 1814; pp. 1405–1608.
- Jones, A.R.; Levy, C.; Hay, S.; Scrutton, N.S. Relating localized protein motions to the reaction coordinate in coenzyme B12-dependent enzymes. FEBS J 2013, 280, 2997–3008. [Google Scholar]
- Eriksson, L.A. Theoretical Biochemistry-Processes and Properties of Biological Systems; Elsevier: Amsterdam, The Netherlands, 2001. [Google Scholar]
- Oliveira, E.F.; Cerqueira, N.M.F.S.A.; Fernandes, P.A.; Ramos, M.J. Mechanism of formation of the internal aldimine in pyridoxal 5′-phosphate-dependent enzymes. J. Am. Chem. Soc 2011, 133, 15496–15505. [Google Scholar]
- Eliot, A.C.; Kirsch, J.F. Pyridoxal phosphate enzymes: Mechanistic, structural, and evolutionary considerations. Annu. Rev. Biochem 2004, 73, 383–415. [Google Scholar]
- Jensen, K.P.; Ryde, U. How the Co–C bond is cleaved in coenzyme B12 enzymes: A theoretical study. J. Am. Chem. Soc 2005, 127, 9117–9128. [Google Scholar]
- Kozlowski, P.M.; Kamachi, T.; Toraya, T.; Yoshizawa, K. Does cob (II) alamin act as a conductor in coenzyme B12 dependent mutases? Angew. Chem. Int. Ed 2007, 46, 980–983. [Google Scholar]
- Bucher, D.; Sandala, G.M.; Durbeej, B.; Radom, L.; Smith, D.M. The elusive 5′-deoxyadenosyl radical in coenzyme-B12-mediated reactions. J. Am. Chem. Soc 2012, 134, 1591–1599. [Google Scholar]
- Sandala, G.M.; Smith, D.M.; Radom, L. Modeling the reactions catalyzed by coenzyme B12-dependent enzymes. Acc. Chem. Res 2010, 43, 642–651. [Google Scholar]
- Buckel, W.; Friedrich, P.; Golding, B.T. Hydrogen bonds guide the short-lived 5′-deoxyadenosyl radical to the place of action. Angew. Chem. Int. Ed 2012, 51, 9974–9976. [Google Scholar]
- Kamachi, T.; Toraya, T.; Yoshizawa, K. Computational mutation analysis of hydrogen abstraction and radical rearrangement steps in the catalysis of coenzyme B12-dependent diol dehydratase. Chem. Eur. J 2007, 13, 7864–7873. [Google Scholar]
- Frey, P.A. Cobalamin coenzymes in enzymology. In Comprehensive Natural Products II Chemistry and Biology; Mander, L., Lui, H.-W., Eds.; Elsevier: Oxford, UK, 2010; Volume 7, pp. 501–546. [Google Scholar]
- Frey, P.A.; Reed, G.H. Pyridoxal-5′-phosphate as the catalyst for radical isomerization in reactions of PLP-dependent aminomutases. Biochim. Biophys. Acta 2011, 1814, 1548–1557. [Google Scholar]
- Dowling, D.P.; Croft, A.K.; Drennan, C.L. Radical use of Rossmann and TIM barrel architectures for controlling coenzyme B12 chemistry. Annu. Rev. Biophys 2012, 41, 403–427. [Google Scholar]
- Baker, J.J.; Stadtman, T.C. Amino Mutases. In B12; Dolphin, D., Ed.; John Wiley & Sons: New York, NY, USA, 1982; Volume 2, pp. 203–232. [Google Scholar]
- Stadtman, T.C. Lysine metabolism by Clostridia. Adv. Enzymol 1973, 38, 413–448. [Google Scholar]
- Morley, C.G.D.; Stadtman, T.C. Studies on the fermentation of d-α-lysine. Purification and properties of an adenosine triphosphate regulated B12-coenzyme-dependent d-α-lysine mutase complex from Clostridium sticklandii. Biochemistry 1970, 9, 4890–4900. [Google Scholar]
- Morley, C.G.D.; Stadtman, T.C. Fermentation of d-α-lysine: On the hydrogen shift catalyzed by the B12 coenzyme dependent D-α-lysine mutase. Biochemistry 1971, 10, 2325–2329. [Google Scholar]
- Morley, C.G.D.; Stadtman, T.C. The role of pyridoxal phosphate in the B12 coenzyme-dependent d-α-lysine mutase reaction. Biochemistry 1972, 11, 600–605. [Google Scholar]
- Baker, J.J.; van der Drift, C.; Stadtman, T.C. Purification and properties of β-lysine mutase, a pyridoxal phosphate and B12 coenzyme dependent enzyme. Biochemistry 1973, 12, 1054–1063. [Google Scholar]
- Frey, P.A.; Chang, C.H. Aminomutases. In Chemistry and Biochemistry of B12; Banerjee, R., Ed.; Wiley: New York, NY, USA, 1999; pp. 835–857. [Google Scholar]
- Dyer, J.K.; Costilow, R.N. 2,4-Diaminovaleric acid: An intermediate in the anaerobic oxidation of ornithine by Clostridium sticklandii. J. Bacteriol 1970, 101, 77–83. [Google Scholar]
- Tsuda, Y.; Friedmann, H.C. Ornithine metabolism by Clostridium sticklandii. Oxidation of ornithine to 2-amino-4-ketopentanoic acid via 2,4-diaminopentanoic acid; participation of B12 coenzyme, pyridoxal phosphate, and pyridine nucleotide. J. Biol. Chem 1970, 245, 5914–5926. [Google Scholar]
- Somack, R.; Costilow, R.N. Purification and properties of a pyridoxal phosphate and coenzyme B12 dependent d-α-ornithine 5,4-aminomutase. Biochemistry 1973, 12, 2597–2604. [Google Scholar]
- Chang, C.H.; Frey, P.A. Cloning, sequencing, heterologous expression, purification, and characterization of adenosylcobalamin-dependent d-lysine 5,6-aminomutase from Clostridium sticklandii. J. Biol. Chem 2000, 275, 106–114. [Google Scholar]
- Tang, K.H.; Chang, C.H.; Frey, P.A. Electron transfer in the substrate-dependent suicide inactivation of lysine 5,6-aminomutase. Biochemistry 2001, 40, 5190–5199. [Google Scholar]
- Tang, K.H.; Harms, A.; Frey, P.A. Identification of a novel pyridoxal 5′-phosphate binding site in adenosylcobalamin-dependent lysine 5,6-aminomutase from Porphyromonas gingivalis. Biochemistry 2002, 41, 8767–8776. [Google Scholar]
- Chen, H.P.; Wu, S.H.; Lin, Y.L.; Chen, C.M.; Tsay, S.S. Cloning, sequencing, heterologous expression, purification, and characterization of adenosylcobalamin-dependent d-ornithine aminomutase from Clostridium sticklandii. J. Biol. Chem 2001, 276, 44744–44750. [Google Scholar]
- Chen, H.P.; Hsui, F.C.; Lin, L.Y.; Ren, C.T.; Wu, S.H. Coexpression, purification and characterization of the E and S subunits of coenzyme B12 and B6 dependent Clostridium sticklandii d-ornithine aminomutase in Escherichia coli. Eur. J. Biochem 2004, 271, 4293–4297. [Google Scholar]
- Tseng, C.H.; Yang, C.H.; Lin, H.J.; Wu, C.H.; Chen, H.P. The S subunit of d-ornithine aminomutase from Clostridium sticklandii is responsible for the allosteric regulation in d-α-lysine aminomutase. FEMS Microbiol. Lett 2007, 274, 148–153. [Google Scholar]
- Tang, K.-H.; Casarez, A.D.; Wu, W.; Frey, P.A. Kinetic and biochemical analysis of the mechanism of action of lysine 5,6-aminomutase. Arch. Biochem. Biophys 2003, 418, 49–54. [Google Scholar]
- Makins, C.; Pickering, A.V.; Mariani, C.; Wolthers, K.R. Mutagenesis of a conserved glutamate reveals the contribution of electrostatic energy to adenosylcobalamin Co–C bond homolysis in ornithine 4,5-aminomutase and methylmalonyl-CoA mutase. Biochemistry 2013, 52, 878–888. [Google Scholar]
- Berkovitch, F.; Behshad, E.; Tang, K.-H.; Enns, E.A.; Frey, P.A.; Drennan, C.L. A locking mechanism preventing radical damage in the absence of substrate, as revealed by the X-ray structure of lysine 5,6-aminomutase. Proc. Natl. Acad. Sci. USA 2004, 101, 15870–15875. [Google Scholar]
- Wolthers, K.R.; Levy, C.; Scrutton, N.S.; Leys, D. Large-scale domain dynamics and adenosylcobalamin reorientation orchestrate radical catalysis in ornithine 4,5-aminomutase. J. Biol. Chem 2010, 285, 13942–13950. [Google Scholar]
- Schneider, G.; Kack, H.; Lindqvist, Y. The manifold of vitamin B6 dependent enzymes. Structure 2000, 8, R1–R6. [Google Scholar]
- Atherton, N.M. Principles of Electron Spin Resonance; Ellis Horwood Limited: London, UK, 1993. [Google Scholar]
- Zelder, O.; Beatrix, B.; Kroll, F.; Buckel, W. Coordination of a histidine residue of the protein-component S to the cobalt atom in coenzyme B12-dependent glutamate mutase from Clostridium cochlearium. FEBS Lett 1995, 369, 252–254. [Google Scholar]
- Lawrence, C.C.; Gerfen, G.J.; Samano, V.; Nitsche, R.; Robins, M.J.; Retey, J.; Stubbe, J. Binding of cob (II) alamin to the adenosylcobalamin-dependent ribonucleotide reductase from Lactobacillus leichmannii—Identification of dimethylbenzimidazole as the axial ligand. J. Biol. Chem 1999, 274, 7039–7042. [Google Scholar]
- Drennan, C.L.; Huang, S.; Drummond, J.T.; Matthews, R.G.; Lidwig, M.L. How a protein binds B12: A 3.0 Å X-ray structure of B12-binding domains of methionine synthase. Science 1994, 266, 1669–1674. [Google Scholar]
- Mancia, F.; Keep, N.H.; Nakagawa, A.; Leadlay, P.F.; McSweeney, S.; Rasmussen, B.; Bosecke, P.; Diat, O.; Evans, P.R. How coenzyme B12 radicals are generated: The crystal structure of methylmalonyl-coenzyme A mutase at 2 Å resolution. Structure 1996, 4, 339–350. [Google Scholar]
- Reitzer, R.; Gruber, K.; Jogl, G.; Wagner, U.G.; Bothe, H.; Buckel, W.; Kratky, C. Glutamate mutase from Clostridium cochlearium: The structure of a coenzyme B12-dependent enzyme provides new mechanistic insights. Structure 1999, 7, 891–902. [Google Scholar]
- Gruber, K.; Reitzer, R.; Kratky, C. Radical shuttling in a protein: Ribose pseudorotation controls alkyl-radical transfer in the coenzyme B12 dependent enzyme glutamate mutase. Angew. Chem. Int. Ed 2001, 40, 3377–3380. [Google Scholar]
- Mancia, F.; Evans, P.R. Conformational changes on substrate binding to methylmalonyl CoA mutase and new insights into the free radical mechanism. Structure 1998, 6, 711–720. [Google Scholar]
- Ouyang, L.Z.; Rulis, P.; Ching, W.Y.; Nardin, G.; Randaccio, L. Accurate redetermination of the X-ray structure and electronic bonding in adenosylcobalamin. Inorg. Chem 2004, 43, 1235–1241. [Google Scholar]
- Champloy, F.; Gruber, K.; Jogl, G.; Kratky, C. XAS spectroscopy reveals X-ray-induced photoreduction of free and protein-bound B12 cofactors. J. Synchrotron Radiat 2000, 7, 267–273. [Google Scholar]
- Boehr, D.D.; Dyson, H.J.; Wright, P.E. An NMR perspective on enzyme dynamics. Chem. Rev 2006, 106, 3055–3079. [Google Scholar]
- Henzler-Wildman, K.A.; Lei, M.; Thai, V.; Kerns, S.J.; Karplus, M.; Kern, D. A hierarchy of timescales in protein dynamics is linked to enzyme catalysis. Nature 2007, 450, 913–916. [Google Scholar]
- Kern, D.; Zuiderweg, E.R. The role of dynamics in allosteric regulation. Curr. Opin. Struct. Biol 2003, 13, 748–757. [Google Scholar]
- Elber, R. Simulations of allosteric transitions. Curr. Opin. Struct. Biol 2011, 21, 167–172. [Google Scholar]
- Agarwal, P.K.; Billeter, S.R.; Rajagopalan, P.T.; Benkovic, S.J.; Hammes-Schiffer, S. Network of coupled promoting motions in enzyme catalysis. Proc. Natl. Acad. Sci. USA 2002, 99, 2794–2799. [Google Scholar]
- Evans, J.C.; Huddler, D.P.; Hilgers, M.T.; Romanchuk, G.; Matthews, R.G.; Ludwig, M.L. Structures of the N-terminal modules imply large domain motions during catalysis by methionine synthase. Proc. Natl. Acad. Sci. USA 2004, 101, 3729–3736. [Google Scholar]
- Bandarian, V.; Ludwig, M.L.; Matthews, R.G. Factors modulating conformational equilibria in large modular proteins: A case study with cobalamin-dependent methionine synthase. Proc. Natl. Acad. Sci. USA 2003, 100, 8156–8163. [Google Scholar]
- Pang, J.Y.; Li, X.; Morokuma, K.; Scrutton, N.S.; Sutcliffe, M.J. Large-scale domain conformational change is coupled to the activation of the Co–C bond in the B12-dependent enzyme ornithine 4,5-aminomutase: A computational study. J. Am. Chem. Soc 2012, 134, 2367–2377. [Google Scholar]
- Ballinger, M.D.; Frey, P.A.; Reed, G.H. Structure of a substrate radical intermediate in the reaction of lysine 2,3-aminomutase. Biochemistry 1992, 31, 10782–10789. [Google Scholar]
- Ballinger, M.D.; Frey, P.A.; Reed, G.H.; LoBrutto, R. Pulsed electron paramagnetic resonance studies of the lysine 2,3-aminomutase substrate radical: Evidence for participation of pyridoxal 5′-phosphate in a radical rearrangement. Biochemistry 1995, 34, 10086–10093. [Google Scholar]
- Wu, W.; Lieder, K.W.; Reed, G.H.; Frey, P.A. Observation of a second substrate radical intermediate in the reaction of lysine 2,3-aminomutase: A radical centered on the β-carbon of the alternative substrate, 4-thia-l-lysine. Biochemistry 1995, 34, 10532–10537. [Google Scholar]
- Han, O.; Frey, P.A. Chemical model for the pyridoxal 5′-phosphate dependent lysine aminomutases. J. Am. Chem. Soc 1990, 112, 8982–8983. [Google Scholar]
- Chen, Y.H.; Maity, A.N.; Frey, P.A.; Ke, S.C. Mechanism-based inhibition reveals transitions between two conformational states in the action of lysine 5,6-aminomutase: A combination of electron paramagnetic resonance spectroscopy, electron nuclear double resonance spectroscopy, and density functional theory study. J. Am. Chem. Soc 2013, 135, 788–794. [Google Scholar]
- Wolthers, K.R.; Rigby, S.E.J.; Scrutton, N.S. Mechanism of radical-based catalysis in the reaction catalyzed by adenosylcobalamin-dependent ornithine 4,5-aminomutase. J. Biol. Chem 2008, 283, 34615–34625. [Google Scholar]
- Thoma, N.H.; Evans, P.R.; Leadlay, P.F. Protection of radical intermediates at the active site of adenosylcobalamin-dependent methylmalonyl-CoA mutase. Biochemistry 2000, 39, 9213–9221. [Google Scholar]
- Toraya, T.; Mori, K. A reactivating factor for coenzyme B12-dependent diol dehydratase. J. Biol. Chem 1999, 274, 3372–3377. [Google Scholar]
- Makins, C.; Miros, F.N.; Scrutton, N.S.; Wolthers, K.R. Role of histidine 225 in adenosylcobalamin-dependent ornithine 4,5-aminomutase. Bioorg. Chem 2012, 40, 39–47. [Google Scholar]
- Friedrich, P.; Baisch, U.; Harrington, R.W.; Lyatuu, F.; Zhou, K.; Zelder, F.; McFarlane, W.; Buckel, W.; Golding, B.T. Experimental study of hydrogen bonding potentially stabilizing the 5′-deoxyadenosyl radical from coenzyme B12. Chem. Eur. J 2012, 18, 16114–16122. [Google Scholar]
- Bencini, A.; Gatteschi, D. EPR of Exchange Coupled Systems; Springer-Verlag: New York, NY, USA, 1990. [Google Scholar]
- Edmondson, D.E. ESR of fine radicals in enzymatic systems. In Biological Magnetic Resonance; Berliner, L.J., Reuben, J., Eds.; Plenum: New York, NY, USA, 1978; Volume 1, pp. 205–237. [Google Scholar]
- Hoffman, B.M. Electron nuclear double resonance (ENDOR) of metalloenzymes. Acc. Chem. Res 1991, 24, 164–170. [Google Scholar]
- Hoffman, B.M. ENDOR of metalloenzymes. Acc. Chem. Res 2003, 36, 522–529. [Google Scholar]
- Lees, N.S.; Chen, D.; Walsby, C.J.; Behshad, E.; Frey, P.A.; Hoffman, B.M. How an enzyme tames reactive intermediates: Positioning of the active-site components of lysine 2,3-aminomutase during enzymatic turnover as determined by ENDOR spectroscopy. J. Am. Chem. Soc 2006, 128, 10145–10154. [Google Scholar]
- Gerfen, G.J. EPR spectroscopy of B12-dependent enzymes. In Chemistry and Biochemistry of B12; Banerjee, R., Ed.; Wiley-Interscience: New York, NY, USA, 1999; pp. 165–195. [Google Scholar]
- Pilbrow, J.R. EPR spectroscopy of B12-dependent enzyme reaction and related systems. In B12; Dolphin, D., Ed.; John Wiley & Sons: New York, NY, USA, 1982; Volume 1, pp. 431–462. [Google Scholar]
- Reed, G.H.; Mansoorabadi, S.O. The positions of radical intermediates in the active sites of adenosylcobalamin-dependent enzymes. Curr. Opin. Struct. Biol 2003, 13, 716–721. [Google Scholar]
- Yoon, M.; Patwardhan, A.; Qiao, C.; Mansoorabadi, S.O.; Menefee, A.L.; Reed, G.H.; Marsh, E.N. Reaction of adenosylcobalamin-dependent glutamate mutase with 2-thiolglutarate. Biochemistry 2006, 45, 11650–11657. [Google Scholar]
- Mansoorabadi, S.O.; Padmakumar, R.; Fazliddinova, N.; Vlasie, M.; Banerjee, R.; Reed, G.H. Characterization of a succinyl-CoA radical-cob (II) alamin spin triplet intermediate in the reaction catalyzed by adenosylcobalamin-dependent methylmalonyl-CoA mutase. Biochemistry 2005, 44, 3153–3158. [Google Scholar]
- Tang, K.H.; Mansoorabadi, S.O.; Reed, G.H.; Frey, P.A. Radical triplets and suicide inhibition in reactions of 4-thia-d- and 4-thia-l-lysine with lysine 5,6-aminomutase. Biochemistry 2009, 48, 8151–8160. [Google Scholar]
- Maity, A.N.; Hsieh, C.P.; Huang, M.H.; Chen, Y.H.; Tang, K.H.; Behshad, E.; Frey, P.A.; Ke, S.C. Evidence for conformational movement and radical mechanism in the reaction of 4-thia-l-lysine with lysine 5,6-aminomutase. J. Phys. Chem 2009, 113, 12161–12163. [Google Scholar]
- Bandarian, V.; Reed, G.H. Analysis of the electron paramagnetic resonance spectrum of a radical intermediate in the coenzyme B12-dependent ethanolamine ammonia-lyase catalyzed reaction of (S)-2-aminopropanol. Biochemistry 2002, 41, 8580–8588. [Google Scholar]
- Ke, S.C. Spin–spin interaction in ethanolamine deaminase. Biochim. Biophys. Acta 2003, 1620, 267–272. [Google Scholar]
- Sun, L.; Groover, O.A.; Canfield, J.M.; Warncke, K. Critical role of arginine 160 of the EutB protein subunit for active site structure and radical catalysis in coenzyme B12-dependent ethanolamine ammonia-lyase. Biochemistry 2008, 47, 5523–5535. [Google Scholar]
- Abend, A.; Bandarian, V.; Reed, G.H.; Frey, P.A. Identification of cis-ethanesemidione as the organic radical derived from glycolaldehyde in the suicide inactivation of dioldehydrase and of ethanolamine ammonia-lyase. Biochemistry 2000, 39, 6250–6257. [Google Scholar]
- Maity, A.N.; Ke, S.-C. Synthesis of 4-thia[5-13C]lysine. J. Label. Compd. Radiopharm 2011, 54, 589–590. [Google Scholar]
- Maity, A.N.; Shaikh, A.C.; Srimurugan, S.; Wu, C.J.; Chen, C.P.; Ke, S.-C. Synthesis of 4-thia-[6-13C]lysine from [2-13C]glycine: Access to site-directed isotopomers of 2-aminoethanol, 2-bromoethylamine and 4-thialysine. Amino Acids 2012, 42, 309–315. [Google Scholar]
- Chen, Y.H.; Maity, A.N.; Pan, Y.C.; Frey, P.A.; Ke, S.-C. Radical stabilization is crucial in the mechanism of action of lysine 5,6-aminomutase: role of tyrosine-263α as revealed by electron paramagnetic resonance spectroscopy. J. Am. Chem. Soc 2011, 133, 17152–17155. [Google Scholar]
- Wetmore, S.D.; Smith, D.M.; Radom, L. Enzyme catalysis of 1,2-amino shifts: The cooperative action of B6, B12, and aminomutases. J. Am. Chem. Soc 2001, 123, 8678–8689. [Google Scholar]
- Wetmore, S.D.; Smith, D.M.; Radom, L. How B6 helps B12: The roles of B6, B12, and the enzymes in aminomutase-catalyzed reactions. J. Am. Chem. Soc 2000, 122, 10208–10209. [Google Scholar]
- Sandala, G.M.; Smith, D.M.; Radom, L. In search of radical intermediates in the reactions catalyzed by lysine 2,3-aminomutase and lysine 5,6-aminomutase. J. Am. Chem. Soc 2006, 128, 16004–16005. [Google Scholar]
- Wu, W.; Booker, S.; Lieder, K.W.; Bandarian, V.; Reed, G.H.; Frey, P.A. Lysine 2,3-aminomutase and trans-4,5-dehydrolysine: Characterization of an allylic analogue of a substrate-based radical in the catalytic mechanism. Biochemistry 2000, 39, 9561–9570. [Google Scholar]
- Danen, W.C.; West, C.T. Nitrogen-centered free radicals. VII. Electron spin resonance investigation of the 1-aziridylcarbinyl and related free radicals. J. Am. Chem. Soc 1974, 96, 2447–2453. [Google Scholar]
- Maity, A.N.; Ke, S.-C. 5-Fluorolysine as alternative substrate of lysine 5,6-aminomutase: A computational study. Comput. Theor. Chem 2013, 1022, 1–5. [Google Scholar]
- Pieper, P.A.; Yang, D.Y.; Zhou, H.Q.; Liu, H.W. 3-Deoxy-3-fluoropyridoxamine 5′-phosphate: Synthesis and chemical and biological properties of a coenzyme B6 analog. J. Am. Chem. Soc 1997, 119, 1809–1817. [Google Scholar]
- Griswold, W.R.; Fisher, A.J.; Toney, M.D. Crystal structures of aspartate aminotransferase reconstituted with 1-deazapyridoxal 5′-phosphate: Internal aldimine and stable l-aspartate external aldimine. Biochemistry 2011, 50, 5918–5924. [Google Scholar]
- Griswold, W.R.; Toney, M.D. Role of the pyridine nitrogen in pyridoxal 5′-phosphate catalysis: Activity of three classes of PLP enzymes reconstituted with deazapyridoxal 5′-phosphate. J. Am. Chem. Soc 2011, 133, 14823–14830. [Google Scholar]
- Griswold, W.R.; Castro, J.N.; Fisher, A.J.; Toney, M.D. Ground-state electronic destabilization via hyperconjugation in aspartate aminotransferase. J. Am. Chem. Soc 2012, 134, 8436–8438. [Google Scholar]
- Griswold, W.R.; Toney, M.D. Chemoenzymatic synthesis of 1-deaza-pyridoxal 5′-phosphate. Bioorg. Med. Chem. Lett 2010, 20, 1352–1354. [Google Scholar]
- Beattie, A.E.; Clarke, D.J.; Wadsworth, J.M.; Lowther, J.; Sin, H.L.; Campopiano, D.J. Reconstitution of the pyridoxal 5′-phosphate (PLP) dependent enzyme serine palmitoyltransferase (SPT) with pyridoxal reveals a crucial role for the phosphate during catalysis. Chem. Commun 2013, 49, 7058–7060. [Google Scholar]
- Wu, B.; Szymanski, W.; Heberling, M.M.; Feringa, B.L.; Janssen, D.B. Aminomutases: Mechanistic diversity, biotechnological applications and future perspectives. Trends Biotech 2011, 29, 352–362. [Google Scholar]
- Guddneppanavar, R.; Choudhury, J.R.; Kheradi, A.R.; Steen, B.D.; Saluta, G.; Kucera, G.L.; Day, C.S.; Bierbach, U. Effect of the diamine nonleaving group in platinum-acridinylthiourea conjugates on DNA damage and cytotoxicity. J. Med. Chem 2007, 50, 2259–2263. [Google Scholar]
- Huang, Y.; Keen, J.C.; Pledgie, A.; Marton, L.J.; Zhu, T.; Sukumar, S.; Park, B.H.; Blair, B.; Brenner, K.; Casero, R.A.; et al. Polyamine analogues down-regulate estrogen receptor α expression in human breast cancer cells. J. Biol. Chem 2006, 281, 19055–19063. [Google Scholar]
- Kunz, F.; Rétey, J.; Arigoni, D.; Tsai, L.; Stadtman, T.C. Die absolute konfiguration der 3,5-diaminohexansäure aus der β-lysin-mutase-reaktion. Helv. Chim. Acta 1978, 61, 1139–1145. [Google Scholar]
- Behshad, E.; Ruzicka, F.J.; Mansoorabadi, S.O.; Chen, D.; Reed, G.H.; Frey, P.A. Enantiomeric free radicals and enzymatic control of stereochemistry in a radical mechanism: The case of lysine 2,3-aminomutases. Biochemistry 2006, 45, 12639–12646. [Google Scholar]
- Hallinan, E.A.; Hagen, T.J.; Bergmanis, A.; Moore, W.M.; Jerome, G.M.; Spangler, D.P.; Stevens, A.M.; Shieh, H.S.; Manning, P.T.; Pitzele, B.S. 5-Fluorinated l-lysine analogues as selective induced nitric oxide synthase inhibitors. J. Med. Chem 2004, 47, 900–906. [Google Scholar]
- Ruzicka, F.J.; Frey, P.A. Kinetic and spectroscopic evidence of negative cooperativity in the action of lysine 2,3-aminomutase. J. Phys. Chem 2010, 114, 16118–16124. [Google Scholar]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
© 2014 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Maity, A.N.; Chen, Y.-H.; Ke, S.-C. Large-Scale Domain Motions and Pyridoxal-5'-Phosphate Assisted Radical Catalysis in Coenzyme B12-Dependent Aminomutases. Int. J. Mol. Sci. 2014, 15, 3064-3087. https://doi.org/10.3390/ijms15023064
Maity AN, Chen Y-H, Ke S-C. Large-Scale Domain Motions and Pyridoxal-5'-Phosphate Assisted Radical Catalysis in Coenzyme B12-Dependent Aminomutases. International Journal of Molecular Sciences. 2014; 15(2):3064-3087. https://doi.org/10.3390/ijms15023064
Chicago/Turabian StyleMaity, Amarendra Nath, Yung-Han Chen, and Shyue-Chu Ke. 2014. "Large-Scale Domain Motions and Pyridoxal-5'-Phosphate Assisted Radical Catalysis in Coenzyme B12-Dependent Aminomutases" International Journal of Molecular Sciences 15, no. 2: 3064-3087. https://doi.org/10.3390/ijms15023064
APA StyleMaity, A. N., Chen, Y.-H., & Ke, S.-C. (2014). Large-Scale Domain Motions and Pyridoxal-5'-Phosphate Assisted Radical Catalysis in Coenzyme B12-Dependent Aminomutases. International Journal of Molecular Sciences, 15(2), 3064-3087. https://doi.org/10.3390/ijms15023064