Computer-Aided Targeting of the PI3K/Akt/mTOR Pathway: Toxicity Reduction and Therapeutic Opportunities

Abstract
:1. The PI3K/Akt/mTOR Pathway
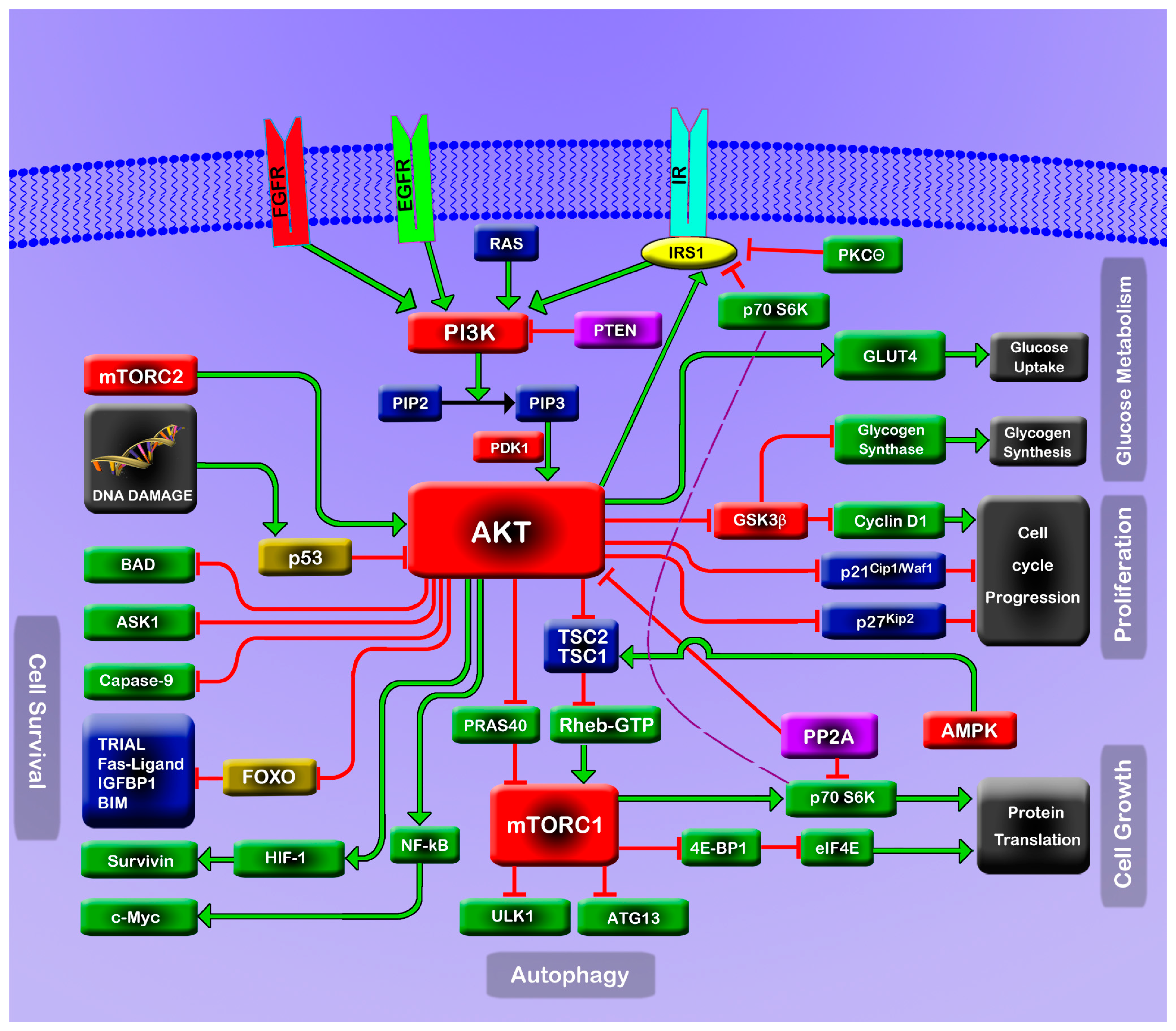
2. Consequences of PI3K/Akt/mTOR Perturbation

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Viral Agents | Mechanism of Action | Reference |
|---|---|---|
| Mouse polyoma virus middle tumor antigen (PyMT) |
| [18] |
| Mouse polyoma virus small tumor antigen (PyST) |
| [19] |
| Simian virus 40 small tumor antigen (SVST) |
| [20] |
| Human papillomaviruses (HPVs) E6 protein |
| [21] [22] |
| Human papillomaviruses (HPVs) E7 protein |
| [23] |
| Hepatitis C virus (HCV) |
| [24,25] |
| Chemical toxicants | Mechanism of Action | Reference |
| Trivalent arsenic |
| [26,27,28,29] [30] [31,32] |
| Cadmium |
| [33] |
| Vanadate |
| [34] |
| Zinc oxide nanoparticles |
| [35] |
| Superparamagnetic iron oxide nanoparticles (SPIONs) |
| [36] |
| Nicotine |
| [37] |
| NNK |
| [37] [38,39] |
| Microcystin-LR |
| [40,41] |
| Physiological products | Mechanism of Action | Reference |
| Free fatty acids (FFAs) |
| [42,43,44] [45] [46] [47] |
| Interleukin (IL)-6 |
| [48] |
| Tumor necrosis factor (TNF)-α |
| [49] |
| Angiotensin II and epinephrine |
| [50] |
2.1. Genetic Alteration
2.2. Viral Usurpation
2.3. Chemical Toxicants
2.4. Physiological Products
3. The PI3K/Akt/mTOR Pathway in Molecular Targeted Therapy: Progress and Worry
4. Computational Modeling for Therapeutic Guidance
- (a)
- Computer-aided targeting in this review refers to the execution of in silico studies that simulate the dynamic changes of certain signaling pathways and using the gleaned information to guide experimental or clinical modulation of the pathway. Such dynamic changes may range from the system level, which refers to the regulatory network of the pathway, to the molecular level, which refers to the structure and conformation of a specific molecule. Guidance gleaned from these computational studies is potentially useful in many facets of therapy, including the formulation of optimized drug combinations to maximize synergistic efficacy with reduced toxicity and designing pharmaceuticals to preferentially target one specific type of molecule to prevent off-target toxicity.
- (b)
- Adequate ex silico data, such as abundant protein-protein interaction evidence or nuclear magnetic resonance (NMR) and X-ray crystallography-illustrated protein structures are necessary for realistic computerized simulation. The methods employed in computer-aided targeting include a collection of software-based modeling applications, each crafted to simulate different levels of pathway and molecular interaction.
- (c)
- For example, in order to formulate an optimized composition of kinase inhibitors in combined therapy, a thorough understanding of the dynamic flow of the signaling pathway should first be acquired from real-world experimental evidence. Then, based on this descriptive experimental data, nodal points (nodes) along the pathway can be defined, and a complicated network may be simplified to a backbone framework for system-level in silico simulation. Mathematical modeling, harnessing prominent biological equations, such as the Michaelis–Menten equation (modeling enzyme kinetics), the Goldbeter–Koshland equation (modeling biological states impacted by both promoting and inhibiting enzymes) and the law of mass action (modeling the behavior of solutions in dynamic equilibrium) can then be used to depict the mechanistic flow of the framework. Graphic output from mathematical models are generated, and the values of different nodes may then be manipulated to mimic different biological states. A review of this graphic output often reveals boundaries formed by combinations of nodal states, highlighting interesting and useful cases of signaling dynamics. Finally, comparing the nodal configurations of pathological states to those of normal states may identify numerous points of transition between the two; analysis of these can identify the most efficient transition from pathological to normal, and this represents an optimized therapeutic strategy.
- (d)
- In silico simulations at the molecular level can also provide guidance on drug design. For example, based on NMR or X-ray crystallographic data, molecular dynamics (MD) simulation applies classical and quantum mechanics to calculate the forces between bonded and non-bonded atoms and, from this, builds functionally useful simulations of whole molecules, especially the receptors of interest as they function and move in three-dimensional space. Powerful software packages, such as AMBER, CHARMM and NAMD, allow sophisticated MD simulations to be achieved. MD simulation can reveal subtle dynamic differences between a targeted molecule and other closely related ones. Furthermore, software packages like FTMAP can be applied to computationally manipulate ligand probes on multiple simulated protein conformations, in order to identify cryptic and allosteric binding sites which might otherwise be easily overlooked by structure-based drug design. Moreover, docking programs, which employ a method known as the relaxed complex scheme (RCS), can be applied to virtually screen the binding sites of drug candidates in a simulated flexible receptor; free-energy calculations can predict the drug-receptor binding efficiency; and techniques, such as alchemical techniques, can greatly simplify the computation. These docking and free-energy analyses are thus particularly advantageous to discover drugs with high specificity to the targeted protein.
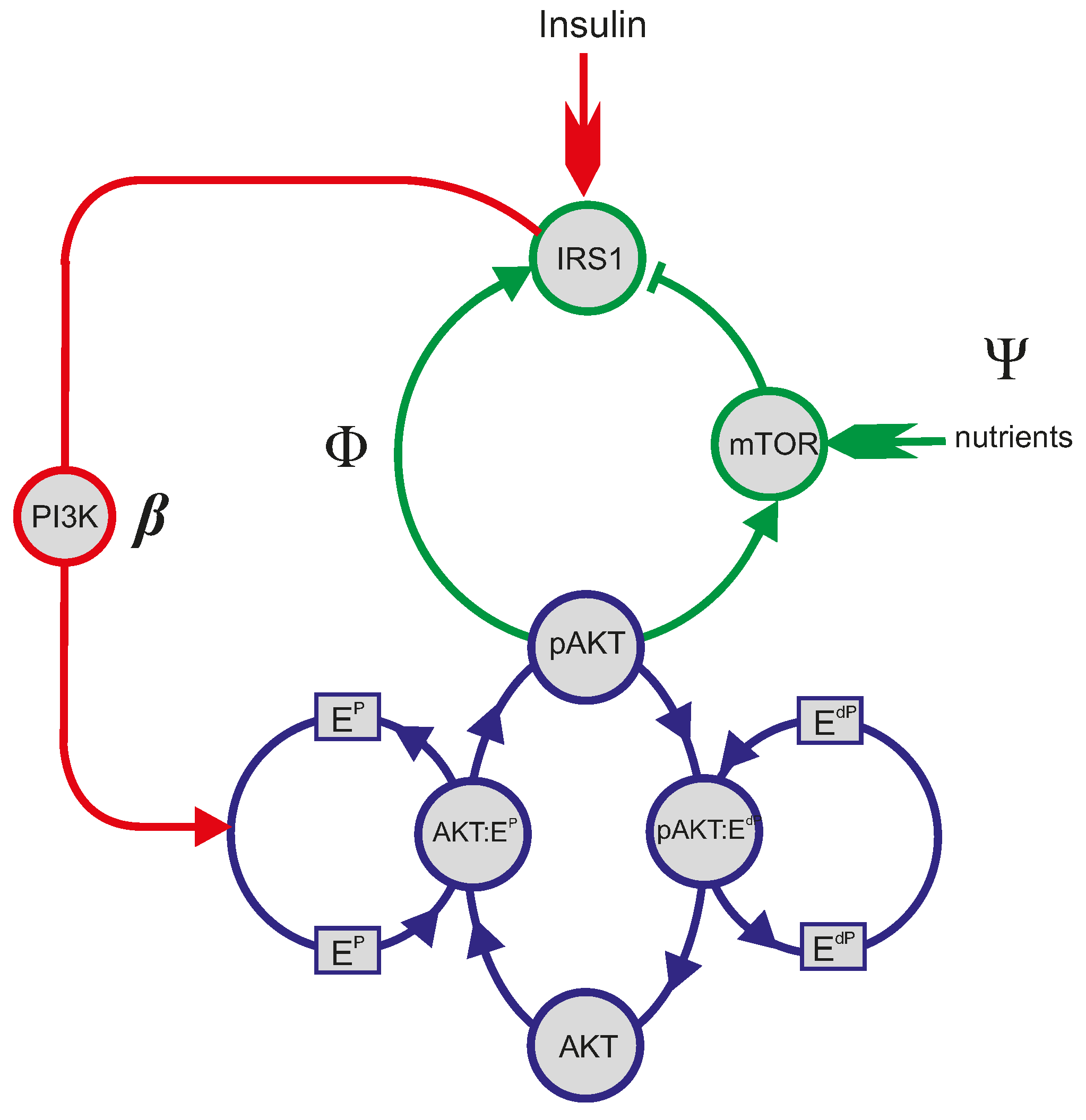
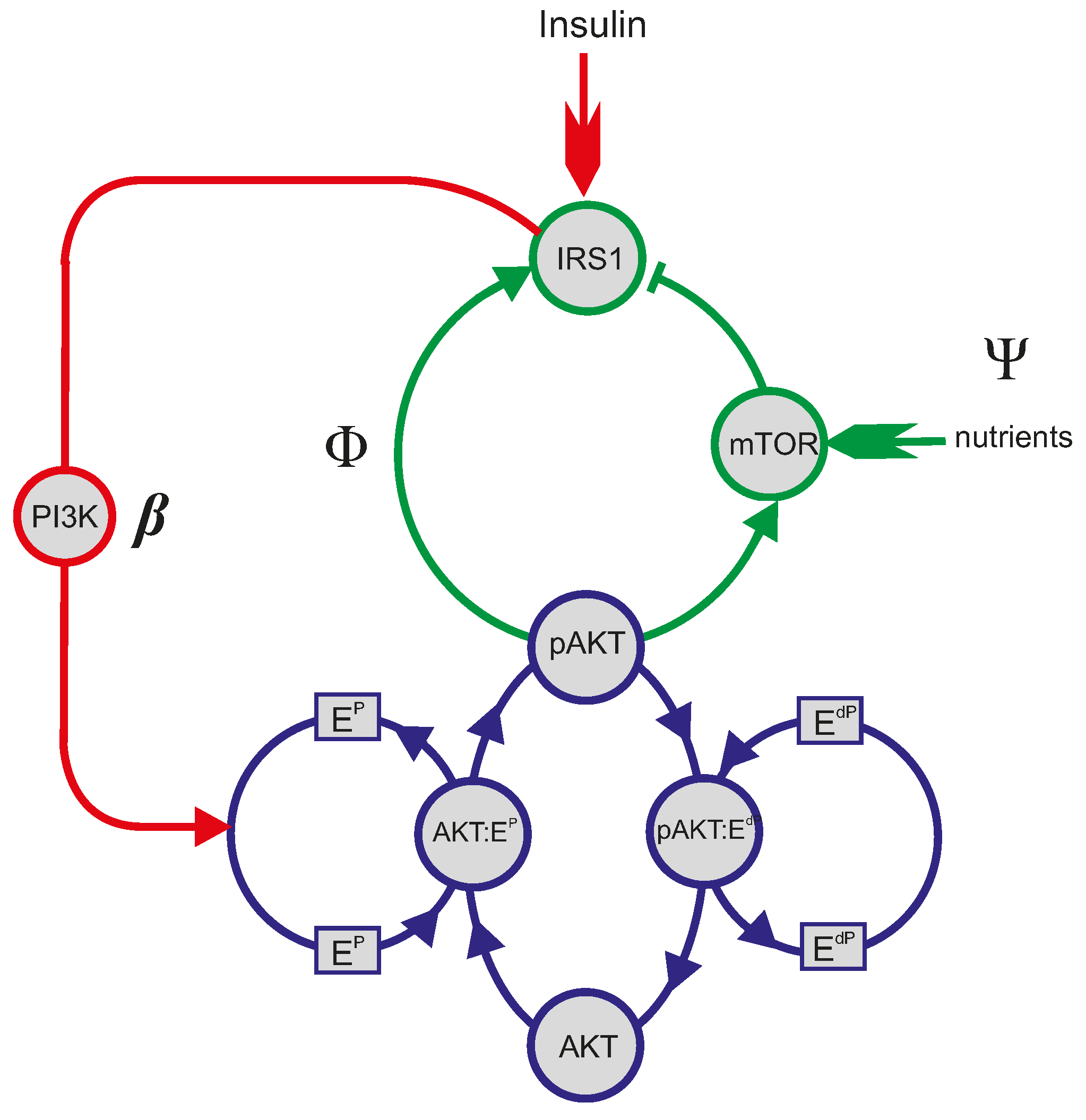
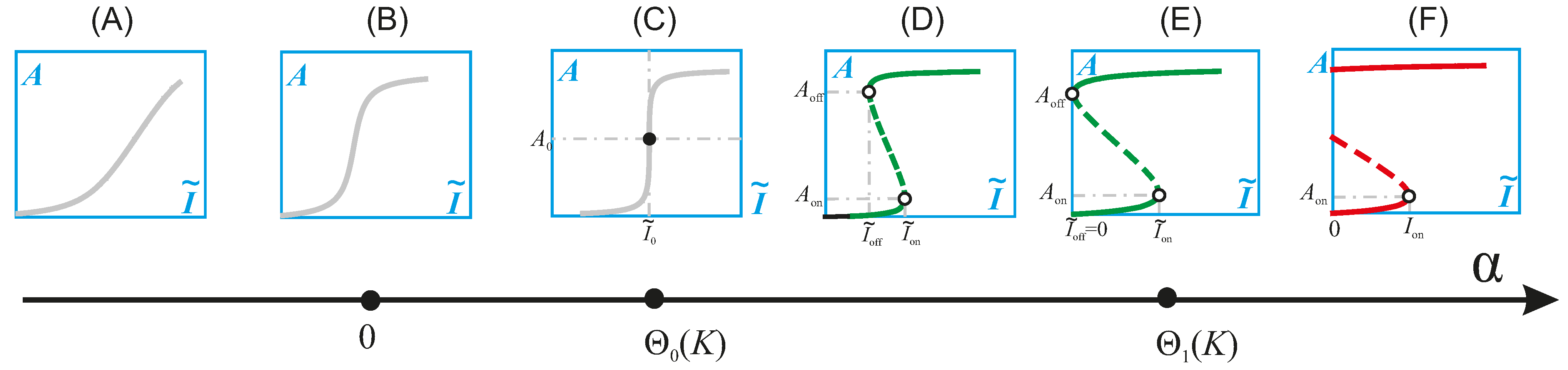
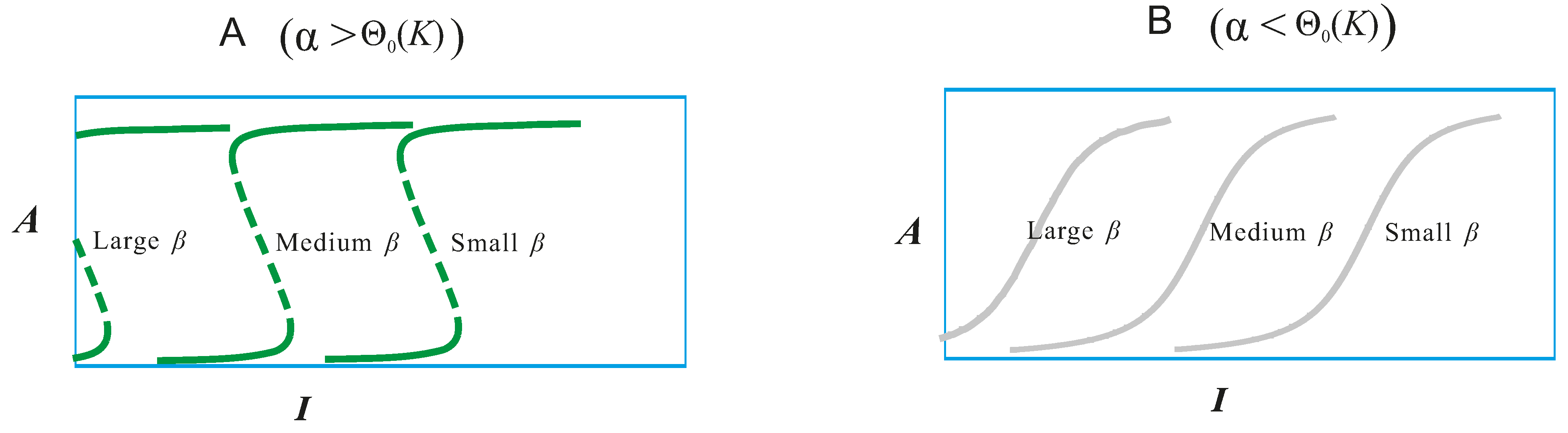
4.1. Mathematical Analysis of the PI3K/Akt/mTOR Pathway
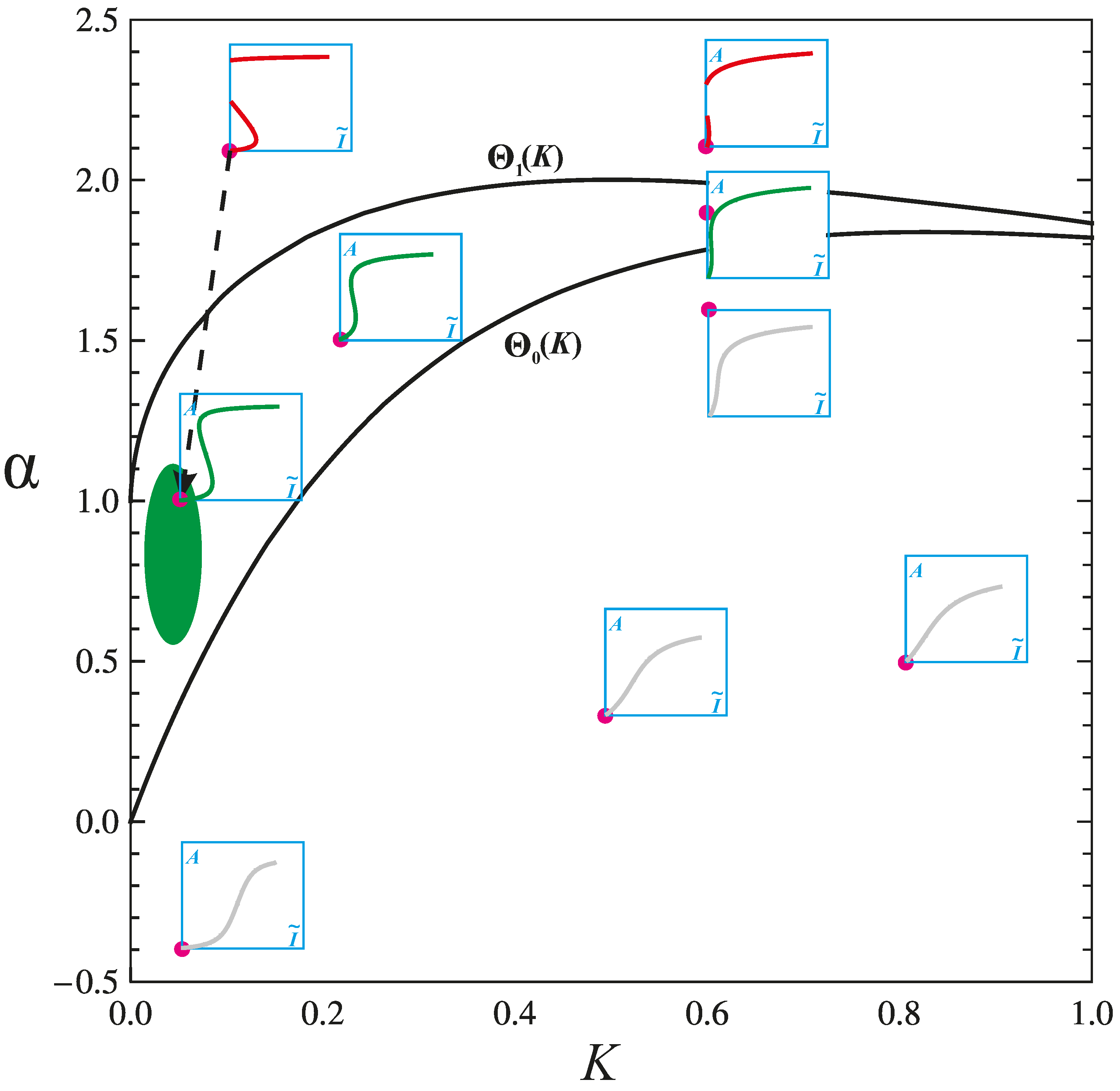
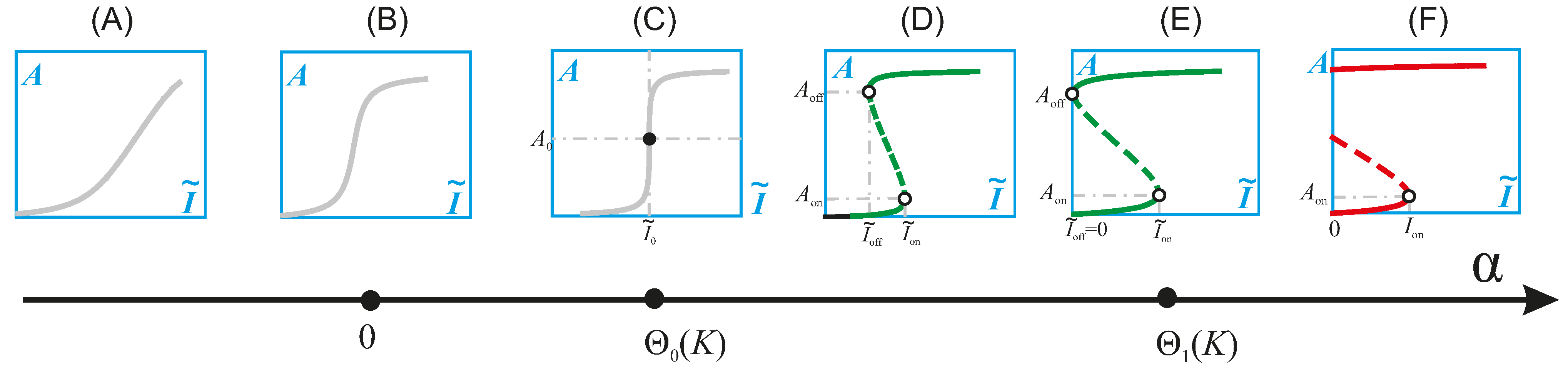

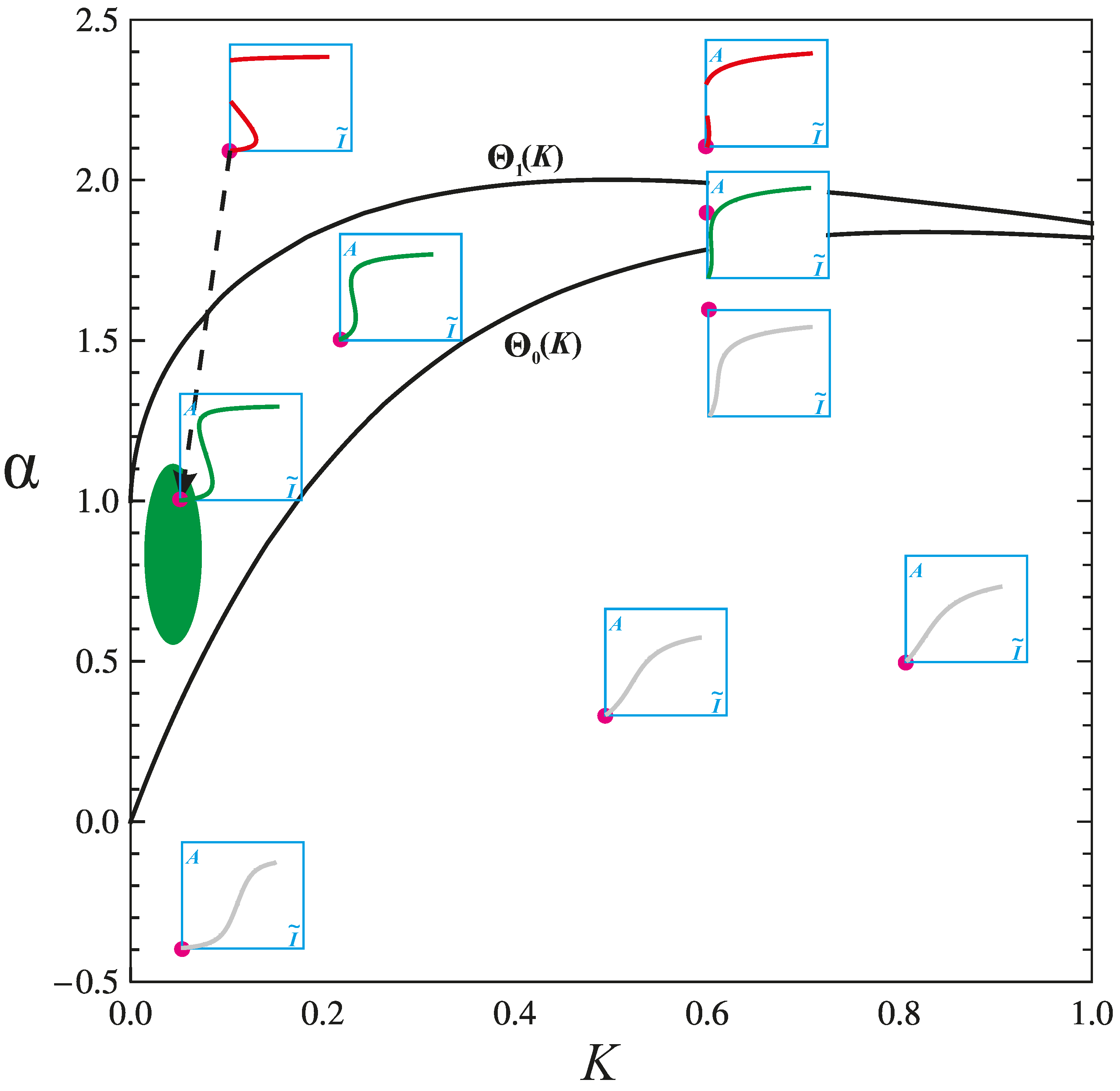
4.2. Physiologic and Pathologic Phenotypes
4.3. Combination Therapy Guided by Computation
4.4. The Yet to Be Explored “EdP” Wing
4.5. Patient Selection Guided by Computation
5. Molecular Dynamics Simulation Inspires a New Generation of Therapeutic Agents
6. Conclusions
Acknowledgments
Conflicts of Interest
References
- Wang, G. Analysis of Complex Diseases: A Mathematical Perspective; CRC Press: Boca Raton, FL, USA, 2013. [Google Scholar]
- Liao, Y.; Hung, M.C. Physiological regulation of Akt activity and stability. Am. J. Transl. Res. 2010, 2, 19–42. [Google Scholar]
- Manning, B.D.; Cantley, L.C. AKT/PKB signaling: Navigating downstream. Cell 2007, 129, 1261–1274. [Google Scholar] [CrossRef]
- Boura-Halfon, S.; Zick, Y. Phosphorylation of IRS proteins, insulin action, and insulin resistance. Am. J. Physiol. Endocrinol. Metab. 2009, 296, E581–E591. [Google Scholar] [CrossRef]
- Luo, M.; Langlais, P.; Yi, Z.; Lefort, N.; de Filippis, E.A.; Hwang, H.; Christ-Roberts, C.Y.; Mandarino, L.J. Phosphorylation of human insulin receptor substrate-1 at Serine 629 plays a positive role in insulin signaling. Endocrinology 2007, 148, 4895–905. [Google Scholar]
- Zoncu, R.; Efeyan, A.; Sabatini, D.M. mTOR: From growth signal integration to cancer, diabetes and ageing. Nat. Rev. Mol. Cell Biol. 2011, 12, 21–35. [Google Scholar] [CrossRef]
- LoPiccolo, J.; Blumenthal, G.M.; Bernstein, W.B.; Dennis, P.A. Targeting the PI3K/Akt/mTOR pathway: Effective combinations and clinical considerations. Drug Resist. Update 2008, 11, 32–50. [Google Scholar]
- Asano, T.; Yao, Y.X.; Zhu, J.J.; Li, D.H.; Abbruzzese, J.L.; Reddy, S.A.G. The PI 3-kinase/Akt signaling pathway is activated due to aberrant Pten expression and targets transcription factors NF-κB and c-Myc in pancreatic cancer cells. Oncogene 2004, 23, 8571–8580. [Google Scholar] [CrossRef]
- Nakanishi, C.; Toi, M. Nuclear factor-kappa B inhibitors as sensitizers to anticancer drugs. Nat. Rev. Cancer 2005, 5, 297–309. [Google Scholar] [CrossRef]
- Porta, C.; Paglino, C.; Mosca, A. Targeting PI3K/Akt/mTOR signaling in cancer. Front. Oncol. 2014, 4. [Google Scholar] [CrossRef]
- Carracedo, A.; Pandolfi, P.P. The PTEN-PI3K pathway: Of feedbacks and cross-talks. Oncogene 2008, 27, 5527–5541. [Google Scholar] [CrossRef]
- Janssens, V.; Rebollo, A. The role and therapeutic potential of Ser/Thr phosphatase PP2A in apoptotic signalling networks in human cancer cells. Curr. Mol. Med. 2012, 12, 268–287. [Google Scholar] [CrossRef]
- Bartholomeusz, C.; Gonzalez-Angulo, A.M. Targeting the PI3K signaling pathway in cancer therapy. Expert Opin. Ther. Targets 2012, 16, 121–130. [Google Scholar] [CrossRef]
- Carpenter, R.L.; Jiang, B.H. Roles of EGFR, PI3K, AKT, and mTOR in heavy metal-induced cancer. Curr. Cancer Drug Targets 2013, 13, 252–266. [Google Scholar] [CrossRef]
- Chitturi, S.; Abeygunasekera, S.; Farrell, G.C.; Holmes-Walker, J.; Hui, J.M.; Fung, C.; Karim, R.; Lin, R.; Samarasinghe, D.; Liddle, C.; et al. NASH and insulin resistance: Insulin hypersecretion and specific association with the insulin resistance syndrome. Hepatology 2002, 35, 373–379. [Google Scholar]
- Gallagher, E.J.; LeRoith, D. Epidemiology and molecular mechanisms tying obesity, diabetes, and the metabolic syndrome with cancer. Diabetes Care 2013, 36 (Suppl. S2), S233–S239. [Google Scholar] [CrossRef]
- Dorn, G.W.; Force, T. Protein kinase cascades in the regulation of cardiac hypertrophy. J. Clin. Investig. 2005, 115, 527–537. [Google Scholar] [CrossRef]
- Utermark, T.; Schaffhausen, B.S.; Roberts, T.M.; Zhao, J.J. The p110α isoform of phosphatidylinositol 3-kinase is essential for polyomavirus middle T antigen-mediated transformation. J. Virol. 2007, 81, 7069–7076. [Google Scholar] [CrossRef]
- Andrabi, S.; Gjoerup, O.V.; Kean, J.A.; Roberts, T.M.; Schaffhausen, B. Protein phosphatase 2A regulates life and death decisions via Akt in a context-dependent manner. Proc. Natl. Acad. Sci. USA 2007, 104, 19011–19016. [Google Scholar] [CrossRef]
- Arroyo, J.D.; Hahn, W.C. Involvement of PP2A in viral and cellular transformation. Oncogene 2005, 24, 7746–7755. [Google Scholar] [CrossRef]
- Lu, Z.; Hu, X.; Li, Y.; Zheng, L.; Zhou, Y.; Jiang, H.; Ning, T.; Basang, Z.; Zhang, C.; Ke, Y. Human papillomavirus 16 E6 oncoprotein interferences with insulin signaling pathway by binding to tuberin. J. Biol. Chem. 2004, 279, 35664–35670. [Google Scholar]
- Marur, S.; D’Souza, G.; Westra, W.H.; Forastiere, A.A. HPV-associated head and neck cancer: A virus-related cancer epidemic. Lancet Oncol. 2010, 11, 781–789. [Google Scholar] [CrossRef]
- Pim, D.; Massimi, P.; Dilworth, S.M.; Banks, L. Activation of the protein kinase B pathway by the HPV-16 E7 oncoprotein occurs through a mechanism involving interaction with PP2A. Oncogene 2005, 24, 7830–7838. [Google Scholar] [CrossRef]
- Mannova, P.; Beretta, L. Activation of the N-Ras-PI3K-Akt-mTOR pathway by hepatitis C virus: Control of cell survival and viral replication. J. Virol. 2005, 79, 8742–8749. [Google Scholar] [CrossRef]
- Bose, S.K.; Shrivastava, S.; Meyer, K.; Ray, R.B.; Ray, R. Hepatitis C virus activates the mTOR/S6K1 signaling pathway in inhibiting IRS-1 function for insulin resistance. J. Virol. 2012, 86, 6315–6322. [Google Scholar]
- Baysan, A.; Yel, L.; Gollapudi, S.; Su, H.; Gupta, S. Arsenic trioxide induces apoptosis via the mitochondrial pathway by upregulating the expression of Bax and Bim in human B cells. Int. J. Oncol. 2007, 30, 313–318. [Google Scholar]
- Choi, Y.J.; Park, J.W.; Suh, S.I.; Mun, K.C.; Bae, J.H.; Song, D.K.; Kim, S.P.; Kwon, T.K. Arsenic trioxide-induced apoptosis in U937 cells involve generation of reactive oxygen species and inhibition of Akt. Int. J. Oncol. 2002, 21, 603–610. [Google Scholar]
- Li, Y.M.; Broome, J.D. Arsenic targets tubulins to induce apoptosis in myeloid leukemia cells. Cancer Res. 1999, 59, 776–780. [Google Scholar]
- Chen, G.Q.; Zhu, J.; Shi, X.G.; Ni, J.H.; Zhong, H.J.; Si, G.Y.; Jin, X.L.; Tang, W.; Li, X.S.; Xong, S.M.; et al. In vitro studies on cellular and molecular mechanisms of arsenic trioxide (As2O3) in the treatment of acute promyelocytic leukemia: As2O3 induces NB4 cell apoptosis with downregulation of Bcl-2 expression and modulation of PML-RAR alpha/PML proteins. Blood 1996, 88, 1052–1061. [Google Scholar]
- Gao, N.; Shen, L.Q.; Zhang, Z.; Leonard, S.S.; He, H.J.; Zhang, X.G.; Shi, X.L.; Jiang, B.H. Arsenite induces HIF-1α and VEGF through PI3K, Akt and reactive oxygen species in DU145 human prostate carcinoma cells. Mol. Cell. Biochem. 2004, 255, 33–45. [Google Scholar] [CrossRef]
- Liu, J.; Chen, B.; Lu, Y.; Guan, Y.; Chen, F. JNK-dependent Stat3 phosphorylation contributes to Akt activation in response to arsenic exposure. Toxicol. Sci. 2012, 129, 363–371. [Google Scholar]
- Beezhold, K.; Liu, J.; Kan, H.; Meighan, T.; Castranova, V.; Shi, X.; Chen, F. miR-190-mediated downregulation of PHLPP contributes to arsenic-induced Akt activation and carcinogenesis. Toxicol. Sci. 2011, 123, 411–420. [Google Scholar] [CrossRef]
- Jing, Y.; Liu, L.Z.; Jiang, Y.; Zhu, Y.X.; Guo, N.L.; Barnett, J.; Rojanasakul, Y.; Agani, F.; Jiang, B.H. Cadmium increases HIF-1 and VEGF expression through ROS, ERK, and AKT signaling pathways and induces malignant transformation of human bronchial epithelial cells. Toxicol. Sci. 2012, 125, 10–19. [Google Scholar] [CrossRef]
- Gao, N.; Ding, M.; Zheng, J.Z.; Zhang, Z.; Leonard, S.S.; Liu, K.J.; Shi, X.L.; Jiang, B.H. Vanadate-induced expression of hypoxia-inducible factor 1α and vascular endothelial growth factor through phosphatidylinositol 3-kinase/Akt pathway and reactive oxygen species. J. Biol. Chem. 2002, 277, 31963–31971. [Google Scholar] [PubMed]
- Roy, R.; Singh, S.K.; Chauhan, K.S.; Das, M.; Tripathi, A.; Dwivedi, P.D. Zinc oxide nanoparticles induce apoptosis by enhancement of autophagy via PI3K/Akt/mTOR inhibition. Toxicol. Lett. 2014, 227, 29–40. [Google Scholar] [CrossRef]
- Rauch, J.; Kolch, W.; Mahmoudi, M. Cell type-specific activation of AKT and ERK signaling pathways by small negatively-charged magnetic nanoparticles. Sci. Rep. 2012, 2. [Google Scholar] [CrossRef]
- West, K.A.; Brognard, J.; Clark, A.S.; Linnoila, I.R.; Yang, X.W.; Swain, S.M.; Harris, C.; Belinsky, S.; Dennis, P.A. Rapid Akt activation by nicotine and a tobacco carcinogen modulates the phenotype of normal human airway epithelial cells. J. Clin. Investig. 2003, 111, 81–90. [Google Scholar] [CrossRef]
- Lonardo, F.; Dragnev, K.H.; Freemantle, S.J.; Ma, Y.; Memoli, N.; Sekula, D.; Knauth, E.A.; Beebe, J.S.; Dmitrovsky, E. Evidence for the epidermal growth factor receptor as a target for lung cancer prevention. Clin. Cancer Res. 2002, 8, 54–60. [Google Scholar]
- Westra, W.H.; Slebos, R.J.; Offerhaus, G.J.; Goodman, S.N.; Evers, S.G.; Kensler, T.W.; Askin, F.B.; Rodenhuis, S.; Hruban, R.H. K-ras oncogene activation in lung adenocarcinomas from former smokers. Evidence that K-ras mutations are an early and irreversible event in the development of adenocarcinoma of the lung. Cancer 1993, 72, 432–438. [Google Scholar]
- Xu, P.F.; Zhang, X.X.; Miao, C.; Fu, Z.Y.; Li, Z.R.; Zhang, G.; Zheng, M.Q.; Liu, Y.F.; Yang, L.Y.; Wang, T. Promotion of melanoma cell invasion and tumor metastasis by microcystin-LR via phosphatidylinositol 3-kinase/AKT pathway. Environ. Sci. Technol. 2013, 47, 8801–8808. [Google Scholar] [PubMed]
- Zhu, Y.L.; Zhong, X.; Zheng, S.; Ge, Z.; Du, Q.; Zhang, S.Z. Transformation of immortalized colorectal crypt cells by microcystin involving constitutive activation of Akt and MAPK cascade. Carcinogenesis 2005, 26, 1207–1214. [Google Scholar]
- Li, Y.; Soos, T.J.; Li, X.; Wu, J.; Degennaro, M.; Sun, X.; Littman, D.R.; Birnbaum, M.J.; Polakiewicz, R.D. Protein kinase C Theta inhibits insulin signaling by phosphorylating IRS1 at Ser1101. J. Biol. Chem. 2004, 279, 45304–45307. [Google Scholar] [PubMed]
- Griffin, M.E.; Marcucci, M.J.; Cline, G.W.; Bell, K.; Barucci, N.; Lee, D.; Goodyear, L.J.; Kraegen, E.W.; White, M.F.; Shulman, G.I. Free fatty acid-induced insulin resistance is associated with activation of protein kinase C theta and alterations in the insulin signaling cascade. Diabetes 1999, 48, 1270–1274. [Google Scholar] [CrossRef]
- Dresner, A.; Laurent, D.; Marcucci, M.; Griffin, M.E.; Dufour, S.; Cline, G.W.; Slezak, L.A.; Andersen, D.K.; Hundal, R.S.; Rothman, D.L.; et al. Effects of free fatty acids on glucose transport and IRS-1-associated phosphatidylinositol 3-kinase activity. J. Clin. Investig. 1999, 103, 253–259. [Google Scholar]
- Wang, X.L.; Zhang, L.; Youker, K.; Zhang, M.X.; Wang, J.; LeMaire, S.A.; Coselli, J.S.; Shen, Y.H. Free fatty acids inhibit insulin signaling-stimulated endothelial nitric oxide synthase activation through upregulating PTEN or inhibiting Akt kinase. Diabetes 2006, 55, 2301–2310. [Google Scholar]
- Zhang, Q.J.; Holland, W.L.; Wilson, L.; Tanner, J.M.; Kearns, D.; Cahoon, J.M.; Pettey, D.; Losee, J.; Duncan, B.; Gale, D.; et al. Ceramide mediates vascular dysfunction in diet-induced obesity by PP2A-mediated dephosphorylation of the eNOS-Akt complex. Diabetes 2012, 61, 1848–1859. [Google Scholar]
- Hsieh, C.T.; Chuang, J.H.; Yang, W.C.; Yin, Y.; Lin, Y.S. Ceramide inhibits insulin-stimulated Akt phosphorylation through activation of Rheb/mTORC1/S6K signaling in skeletal muscle. Cell Signal. 2014, 26, 1400–1408. [Google Scholar] [CrossRef] [PubMed]
- Nieto-Vazquez, I.; Fernandez-Veledo, S.; de Alvaro, C.; Lorenzo, M. Dual role of interleukin-6 in regulating insulin sensitivity in murine skeletal muscle. Diabetes 2008, 57, 3211–3221. [Google Scholar] [CrossRef]
- Zhang, J.; Gao, Z.G.; Yin, J.; Quon, M.J.; Ye, J.P. S6K directly phosphorylates IRS-1 on Ser-270 to promote insulin resistance in response to TNF-α signaling through IKK2. J. Biol. Chem. 2008, 283, 35375–35382. [Google Scholar] [PubMed]
- Oudit, G.Y.; Penninger, J.M. Cardiac regulation by phosphoinositide 3-kinases and PTEN. Cardiovasc. Res. 2009, 82, 250–260. [Google Scholar]
- Yin, Y.; Shen, W.H. PTEN: A new guardian of the genome. Oncogene 2008, 27, 5443–5453. [Google Scholar]
- Samuels, Y.; Wang, Z.H.; Bardelli, A.; Silliman, N.; Ptak, J.; Szabo, S.; Yan, H.; Gazdar, A.; Powell, D.M.; Riggins, G.J.; et al. High frequency of mutations of the PIK3CA gene in human cancers. Science 2004, 304, 554–554. [Google Scholar]
- Courtney, K.D.; Corcoran, R.B.; Engelman, J.A. The PI3K pathway as drug target in human cancer. J. Clin. Oncol. 2010, 28, 1075–1083. [Google Scholar] [CrossRef]
- Grabiner, B.C.; Nardi, V.; Birsoy, K.; Possemato, R.; Shen, K.; Sinha, S.; Jordan, A.; Beck, A.H.; Sabatini, D.M. A diverse array of cancer-associated MTOR mutations are hyperactivating and can predict rapamycin sensitivity. Cancer Discov. 2014, 4, 554–563. [Google Scholar] [CrossRef] [PubMed]
- Westermarck, J.; Hahn, W.C. Multiple pathways regulated by the tumor suppressor PP2A in transformation. Trends Mol. Med. 2008, 14, 152–160. [Google Scholar] [CrossRef]
- Calin, G.A.; di Iasio, M.G.; Caprini, E.; Vorechovsky, I.; Natali, P.G.; Sozzi, G.; Croce, C.M.; Barbanti-Brodano, G.; Russo, G.; Negrini, M. Low frequency of alterations of the α (PPP2R1A) and β (PPP2R1B) isoforms of the subunit A of the serine-threonine phosphatase 2A in human neoplasms. Oncogene 2000, 19, 1191–1195. [Google Scholar] [CrossRef]
- Laine, A.; Sihto, H.; Come, C.; Rosenfeldt, M.T.; Zwolinska, A.; Niemela, M.; Khanna, A.; Chan, E.K.; Kahari, V.M.; Kellokumpu-Lehtinen, P.L.; et al. Senescence Sensitivity of Breast Cancer Cells Is Defined by Positive Feedback Loop between CIP2A and E2F1. Cancer Discov. 2013, 3, 182–197. [Google Scholar]
- Chen, K.F.; Liu, C.Y.; Lin, Y.C.; Yu, H.C.; Liu, T.H.; Hou, D.R.; Chen, P.J.; Cheng, A.L. CIP2A mediates effects of bortezomib on phospho-Akt and apoptosis in hepatocellular carcinoma cells. Oncogene 2010, 29, 6257–6266. [Google Scholar] [PubMed]
- Weinstein, I.B. Cancer. Addiction to oncogenes—The Achilles heal of cancer. Science 2002, 297, 63–64. [Google Scholar]
- Cooray, S. The pivotal role of phosphatidylinositol 3-kinase-Akt signal transduction in virus survival. J. Gen. Virol. 2004, 85, 1065–1076. [Google Scholar] [CrossRef] [PubMed]
- Buchkovich, N.J.; Yu, Y.; Zampieri, C.A.; Alwine, J.C. The TORrid affairs of viruses: Effects of mammalian DNA viruses on the PI3K-Akt-mTOR signalling pathway. Nat. Rev. Microbiol. 2008, 6, 265–275. [Google Scholar] [CrossRef]
- Diehl, N.; Schaal, H. Make yourself at home: Viral hijacking of the PI3K/Akt signaling pathway. Viruses 2013, 5, 3192–3212. [Google Scholar] [CrossRef]
- Chami, M.; Oules, B.; Paterlini-Brechot, P. Cytobiological consequences of calcium-signaling alterations induced by human viral proteins. Biochim. Biophys. Acta 2006, 1763, 1344–1362. [Google Scholar]
- DeAngelis, T.; Chen, J.; Wu, A.; Prisco, M.; Baserga, R. Transformation by the simian virus 40 T antigen is regulated by IGF-I receptor and IRS-1 signaling. Oncogene 2006, 25, 32–42. [Google Scholar]
- Schiffman, M.; Castle, P.E.; Jeronimo, J.; Rodriguez, A.C.; Wacholder, S. Human papillomavirus and cervical cancer. Lancet 2007, 370, 890–907. [Google Scholar] [CrossRef]
- Ouyang, W.; Li, J.X.; Ma, Q.; Huang, C.S. Essential roles of PI-3K/Akt/IKKβ/NFκB pathway in cyclin D1 induction by arsenite in JB6 Cl41 cells. Carcinogenesis 2006, 27, 864–873. [Google Scholar] [CrossRef]
- Wang, L.; Jiang, H.Y.; Yin, Z.B.; Aschner, M.; Cai, J.Y. Methylmercury toxicity and Nrf2-dependent detoxification in astrocytes. Toxicol. Sci. 2009, 107, 135–143. [Google Scholar]
- Cai, B.B.; Xia, Z.G. p38 MAP kinase mediates arsenite-induced apoptosis through FOXO3a activation and induction of Bim transcription. Apoptosis 2008, 13, 803–810. [Google Scholar] [CrossRef]
- Ohnishi, K.; Yoshida, H.; Shigeno, K.; Nakamura, S.; Fujisawa, S.; Naito, K.; Shinjo, K.; Fujita, Y.; Matsui, H.; Takeshita, A.; et al. Prolongation of the QT interval and ventricular tachycardia in patients treated with arsenic trioxide for acute promyelocytic leukemia. Ann. Intern. Med. 2000, 133, 881–885. [Google Scholar]
- Kitamura, Y.; Koide, M.; Akakabe, Y.; Matsuo, K.; Shimoda, Y.; Soma, Y.; Ogata, T.; Ueyama, T.; Matoba, S.; Yamada, H.; et al. Manipulation of cardiac phosphatidylinositol 3-kinase (PI3K)/Akt signaling by apoptosis regulator through modulating IAP expression (ARIA) regulates cardiomyocyte death during doxorubicin-induced cardiomyopathy. J. Biol. Chem. 2014, 289, 2788–2800. [Google Scholar]
- Wang, M.; Sun, G.B.; Sun, X.; Wang, H.W.; Meng, X.B.; Qin, M.; Sun, J.; Luo, Y.; Sun, X.B. Cardioprotective effect of salvianolic acid B against arsenic trioxide-induced injury in cardiac H9c2 cells via the PI3K/Akt signal pathway. Toxicol. Lett. 2013, 216, 100–107. [Google Scholar] [CrossRef]
- Tsurutani, J.; Castillo, S.S.; Brognard, J.; Granville, C.A.; Zhang, C.; Gills, J.J.; Sayyah, J.; Dennis, P.A. Tobacco components stimulate Akt-dependent proliferation and NFκB-dependent survival in lung cancer cells. Carcinogenesis 2005, 26, 1182–1195. [Google Scholar]
- Jin, Z.; Gao, F.; Flagg, T.; Deng, X. Nicotine induces multi-site phosphorylation of Bad in association with suppression of apoptosis. J. Biol. Chem. 2004, 279, 23837–23844. [Google Scholar] [PubMed]
- Xin, M.; Deng, X. Nicotine inactivation of the proapoptotic function of Bax through phosphorylation. J. Biol. Chem. 2005, 280, 10781–1089. [Google Scholar] [PubMed]
- Matzinger, S.A.; Crist, K.A.; Stoner, G.D.; Anderson, M.W.; Pereira, M.A.; Steele, V.E.; Kelloff, G.J.; Lubet, R.A.; You, M. K-ras mutations in lung tumors from A/J and A/JxTSG-p53 F1 mice treated with 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone and phenethyl isothiocyanate. Carcinogenesis 1995, 16, 2487–2492. [Google Scholar] [CrossRef] [PubMed]
- Memmott, R.M.; Dennis, P.A. The role of the Akt/mTOR pathway in tobacco carcinogen-induced lung tumorigenesis. Clin. Cancer Res. 2010, 16, 4–10. [Google Scholar] [CrossRef] [PubMed]
- Takumi, S.; Komatsu, M.; Furukawa, T.; Ikeda, R.; Sumizawa, T.; Akenaga, H.; Maeda, Y.; Aoyama, K.; Arizono, K.; Ando, S.; et al. p53 Plays an important role in cell fate determination after exposure to microcystin-LR. Environ. Health Perspect. 2010, 118, 1292–1298. [Google Scholar]
- Fu, W.Y.; Chen, J.P.; Wang, M.M.; Xu, L.H. Altered expression of p53, Bcl-2 and Bax induced by microcystin-LR in vivo and in vitro. Toxicon 2005, 46, 171–177. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Ying, L.; Wang, H.; Li, N.; Fu, W.; Guo, Z.; Xu, L. Microcystin-LR induces ceramide to regulate PP2A and destabilize cytoskeleton in HEK293 cells. Toxicol. Sci. 2012, 128, 147–157. [Google Scholar]
- Cai, B.; Chang, S.H.; Becker, E.B.; Bonni, A.; Xia, Z. p38 MAP kinase mediates apoptosis through phosphorylation of BimEL at Ser-65. J. Biol. Chem. 2006, 281, 25215–25222. [Google Scholar] [PubMed]
- Namgung, U.; Xia, Z. Arsenite-induced apoptosis in cortical neurons is mediated by c-Jun N-terminal protein kinase 3 and p38 mitogen-activated protein kinase. J. Neurosci. 2000, 20, 6442–6451. [Google Scholar] [PubMed]
- Li, T.; Wang, W.; Pan, Y.W.; Xu, L.; Xia, Z. A hydroxylated metabolite of flame-retardant PBDE-47 decreases the survival, proliferation, and neuronal differentiation of primary cultured adult neural stem cells and interferes with signaling of ERK5 MAP kinase and neurotrophin 3. Toxicol. Sci. 2013, 134, 111–124. [Google Scholar] [CrossRef]
- Chen, F.; Demers, L.M.; Shi, X. Upstream signal transduction of NF-κB activation. Curr. Drug Targets Inflamm. Allergy 2002, 1, 137–149. [Google Scholar]
- Pacurari, M.; Yin, X.J.; Zhao, J.S.; Ding, M.; Leonard, S.S.; Schwegier-Berry, D.; Ducatman, B.S.; Sbarra, D.; Hoover, M.D.; Castranova, V.; et al. Raw single-wall carbon nanotubes induce oxidative stress and activate MAPKs, AP-1, NF-κB, and Akt in normal and malignant human mesothelial cells. Environ. Health Perspect. 2008, 116, 1211–1217. [Google Scholar]
- Arredondo, J.; Chernyavsky, A.I.; Jolkovsky, D.L.; Pinkerton, K.E.; Grando, S.A. Receptor-mediated tobacco toxicity: Cooperation of the Ras/Raf-1/MEK1/ERK and JAK-2/STAT-3 pathways downstream of α7 nicotinic receptor in oral keratinocytes. FASEB J. 2006, 20, 2093–2101. [Google Scholar] [CrossRef] [PubMed]
- Hsuan, S.L.; Klintworth, H.M.; Xia, Z.G. Basic fibroblast growth factor protects against rotenone-induced dopaminergic cell death through activation of extracellular signal-regulated kinases 1/2 and phosphatidylinositol-3 kinase pathways. J. Neurosci. 2006, 26, 4481–4491. [Google Scholar] [CrossRef] [PubMed]
- Shaik, Z.P.; Fifer, E.K.; Nowak, G. Akt activation improves oxidative phosphorylation in renal proximal tubular cells following nephrotoxicant injury. Am. J. Physiol. Ren. Physiol. 2008, 294, F423–F432. [Google Scholar]
- Sudarsanam, S.; Johnson, D.E. Functional consequences of mTOR inhibition. Curr. Opin. Drug Discov. 2010, 13, 31–40. [Google Scholar]
- Wang, G.Y. Singularity analysis of the AKT signaling pathway reveals connections between cancer and metabolic diseases. Phys. Biol. 2010, 7. [Google Scholar] [CrossRef]
- Mclean, B.A.; Zhabyeyev, P.; Pituskin, E.; Paterson, I.; Haykowsky, M.J.; Oudit, G.Y. PI3K Inhibitors as Novel Cancer Therapies: Implications for Cardiovascular Medicine. J. Card. Fail. 2013, 19, 268–282. [Google Scholar] [CrossRef] [PubMed]
- Suh, J.M.; Jonker, J.W.; Ahmadian, M.; Goetz, R.; Lackey, D.; Osborn, O.; Huang, Z.; Liu, W.; Yoshihara, E.; van Dijk, T.H.; et al. Endocrinization of FGF1 produces a neomorphic and potent insulin sensitizer. Nature 2014, 513, 436–439. [Google Scholar]
- Crackower, M.A.; Oudit, G.Y.; Kozieradzki, I.; Sarao, R.; Sun, H.; Sasaki, T.; Hirsch, E.; Suzuki, A.; Shioi, T.; Irie-Sasaki, J.; et al. Regulation of myocardial contractility and cell size by distinct PI3K-PTEN signaling pathways. Cell 2002, 110, 737–749. [Google Scholar]
- Schwartzbauer, G.; Robbins, J. The tumor suppressor gene PTEN can regulate cardiac hypertrophy and survival. J. Biol. Chem. 2001, 276, 35786–35793. [Google Scholar] [CrossRef] [PubMed]
- McMullen, J.R.; Jennings, G.L. Differences between pathological and physiological cardiac hypertrophy: Novel therapeutic strategies to treat heart failure. Clin. Exp. Pharmacol. Physiol. 2007, 34, 255–262. [Google Scholar] [CrossRef]
- McMullen, J.R.; Amirahmadi, F.; Woodcock, E.A.; Schinke-Braun, M.; Bouwman, R.D.; Hewitt, K.A.; Mollica, J.P.; Zhang, L.; Zhang, Y.Y.; Shioi, T.; et al. Protective effects of exercise and phosphoinositide 3-kinase(p110 α) signaling in dilated and hypertrophic cardiomyopathy. Proc. Natl. Acad. Sci. USA 2007, 104, 612–617. [Google Scholar]
- Ellison, G.M.; Waring, C.D.; Vicinanza, C.; Torella, D. Physiological cardiac remodelling in response to endurance exercise training: Cellular and molecular mechanisms. Heart 2012, 98, 5–10. [Google Scholar] [CrossRef] [PubMed]
- McMullen, J.R.; Shioi, T.; Zhang, L.; Tarnavski, O.; Sherwood, M.C.; Kang, P.M.; Izumo, S. Phosphoinositide 3-kinase(p110α) plays a critical role for the induction of physiological, but not pathological, cardiac hypertrophy. Proc. Natl. Acad. Sci. USA 2003, 100, 12355–12360. [Google Scholar] [CrossRef] [PubMed]
- Bernardo, B.C.; Weeks, K.L.; Pretorius, L.; McMullen, J.R. Molecular distinction between physiological and pathological cardiac hypertrophy: Experimental findings and therapeutic strategies. Pharmacol. Ther. 2010, 128, 191–227. [Google Scholar] [CrossRef]
- Heineke, J.; Molkentin, J.D. Regulation of cardiac hypertrophy by intracellular signalling pathways. Nat. Rev. Mol. Cell Biol. 2006, 7, 589–600. [Google Scholar] [CrossRef]
- Eifert, C.; Powers, R.S. From cancer genomes to oncogenic drivers, tumour dependencies and therapeutic targets. Nat. Rev. Cancer 2012, 12, 572–578. [Google Scholar] [CrossRef]
- Yap, T.A.; Garrett, M.D.; Walton, M.I.; Raynaud, F.; de Bono, J.S.; Workman, P. Targeting the PI3K-AKT-mTOR pathway: Progress, pitfalls, and promises. Curr. Opin. Pharmacol. 2008, 8, 393–412. [Google Scholar]
- Granville, C.A.; Memmott, R.M.; Gills, J.J.; Dennis, P.A. Handicapping the race to develop inhibitors of the phosphoinositide 3-kinase/Akt/mammalian target of rapamycin pathway. Clin. Cancer Res. 2006, 12, 679–689. [Google Scholar] [CrossRef] [PubMed]
- Rodon, J.; Dienstmann, R.; Serra, V.; Tabemero, J. Development of PI3K inhibitors: Lessons learned from early clinical trials. Nat. Rev. Clin. Oncol. 2013, 10, 143–153. [Google Scholar] [CrossRef]
- Fruman, D.A.; Rommel, C. PI3K and cancer: Lessons, challenges and opportunities. Nat. Rev. Drug Discov. 2014, 13, 140–156. [Google Scholar] [CrossRef]
- Bendell, J.C.; Rodon, J.; Burris, H.A.; de Jonge, M.; Verweij, J.; Birle, D.; Demanse, D.; de Buck, S.S.; Ru, Q.H.C.; Peters, M.; et al. Phase I, dose-escalation study of BKM120, an oral pan-class I PI3K inhibitor, in patients with advanced solid tumors. J. Clin. Oncol. 2012, 30, 282–290. [Google Scholar]
- Shapiro, G.I.; Rodon, J.; Bedell, C.; Kwak, E.L.; Baselga, J.; Brana, I.; Pandya, S.S.; Scheffold, C.; Laird, A.D.; Nguyen, L.T.; et al. Phase I safety, pharmacokinetic, and pharmacodynamic study of SAR245408 (XL147), an oral pan-class I PI3K inhibitor, in patients with advanced solid tumors. Clin. Cancer Res. 2014, 20, 233–245. [Google Scholar]
- Dienstmann, R.; Rodon, J.; Serra, V.; Tabernero, J. Picking the point of inhibition: A comparative review of PI3K/AKT/mTOR pathway inhibitors. Mol. Cancer Ther. 2014, 13, 1021–1031. [Google Scholar] [CrossRef]
- Busaidy, N.L.; Farooki, A.; Dowlati, A.; Perentesis, J.P.; Dancey, J.E.; Doyle, L.A.; Brell, J.M.; Siu, L.L. Management of metabolic effects associated with anticancer agents targeting the PI3K-Akt-mTOR pathway. J. Clin. Oncol. 2012, 30, 2919–2928. [Google Scholar] [CrossRef]
- Hudes, G.; Carducci, M.; Tomczak, P.; Dutcher, J.; Figlin, R.; Kapoor, A.; Staroslawska, E.; Sosman, J.; McDermott, D.; Bodrogi, I.; et al. Temsirolimus, interferon alfa, or both for advanced renal-cell carcinoma. N. Engl. J. Med. 2007, 356, 2271–2281. [Google Scholar]
- Crouthamel, M.C.; Kahana, J.A.; Korenchuk, S.; Zhang, S.Y.; Sundaresan, G.; Eberwein, D.J.; Brown, K.K.; Kumar, R. Mechanism and management of AKT inhibitor-induced hyperglycemia. Clin. Cancer Res. 2009, 15, 217–225. [Google Scholar] [CrossRef] [PubMed]
- Vanneman, M.; Dranoff, G. Combining immunotherapy and targeted therapies in cancer treatment. Nat. Rev. Cancer 2012, 12, 237–251. [Google Scholar] [CrossRef]
- Howe, L.R.; Subbaramaiah, K.; Hudis, C.A.; Dannenberg, A.J. Molecular pathways: Adipose inflammation as a mediator of obesity-associated cancer. Clin. Cancer Res. 2013, 19, 6074–6083. [Google Scholar] [CrossRef]
- Cnop, M.; Welsh, N.; Jonas, J.C.; Jorns, A.; Lenzen, S.; Eizirik, D.L. Mechanisms of pancreatic β-cell death in type 1 and type 2 diabetes: Many differences, few similarities. Diabetes 2005, 54, S97–S107. [Google Scholar] [CrossRef]
- Cheng, H.; Force, T. Molecular mechanisms of cardiovascular toxicity of targeted cancer therapeutics. Circ. Res. 2010, 106, 21–34. [Google Scholar]
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef]
- Yung, T.K.; Chan, K.C.; Mok, T.S.; Tong, J.; To, K.F.; Lo, Y.M. Single-molecule detection of epidermal growth factor receptor mutations in plasma by microfluidics digital PCR in non-small cell lung cancer patients. Clin. Cancer Res. 2009, 15, 2076–2084. [Google Scholar] [CrossRef] [PubMed]
- Rini, B.I. New strategies in kidney cancer: Therapeutic advances through understanding the molecular basis of response and resistance. Clin. Cancer Res. 2010, 16, 1348–1354. [Google Scholar] [CrossRef] [PubMed]
- Naing, A. Overcoming resistance to mTOR inhibition for enhanced strategies in clinical trials. Expert Opin. Inv. Drugs 2013, 22, 679–685. [Google Scholar] [CrossRef]
- Klempner, S.J.; Myers, A.P.; Cantley, L.C. What a tangled web we weave: Emerging resistance mechanisms to inhibition of the phosphoinositide 3-kinase pathway. Cancer Discov. 2013, 3, 1345–1354. [Google Scholar] [CrossRef]
- O’Reilly, K.E.; Rojo, F.; She, Q.B.; Solit, D.; Mills, G.B.; Smith, D.; Lane, H.; Hofmann, F.; Hicklin, D.J.; Ludwig, D.L.; et al. mTOR inhibition induces upstream receptor tyrosine kinase signaling and activates Akt. Cancer Res. 2006, 66, 1500–1508. [Google Scholar]
- Shi, Y.J.; Yan, H.J.; Frost, P.; Gera, J.; Lichtenstein, A. Mammalian target of rapamycin inhibitors activate the AKT kinase in multiple myeloma cells by up-regulating the insulin-like growth factor receptor/insulin receptor substrate-1/phosphatidylinositol 3-kinase cascade. Mol. Cancer Ther. 2005, 4, 1533–1540. [Google Scholar] [CrossRef] [PubMed]
- Rodrik-Outmezguine, V.S.; Chandarlapaty, S.; Pagano, N.C.; Poulikakos, P.I.; Scaltriti, M.; Moskatel, E.; Baselga, J.; Guichard, S.; Rosen, N. mTOR kinase inhibition causes feedback-dependent biphasic regulation of AKT signaling. Cancer Discov. 2011, 1, 248–259. [Google Scholar] [CrossRef]
- Zakikhani, M.; Blouin, M.J.; Piura, E.; Pollak, M.N. Metformin and rapamycin have distinct effects on the AKT pathway and proliferation in breast cancer cells. Breast Cancer Res. Treat. 2010, 123, 271–279. [Google Scholar] [CrossRef]
- Dowling, R.J.O.; Goodwin, P.J.; Stambolic, V. Understanding the benefit of metformin use in cancer treatment. BMC Med. 2011, 9. [Google Scholar] [CrossRef]
- Larson, H.; Chan, E.; Sudarsanam, S.; Johnson, D.E. Biomarkers. Methods Mol. Biol. 2013, 930, 253–273. [Google Scholar]
- Andersen, J.N.; Sathyanarayanan, S.; di Bacco, A.; Chi, A.; Zhang, T.; Chen, A.H.; Dolinski, B.; Kraus, M.; Roberts, B.; Arthur, W.; et al. Pathway-based identification of biomarkers for targeted therapeutics: Personalized oncology with PI3K pathway inhibitors. Sci. Transl. Med. 2010, 2. [Google Scholar] [CrossRef]
- Sedaghat, A.R.; Sherman, A.; Quon, M.J. A mathematical model of metabolic insulin signaling pathways. Am. J. Physiol. Endocrinol. Metab. 2002, 283, E1084–E1101. [Google Scholar] [PubMed]
- Wang, G.Y.; Krueger, G.R.F. Computational analysis of mTOR signaling pathway: Bifurcation, carcinogenesis, and drug discovery. Anticancer Res. 2010, 30, 2683–2688. [Google Scholar] [PubMed]
- Wang, G.Y. Optimal homeostasis necessitates bistable control. J. R. Soc. Interface 2012, 9, 2723–2734. [Google Scholar]
- Wendel, H.G.; de Stanchina, E.; Fridman, J.S.; Malina, A.; Ray, S.; Kogan, S.; Cordon-Cardo, C.; Pelletier, J.; Lowe, S.W. Survival signalling by Akt and eIF4E in oncogenesis and cancer therapy. Nature 2004, 428, 332–337. [Google Scholar] [CrossRef] [PubMed]
- Ao, P.; Galas, D.; Hood, L.; Zhu, X.M. Cancer as robust intrinsic state of endogenous molecular-cellular network shaped by evolution. Med. Hypotheses 2008, 70, 678–684. [Google Scholar] [CrossRef]
- Huang, S. Gene expression profiling, genetic networks, and cellular states: An integrating concept for tumorigenesis and drug discovery. J. Mol. Med. 1999, 77, 469–480. [Google Scholar]
- Zhang, J.; Zhang, L.L.; Shen, L.; Xu, X.M.; Yu, H.G. Regulation of AKT gene expression by cisplatin. Oncol. Lett. 2013, 5, 756–760. [Google Scholar]
- Junttila, M.R.; Puustinen, P.; Niemela, M.; Ahola, R.; Arnold, H.; Bottzauw, T.; Ala-Aho, R.; Nielsen, C.; Ivaska, J.; Taya, Y.; et al. CIP2A inhibits PP2A in human malignancies. Cell 2007, 130, 51–62. [Google Scholar]
- Sents, W.; Ivanova, E.; Lambrecht, C.; Haesen, D.; Janssens, V. The biogenesis of active protein phosphatase 2A holoenzymes: A tightly regulated process creating phosphatase specificity. FEBS J. 2013, 280, 644–661. [Google Scholar] [CrossRef] [PubMed]
- Janghorban, M.; Farrell, A.S.; Allen-Petersen, B.L.; Pelz, C.; Daniel, C.J.; Oddo, J.; Langer, E.M.; Christensen, D.J.; Sears, R.C. Targeting c-MYC by antagonizing PP2A inhibitors in breast cancer. Proc. Natl. Acad. Sci. USA 2014, 111, 9157–9162. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.P.; Lai, Y.D.; Li, D.C.; Zhu, X.N.; Yang, P.; Li, W.X.; Zhu, W.; Zhao, J.; Li, X.D.; Xiao, Y.M.; et al. α4 is highly expressed in carcinogen-transformed human cells and primary human cancers. Oncogene 2011, 30, 2943–2953. [Google Scholar]
- Li, T.; Huang, P.; Liang, J.; Fu, W.Y.; Guo, Z.L.; Xu, L.H. Microcystin-LR (MCLR) Induces a Compensation of PP2A Activity Mediated by α4 Protein in HEK293 Cells. Int. J. Biol. Sci. 2011, 7, 740–752. [Google Scholar] [PubMed]
- Mumby, M. PP2A: Unveiling a reluctant tumor suppressor. Cell 2007, 130, 21–24. [Google Scholar] [CrossRef]
- Khanna, A.; Pimanda, J.E.; Westermarck, J. Cancerous inhibitor of protein phosphatase 2A, an emerging human oncoprotein and a potential cancer therapy target. Cancer Res. 2013, 73, 6548–6553. [Google Scholar]
- Wei, L.; Qu, W.; Sun, J.; Wang, X.; Lv, L.; Xie, L.; Song, X. Knockdown of cancerous inhibitor of protein phosphatase 2A may sensitize NSCLC cells to cisplatin. Cancer Gene Ther. 2014, 21, 194–199. [Google Scholar] [CrossRef]
- Dias-Santagata, D.; Akhavanfard, S.; David, S.S.; Vernovsky, K.; Kuhlmann, G.; Boisvert, S.L.; Stubbs, H.; McDermott, U.; Settleman, J.; Kwak, E.L.; et al. Rapid targeted mutational analysis of human tumours: A clinical platform to guide personalized cancer medicine. EMBO Mol. Med. 2010, 2, 146–158. [Google Scholar]
- Amzel, L.M.; Huang, C.H.; Mandelker, D.; Lengauer, C.; Gabelli, S.B.; Vogelstein, B. Structural comparisons of class I phosphoinositide 3-kinases. Nat. Rev. Cancer 2008, 8, 665–669. [Google Scholar] [CrossRef]
- Borhani, D.W.; Shaw, D.E. The future of molecular dynamics simulations in drug discovery. J. Comput. Aided Mol. Des. 2012, 26, 15–26. [Google Scholar] [CrossRef]
- Durrant, J.D.; McCammon, J.A. Molecular dynamics simulations and drug discovery. BMC Biol. 2011, 9. [Google Scholar] [CrossRef]
- Lahti, J.L.; Tang, G.W.; Capriotti, E.; Liu, T.; Altman, R.B. Bioinformatics and variability in drug response: A protein structural perspective. J. R. Soc. Interface 2012, 9, 1409–1437. [Google Scholar] [CrossRef]
- Foukas, L.C.; Berenjeno, I.M.; Gray, A.; Khwaja, A.; Vanhaesebroeck, B. Activity of any class IA PI3K isoform can sustain cell proliferation and survival. Proc. Natl. Acad. Sci. USA 2010, 107, 11381–11386. [Google Scholar] [CrossRef] [PubMed]
- Virone-Oddos, A.; Bonnevaux, H.; Lemaitre, O.; Vincent, L.; Halley, F.; Demers, B.; Charrier, V.; Courtin, O.; Guerif, S.; Besret, L. Discovery and characterization of SAR260301, a novel PI3Kβ-selective inhibitor in clinical development for the treatment of PTEN-deficient tumors. Cancer Res. 2013, 73. [Google Scholar] [CrossRef]
- Engelman, J.A. Targeting PI3K signalling in cancer: Opportunities, challenges and limitations. Nat. Rev. Cancer 2009, 9, 550–562. [Google Scholar] [CrossRef]
- So, L.; Yea, S.S.; Oak, J.S.; Lu, M.; Manmadhan, A.; Ke, Q.H.; Janes, M.R.; Kessler, L.V.; Kucharski, J.M.; Li, L.S.; et al. Selective inhibition of phosphoinositide 3-kinase p110α preserves lymphocyte function. J. Biol. Chem. 2013, 288, 5718–5731. [Google Scholar]
- Zheng, Z.H.; Amran, S.I.; Thompson, P.E.; Jennings, I.G. Isoform-selective inhibition of phosphoinositide 3-kinase: Identification of a new region of nonconserved amino acids critical for p110α inhibition. Mol. Pharmacol. 2011, 80, 657–664. [Google Scholar] [CrossRef]
- Wang, J.N.; Wang, F.F.; Xiao, Z.T.; Sheng, G.W.; Li, Y.; Wang, Y.H. Molecular simulation of a series of benzothiazole PI3Kα inhibitors: Probing the relationship between structural features, anti-tumor potency and selectivity. J. Mol. Model. 2012, 18, 2943–2958. [Google Scholar]
- Li, Y.P.; Zhang, J.Y.; He, D.L.; Liang, Q.; Wang, Y.W. Characterization of molecular recognition of Phosphoinositide-3-kinase alpha inhibitor through molecular dynamics simulation. J. Mol. Model. 2012, 18, 1907–1916. [Google Scholar]
- Zhu, J.Y.; Pan, P.C.; Li, Y.Y.; Wang, M.; Li, D.; Cao, B.Y.; Mao, X.L.; Hou, T.J. Theoretical studies on beta and delta isoform-specific binding mechanisms of phosphoinositide 3-kinase inhibitors. Mol. Biosyst. 2014, 10, 454–466. [Google Scholar] [PubMed]
- Fritsch, C.M.; Schnell, C.; Chatenay-Rivauday, C.; Guthy, D.A.; Pover, A.D.; Wartmann, M.; Brachmann, S.; Maira, S.-M.; Huang, A.; Quadt, C. NVP-BYL719, a novel PI3Kα selective inhibitor with all the characteristics required for clinical development as an anti-cancer agent. Cancer Res. 2012, 72 (Suppl. S8), 3748. [Google Scholar] [CrossRef]
- Jessen, K.; Kessler, L.; Kucharski, J.; Guo, X.; Staunton, J.; Janes, M.; Elia, M.; Banerjee, U.; Lan, L.; Wang, S. Abstract A171: A potent and selective PI3K inhibitor, INK1117, targets human cancers harboring oncogenic PIK3CA mutations. Mol. Cancer Ther. 2011, 10. [Google Scholar] [CrossRef]
- Yang, Q.; Chen, L.S.; Neelapu, S.S.; Lanutti, B.J.; Gandhi, V. PI3Kδ inhibitor, GS-1101, impacts transcription and translation in mantle cell lymphoma. In Proceedings of the 104th Annual Meeting of the American Association for Cancer Research, Washington, DC, USA, 6–10 April 2013. Abstract nr 3265.
- Macias-Perez, I.M.; Flinn, I.W. GS-1101: A Delta-Specific PI3K Inhibitor in Chronic Lymphocytic Leukemia. Curr. Hematol. Malig. Rep. 2013, 8, 22–27. [Google Scholar] [CrossRef]
- Wong, M.H.; Xue, A.; Julovi, S.M.; Pavlakis, N.; Samra, J.S.; Hugh, T.J.; Gill, A.J.; Peters, L.; Baxter, R.C.; Smith, R.C. Cotargeting of epidermal growth factor receptor and pI3K overcomes PI3K-Akt oncogenic dependence in pancreatic ductal adenocarcinoma. Clin. Cancer Res. 2014, 20, 4047–4058. [Google Scholar] [CrossRef] [PubMed]
- Brake, R.L.; Fabrey, R.; Szwaya, J.; Fitzgerald, M.; Iartchouk, N.; Guo, X.; K., K.; Zohren, F.; Manfredi, M. The combination of mTORC1/2 and PI3Kα inhibition alleviates PI3K pathway reactivation and leads to significant antitumor activity in multiple preclinical xenograft models. In Proceedings of the AACR-NCI-EORTC International Conference: Molecular Targets and Cancer Therapeutics, Boston, MA, USA, 19–23 October 2013. Abstract nr C176.
- Sabbah, D.A.; Vennerstrom, J.L.; Zhong, H.Z. Docking studies on isoform-specific inhibition of phosphoinositide-3-kinases. J. Chem. Inf. Model. 2010, 50, 1887–1898. [Google Scholar] [CrossRef]
- Ohwada, J.; Ebiike, H.; Kawada, H.; Tsukazaki, M.; Nakamura, M.; Miyazaki, T.; Morikami, K.; Yoshinari, K.; Yoshida, M.; Kondoh, O.; et al. Discovery and biological activity of a novel class I PI3K inhibitor, CH5132799. Bioorg. Med. Chem. Lett. 2011, 21, 1767–1772. [Google Scholar]
- Liu, J.L.; Gao, G.R.; Zhang, X.; Cao, S.F.; Guo, C.L.; Wang, X.; Tong, L.J.; Ding, J.; Duan, W.H.; Meng, L.H. DW09849, a selective phosphatidylinositol 3-kinase (PI3K) inhibitor, prevents PI3K signaling and preferentially inhibits proliferation of cells containing the oncogenic mutation p110α (H1047R). J. Pharmacol. Exp. Ther. 2014, 348, 432–441. [Google Scholar] [CrossRef]
- Tanaka, H.; Yoshida, M.; Tanimura, H.; Fujii, T.; Sakata, K.; Tachibana, Y.; Ohwada, J.; Ebiike, H.; Kuramoto, S.; Morita, K.; et al. The selective class I PI3K inhibitor CH5132799 targets human cancers harboring oncogenic PIK3CA mutations. Clin. Cancer Res. 2011, 17, 3272–3281. [Google Scholar]
- Chen, S.F.; Cao, Y.; Chen, J.J.; Chen, J.Z. Binding selectivity studies of PKBα using molecular dynamics simulation and free energy calculations. J. Mol. Model. 2013, 19, 5097–5112. [Google Scholar]
- Wu, F.; Hou, X.Y.; Luo, H.; Zhou, M.; Zhang, W.J.; Ding, Z.Y.; Li, R. Exploring the selectivity of PI3Kα and mTOR inhibitors by 3D-QSAR, molecular dynamics simulations and MM/GBSA binding free energy decomposition. Medchemcomm 2013, 4, 1482–1496. [Google Scholar]
- Cheng, S.; Niv, M.Y. Molecular dynamics simulations and elastic network analysis of protein kinase B (Akt/PKB) inactivation. J. Chem. Inf. Model. 2010, 50, 1602–1610. [Google Scholar] [CrossRef]
- Calleja, V.; Laguerre, M.; Parker, P.J.; Larijani, B. Role of a novel PH-kinase domain interface in PKB/Akt regulation: Structural mechanism for allosteric inhibition. PLoS Biol. 2009, 7, e17. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, T.; Wang, G. Computer-Aided Targeting of the PI3K/Akt/mTOR Pathway: Toxicity Reduction and Therapeutic Opportunities. Int. J. Mol. Sci. 2014, 15, 18856-18891. https://doi.org/10.3390/ijms151018856
Li T, Wang G. Computer-Aided Targeting of the PI3K/Akt/mTOR Pathway: Toxicity Reduction and Therapeutic Opportunities. International Journal of Molecular Sciences. 2014; 15(10):18856-18891. https://doi.org/10.3390/ijms151018856
Chicago/Turabian StyleLi, Tan, and Guanyu Wang. 2014. "Computer-Aided Targeting of the PI3K/Akt/mTOR Pathway: Toxicity Reduction and Therapeutic Opportunities" International Journal of Molecular Sciences 15, no. 10: 18856-18891. https://doi.org/10.3390/ijms151018856
APA StyleLi, T., & Wang, G. (2014). Computer-Aided Targeting of the PI3K/Akt/mTOR Pathway: Toxicity Reduction and Therapeutic Opportunities. International Journal of Molecular Sciences, 15(10), 18856-18891. https://doi.org/10.3390/ijms151018856