Metabolomics of Genetically Modified Crops





Abstract
:1. Introduction

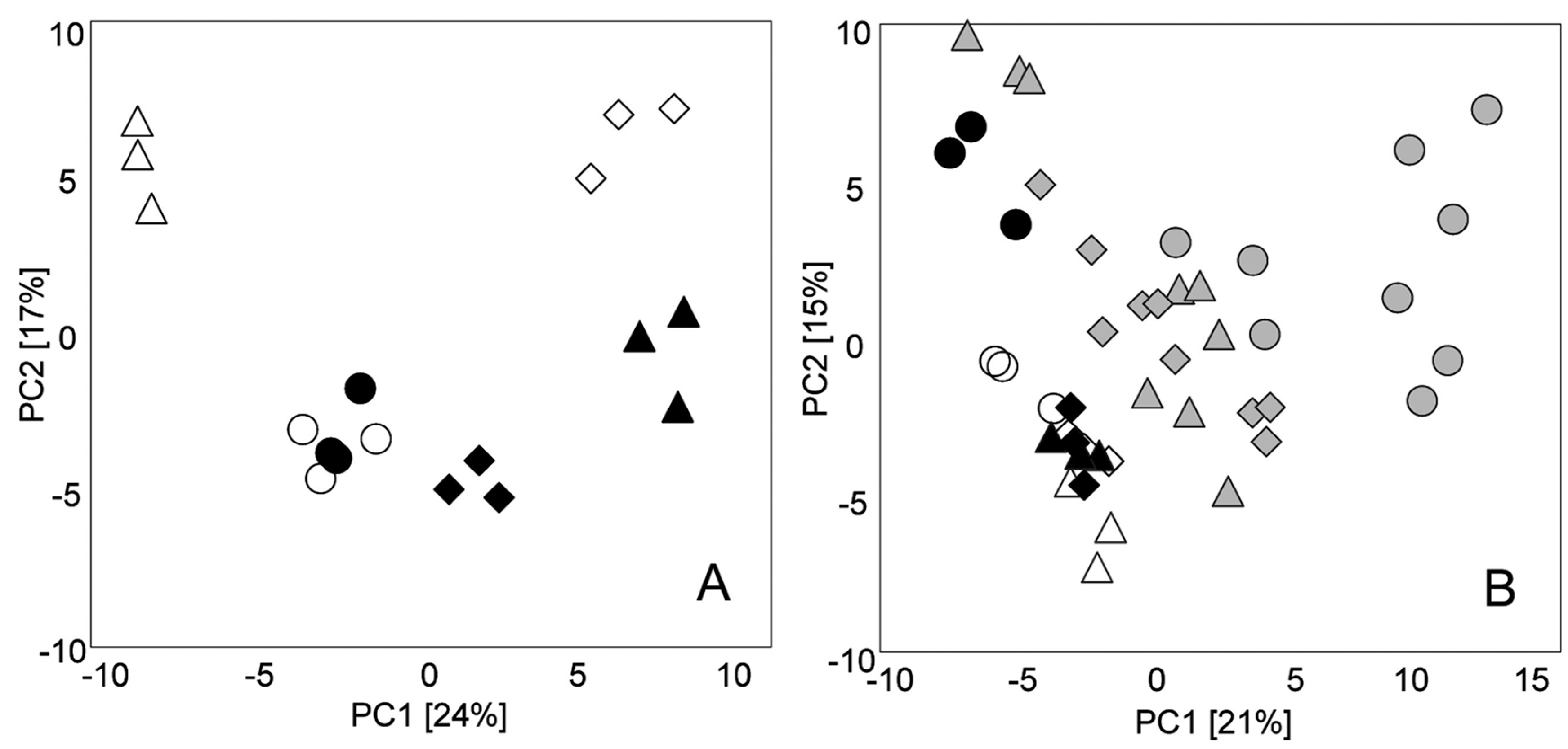{kind=link}
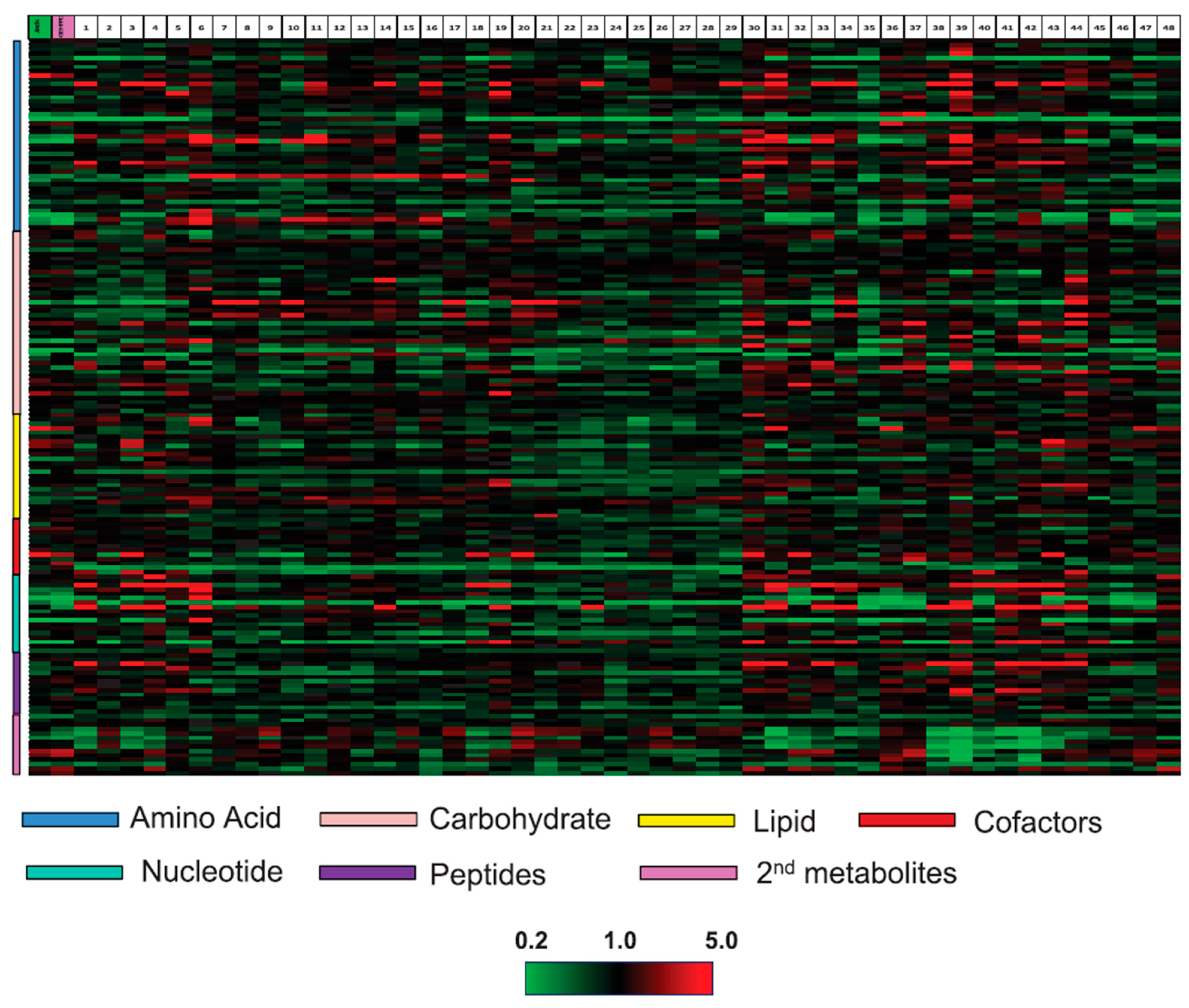{kind=link}
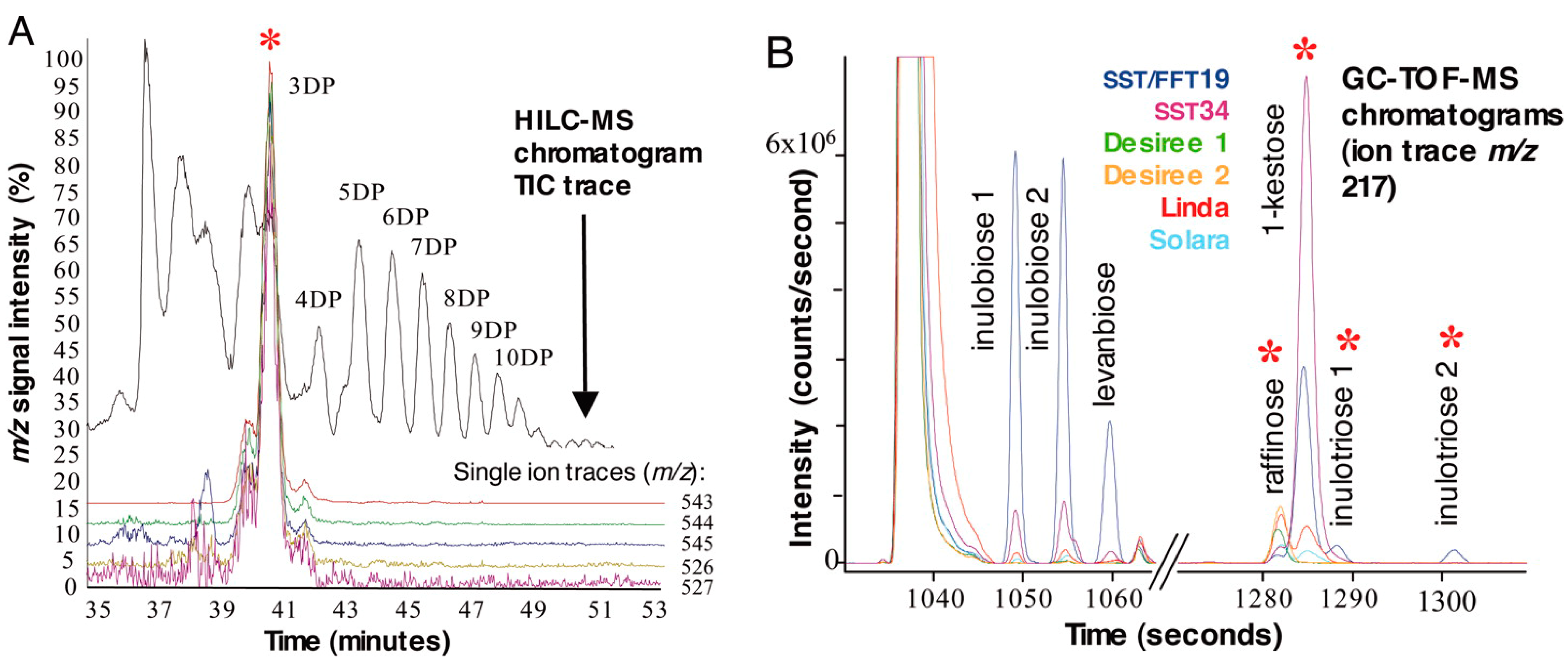{kind=link}
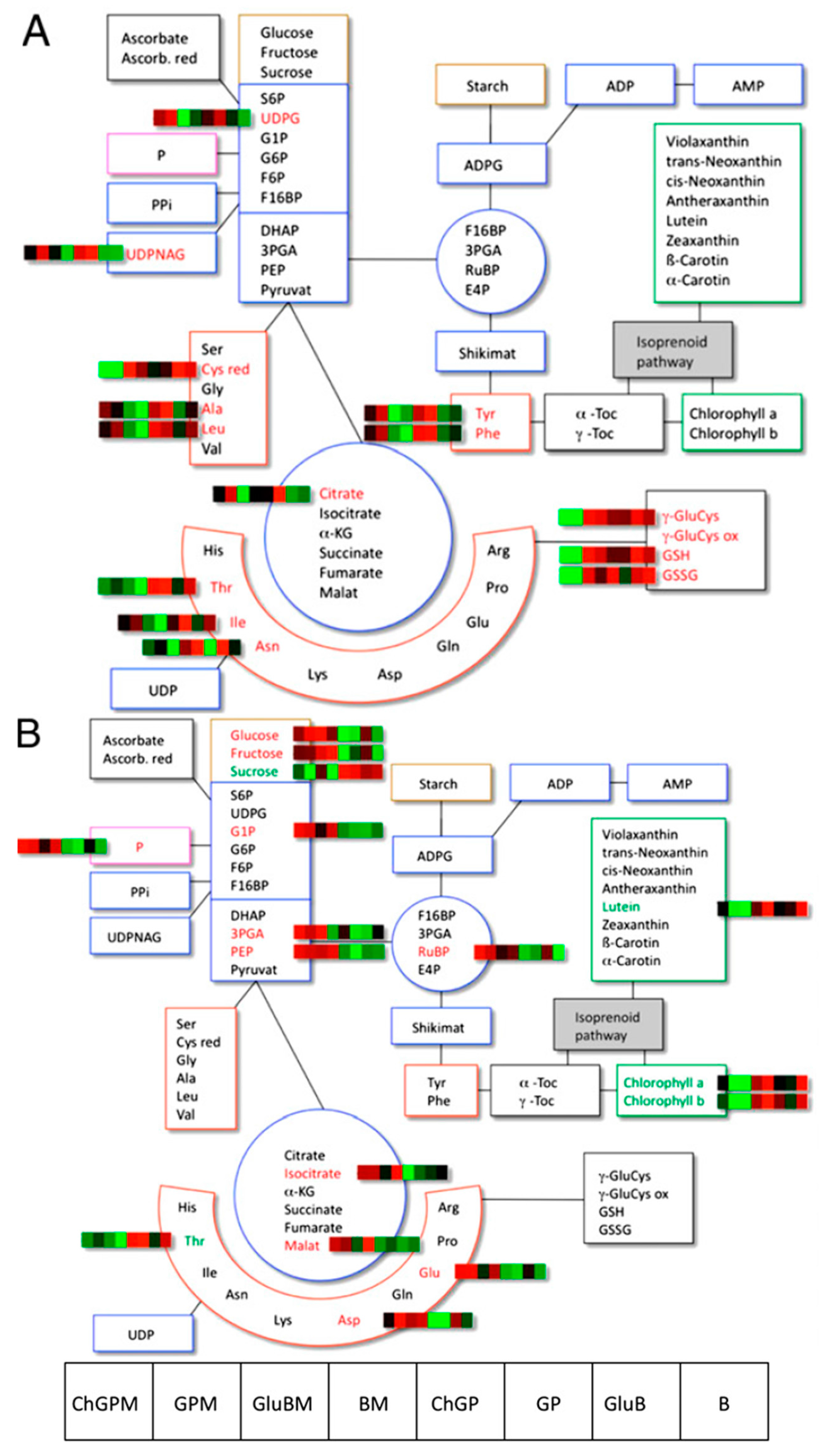{kind=link}
| GM Crop | Tissue | Donor Specie | Genetic Modification | Phenotype | Analytical Technique | References |
|---|---|---|---|---|---|---|
| Rice | Seed | B. thuringiensis | Cry1Ab | Insect resistance | FTIR MS, NMR | [25] |
| Seed | B. thuringiensis | Cry1Ac, sck | Insect resistance | GC-FID, GC-EI-Q MS | [26] | |
| Leaf | Z. mays | C1, R-S | Flavonoid production | LC-ESI-Q MS, LC-DAD | [27] | |
| Leaf, seed, root | O. sativa | YK1 | Stress tolerance | CE-ESI-Q MS | [28] | |
| Seed | O. sativa | RCH10, RAC22, β-Glu, B-RIP | Antifungal activity | NIRS, GC-EI-Q MS, LC-DAD, ICP-AES | [29] | |
| Seed | O. sativa | Mod. (Xa23, Xa21 genes) | Insect resistance | GC-EI-Q MS | [30] | |
| Seed | B. thuringiensis | Cry1Ac, sck | Insect Resistance | LC-ESI-Q/TOF MS | [31] | |
| Seed | E. coli | GlgC-TM | Nutritionally enhanced | LC-ESI-Q MS | [32] | |
| Seed | N. tabacum | ASA2 | Nutritionally enhanced | LC-ESI-Q MS | [33] | |
| Seed | A. tumefaciens | Psy-2A-CrtI Bar | Nutritionally enhanced Herbicide tolerance | GC-EI-TOF MS | [34] | |
| Leaf, seed | E. coli/O. sativa | LysC, dapA/LKR/SDH | Nutritionally enhanced | LC-FTIR MS, GC-EI-Q MS | [35] | |
| Maize | Grain | B. thuringiensis | Cry1Ab | Insect resistance | NMR | [36,37] |
| Grain | Z. mays | Mod. (Rpd3 gene) | Seed development | NMR | [38] | |
| Grain | B. thuringiensis | Cry1Ab | Insect resistance | NMR | [39] | |
| Grain | B. thuringiensis | Cry1Ab | Insect resistance | CE-ESI-TOF MS | [40] | |
| Grain | B. thuringiensis | Cry1Ab | Insect resistance | FT-ICR MS | [41] | |
| Grain | B. thuringiensis | Bt toxin | Insect resistance | GC-EI-Q MS | [42] | |
| Grain | B. thuringiensis | Cry1Ab | Insect resistance | GC-EI-Q MS | [43] | |
| Grain | B. thuringiensis A. tumefaciens | Cry1Ab CP4 EPSPS | Insect resistance Herbicide tolerance | GC-EI-Q MS | [44] | |
| Grain | Z. mays | Mod. (Zmpsy1, Pacrtl, Gllycb, Glbch, ParacrtW genes) | Nutritionally enhanced | LC-DA, LC-ESI-APCI MS | [45] | |
| Grain | B. thuringiensis | Bt toxin | Herbicide tolerance Insect resistance | NMR, GC-EI-Q-MS | [46] | |
| Soybean | Seed | A. tumefaciens | CP4 EPSPS | Herbicide tolerance | GC-EI-Q MS | [42] |
| Seed | Agrobacterium spp. | 837ASDIS | Herbicide tolerance | GC-EI-Q MS | [43] | |
| Seed | A. tumefaciens | CP4 EPSPS | Herbicide tolerance | CE-ESI-TOF MS | [47] | |
| Leaf, EC, seed | N. tabacum | ASA2 | Nutritionally enhanced | GC-EI-Q MS | [48] | |
| Seed | A. tumefaciens | CP4 EPSPS | Herbicide tolerance | CE-ESI-TOF MS | [49] | |
| Seed | Avena spp | Mod. (HPPD gene) | Herbicide tolerance | LC-ESI-Q MS, GC-EI-Q MS | [50] | |
| Seed | A. tumefaciens | CP4 EPSPS | Herbicide tolerance | CE-ESI-TOF MS, GC-EI-TOF MS, LC-ESI-Q/TOF MS, ICP MS | [51] | |
| Alfalfa | Stem, leaf | N. tabacum | PAL2 | Nutritionally enhanced | LC-UV | [52] |
| Pea | Leaf | S. hygroscopicus | Bar | Herbicide tolerance | NMR | [53] |
| Wheat | Leaf | U. maydis | Chit/Gluc, RIP, Mod. (KP4 gene) | Fungal resistance | LC-DAD, LC-ESI-Q MS | [5] |
| Seed | T. aestivum | Glu-A1, Glu-D1 | Nutritionally enhanced | NMR | [54] | |
| Potato | Tuber | A. pullulans, S. tuberosum | W2, FK, Mal1, SamDC | Starch biosynthesis, leaf morphology, ethylene production | GC-EI-Q MS | [6] |
| Tuber | S. tuberosum | AGPase, StcPGM, StpPGM | Altered starch composition | GC-EI-Q MS | [55,56.57] | |
| Tuber | C. scolymus | 1-SST, 1-FFT | Inulin synthesis | GC-EI-TOF MS, LC-ESI-Q MS | [58] | |
| Tuber | A. tumefaciens | Potato virus Y | Virus resistance | CE-ESI-IT-MS/MS | [59] | |
| Tuber | A. thaliana | DREB1A | Stress tolerance | GC-EI-TOF MS, LC-ESI-Q MS | [60] | |
| Tuber | A. pullulans | W2 | Waxy phenotype | LC-UV, NMR | [61] | |
| Leaf | S. cerevisiae | TPS1 | Drought resistance | GC-EI-Q MS | [62] | |
| Tomato | Fruit | A. tumafaciens | LBA4404 | Improved texture, mouthfeel, colour | NMR | [63] |
| Leaf, fruit | A. thaliana | AtHXK1 | Altered carbohydrate metabolism | GC-EI-Q MS | [64] | |
| Fruit | Z. mays | LC1, C1 | Increased flavonol content | NMR | [65,66] | |
| Fruit | E. coli | DXS | Increased carotenoid content | LC-DAD | [67] | |
| Fruit | V. vinifera | Stilbene synthase | Resveratrol synthesis | LC-ESI-Q MS | [68] | |
| Fruit | R. dulcifica | Miraculin | Sweet flavor | GC-EI-TOF MS, LC-ESI-Q/TOF MS, CE-ESI/TOF MS | [69] | |
| Tobacco | Leaf | E. coli/P. fluorencens | Ent/CpmsB | Salicylic acid producing plants | NMR | [70] |
| Lettuce | Leaf | E. coli | Asn A | Growth enhanced | NMR, GC-FID | [71,72,73] |
| Cucumber | Fruit | T. daniellii | Thaumatin-II | Sweet flavor | GC-EI-TOF MS | [74] |
| Fruit | T. daniellii | Preprothaumatin-II | Aroma, sweet flavor | GC-EI-Q/TOF MS | [75] | |
| Raspberry | Fruit | RBDV | Virus movement protein | Virus resistance | GC-EI-Q MS | [76] |
| Grapevine | Leaf | E. coli | Adh | Abiotic stress | GC-EI-Q MS, LC-ESI-IT MS, LC-DAD | [77] |
| Peppermint | Leaf | Mentha x piperita | Mod. (MFS gene), DXR | Essential oils content | GC-FID | [78] |
| Cabbage | Leaf | A. tumafaciens | Bar | Herbicide tolerance | LC-DAD, LC-ESI/Q MS | [79] |
| Papaya | Fruit | C. papaya | Mod. (55-1 gene) | Virus resistance | LC-DAD, GC-FID | [80] |
| Pulp, Leaf | C. papaya | Mod. (rep gene) | Virus resistance | GC-EI-Q MS, LC-DAD, LC-ESI-Q MS | [81] | |
| Poplar | Cambial region | P. trichocarpa | Mod. (hipI-SOD gene) | Superoxide production | GC-EI-TOF MS, LC-ESI/TOF MS | [82] |
| Barley | Seed | B. amyloliquefaciens | GluB, ChGP | Antifungal activity | LC-ESI-IT MS | [83] |
2. Metabolomics and GM (Genetically Modified) Crops: Case Studies
2.1. Rice
2.2. Maize

2.3. Soybean and Other Legumes
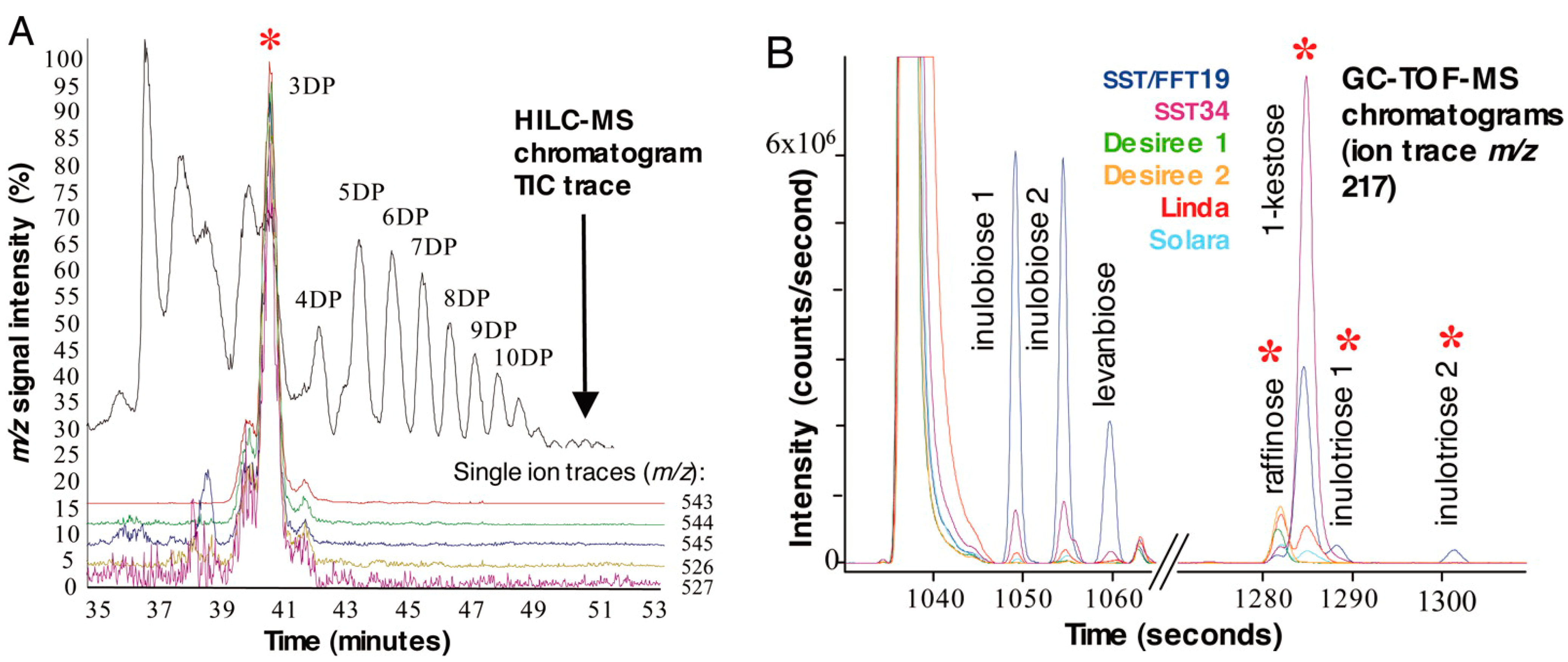
2.4. Wheat
2.5. Potato
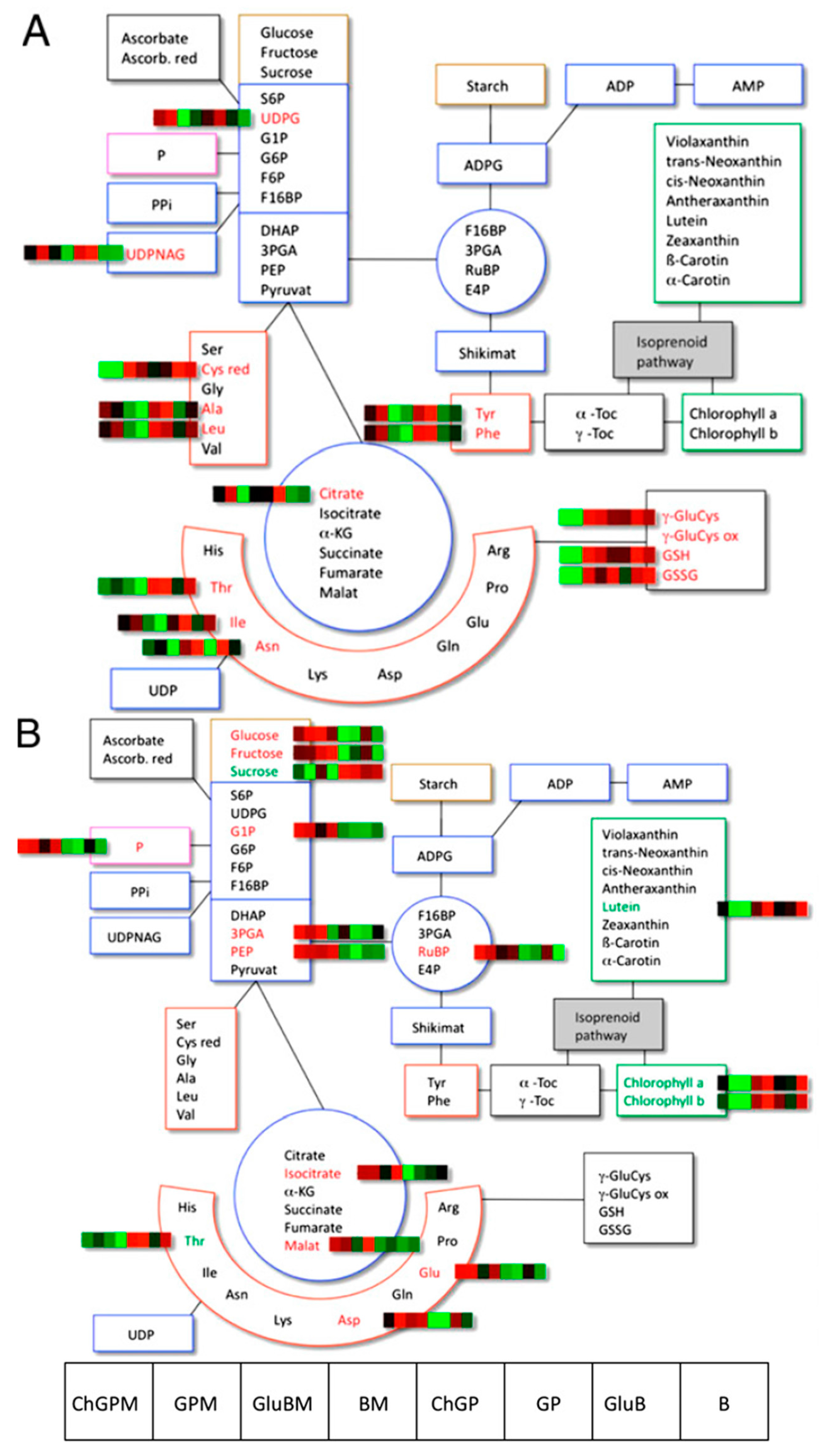
2.6. Tomato
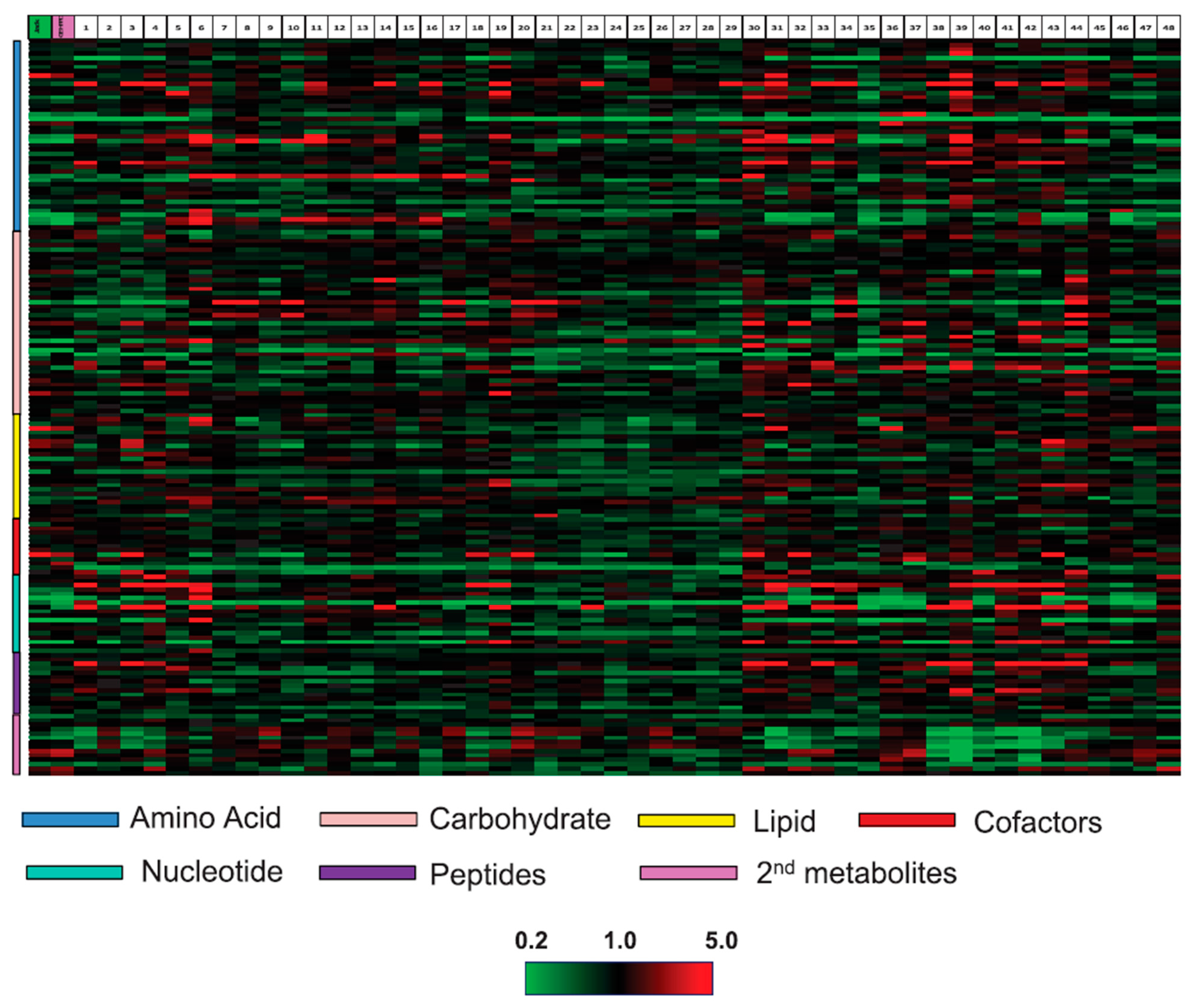
2.7. Other GM Crops
3. Concluding Remarks
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Lönnerdal, B. Genetically modified plants for improved trace element nutrition. J. Nutr. 2003, 133, 1490S–1493S. [Google Scholar]
- McGloughlin, M.N. Modifying agricultural crops for improved nutrition. New Biotechnol. 2010, 30, 494–504. [Google Scholar]
- Dunwell, J.M. Transgenic cereals: Current status and future prospects. J. Cereal Sci. 2014, 59, 419–434. [Google Scholar]
- Cellini, F.; Chesson, A.; Colquhoun, I.; Constable, A.; Davies, H.V.; Engel, K.H.; Gatehouse, A.M.; Kärenlampi, S.; Kok, E.J.; Leguay, J.J.; et al. Unintended effects and their detection in genetically modified crops. Food Chem. Toxicol. 2004, 42, 1089–1125. [Google Scholar]
- Ioset, J.R.; Urbaniak, B.; Ndjoko-Ioset, K.; Wirth, J.; Martin, F.; Gruissem, W.; Hostettmann, K.; Sautter, C. Flavonoid profiling among wild type and related GM wheat varieties. Plant Mol. Biol. 2007, 65, 645–654. [Google Scholar]
- Shepherd, L.V.T.; McNicol, J.W.; Razzo, R.; Taylor, M.A.; Davies, H.V. Assessing the potential for unintended effects in genetically modified potatoes perturbed in metabolic and developmental processes. Targeted analysis of key nutrients and anti-nutrients. Transgenic Res. 2006, 15, 409–425. [Google Scholar]
- Millstone, E.; Brunner, E.; Mayer, S. Beyond “substantial equivalence”. Nature 1999, 401, 525–526. [Google Scholar]
- EFSA. Guidance document of the Scientific Panel on Genetically Modified Organisms for the risk assessment of genetically modified plants and derived food and feed. EFSA J. 2006, 99, 1–100.
- Ad Hoc Technical Expert Group (AHTEG). Final report of the ad hoc technical expert group on risk assessment and risk management under the Cartagena protocol on biosafety. In Proceedings of the United Nations Environment Programme Convention for Biodiversity, Ljubljana, Slovenia, 20–23 April 2010; Available online: http://www.cbd.int/doc/meetings/bs/bsrarm-02/official/bsrarm-02-05-en.pdf (accessed on 15 October 2014).
- Chassy, B.M. Can –omics inform a food safety assessment? Regul. Toxicol. Pharmacol. 2010, 58, S62–S70. [Google Scholar]
- Valdés, A.; Simó, C.; Ibáñez, C.; García-Cañas, V. Foodomics strategies for the analysis of transgenic foods. TrAC Trends Anal. Chem. 2013, 52, 2–15. [Google Scholar]
- Schauer, N.; Fernie, A.R. Plant metabolomics: Towards biological function and mechanism. Trends Plant Sci. 2006, 11, 508–516. [Google Scholar]
- Hall, R.D. Plant metabolomics: From holistic hope, to hype, to hot topic. New Phytol. 2006, 169, 453–468. [Google Scholar]
- Harrigan, G.G.; Martino-Catt, S.; Glenn, K.C. Metabolomics, metabolic diversity and genetic variation in crops. Metabolomics 2007, 3, 259–272. [Google Scholar]
- Dixon, R.A.; Gang, D.R.; Charlton, A.J.; Fiehn, O.; Kuiper, H.A.; Reynolds, T.L.; Tjeerdema, R.S.; Jeffery, E.H.; German, J.B.; Ridley, W.P.; et al. Applications of metabolomics in agriculture. J. Agric. Food Chem. 2006, 54, 8984–8994. [Google Scholar]
- Seger, C.; Sturm, S. Analytical aspects of plant metabolite profiling platforms: Current standings and future aims. J. Proteome Res. 2007, 6, 480–497. [Google Scholar]
- Hegeman, A.D. Plant metabolomics-meeting the analytical challenges of comprehensive metabolite analysis. Brief. Funct. Genomics 2010, 9, 139–148. [Google Scholar]
- Eisenreich, W.; Bacher, A. Advances of high-resolution NMR techniques in the structural and metabolic analysis of plant biochemistry. Phytochemistry 2007, 68, 2799–2815. [Google Scholar]
- Pan, Z.; Raftery, D. Comparing and combining NMR spectroscopy and mass spectrometry in metabolomics. Anal. Bioanal. Chem. 2007, 387, 525–527. [Google Scholar]
- García-Cañas, V.; Simó, C.; León, C.; Ibáñez, E.; Cifuentes, A. MS-based analytical methodologies to characterize genetically modified crops. Mass Spectrom. Rev. 2011, 30, 396–416. [Google Scholar]
- Herrero, M.; Simó, C.; García-Cañas, V.; Ibáñez, E.; Cifuentes, A. Foodomics: MS-based strategies in modern food science and nutrition. Mass Spectrom. Rev. 2012, 31, 49–69. [Google Scholar]
- Ibáñez, C.; García-Cañas, V.; Valdés, A.; Simó, C. Novel MS-based approaches and applications in food metabolomics. TrAC Trends Anal. Chem. 2013, 52, 100–111. [Google Scholar]
- Takats, z.; Wiseman, J.M.; Cooks, R.G. Ambient mass spectrometry using desorption electrospray ionization (DESI): Instrumentation, mechanisms and applications in forensics, chemistry, and biology. J. Mass Spectrom. 2005, 40, 1261–1275. [Google Scholar]
- Hoekenga, O.A. Using metabolomics to estimate unintended effects in transgenic crop plants: Problems, promises, and opportunities. J. Biomol. Tech. 2008, 19, 159–166. [Google Scholar]
- Keymanesh, K.; Darvishi, M.H.; Sardari, S. Metabolome comparison of transgenic and non-transgenic rice by statistical analysis of FTIR and NMR spectra. Rice Sci. 2009, 16, 119–123. [Google Scholar]
- Zhou, J.; Ma, C.; Xu, H.; Yuan, K.; Lu, X.; Zhu, Z.; Wu, Y.; Xu, G. Metabolic profiling of transgenic rice with cryIAc and sck genes: An evaluation of unintended effects at metabolic level by using GC-FID and GC-MS. J. Chromatogr. B 2009, 877, 725–732. [Google Scholar]
- Shin, Y.M.; Park, H.J.; Yim, S.D.; Baek, N.I.; Lee, C.H.; An, G.; Woo, Y.M. Transgenic rice lines expressing maize C1 and R-S regulatory genes produce various flavonoids in the endosperm. Plant Biol. 2006, 4, 303–315. [Google Scholar]
- Takahashi, H.; Hayashi, M.; Goto, F.; Sato, S.; Soga, T.; Nishioka, T.; Tomita, M.; Kawai-Yamada, M.; Uchimiya, H. Evaluation of metabolic alteration in transgenic rice overexpressing dihydroflavonol-4-reductase. Ann. Bot. 2006, 98, 819–825. [Google Scholar]
- Jiao, Z.; Si, X.X.; Li, G.K.; Zhang, Z.M.; Xu, X.P. Unintended compositional changes in transgenic rice seeds (Oryza sativa L.) studied by spectral and chromatographic analysis coupled with chemometrics methods. J. Agric. Food Chem. 2010, 58, 1746–1754. [Google Scholar]
- Wu, J.; Yu, H.C.; Dai, H.F.; Mei, W.L.; Huang, X.; Zhu, S.F.; Peng, M. Metabolite profiles of rice cultivars containing bacterial blight-resistant genes are distinctive from susceptible rice. Acta Biochim. Biophys. Sin. 2012, 44, 650–659. [Google Scholar]
- Chang, Y.; Zhao, C.; Zhu, Z.; Wu, Z.; Zhou, J.; Zhao, Y.; Lu, X.; Xu, G. Metabolic profiling based on LC/MS to evaluate unintended effects of transgenic rice with cry1Ac and sck genes. Plant Mol. Biol. 2012, 78, 477–487. [Google Scholar]
- Nagai, Y.S.; Sakulsinghroj, C.; Edwards, G.E.; Satoh, H.; Greene, T.W.; Blakeslee, B.; Okita, T.W. Control of starch synthesis in cereals: Metabolite analysis of transgenic rice expressing an up-regulated cytoplasmic ADP-Glucose pyrophosphorylase in developing seeds. Cell Physiol. 2009, 50, 635–643. [Google Scholar]
- Matsuda, F.; Ishihara, A.; Takanashi, K.; Morino, K.; Miyazawa, H.; Wakasa, K.; Miyagawa, H. Metabolic profiling analysis of genetically modified rice seedlings that overproduce tryptophan reveals the occurrence of its inter-tissue translocation. Plant Biotechnol. 2010, 27, 17–27. [Google Scholar]
- Kim, J.K.; Park, S.Y.; Lee, S.M.; Lim, S.-H.; Kim, H.J.; Oh, S.-D.; Yeo, Y.; Cho, H.S.; Ha, S.H. Unintended polar metabolite profiling of carotenoid-biofortified transgenic rice reveals substantial equivalence to its non-transgenic counterpart. Plant Biotechnol. Rep. 2013, 7, 121–128. [Google Scholar]
- Long, X.; Liu, Q.; Chan, M.; Wang, Q.; Sun, S.S. Metabolic engineering and profiling of rice with increased lysine. Plant Biotechnol. J. 2013, 11, 490–501. [Google Scholar]
- Manetti, C.; Bianchetti, C.; Bizzarri, M.; Casciani, L.; Castro, C.; D’Ascenzo, G.; Delfini, M.; di Cocco, M.E.; Lagana, A.; Miccheli, A.; et al. NMR-based metabonomic study of transgenic maize. Phytochemistry 2004, 65, 3187–3198. [Google Scholar]
- Manetti, C.; Bianchetti, C.; Casciani, L.; Castro, C.; di Cocco, M.E.; Miccheli, A.; Motto, M.; Conti, F. A metabonomic study of transgenic maize (Zea mays) seeds revealed variations in osmolytes and branched amino acids. J. Exp. Bot. 2006, 57, 2613–2625. [Google Scholar]
- Castro, C.; Manetti, C. A multiway approach to analyze metabonomic data: A study of maize seeds development. Anal. Biochem. 2007, 371, 194–200. [Google Scholar]
- Piccioni, F.; Capitani, D.; Zolla, L.; Mannina, L. NMR metabolic profiling of transgenic maize with the Cry1A(b) gene. J. Agric. Food Chem. 2009, 57, 6041–6049. [Google Scholar]
- Levandi, T.; Leon, C.; Kaljurand, M.; García-Cañas, V.; Cifuentes, A. Capillary electrophoresis time-of-flight mass spectrometry for comparative metabolomics of transgenic versus conventional maize. Anal. Chem. 2008, 80, 6329–6335. [Google Scholar]
- Leon, C.; Rodriguez-Meizoso, I.; Lucio, M.; Garcia-Cañas, V.; Ibañez, E.; Schmitt-Kopplin, P.; Cifuentes, A. Metabolomics of transgenic maize combining Fourier transform-ion cyclotron resonance-mass spectrometry, capillary electrophoresis-mass spectrometry and pressurized liquid extraction. J. Chromatogr. A 2009, 1216, 7314–7323. [Google Scholar]
- Bernal, J.L.; Nozal, M.J.; Toribio, L.; Diego, C.; Mayo, R.; Maestre, R. Use of supercritical fluid extraction and gas chromatography-mass spectrometry to obtain amino acid profiles from several genetically modified varieties of maize and soybean. J. Chromatogr. A 2008, 1192, 266–272. [Google Scholar]
- Jimenez, J.J.; Bernal, J.L.; Nozal, M.J.; Toribio, L.; Bernal, J. Profile and relative concentrations of fatty acids in corn and soybean seeds from transgenic and isogenic crops. J. Chromatogr. A 2009, 1216, 7288–7295. [Google Scholar]
- Frank, T.; Röhlig, R.M.; Davies, H.V.; Barros, E.; Engel, K. Metabolite profiling of maize kernels—genetic modification versus environmental influence. J. Agric. Food Chem. 2012, 60, 3005–3012. [Google Scholar]
- Rivera, S.M.; Vilaró, F.; Zhu, C.; Bai, C.; Farré, G.; Christou, P.; Canela-Garayoa, R. Fast quantitative method for the analysis of carotenoids in transgenic maize. J. Agric. Food Chem. 2013, 61, 5279–5285. [Google Scholar]
- Barros, E.; Lezar, S.; Anttonen, M.J.; van Dijk, J.P.; Röhlig, R.M.; Kok, E.J.; Engel, K.H. Comparison of two GM maize varieties with a nearisogenic non-GM variety using transcriptomics, proteomics and metabolomics. Plant Biotechnol. J. 2010, 8, 436–451. [Google Scholar]
- García-Villalba, R.; León, C.; Dinelli, G.; Segura-Carretero, A.; Fernández-Gutiérrez, A.; Garcia-Cañas, V.; Cifuentes, A. Comparative metabolomic study of transgenic versus conventional soybean using capillary electrophoresis-time-of-flight mass spectrometry. J. Chromatogr. A 2008, 1195, 164–173. [Google Scholar]
- Inaba, Y.; Brotherton, J.E.; Ulanov, A.; Widholm, J.M. Expression of a feedback insensitive anthranilate synthase gene from tobacco increases free tryptophan in soybean plants. Plant Cell Rep. 2007, 26, 1763–1771. [Google Scholar]
- Giuffrida, A.; León, C.; García-Cañas, V.; Cucinotta, V.; Cifuentes, A. Modified cyclodextrins for fast and sensitive chiral-capillary electrophoresis-mass spectrometry. Electrophoresis 2009, 30, 1734–1742. [Google Scholar]
- Clarke, J.D.; Alexander, D.C.; Ward, D.P.; Ryals, J.A.; Mitchell, M.W.; Wulff, J.E.; Guo, L. Asessment of genetically modified soybean in relation to natural variation in the soybean seed metabolome. Sci. Rep. 2013, 3. [Google Scholar] [CrossRef]
- Kusano, M.; Baxter, I.; Fukushima, A.; Oikawa, A.; Okazaki, Y.; Nakabayashi, R.; Bouvrette, D.J.; Achard, F.; Jakubowski, A.R.; Ballam, J.M.; et al. Assessing metabolomic and chemical diversity of a soybean lineage representing 35 years of breeding. Metabolomics 2014. [Google Scholar] [CrossRef]
- Chen, F.; Duran, A.L.; Blount, J.W.; Sumner, L.W.; Dixon, R.A. Profiling phenolic metabolites in transgenic alfalfa modified in lignin biosynthesis. Phytochemistry 2003, 64, 1013–1021. [Google Scholar]
- Charlton, A.; Allnutt, T.; Holmes, S.; Chisholm, J.; Bean, S.; Ellis, N.; Mullineaux, P.; Oehlschlager, S. NMR profiling of transgenic peas. Plant Biotechnol. J. 2004, 2, 27–35. [Google Scholar]
- Baker, J.M.; Hawkins, N.D.; Ward, J.L.; Lovegrove, A.; Napier, J.A.; Shewry, P.R.; Beale, M.H. A metabolomic study of substantial equivalence of field-grown genetically modified wheat. Plant Biotechnol. J. 2006, 4, 381–392. [Google Scholar]
- Roessner, U.; Wagner, C.; Kopka, J.; Trethewney, R.N.; Willmitzer, L. Simultaneous analysis of metabolites in potato tuber by gas chromatography-mass spectrometry. Plant J. 2000, 23, 131–142. [Google Scholar]
- Roessner, U.; Willmitzer, L.; Fernie, A.R. High-Resolution metabolic phenotyping of genetically and environmentally diverse potato tuber systems. Identification of phenocopies. Plant Physiol. 2001, 127, 749–764. [Google Scholar]
- Roessner, U.; Luedemann, A.; Brust, D.; Fiehn, O.; Linke, T.; Willmitzer, L.; Fernie, A.R. Metabolic profiling allows comprehensive phenotyping of genetically or environmentally modified plant systems. Plant Cell 2001, 13, 11–29. [Google Scholar]
- Catchpole, G.S.; Beckmann, M.; Enot, D.P.; Mondhe, M.; Zywicki, B.; Taylor, J.; Hardy, N.; Smith, A.; King, R.D.; Kell, D.B.; et al. Hierarchical metabolomics demonstrates substantial compositional similarity between genetically modified and conventional potato crops. Proc. Natl. Acad. Sci. USA 2005, 102, 14458–14462. [Google Scholar]
- Bianco, G.; Schmitt-Kopplin, P.; Crescenzi, A.; Comes, S.; Kettrup, A.; Cataldi, T.R. Evaluation of glycoalkaloids in tubers of genetically modified virus Y-resistant potato plants (var. Désirée) by non-aqueous capillary electrophoresis coupled with electrospray ionization mass spectrometry (NACE-ESI-MS). Anal. Bioanal. Chem. 2003, 375, 799–804. [Google Scholar]
- Iwaki, T.; Guo, L.; Ryals, J.A.; Yasuda, S.; Shimazaki, T.; Kikuchi, A.; Watanabe, K.N.; Kasuga, M.; Yamaguchi-Shinozaki, K.; Ogawa, T.; et al. Metabolic profiling of transgenic potato tubers expressing Arabidopsis dehydration response element-binding protein 1A (DREB1A). J. Agric. Food Chem. 2013, 61, 893–900. [Google Scholar]
- Defernez, M.; Gunning, Y.M.; Parr, A.J.; Shepherd, L.V.; Davies, H.V.; Colquhoun, I.J. NMR and HPLC-UV profiling of potatoes with genetic modifications to metabolic pathways. J. Agric. Food Chem. 2004, 52, 6075–6085. [Google Scholar]
- Kondrák, M.; Marincs, F.; Antal, F.; Juhász, Z.; Bánfalvi, Z. Effects of yeast trehalose-6-phosphate synthase 1 on gene expression and carbohydrate contents of potato leaves under drought stress conditions. BMC Plant Biol. 2012, 12. [Google Scholar] [CrossRef]
- Noteborn, H.P.; Lommen, A.; van der Jagt, R.C.; Weseman, J.M. Chemical fingerprinting for the evaluation of unintended secondary metabolic changes in transgenic food crops. J. Biotechnol. 2000, 77, 103–114. [Google Scholar]
- Roessner-Tunali, U.; Hegemann, B.; Lytovchenko, A.; Carrari, F.; Bruedigam, C.; Granot, D.; Fernie, A.R. Metabolic profiling of transgenic tomato plants overexpressing hexokinase reveals that the influence of hexose phosphorylation diminishes during fruit development. Plant Physiol. 2003, 133, 84–99. [Google Scholar]
- Le Gall, G.; Colquhoun, I.J.; Davis, A.L.; Collins, G.J.; Verhoeyen, M.E. Metabolite profiling of tomato (Lycopersicon esculentum) using 1H NMR spectroscopy as a tool to detect potential unintended effects following a genetic modification. J. Agric. Food Chem. 2003, 51, 2447–2456. [Google Scholar]
- Le Gall, G.; DuPont, M.S.; Mellon, F.A.; Davis, A.L.; Collins, G.J.; Verhoeyen, M.E.; Colquhoun, I.J. Characterization and content of flavonoid glycosides in genetically modified tomato (Lycopersicon esculentum) fruits. J. Agric. Food Chem. 2003, 51, 2438–2446. [Google Scholar]
- Long, M.; Millar, D.J.; Kimura, Y.; Donovan, G.; Rees, J.; Fraser, P.D.; Bramley, P.M.; Bolwell, G.P. Metabolite profiling of carotenoid and phenolic pathways in mutant and transgenic lines of tomato: Identification of a high antioxidant fruit line. Phytochemistry 2006, 67, 1750–1757. [Google Scholar]
- Nicoletti, I.; de Rossi, A.; Giovinazzo, G.; Corradini, D. Identification and quantification of stilbenes in fruits of transgenic tomato plants (Lycopersicon esculentum Mill.) by reversed phase HPLC with photodiode array and mass spectrometry detection. J. Agric. Food Chem. 207, 55, 3304–3311. [Google Scholar]
- Kusano, M.; Redestig, H.; Hirai, T.; Oikawa, A.; Matsuda, F.; Fukushima, A.; Arita, M.; Watanabe, S.; Yano, M.; Hiwasa-Tanase, K.; et al. Covering chemical diversity of genetically-modified tomatoes using metabolomics for objective substantial equivalence assessment. PLoS One 2011, 16, e16989. [Google Scholar]
- Choi, H.K.; Choi, Y.H.; Verberne, M.; Lefeber, A.W.; Erkelens, C.; Verpoorte, R. Metabolic fingerprinting of wild type and transgenic tobacco plants by 1H NMR and multivariate analysis technique. Phytochemistry 2004, 65, 857–864. [Google Scholar]
- Sobolev, A.P.; Segre, A.L.; Giannino, D.; Mariotti, D.; Nicolodi, C.; Brosio, E.; Amato, M.E. Strong increase of foliar inulin occurs in transgenic lettuce plants (Lactuca sativa L.) overexpressing the Asparagine Synthetase A gene from Escherichia coli. J. Agric. Food Chem. 2007, 55, 10827–10831. [Google Scholar]
- Sobolev, A.P.; Capitani, D.; Giannino, D.; Nicolodi, C.; Testone, G.; Santoro, F.; Frugis, G.; Iannelli, M.A.; Mattoo, A.K.; Brosio, E.; et al. NMR-metabolic methodology in the study of GM foods. Nutrients 2010, 2, 1–15. [Google Scholar]
- Sobolev, A.P.; Testone, G.; Santoro, F.; Nicolodi, C.; Iannelli, M.A.; Amato, M.E.; Ianniello, A.; Brosio, E.; Giannino, D.; Mannina, L. Quality traits of conventional and transgenic lettuce (Lactuca sativa L.) at harvesting by NMR metabolic profiling. J. Agric. Food Chem. 2010, 58, 6928–6936. [Google Scholar]
- Tagashira, N.; Plader, W.; Filipecki, M.; Yin, Z.; Wiśniewska, A.; Gaj, P.; Szwacka, M.; Fiehn, O.; Hoshi, Y.; Kondo, K.; et al. The metabolic profiles of transgenic cucumber lines vary with different chromosomal locations of the transgene. Cell Mol. Biol. Lett. 2005, 10, 697–710. [Google Scholar]
- Zawirska-Wojtasiak, R.; Gośliński, M.; Szwacka, M.; Gajc-Wolska, J.; Mildner-Szkudlarz, S. Aroma evaluation of transgenic, thaumatin II-producing cucumber fruits. J. Food Sci. 2009, 74, C204–C210. [Google Scholar]
- Malowicki, S.M.M.; Martin, R.; Qian, M.C. Comparison of sugar, acids, and volatile composition in raspberry bushy dwarf virus-resistant transgenic raspberries and the wild type “meeker” (Rubus Idaeus L.). J. Agric. Food Chem. 2008, 56, 6648–6655. [Google Scholar]
- Tesniere, C.; Torregrosa, L.; Pradal, M.; Souquet, J.M.; Gilles, C.; Dos Santos, K.; Chatelet, P.; Gunata, Z. Effects of genetic manipulation of alcohol dehydrogenase levels on the response to stress and the synthesis of secondary metabolites in grapevine leaves. J. Exp. Bot. 2006, 57, 91–99. [Google Scholar]
- Lange, B.M.; Mahmoud, S.S.; Wildung, M.R.; Turner, G.W.; Davis, E.M.; Lange, I.; Bake, R.C.; Boydston, R.A.; Croteau, R.B. Improving peppermint essential oil yield and composition by metabolic engineering. Proc. Natl. Acad. Sci. USA 2011, 108, 16944–16949. [Google Scholar]
- Kim, J.K.; Ryu, T.H.; Sohn, S.I.; Kim, J.H.; Chu, S.M.; Yu, C.Y.; Baek, H.J. Metabolic fingerprinting study on the substantial equivalence of Genetically Modified (GM) Chinese cabbage to non-gm cabbage. J. Korean Soc. Appl. Biol. 2009, 52, 186–192. [Google Scholar]
- Tripathi, S.; Suzuki, J.Y.; Carr, J.B.; McQuate, G.T.; Ferreira, S.A.; Manshardt, R.M.; Pitz, K.Y.; Wall, M.M.; Gonsalves, D. Nutritional composition of Rainbow papaya, the first commercialized transgenic fruit crop. J. Food Compos. Anal. 2011, 24, 140–147. [Google Scholar]
- Jiao, Z.; Deng, J.C.; Li, G.K.; Zhang, Z.M.; Cai, Z.W. Study on the compositional differences between transgenic and non-transgenic papaya. J. Food Compos. Anal. 2010, 23, 640–647. [Google Scholar]
- Srivastava, V.; Obudulu, O.; Bygdell, J.; Löfstedt, T.; Rydén, P.; Nilsson, R.; Ahnlund, M.; Johansson, A.; Jonsson, P.; Freyhult, E.; et al. OnPLS integration of transcriptomic, proteomic and metabolomic data shows multi-level oxidative stress responses in the cambium of transgenic hipI- superoxide dismutase Populus plants. BMC Genomics 2013, 14. [Google Scholar] [CrossRef]
- Kogel, K.H.; Voll, L.M.; Schäfer, P.; Jansen, C.; Wu, Y.; Langen, G.; Imani, J.; Hofmann, J.; Schmiedl, A.; Sonnewald, S.; et al. Transcriptome and metabolome profiling of field-grown transgenic barley lack induced differences but show cultivar-specific variances. Proc. Natl. Acad. Sci. USA 2010, 107, 6198–6203. [Google Scholar]
- Cantrell, R.P.; Reeves, T.G. The rice genome. The cereal of the world’s poor takes center stage. Science 2002, 296. [Google Scholar] [CrossRef]
- Goff, S.A.; Ricke, D.; Lan, T.H.; Presting, G.; Wang, R.; Dunn, M.; Glazebrook, J.; Sessions, A.; Oeller, P.; Varma, H.; et al. A draft sequence of the rice genome (Oryza sativa L. ssp. japonica). Science 2002, 296, 92–100. [Google Scholar]
- Oikawa, A.; Matsuda, F.; Kusano, M.; Okazaki, Y.; Saito, K. Rice metabolomics. Rice 2008, 1, 63–71. [Google Scholar]
- Choudhary, V.K.; Suresh-Kumar, P. Maize production, economics and soil productivity under different organic source of nutrients in eastern himalayan region, India. Int. J. Plant Prod. 2013, 7, 167–186. [Google Scholar]
- Skogerson, K.; Harrigan, G.G.; Reynolds, T.L.; Halls, S.C.; Ruebelt, M.; Iandolino, A.; Pandravada, A.; Glenn, K.C.; Fiehn, O. Impact of genetics and environment on the metabolite composition of maize grain. J. Agric. Food Chem. 2010, 58, 3600–3610. [Google Scholar]
- Asiago, V.M.; Hazebroek, J.; Harp, T.; Zhong, C. Effects of genetics and environment on the metabolome of commercial maize hybrids: A multisite study. J. Agric. Food Chem. 2012, 60, 11498–11508. [Google Scholar]
- Baniasadi, H.; Vlahakis, C.; Hazebroek, J.; Zhong, C.; Asiago, V. Effect of environment and genotype on commercial maize hybrids using LC/MS-based metabolomics. J. Agric. Food Chem. 2014, 62, 1412–1422. [Google Scholar]
- Duvernaya, W.H.; Chinna, M.S.; Yencho, G.C. Hydrolysis and fermentation of sweetpotatoes for production of fermentable sugars and ethanol. Ind. Crop Prod. 2013, 42, 527–537. [Google Scholar]
- Urbanczyk-Wochniak, E.; Baxter, C.; Kolbe, A.; Kopka, J.; Sweetlove, L.J.; Fernie, A.R. Profiling of diurnal patterns of metabolite and transcript abundance in potato (Solanum tuberosum) leaves. Planta 2005, 221, 891–903. [Google Scholar]
- García-Cañas, V.; Simó, C.; Herrero, M.; Ibáñez, E.; Cifuentes, A. Present and future challenges in food analysis: Foodomics. Anal. Chem. 2012, 84, 10150–10159. [Google Scholar]
- Fukushima, A.; Saito, K. Development of the Arabidopsis metabolome database. Plant Physiol. 2014. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Simó, C.; Ibáez, C.; Valdés, A.; Cifuentes, A.; García-Cañas, V. Metabolomics of Genetically Modified Crops. Int. J. Mol. Sci. 2014, 15, 18941-18966. https://doi.org/10.3390/ijms151018941
Simó C, Ibáez C, Valdés A, Cifuentes A, García-Cañas V. Metabolomics of Genetically Modified Crops. International Journal of Molecular Sciences. 2014; 15(10):18941-18966. https://doi.org/10.3390/ijms151018941
Chicago/Turabian StyleSimó, Carolina, Clara Ibáez, Alberto Valdés, Alejandro Cifuentes, and Virginia García-Cañas. 2014. "Metabolomics of Genetically Modified Crops" International Journal of Molecular Sciences 15, no. 10: 18941-18966. https://doi.org/10.3390/ijms151018941
APA StyleSimó, C., Ibáez, C., Valdés, A., Cifuentes, A., & García-Cañas, V. (2014). Metabolomics of Genetically Modified Crops. International Journal of Molecular Sciences, 15(10), 18941-18966. https://doi.org/10.3390/ijms151018941