Small Molecule Inhibitors of AI-2 Signaling in Bacteria: State-of-the-Art and Future Perspectives for Anti-Quorum Sensing Agents

Abstract
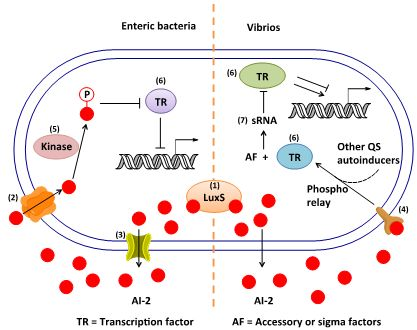
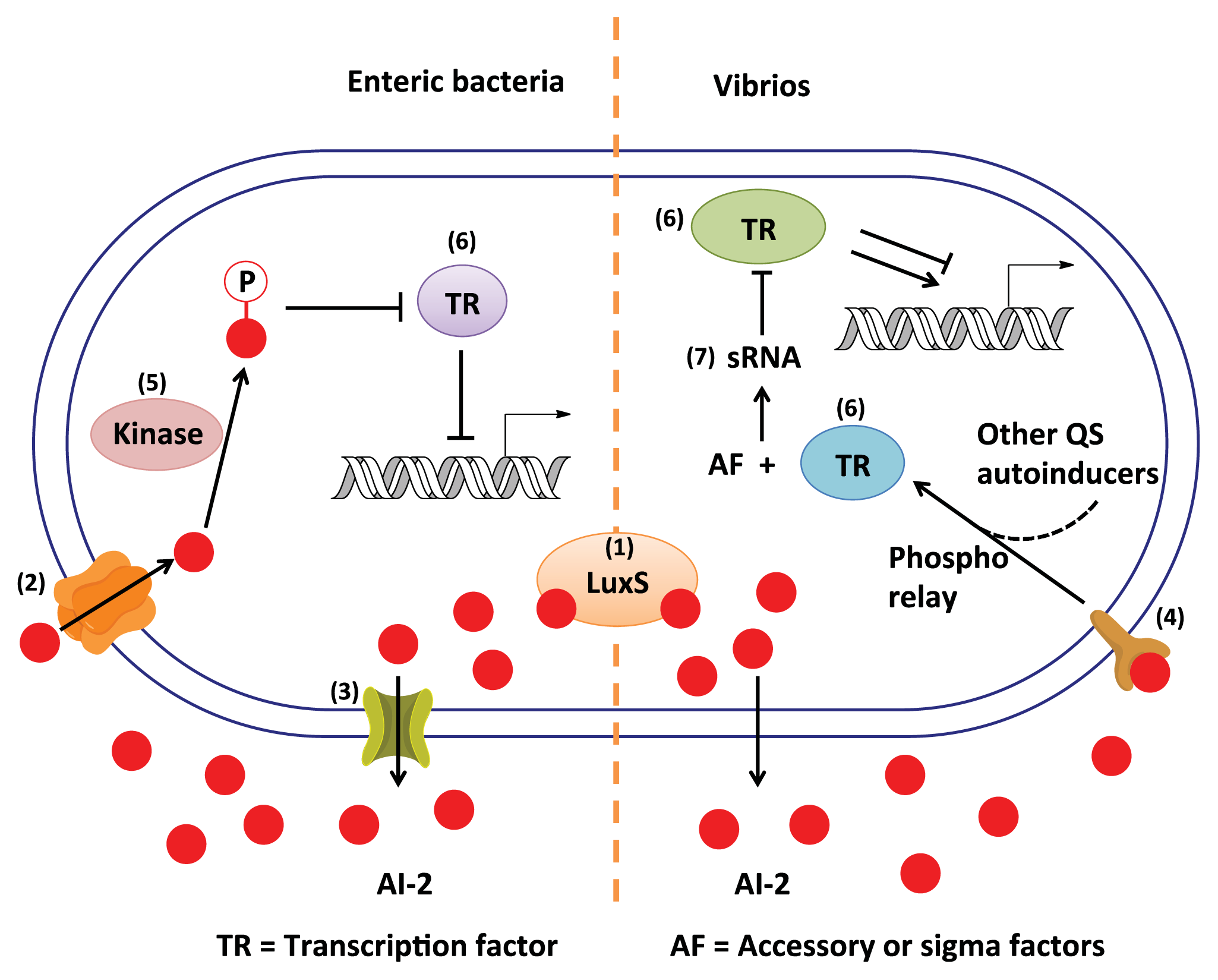
:
1. Introduction
1.1. A Paradigm Shift from Bactericidal and Bacteriostatic Agents to Anti-Virulence Agents
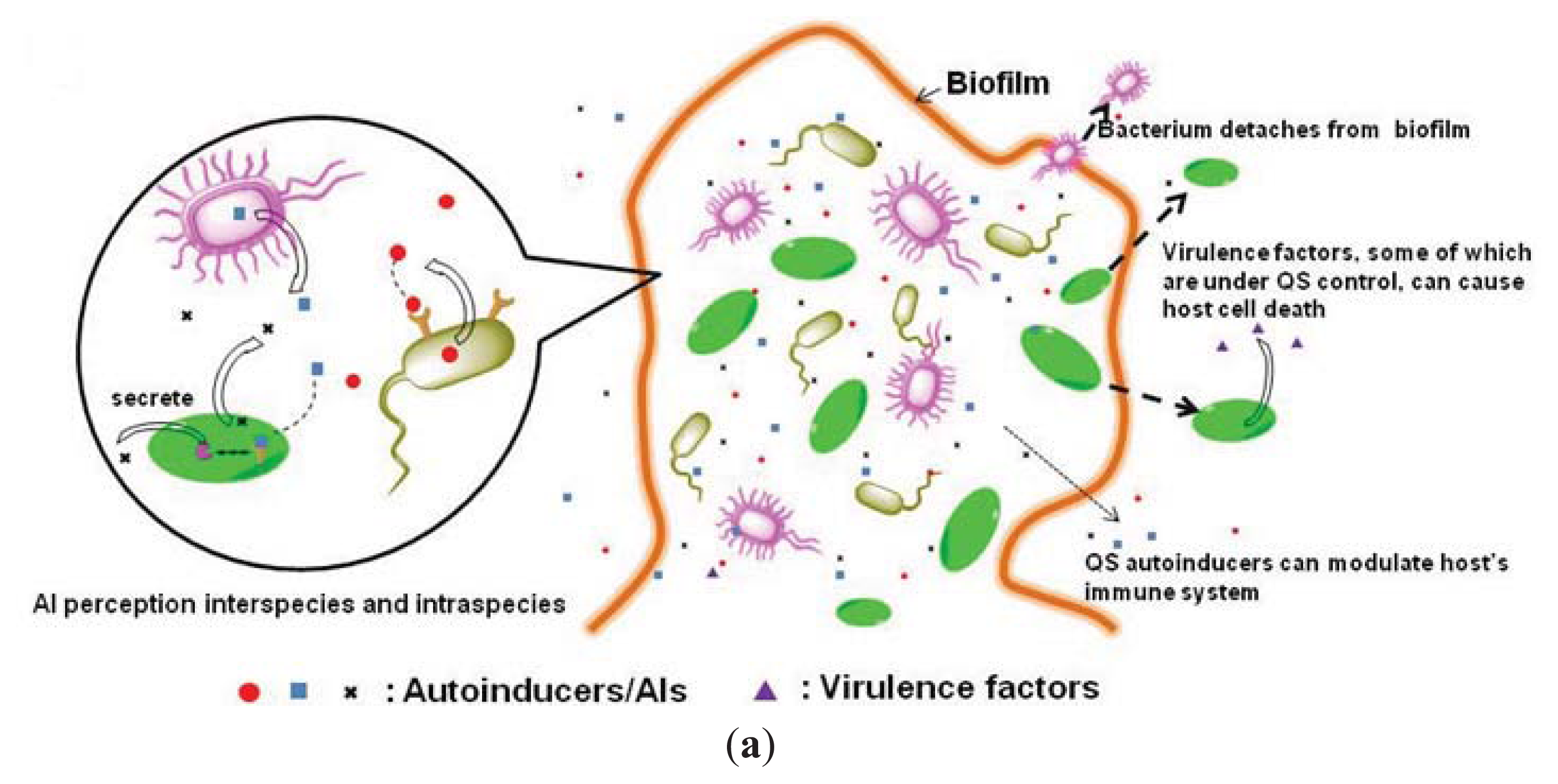
1.2. Inhibition of Quorum-Sensing as an Anti-Virulence Strategy
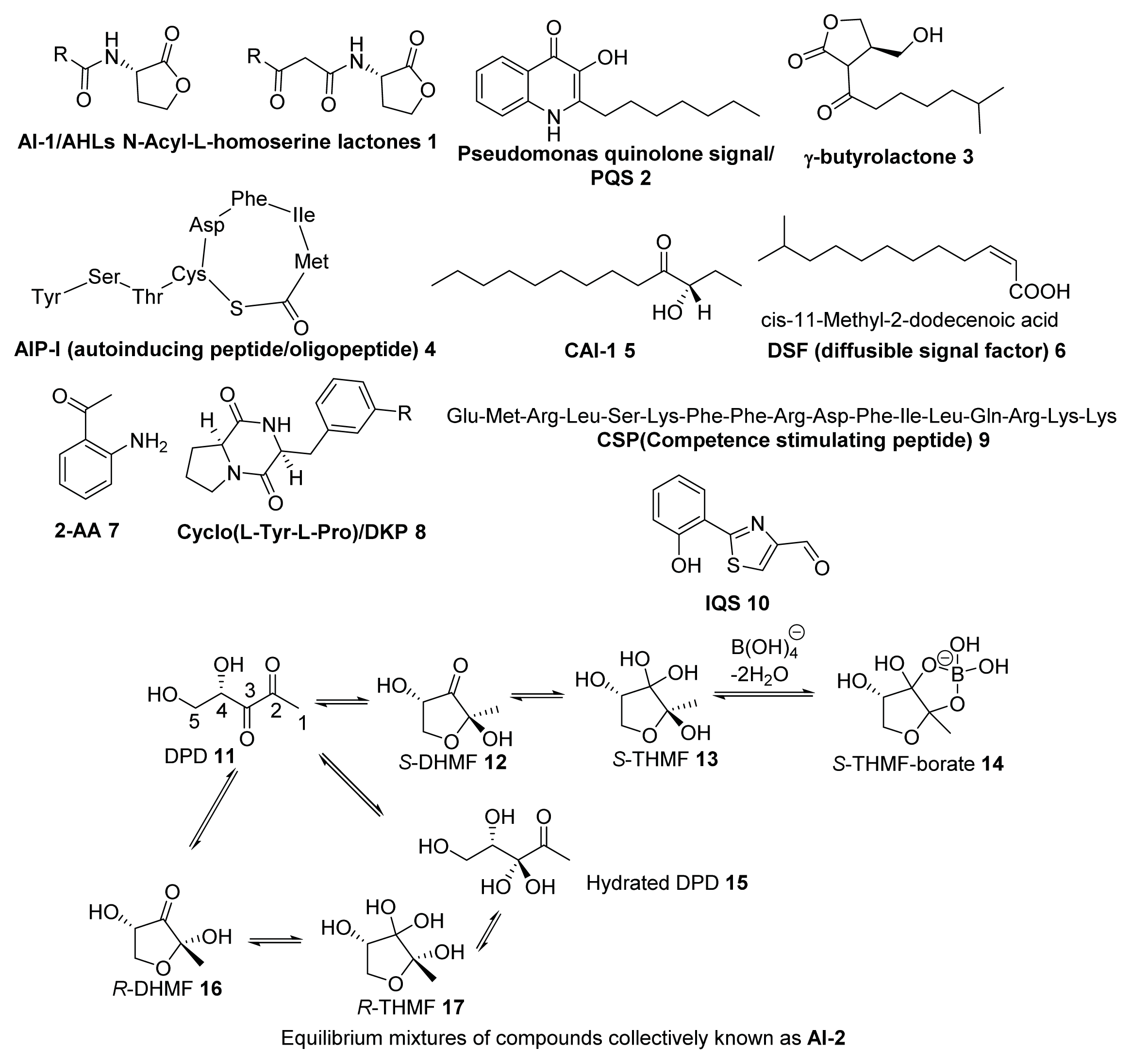
2. Synthesis of AI-2
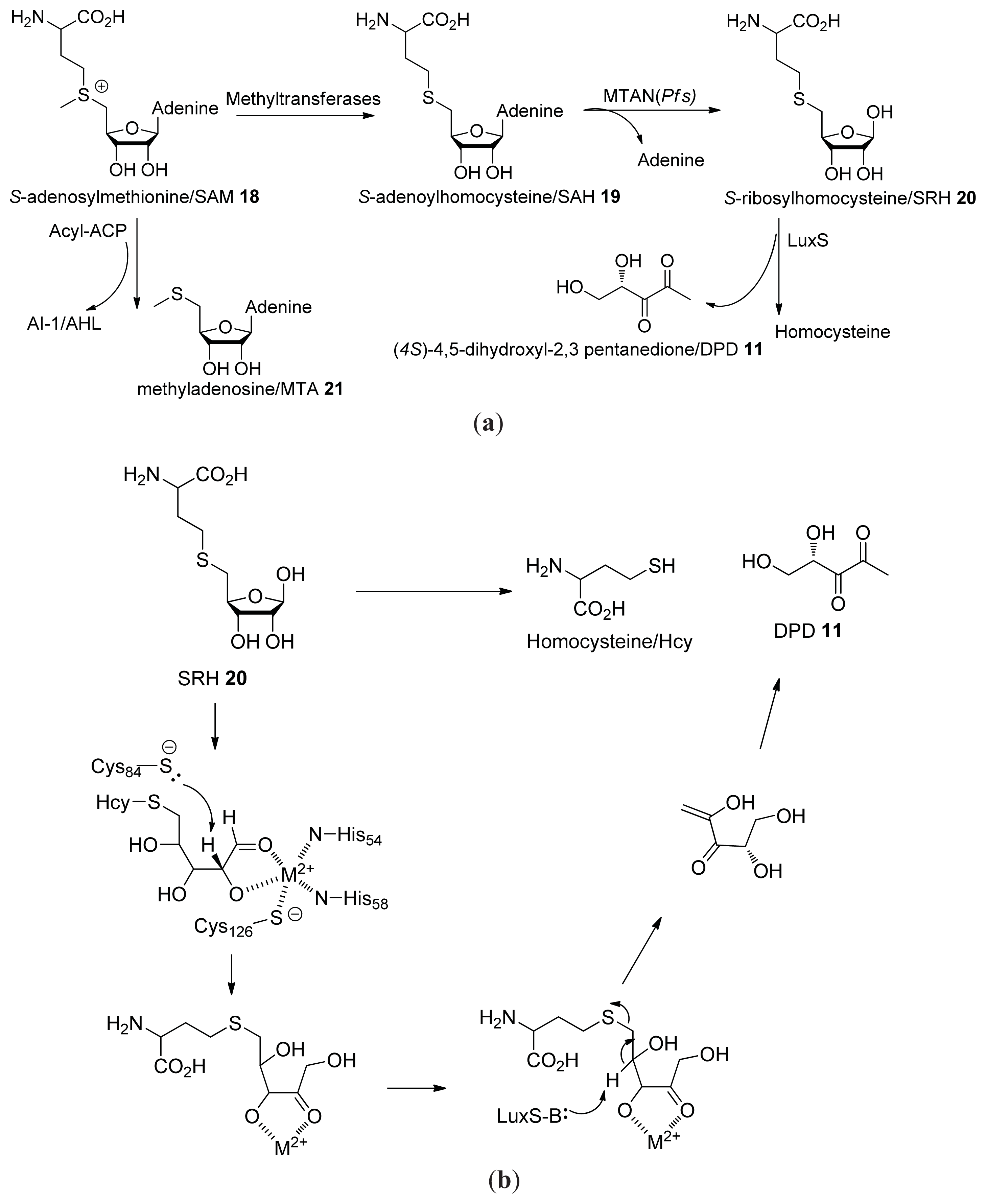
2.1. Biosynthesis of AI-2
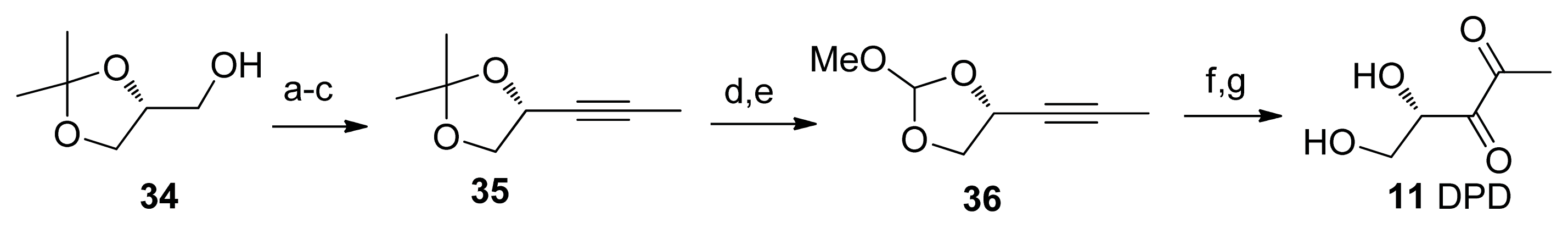
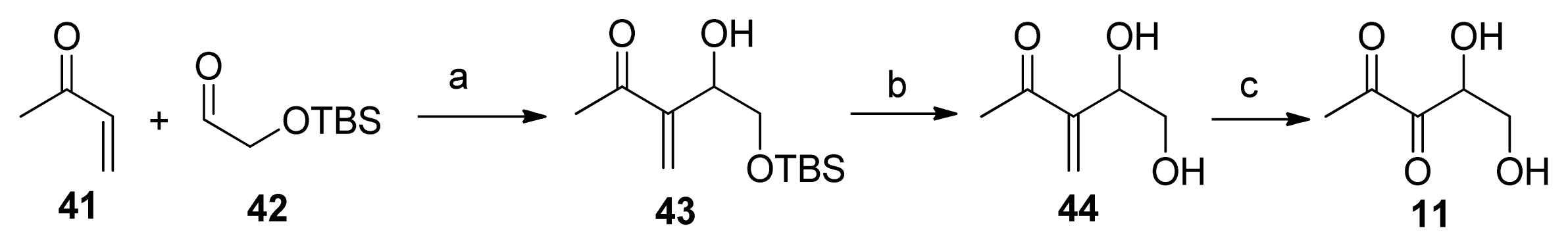
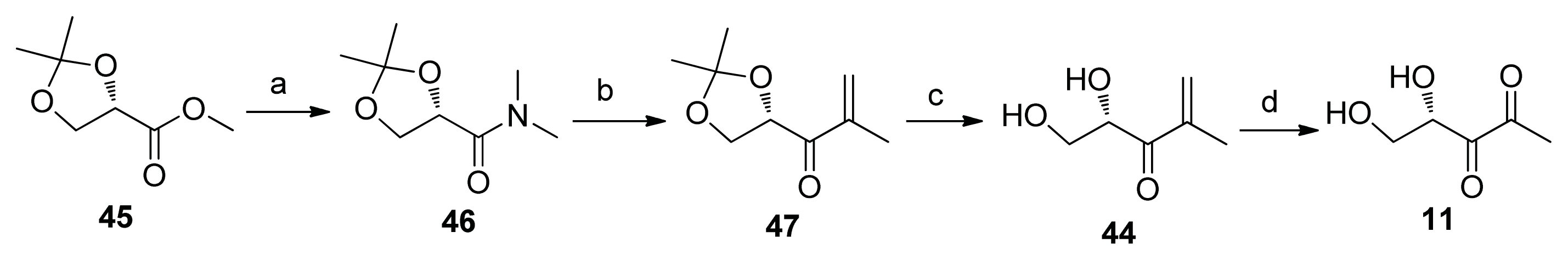
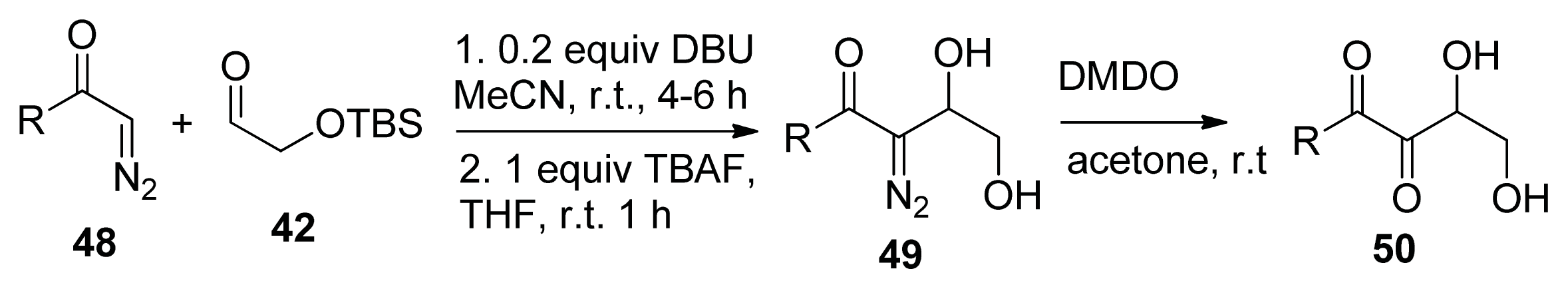
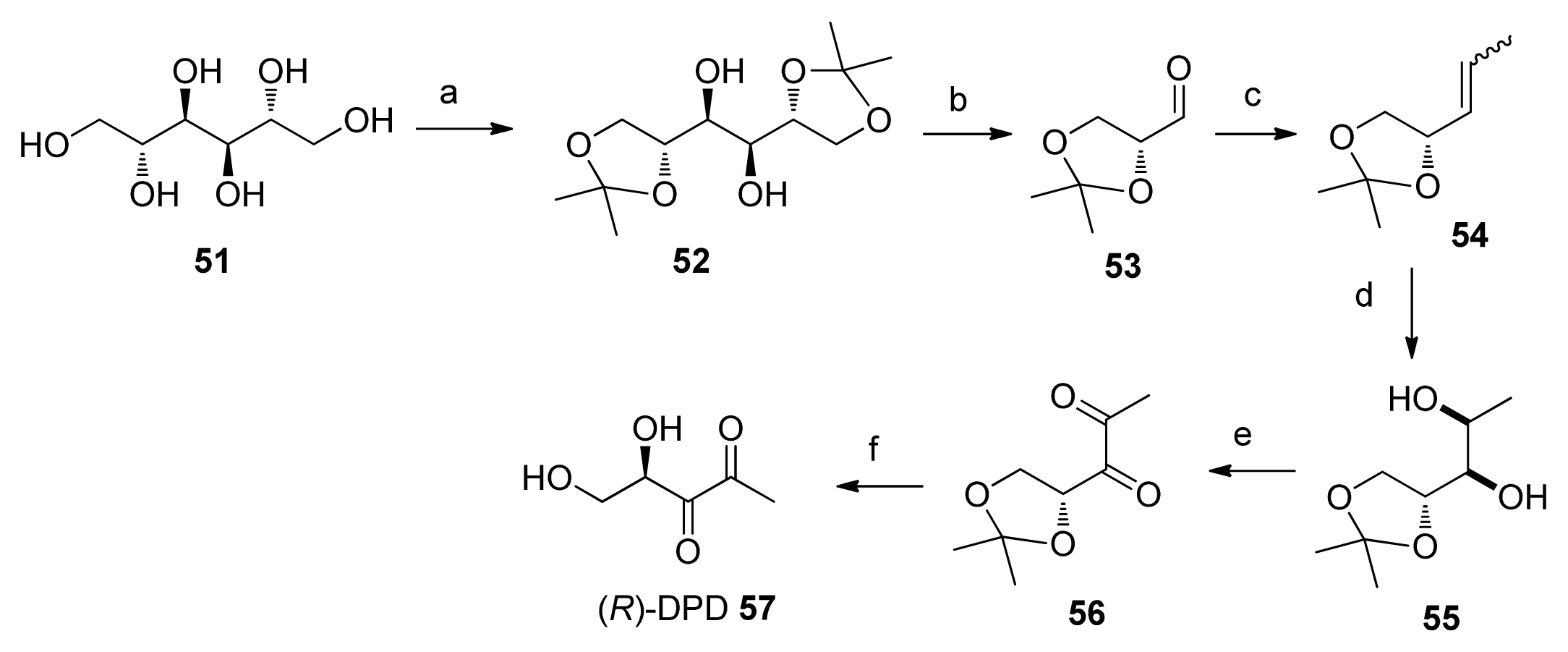
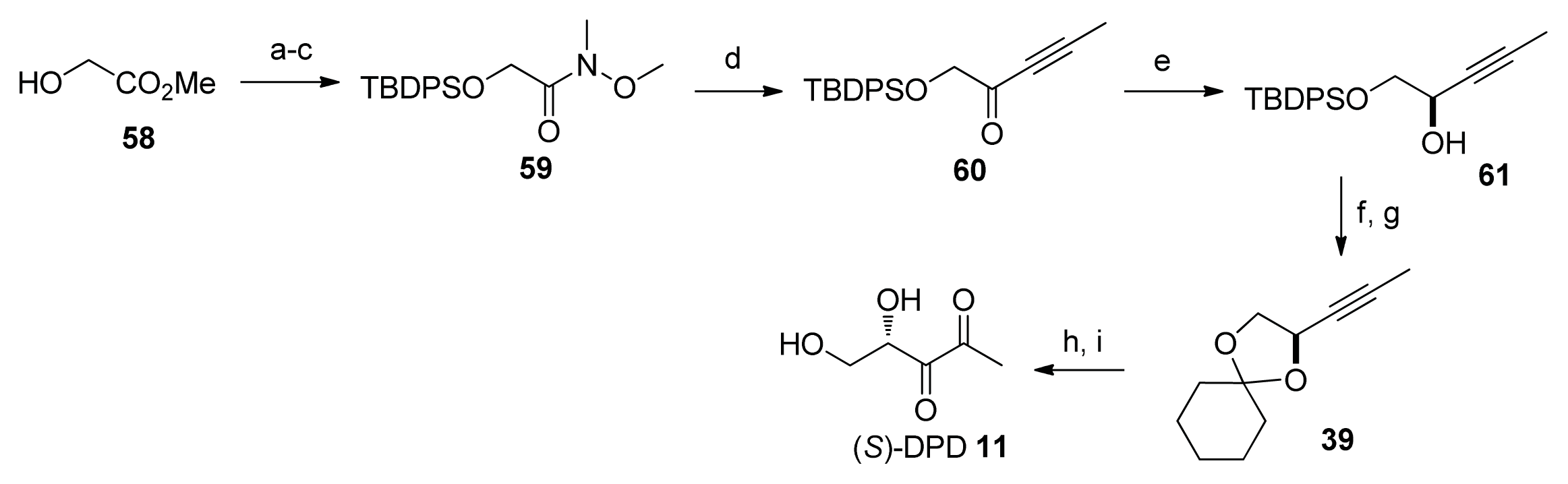
2.2. Chemical Synthesis of AI-2
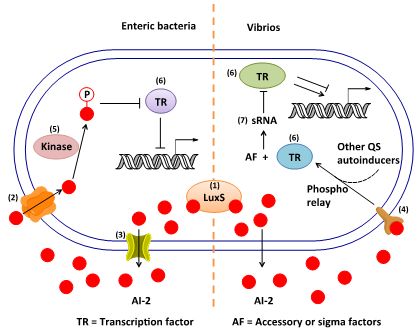
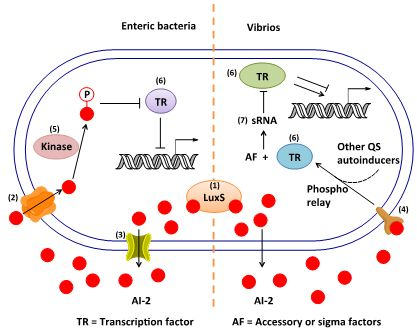
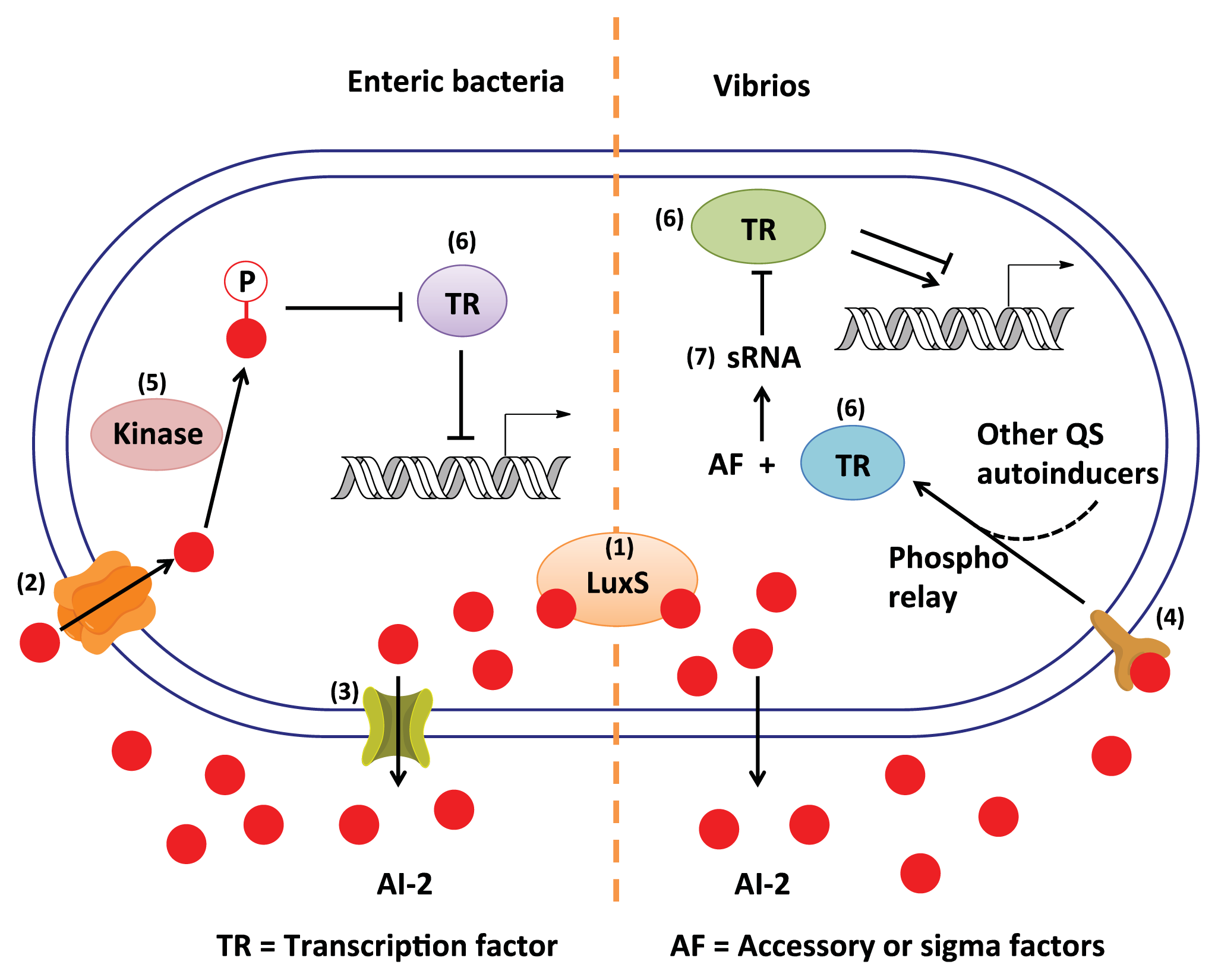
3. AI-2 Signaling Pathway
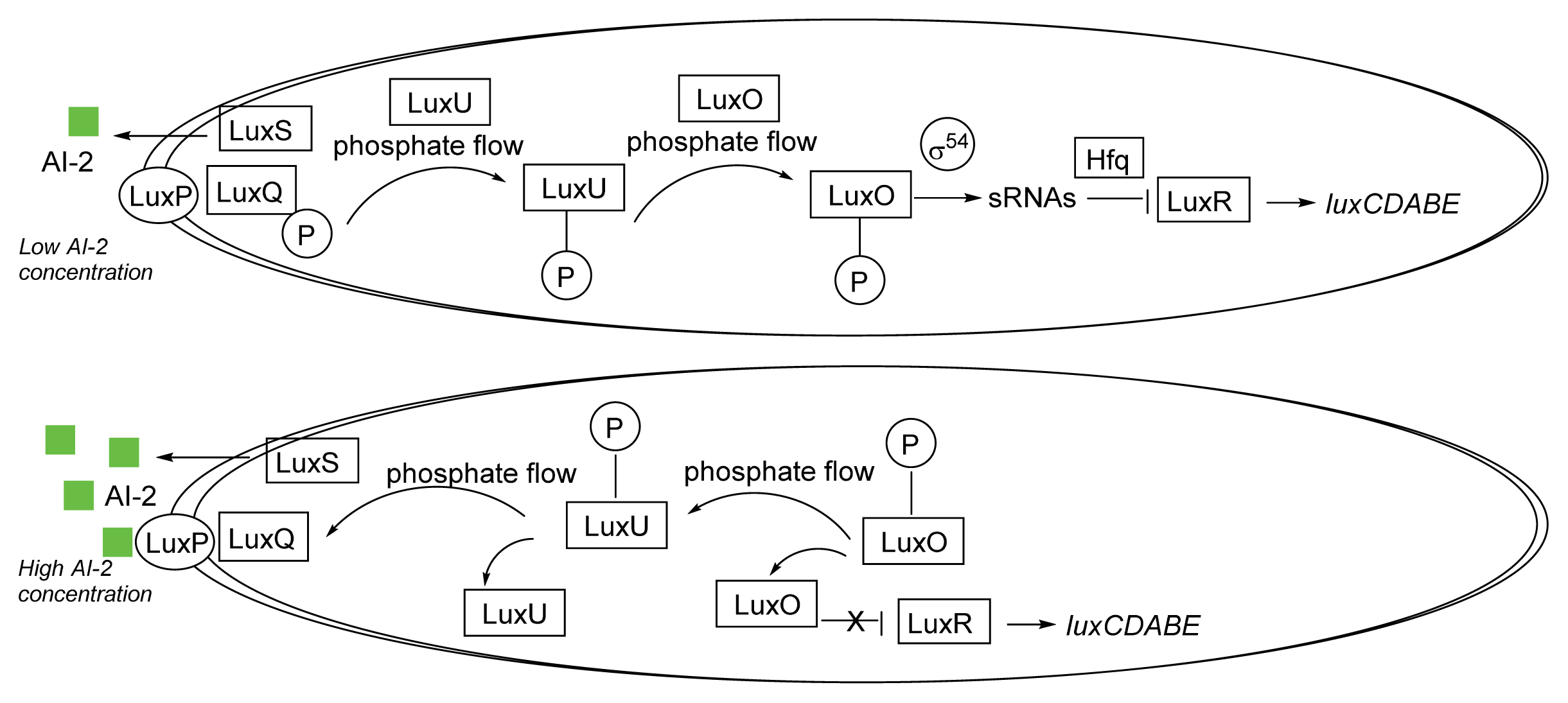
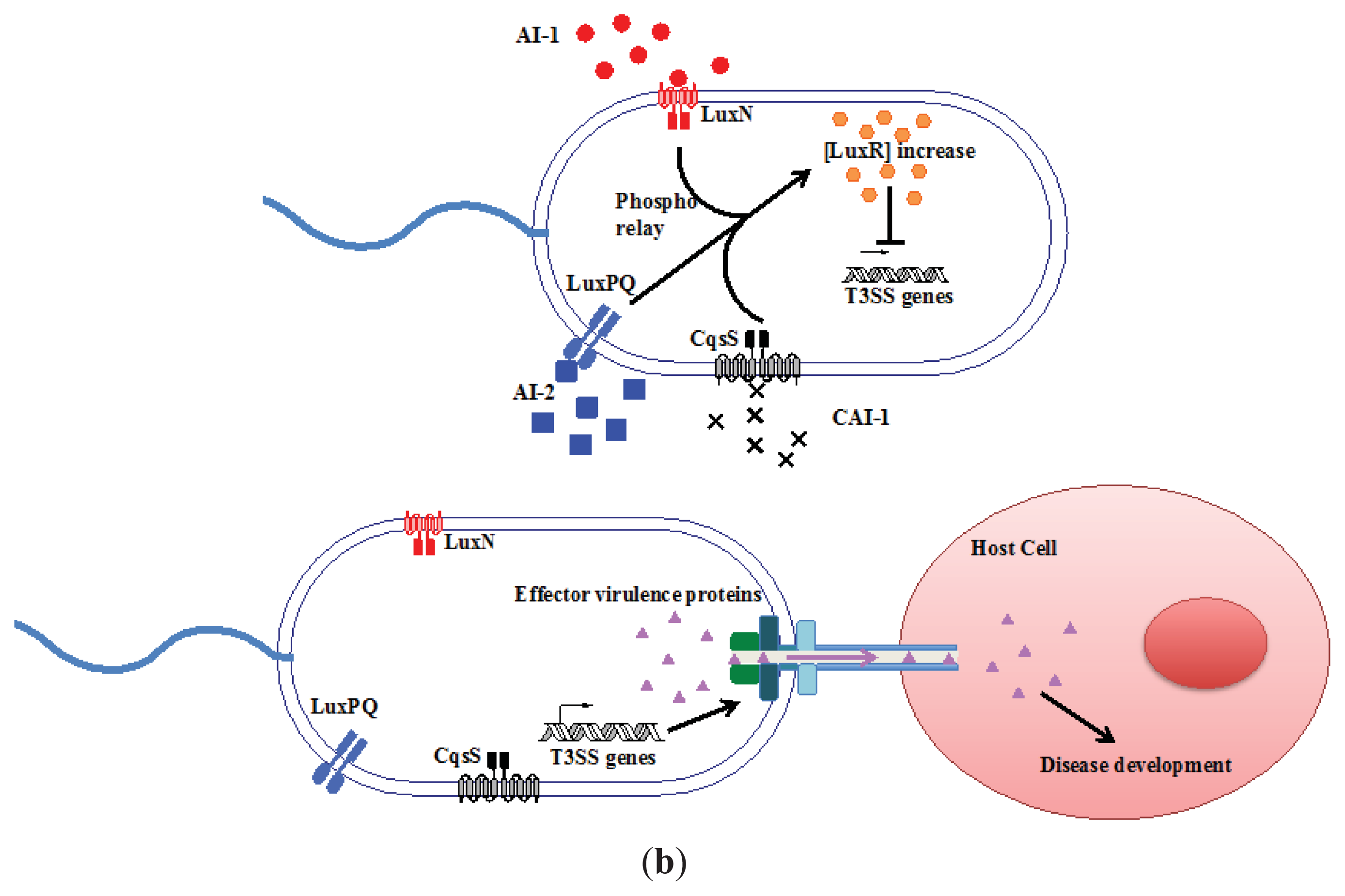
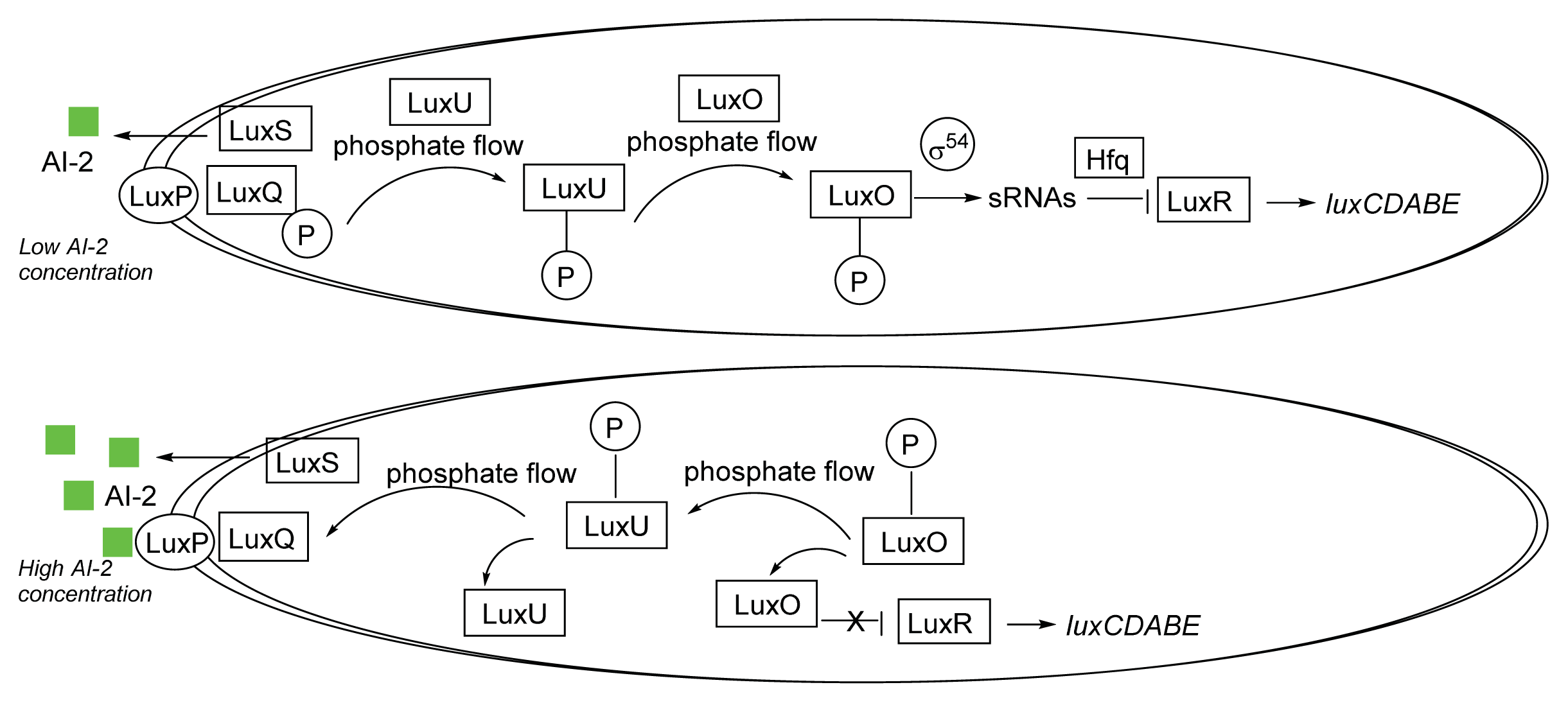
3.1. AI-2 Mediated QS Circuit in V. harveyi and V. cholerae
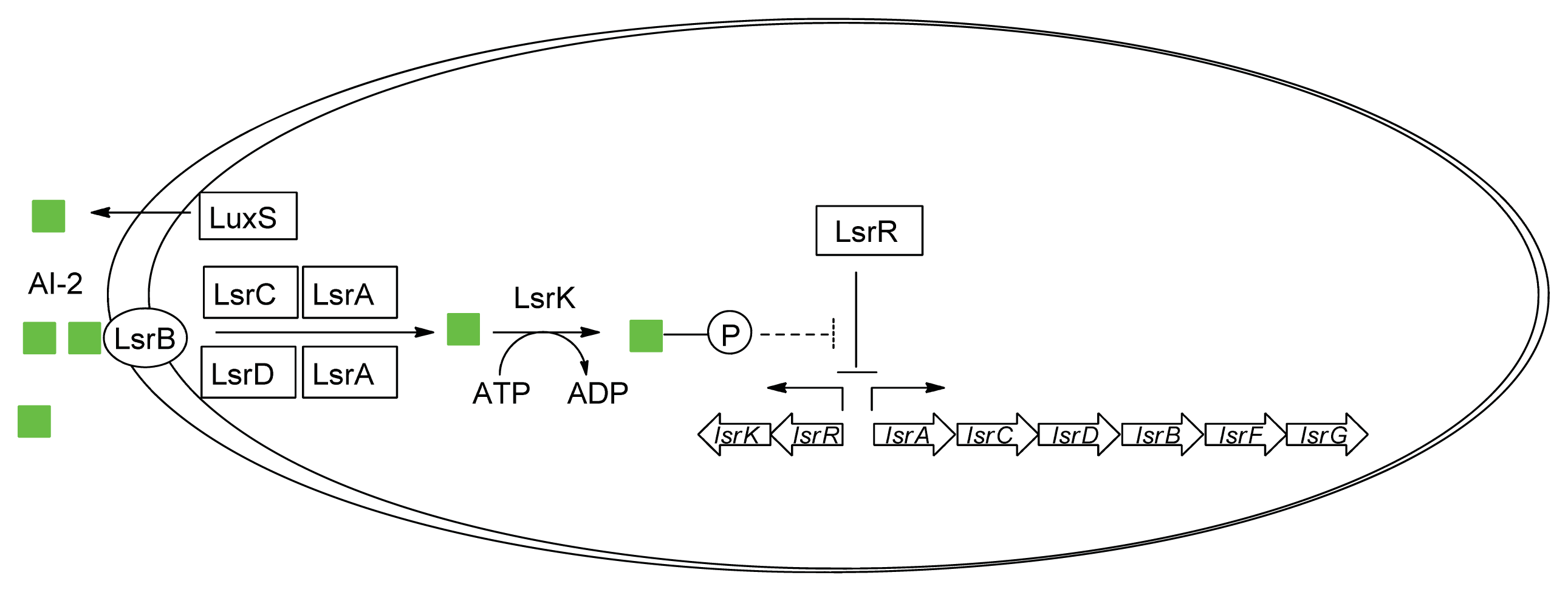
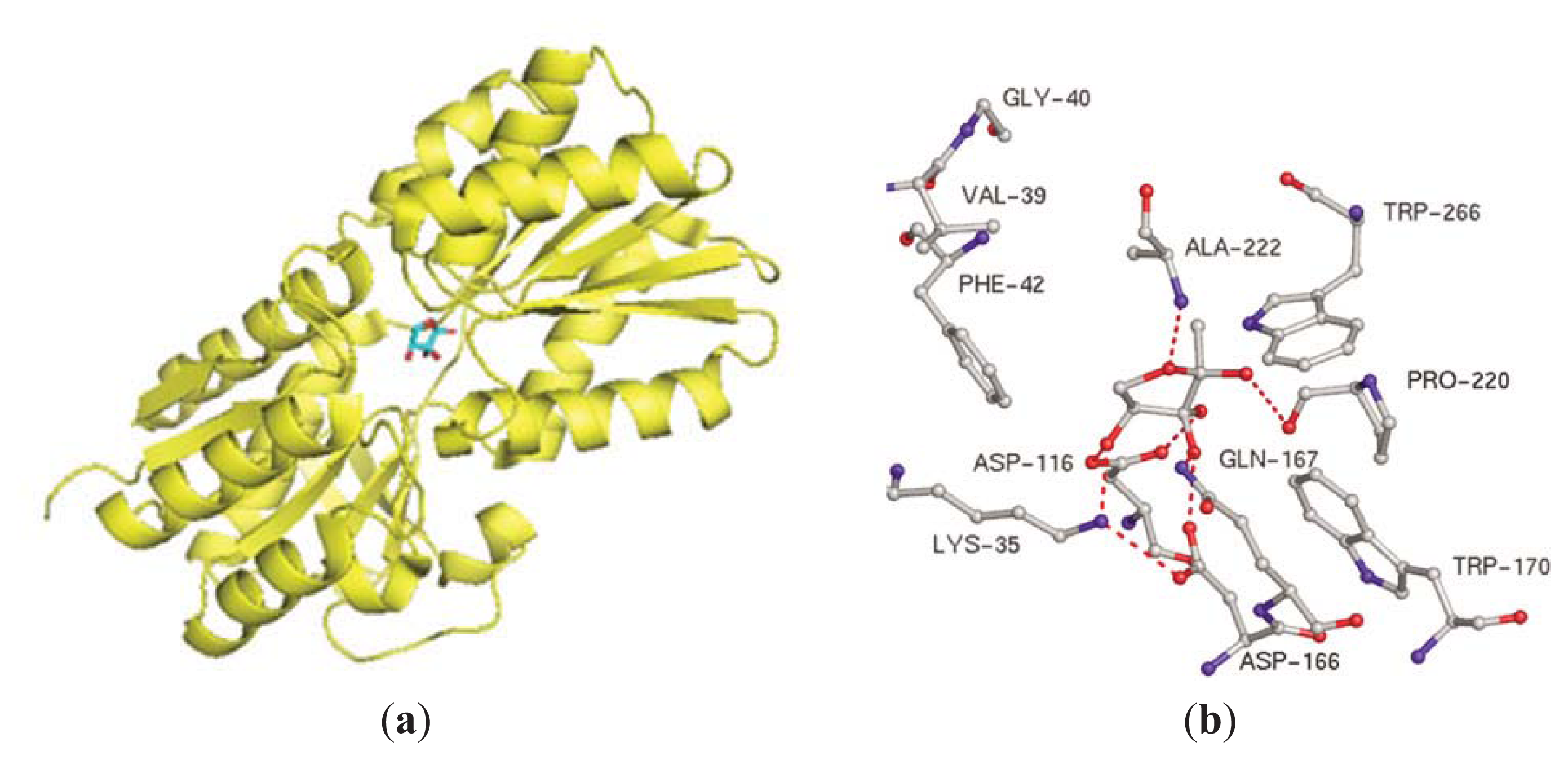
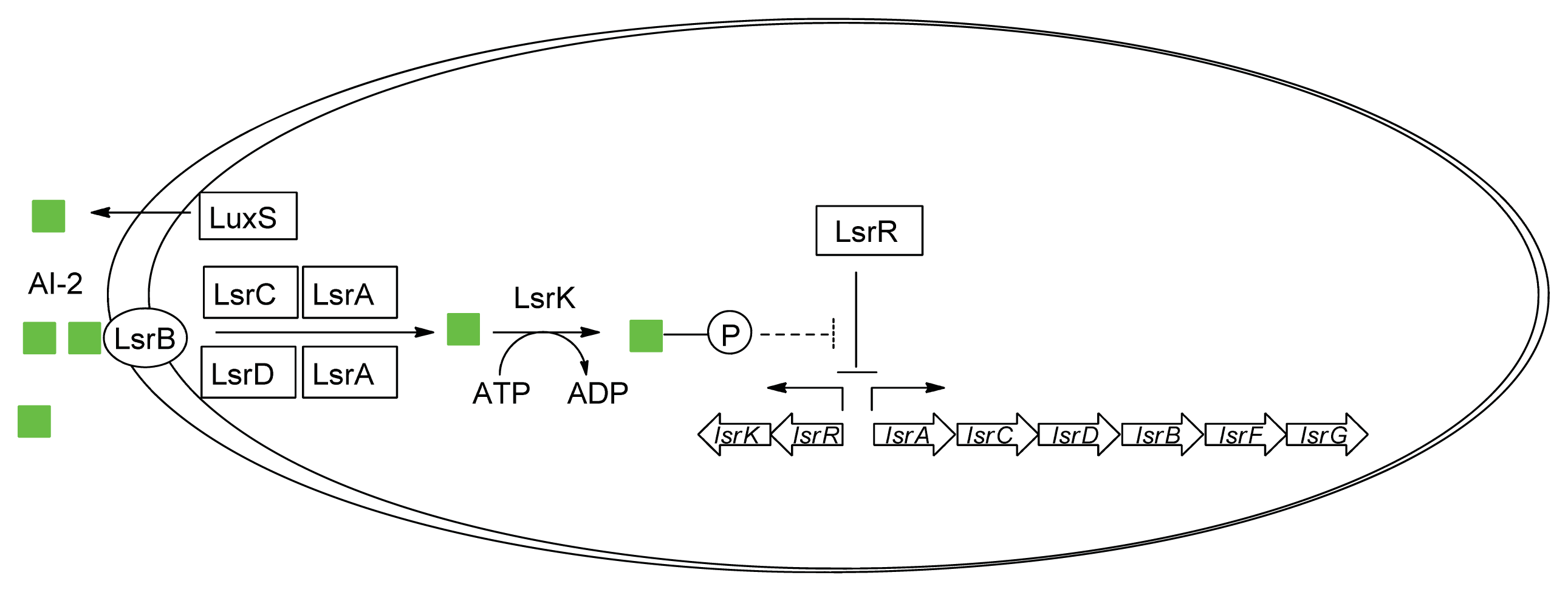
3.2. AI-2 Mediated QS Circuit in E. coli or S. typhimurium
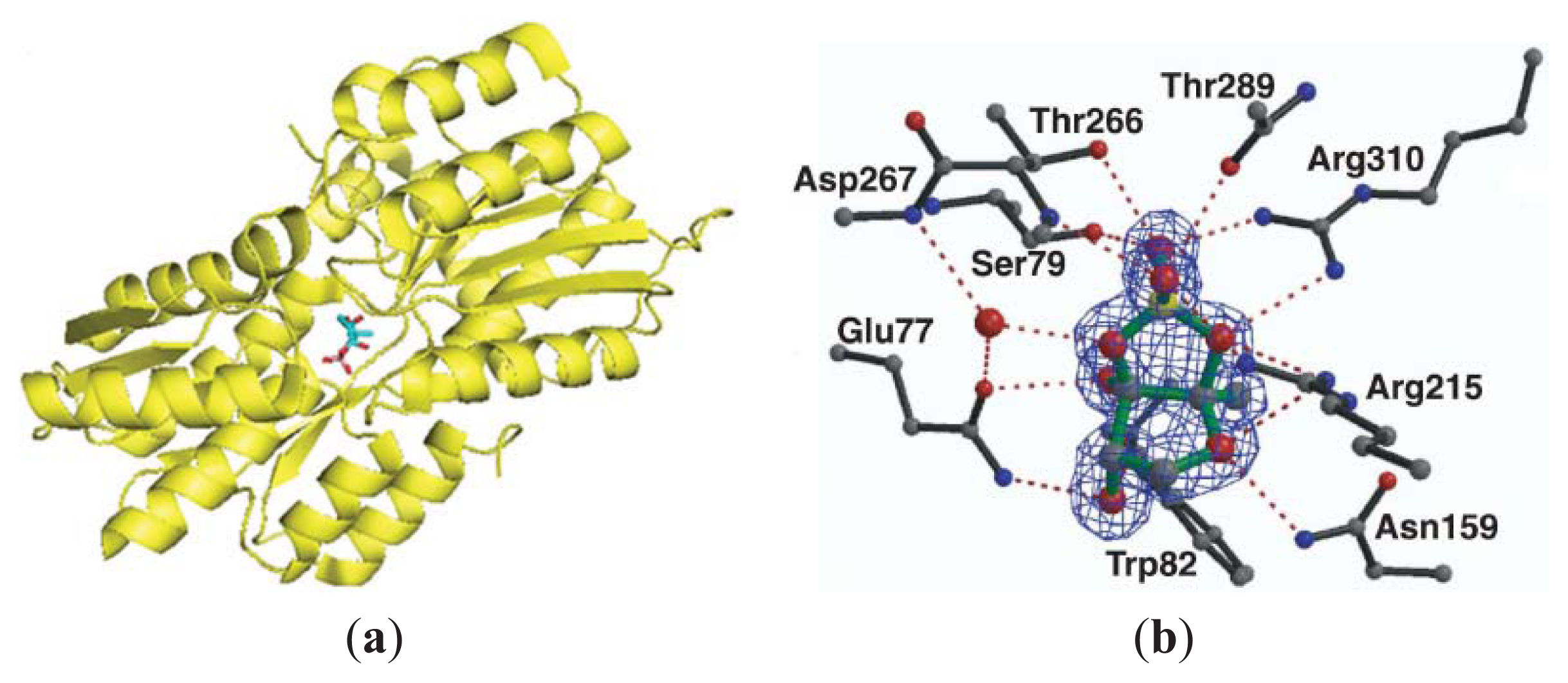
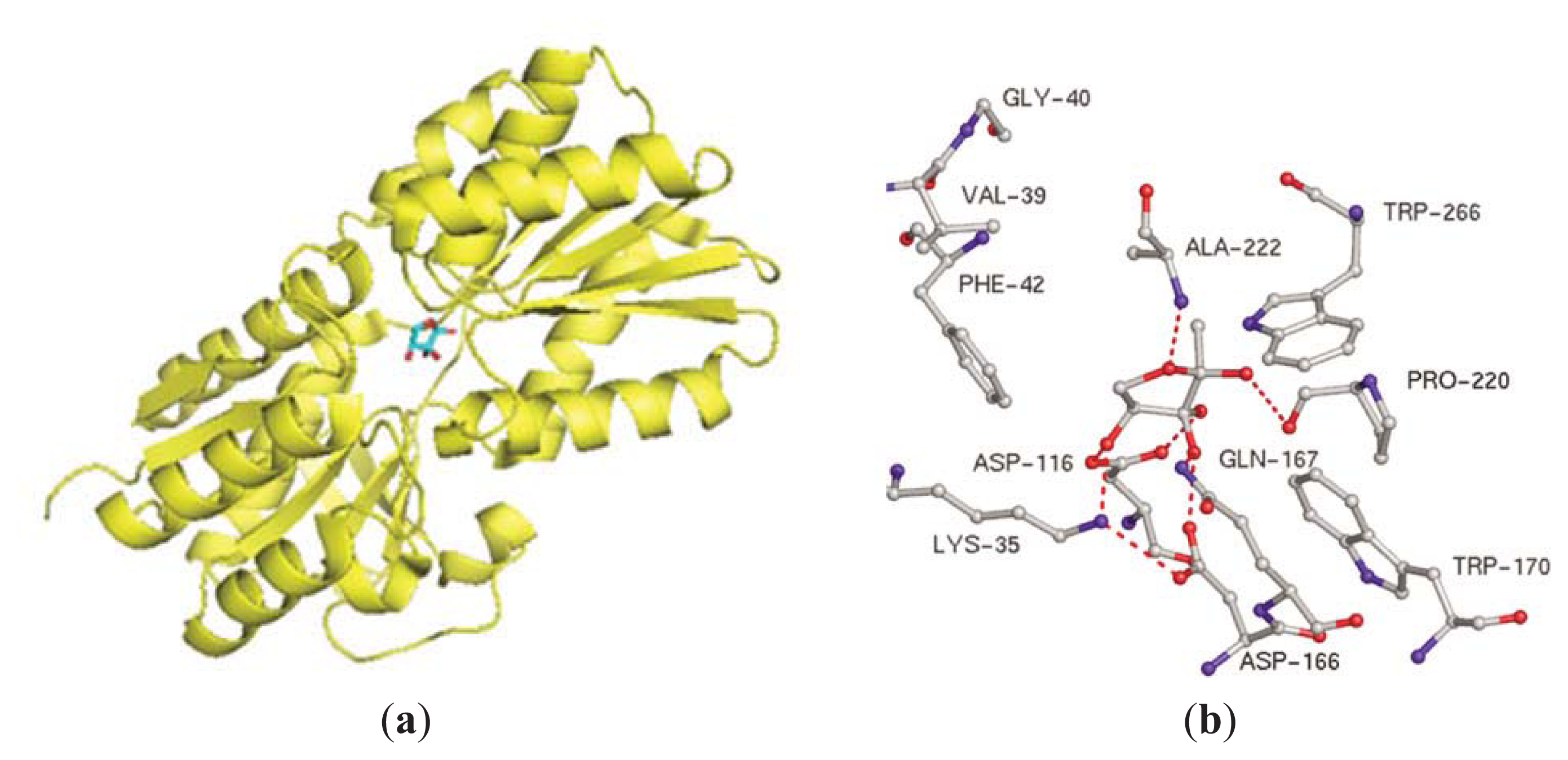
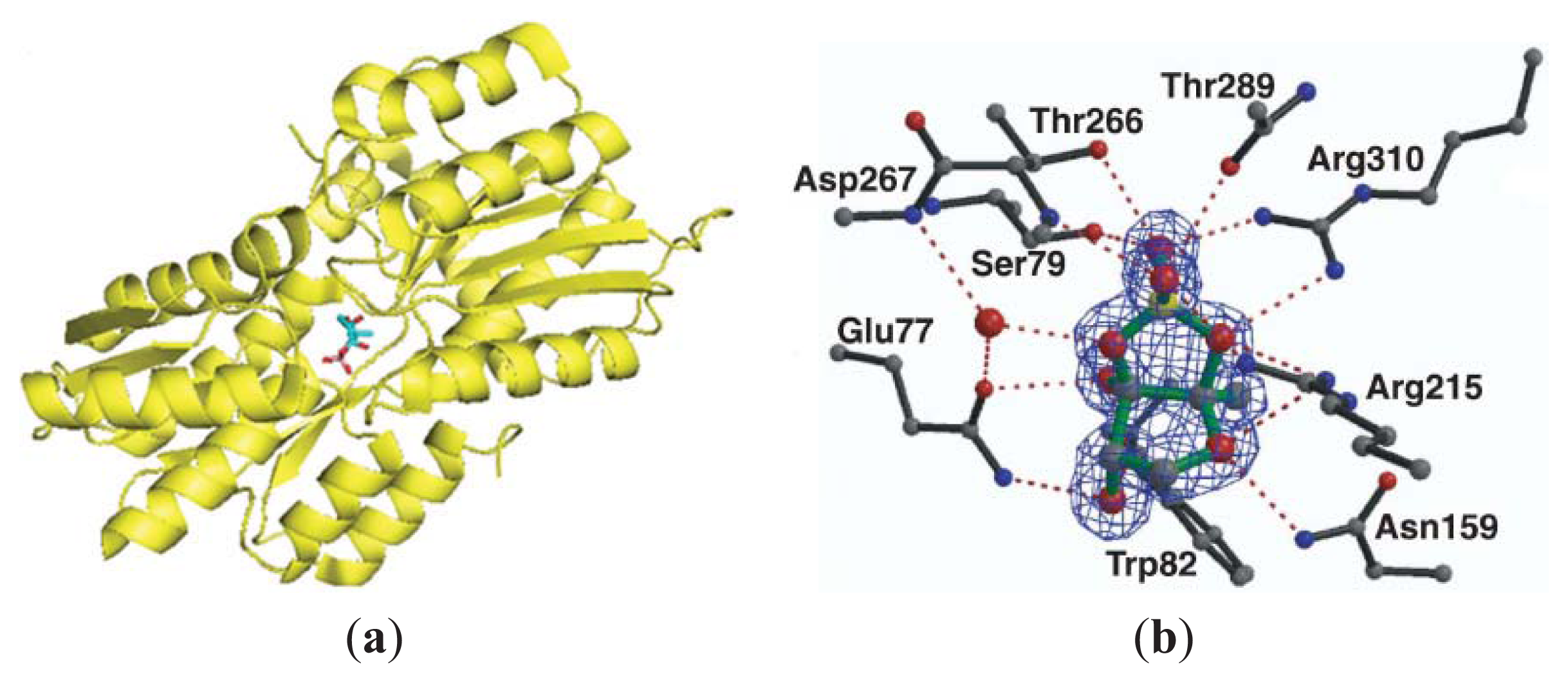
3.3. Other Possible AI-2 Receptors
4. Small Molecule Inhibitors of AI-2 Signaling
4.1. AI-2 Synthase Inhibitors
4.2. AI-2 Receptor QS Inhibitors
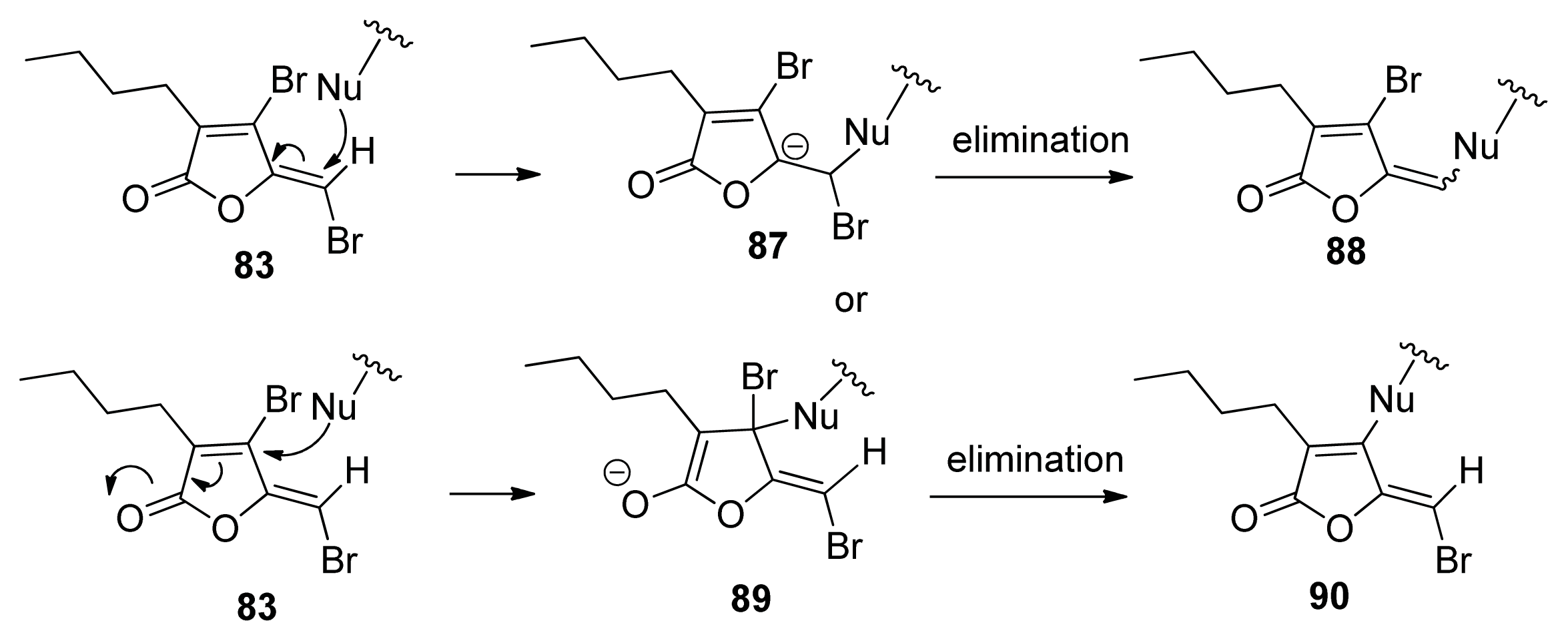
4.3. AI-2 Analogs as QS Inhibitors
4.4. Inhibition of AI-2 QS by Dietary Compounds
5. Conclusions
Acknowledgements
Conflicts of Interest
References
- Mullard, A. 2010 FDA drug approvals. Nat. Rev. Drug Discov 2011, 10, 82–85. [Google Scholar]
- Mullard, A. 2011 FDA drug approvals. Nat. Rev. Drug Discov 2012, 11, 91–94. [Google Scholar]
- Mullard, A. 2012 FDA drug approvals. Nat. Rev. Drug Discov 2013, 12, 87–90. [Google Scholar]
- Sintim, H.O.; Smith, J.A.; Wang, J.; Nakayama, S.; Yan, L. Paradigm shift in discovering next-generation anti-infective agents: Targeting quorum sensing, c-di-GMP signaling and biofilm formation in bacteria with small molecules. Future Med. Chem 2010, 2, 1005–1035. [Google Scholar]
- Bassler, B.L. How bacteria talk to each other: Regulation of gene expression by quorum sensing. Curr. Opin. Microbiol 1999, 2, 582–587. [Google Scholar]
- Antunes, L.C.; Ferreira, R.B.; Buckner, M.M.; Finlay, B.B. Quorum sensing in bacterial virulence. Microbiology 2010, 156, 2271–2282. [Google Scholar]
- Fuqua, W.C.; Winans, S.C.; Greenberg, E.P. Quorum sensing in bacteria: The LuxR-LuxI family of cell density-responsive transcriptional regulators. J. Bacteriol 1994, 176, 269–275. [Google Scholar]
- Tomasz, A. Control of the competent state in Pneumococcus by a hormone-like cell product: An example for a new type of regulatory mechanism in bacteria. Nature 1965, 208, 155–159. [Google Scholar]
- Lazazzera, B.A.; Grossman, A.D. The ins and outs of peptide signaling. Trends Microbiol 1998, 6, 288–294. [Google Scholar]
- Nealson, K.H.; Platt, T.; Hastings, J.W. Cellular control of the synthesis and activity of the bacterial luminescent system. J. Bacteriol 1970, 104, 313–322. [Google Scholar]
- Nealson, K.H. Autoinduction of bacterial luciferase. Occurrence, mechanism and significance. Arch. Microbiol 1977, 112, 73–79. [Google Scholar]
- Henke, J.M.; Bassler, B.L. Quorum sensing regulates type III secretion in Vibrio harveyi and Vibrio parahaemolyticus. J. Bacteriol 2004, 186, 3794–3805. [Google Scholar]
- Eberhard, A.; Burlingame, A.L.; Eberhard, C.; Kenyon, G.L.; Nealson, K.H.; Oppenheimer, N.J. Structural identification of autoinducer of Photobacterium fischeri luciferase. Biochemistry 1981, 20, 2444–2449. [Google Scholar]
- Magnuson, R.; Solomon, J.; Grossman, A.D. Biochemical and genetic characterization of a competence pheromone from B. subtilis. Cell 1994, 77, 207–216. [Google Scholar]
- Chen, X.; Schauder, S.; Potier, N.; Van Dorsselaer, A.; Pelczer, I.; Bassler, B.L.; Hughson, F.M. Structural identification of a bacterial quorum-sensing signal containing boron. Nature 2002, 415, 545–549. [Google Scholar]
- Pesci, E.C.; Milbank, J.B.; Pearson, J.P.; McKnight, S.; Kende, A.S.; Greenberg, E.P.; Iglewski, B.H. Quinolone signaling in the cell-to-cell communication system of Pseudomonas aeruginosa. Proc. Natl. Acad. Sci. USA 1999, 96, 11229–11234. [Google Scholar]
- Takano, E. Gamma-butyrolactones: Streptomyces signalling molecules regulating antibiotic production and differentiation. Curr. Opin. Microbiol 2006, 9, 287–294. [Google Scholar]
- Khokhlov, A.S.; Tovarova, I.I.; Borisova, L.N.; Pliner, S.A.; Shevchenko, L.N.; Kornitskaia, E.; Ivkina, N.S.; Rapoport, I.A. The A-factor, responsible for streptomycin biosynthesis by mutant strains of Actinomyces streptomycini. Dokl. Akad. Nauk SSSR 1967, 177, 232–235. [Google Scholar]
- Miller, M.B.; Skorupski, K.; Lenz, D.H.; Taylor, R.K.; Bassler, B.L. Parallel quorum sensing systems converge to regulate virulence in Vibrio cholerae. Cell 2002, 110, 303–314. [Google Scholar]
- Higgins, D.A.; Pomianek, M.E.; Kraml, C.M.; Taylor, R.K.; Semmelhack, M.F.; Bassler, B.L. The major Vibrio cholerae autoinducer and its role in virulence factor production. Nature 2007, 450, 883–886. [Google Scholar]
- Barber, C.E.; Tang, J.L.; Feng, J.X.; Pan, M.Q.; Wilson, T.J.; Slater, H.; Dow, J.M.; Williams, P.; Daniels, M.J. A novel regulatory system required for pathogenicity of Xanthomonas campestris is mediated by a small diffusible signal molecule. Mol. Microbiol 1997, 24, 555–566. [Google Scholar]
- Kesarwani, M.; Hazan, R.; He, J.; Que, Y.A.; Apidianakis, Y.; Lesic, B.; Xiao, G.; Dekimpe, V.; Milot, S.; Deziel, E.; et al. A quorum sensing regulated small volatile molecule reduces acute virulence and promotes chronic infection phenotypes. PLoS Pathog 2011, 7, e1002192. [Google Scholar]
- Holden, M.T.; Ram Chhabra, S.; de Nys, R.; Stead, P.; Bainton, N.J.; Hill, P.J.; Manefield, M.; Kumar, N.; Labatte, M.; England, D.; et al. Quorum-sensing cross talk: Isolation and chemical characterization of cyclic dipeptides from Pseudomonas aeruginosa and other gram-negative bacteria. Mol. Microbiol 1999, 33, 1254–1266. [Google Scholar]
- Lee, J.; Wu, J.; Deng, Y.; Wang, J.; Wang, C.; Chang, C.; Dong, Y.; Williams, P.; Zhang, L.H. A cell-cell communication signal integrates quorum sensing and stress response. Nat. Chem. Biol 2013, 9, 339–343. [Google Scholar]
- Havarstein, L.S.; Coomaraswamy, G.; Morrison, D.A. An unmodified heptadecapeptide pheromone induces competence for genetic transformation in Streptococcus pneumoniae. Proc. Natl. Acad. Sci. USA 1995, 92, 11140–11144. [Google Scholar]
- Taga, M.E.; Bassler, B.L. Chemical communication among bacteria. Proc. Natl. Acad. Sci. USA 2003, 100, 14549–14554. [Google Scholar]
- Federle, M.J.; Bassler, B.L. Interspecies communication in bacteria. J. Clin. Invest 2003, 112, 1291–1299. [Google Scholar]
- McKnight, S.L.; Iglewski, B.H.; Pesci, E.C. The Pseudomonas quinolone signal regulates rhl quorum sensing in Pseudomonas aeruginosa. J. Bacteriol 2000, 182, 2702–2708. [Google Scholar]
- Kelly, R.C.; Bolitho, M.E.; Higgins, D.A.; Lu, W.; Ng, W.L.; Jeffrey, P.D.; Rabinowitz, J.D.; Semmelhack, M.F.; Hughson, F.M.; Bassler, B.L. The Vibrio cholerae quorum-sensing autoinducer CAI-1: Analysis of the biosynthetic enzyme CqsA. Nat. Chem. Biol 2009, 5, 891–895. [Google Scholar]
- Schauder, S.; Bassler, B.L. The languages of bacteria. Genes Dev 2001, 15, 1468–1480. [Google Scholar]
- Roy, V.; Meyer, M.T.; Smith, J.A.; Gamby, S.; Sintim, H.O.; Ghodssi, R.; Bentley, W.E. AI-2 analogs and antibiotics: A synergistic approach to reduce bacterial biofilms. Appl. Microbiol. Biotechnol 2013, 97, 2627–2638. [Google Scholar]
- Pereira, C.S.; Thompson, J.A.; Xavier, K.B. AI-2-mediated signalling in bacteria. FEMS Microbiol. Rev 2013, 37, 156–181. [Google Scholar]
- Sperandio, V.; Torres, A.G.; Giron, J.A.; Kaper, J.B. Quorum sensing is a global regulatory mechanism in enterohemorrhagic Escherichia coli O157:H7. J. Bacteriol 2001, 183, 5187–5197. [Google Scholar]
- Wood, T.K. Insights on Escherichia coli biofilm formation and inhibition from whole-transcriptome profiling. Environ. Microbiol 2009, 11, 1–15. [Google Scholar]
- Choi, J.; Shin, D.; Ryu, S. Implication of quorum sensing in Salmonella enterica serovar typhimurium virulence: The luxS gene is necessary for expression of genes in pathogenicity island 1. Infect. Immun 2007, 75, 4885–4890. [Google Scholar]
- Yarwood, J.M.; Bartels, D.J.; Volper, E.M.; Greenberg, E.P. Quorum sensing in Staphylococcus aureus biofilms. J. Bacteriol 2004, 186, 1838–1850. [Google Scholar]
- Ahmed, N.A.; Petersen, F.C.; Scheie, A.A. AI-2 quorum sensing affects antibiotic susceptibility in Streptococcus anginosus. J. Antimicrob. Chemother 2007, 60, 49–53. [Google Scholar]
- Armbruster, C.E.; Hong, W.; Pang, B.; Weimer, K.E.; Juneau, R.A.; Turner, J.; Swords, W.E. Indirect pathogenicity of Haemophilus influenzae and Moraxella catarrhalis in polymicrobial otitis media occurs via interspecies quorum signaling. MBio 2010, 1, e00102–10. [Google Scholar]
- Rader, B.A.; Wreden, C.; Hicks, K.G.; Sweeney, E.G.; Ottemann, K.M.; Guillemin, K. Helicobacter pylori perceives the quorum-sensing molecule AI-2 as a chemorepellent via the chemoreceptor TlpB. Microbiology 2011, 157, 2445–2455. [Google Scholar]
- Hammer, B.K.; Bassler, B.L. Quorum sensing controls biofilm formation in Vibrio cholerae. Mol. Microbiol 2003, 50, 101–104. [Google Scholar]
- Bassler, B.L.; Wright, M.; Silverman, M.R. Multiple signalling systems controlling expression of luminescence in Vibrio harveyi: Sequence and function of genes encoding a second sensory pathway. Mol. Microbiol 1994, 13, 273–286. [Google Scholar]
- Waters, C.M.; Bassler, B.L. The Vibrio harveyi quorum-sensing system uses shared regulatory components to discriminate between multiple autoinducers. Genes Dev 2006, 20, 2754–2767. [Google Scholar]
- Engebrecht, J.; Nealson, K.; Silverman, M. Bacterial bioluminescence: Isolation and genetic analysis of functions from Vibrio fischeri. Cell 1983, 32, 773–781. [Google Scholar]
- Gelhaus, H.C.; Rozak, D.A.; Nierman, W.C.; Chen, D.; Varga, J.J.; Zadeh, M.; Ulrich, R.L.; Adamovicz, J.J. Exogenous Yersinia pestis quorum sensing molecules N-octanoyl-homoserine lactone and N-(3-oxooctanoyl)-homoserine lactone regulate the LcrV virulence factor. Microb. Pathog 2009, 46, 283–287. [Google Scholar]
- James, D.; Shao, H.; Lamont, R.J.; Demuth, D.R. The Actinobacillus actinomycetemcomitans ribose binding protein RbsB interacts with cognate and heterologous autoinducer 2 signals. Infect. Immun 2006, 74, 4021–4029. [Google Scholar]
- Geske, G.D.; O’Neill, J.C.; Blackwell, H.E. Expanding dialogues: From natural autoinducers to non-natural analogues that modulate quorum sensing in Gram-negative bacteria. Chem. Soc. Rev 2008, 37, 1432–1447. [Google Scholar]
- Galloway, W.R.; Hodgkinson, J.T.; Bowden, S.D.; Welch, M.; Spring, D.R. Quorum sensing in Gram-negative bacteria: Small-molecule modulation of AHL and AI-2 quorum sensing pathways. Chem. Rev 2011, 111, 28–67. [Google Scholar]
- LaSarre, B.; Federle, M.J. Exploiting quorum sensing to confuse bacterial pathogens. Microbiol. Mol. Biol. Rev 2013, 77, 73–111. [Google Scholar]
- Schauder, S.; Shokat, K.; Surette, M.G.; Bassler, B.L. The LuxS family of bacterial autoinducers: Biosynthesis of a novel quorum-sensing signal molecule. Mol. Microbiol 2001, 41, 463–476. [Google Scholar]
- Winzer, K.; Hardie, K.R.; Burgess, N.; Doherty, N.; Kirke, D.; Holden, M.T.; Linforth, R.; Cornell, K.A.; Taylor, A.J.; Hill, P.J.; et al. LuxS: Its role in central metabolism and the in vitro synthesis of 4-hydroxy-5-methyl-3(2H)-furanone. Microbiology 2002, 148, 909–922. [Google Scholar]
- Surette, M.G.; Miller, M.B.; Bassler, B.L. Quorum sensing in Escherichia coli, Salmonella typhimurium, and Vibrio harveyi: A new family of genes responsible for autoinducer production. Proc. Natl. Acad. Sci. USA 1999, 96, 1639–1644. [Google Scholar]
- Lerat, E.; Moran, N.A. The evolutionary history of quorum-sensing systems in bacteria. Mol. Biol. Evol 2004, 21, 903–913. [Google Scholar]
- Hardie, K.R.; Heürlier, K. Establishing bacterial communities by “word of mouth”: LuxS and autoinducer 2 in biofilm development. Nat. Rev. Microbiol 2008, 6, 635–643. [Google Scholar]
- Hauck, T.; Hubner, Y.; Bruhlmann, F.; Schwab, W. Alternative pathway for the formation of 4,5-dihydroxy-2,3-pentanedione, the proposed precursor of 4-hydroxy-5-methyl-3(2H)-furanone as well as autoinducer-2, and its detection as natural constituent of tomato fruit. Biochim. Biophys. Acta 2003, 1623, 109–119. [Google Scholar]
- Zang, T.; Lee, B.W.; Cannon, L.M.; Ritter, K.A.; Dai, S.; Ren, D.; Wood, T.K.; Zhou, Z.S. A naturally occurring brominated furanone covalently modifies and inactivates LuxS. Bioorg. Med. Chem. Lett 2009, 19, 6200–6204. [Google Scholar]
- Globisch, D.; Lowery, C.A.; McCague, K.C.; Janda, K.D. Uncharacterized 4,5-dihydroxy-2,3-pentanedione (DPD) molecules revealed through NMR spectroscopy: Implications for a greater signaling diversity in bacterial species. Angew. Chem. Int. Ed. Engl 2012, 51, 4204–4208. [Google Scholar]
- Tavender, T.J.; Halliday, N.M.; Hardie, K.R.; Winzer, K. LuxS-independent formation of AI-2 from ribulose-5-phosphate. BMC Microbiol 2008, 8, 98, :1–98:8.. [Google Scholar]
- Kong, P.; Tyler, B.M.; Richardson, P.A.; Lee, B.W.; Zhou, Z.S.; Hong, C. Zoospore interspecific signaling promotes plant infection by Phytophthora. BMC Microbiol 2010, 10, 313, :1–313:9.. [Google Scholar]
- Nichols, J.D.; Johnson, M.R.; Chou, C.J.; Kelly, R.M. Temperature, not LuxS, mediates AI-2 formation in hydrothermal habitats. FEMS Microbiol. Ecol 2009, 68, 173–181. [Google Scholar]
- Gonzalez, J.E.; Keshavan, N.D. Messing with bacterial quorum sensing. Microbiol. Mol. Biol. Rev 2006, 70, 859–875. [Google Scholar]
- Meijler, M.M.; Hom, L.G.; Kaufmann, G.F.; McKenzie, K.M.; Sun, C.; Moss, J.A.; Matsushita, M.; Janda, K.D. Synthesis and biological validation of a ubiquitous quorum-sensing molecule. Angew. Chem. Int. Ed. Engl 2004, 43, 2106–2108. [Google Scholar]
- Lowery, C.A.; Park, J.; Kaufmann, G.F.; Janda, K.D. An unexpected switch in the modulation of AI-2-based quorum sensing discovered through synthetic 4,5-dihydroxy-2,3-pentanedione analogues. J. Am. Chem. Soc 2008, 130, 9200–9201. [Google Scholar]
- Tsuchikama, K.; Zhu, J.; Lowery, C.A.; Kaufmann, G.F.; Janda, K.D. C4-alkoxy-HPD: A potent class of synthetic modulators surpassing nature in AI-2 quorum sensing. J. Am. Chem. Soc 2012, 134, 13562–13564. [Google Scholar]
- Tsuchikama, K.; Lowery, C.A.; Janda, K.D. Probing autoinducer-2 based quorum sensing: The biological consequences of molecules unable to traverse equilibrium states. J. Org. Chem 2011, 76, 6981–6989. [Google Scholar]
- Smith, J.A.; Wang, J.; Nguyen-Mau, S.M.; Lee, V.; Sintim, H.O. Biological screening of a diverse set of AI-2 analogues in Vibrio harveyi suggests that receptors which are involved in synergistic agonism of AI-2 and analogues are promiscuous. Chem. Commun (Camb) 2009, 45, 7033–7035. [Google Scholar]
- Roy, V.; Smith, J.A.; Wang, J.; Stewart, J.E.; Bentley, W.E.; Sintim, H.O. Synthetic analogs tailor native AI-2 signaling across bacterial species. J. Am. Chem. Soc 2010, 132, 11141–11150. [Google Scholar]
- Gamby, S.; Roy, V.; Guo, M.; Smith, J.A.; Wang, J.; Stewart, J.E.; Wang, X.; Bentley, W.E.; Sintim, H.O. Altering the communication networks of multispecies microbial systems using a diverse toolbox of AI-2 analogues. ACS Chem. Biol 2012, 7, 1023–1030. [Google Scholar]
- Guo, M.; Gamby, S.; Nakayama, S.; Smith, J.; Sintim, H.O. A pro-drug approach for selective modulation of AI-2-mediated bacterial cell-to-cell communication. Sensors (Basel) 2012, 12, 3762–3772. [Google Scholar]
- Frezza, M.; Balestrino, D.; Soulère, L.; Reverchon, S.; Queneau, Y.; Forestier, C.; Doutheau, A. Synthesis and biological evaluation of the trifluoromethyl analog of (4S)-4,5-Dihydroxy-2,3-pentanedione (DPD). Eur. J. Org. Chem 2006, 2006, 4731–4736. [Google Scholar]
- Ganin, H.; Tang, X.; Meijler, M.M. Inhibition of Pseudomonas aeruginosa quorum sensing by AI-2 analogs. Bioorg. Med. Chem. Lett 2009, 19, 3941–3944. [Google Scholar]
- Rui, F.; Marques, J.C.; Miller, S.T.; Maycock, C.D.; Xavier, K.B.; Ventura, M.R. Stereochemical diversity of AI-2 analogs modulates quorum sensing in Vibrio harveyi and Escherichia coli. Bioorg. Med. Chem 2012, 20, 249–256. [Google Scholar]
- Semmelhack, M.F.; Campagna, S.R.; Federle, M.J.; Bassler, B.L. An expeditious synthesis of DPD and boron binding studies. Org. Lett 2005, 7, 569–572. [Google Scholar]
- De Keersmaecker, S.C.; Varszegi, C.; van Boxel, N.; Habel, L.W.; Metzger, K.; Daniels, R.; Marchal, K.; De Vos, D.; Vanderleyden, J. Chemical synthesis of (S)-4,5-dihydroxy-2,3-pentanedione, a bacterial signal molecule precursor, and validation of its activity in Salmonella typhimurium. J. Biol. Chem 2005, 280, 19563–19568. [Google Scholar]
- Frezza, M.; Soulère, L.; Queneau, Y.; Doutheau, A. A Baylis–Hillman/ozonolysis route towards (±) 4,5-dihydroxy-2,3-pentanedione (DPD) and analogues. Tetrahedron Lett 2005, 46, 6495–6498. [Google Scholar]
- Trost, B.M.; Malhotra, S.; Fried, B.A. Magnesium-catalyzed asymmetric direct aldol addition of ethyl diazoacetate to aromatic, aliphatic, and alpha, beta-unsaturated aldehydes. J. Am. Chem. Soc 2009, 131, 1674–1675. [Google Scholar]
- Yao, W.; Wang, J. Direct catalytic asymmetric aldol-type reaction of aldehydes with ethyl diazoacetate. Org. Lett 2003, 5, 1527–1530. [Google Scholar]
- Kadirvel, M.; Stimpson, W.T.; Moumene-Afifi, S.; Arsic, B.; Glynn, N.; Halliday, N.; Williams, P.; Gilbert, P.; McBain, A.J.; Freeman, S.; et al. Synthesis and bioluminescence-inducing properties of autoinducer (S)-4,5-dihydroxypentane-2,3-dione and its enantiomer. Bioorg. Med. Chem. Lett 2010, 20, 2625–2628. [Google Scholar]
- Ascenso, O.S.; Marques, J.C.; Santos, A.R.; Xavier, K.B.; Ventura, M.R.; Maycock, C.D. An efficient synthesis of the precursor of AI-2, the signalling molecule for inter-species quorum sensing. Bioorg. Med. Chem 2011, 19, 1236–1241. [Google Scholar]
- Ng, W.L.; Bassler, B.L. Bacterial quorum-sensing network architectures. Annu. Rev. Genet 2009, 43, 197–222. [Google Scholar]
- Neiditch, M.B.; Federle, M.J.; Pompeani, A.J.; Kelly, R.C.; Swem, D.L.; Jeffrey, P.D.; Bassler, B.L.; Hughson, F.M. Ligand-induced asymmetry in histidine sensor kinase complex regulates quorum sensing. Cell 2006, 126, 1095–1108. [Google Scholar]
- Lenz, D.H.; Mok, K.C.; Lilley, B.N.; Kulkarni, R.V.; Wingreen, N.S.; Bassler, B.L. The small RNA chaperone Hfq and multiple small RNAs control quorum sensing in Vibrio harveyi and Vibrio cholerae. Cell 2004, 118, 69–82. [Google Scholar]
- Herzberg, M.; Kaye, I.K.; Peti, W.; Wood, T.K. YdgG (TqsA) controls biofilm formation in Escherichia coli K-12 through autoinducer 2 transport. J. Bacteriol 2006, 188, 587–598. [Google Scholar]
- Taga, M.E.; Semmelhack, J.L.; Bassler, B.L. The LuxS-dependent autoinducer AI-2 controls the expression of an ABC transporter that functions in AI-2 uptake in Salmonella typhimurium. Mol. Microbiol 2001, 42, 777–793. [Google Scholar]
- Miller, S.T.; Xavier, K.B.; Campagna, S.R.; Taga, M.E.; Semmelhack, M.F.; Bassler, B.L.; Hughson, F.M. Salmonella typhimurium recognizes a chemically distinct form of the bacterial quorum-sensing signal AI-2. Mol. Cell 2004, 15, 677–687. [Google Scholar]
- Xavier, K.B.; Bassler, B.L. Regulation of uptake and processing of the quorum-sensing autoinducer AI-2 in Escherichia coli. J. Bacteriol 2005, 187, 238–248. [Google Scholar]
- Taga, M.E.; Miller, S.T.; Bassler, B.L. Lsr-mediated transport and processing of AI-2 in Salmonella typhimurium. Mol. Microbiol 2003, 50, 1411–1427. [Google Scholar]
- Li, J.; Attila, C.; Wang, L.; Wood, T.K.; Valdes, J.J.; Bentley, W.E. Quorum sensing in Escherichia coli is signaled by AI-2/LsrR: Effects on small RNA and biofilm architecture. J. Bacteriol 2007, 189, 6011–6020. [Google Scholar]
- Wang, L.; Li, J.; March, J.C.; Valdes, J.J.; Bentley, W.E. luxS-dependent gene regulation in Escherichia coli K-12 revealed by genomic expression profiling. J. Bacteriol 2005, 187, 8350–8360. [Google Scholar]
- Gonzalez Barrios, A.F.; Zuo, R.; Hashimoto, Y.; Yang, L.; Bentley, W.E.; Wood, T.K. Autoinducer 2 controls biofilm formation in Escherichia coli through a novel motility quorum-sensing regulator (MqsR, B3022). J. Bacteriol 2006, 188, 305–316. [Google Scholar]
- Wu, M.; Tao, Y.; Liu, X.; Zang, J. Structural basis for phosphorylated autoinducer-2 modulation of the oligomerization state of the global transcription regulator LsrR from Escherichia coli. J. Biol. Chem 2013, 288, 15878–15887. [Google Scholar]
- Pereira, C.S.; de Regt, A.K.; Brito, P.H.; Miller, S.T.; Xavier, K.B. Identification of functional LsrB-like autoinducer-2 receptors. J. Bacteriol 2009, 191, 6975–6987. [Google Scholar]
- Ferro, A.J.; Barrett, A.; Shapiro, S.K. Kinetic properties and the effect of substrate analogues on 5′-methylthioadenosine nucleosidase from Escherichia coli. Biochim. Biophys. Acta 1976, 438, 487–494. [Google Scholar]
- Singh, V.; Shi, W.; Almo, S.C.; Evans, G.B.; Furneaux, R.H.; Tyler, P.C.; Painter, G.F.; Lenz, D.H.; Mee, S.; Zheng, R.; et al. Structure and inhibition of a quorum sensing target from Streptococcus pneumoniae. Biochemistry 2006, 45, 12929–12941. [Google Scholar]
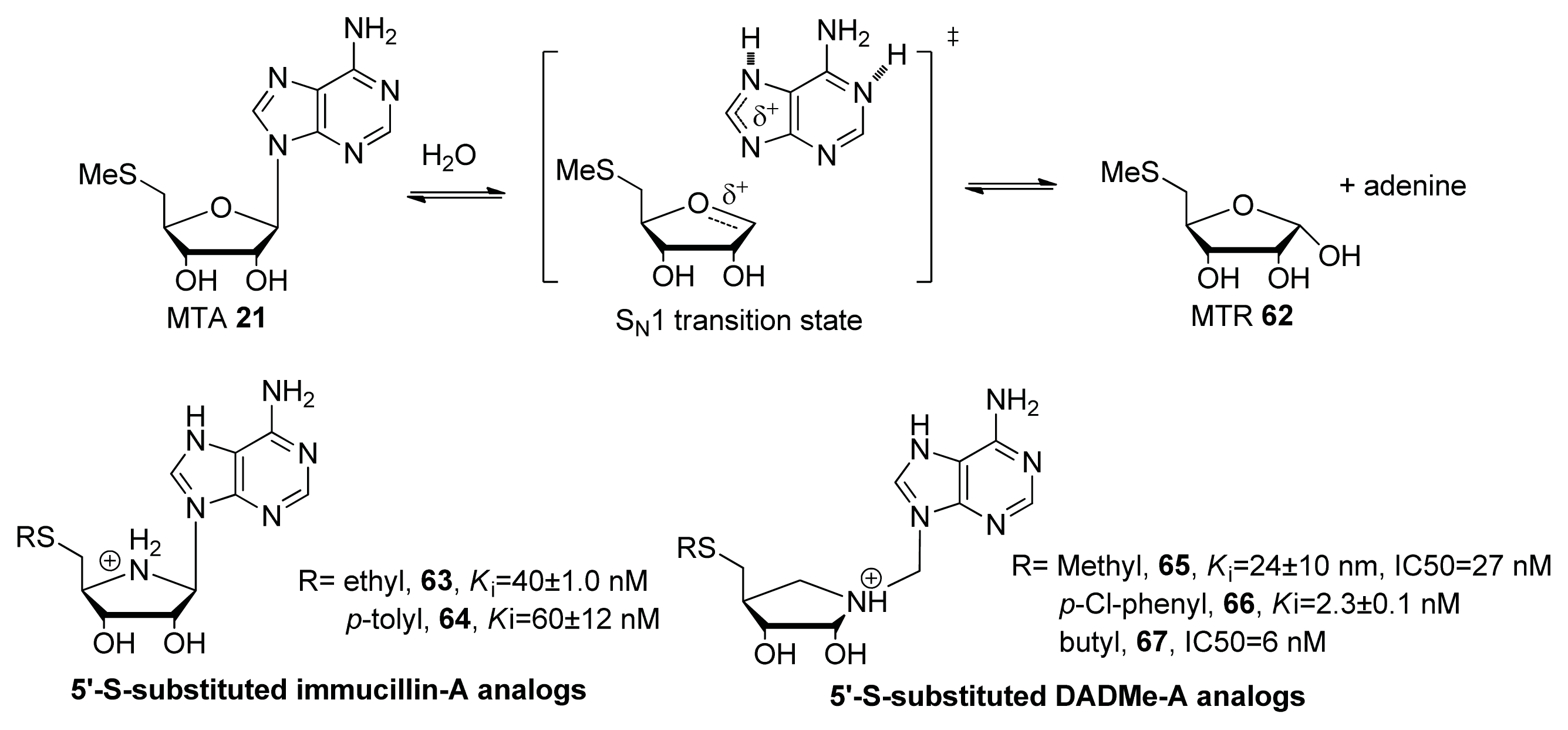
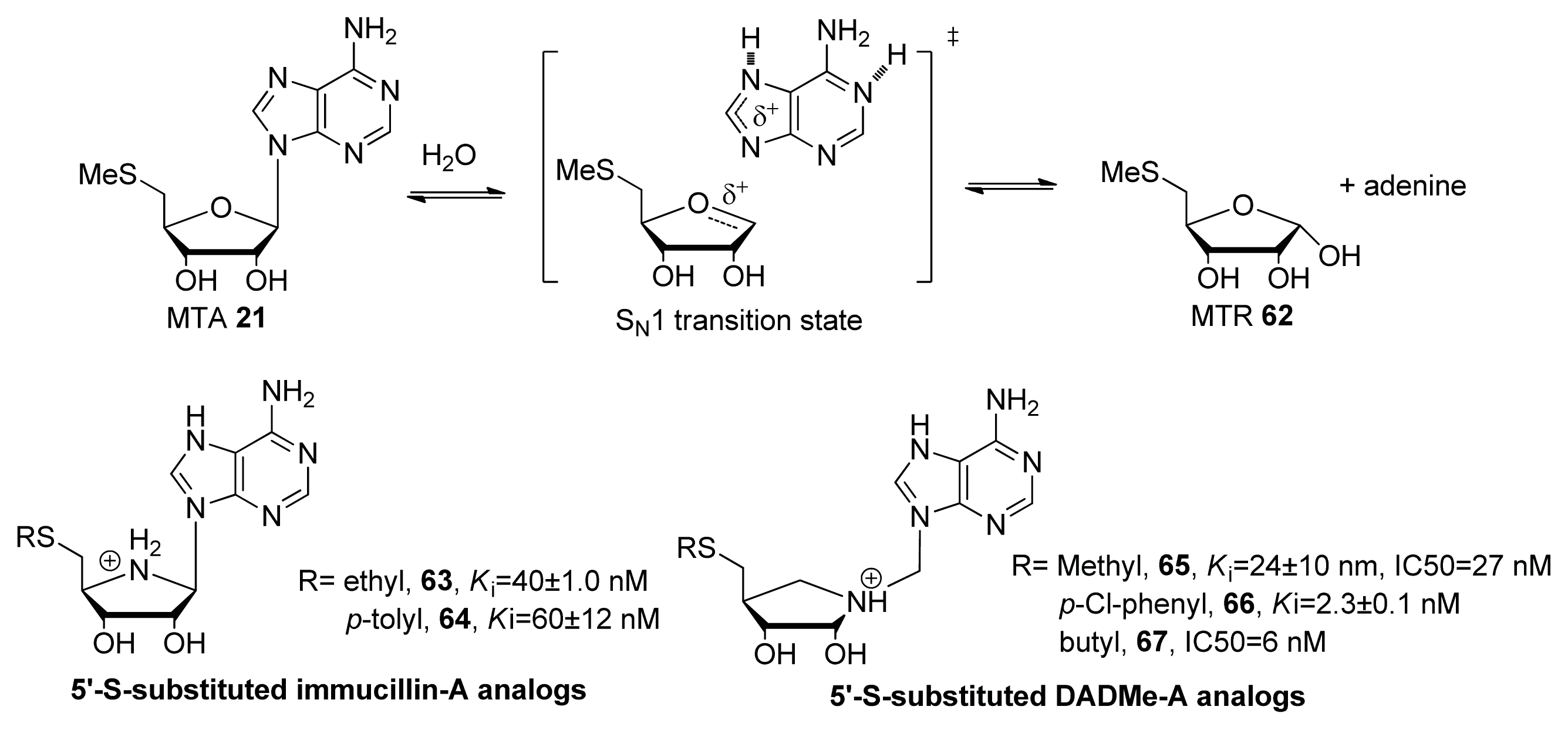
- Gutierrez, J.A.; Crowder, T.; Rinaldo-Matthis, A.; Ho, M.C.; Almo, S.C.; Schramm, V.L. Transition state analogs of 5′-methylthioadenosine nucleosidase disrupt quorum sensing. Nat. Chem. Biol 2009, 5, 251–257. [Google Scholar]
- Lee, J.E.; Settembre, E.C.; Cornell, K.A.; Riscoe, M.K.; Sufrin, J.R.; Ealick, S.E.; Howell, P.L. Structural comparison of MTA phosphorylase and MTA/AdoHcy nucleosidase explains substrate preferences and identifies regions exploitable for inhibitor design. Biochemistry 2004, 43, 5159–5169. [Google Scholar]
- Longshaw, A.I.; Adanitsch, F.; Gutierrez, J.A.; Evans, G.B.; Tyler, P.C.; Schramm, V.L. Design and synthesis of potent “sulfur-free” transition state analogue inhibitors of 5′-methylthioadenosine nucleosidase and 5′-methylthioadenosine phosphorylase. J. Med. Chem 2010, 53, 6730–6746. [Google Scholar]
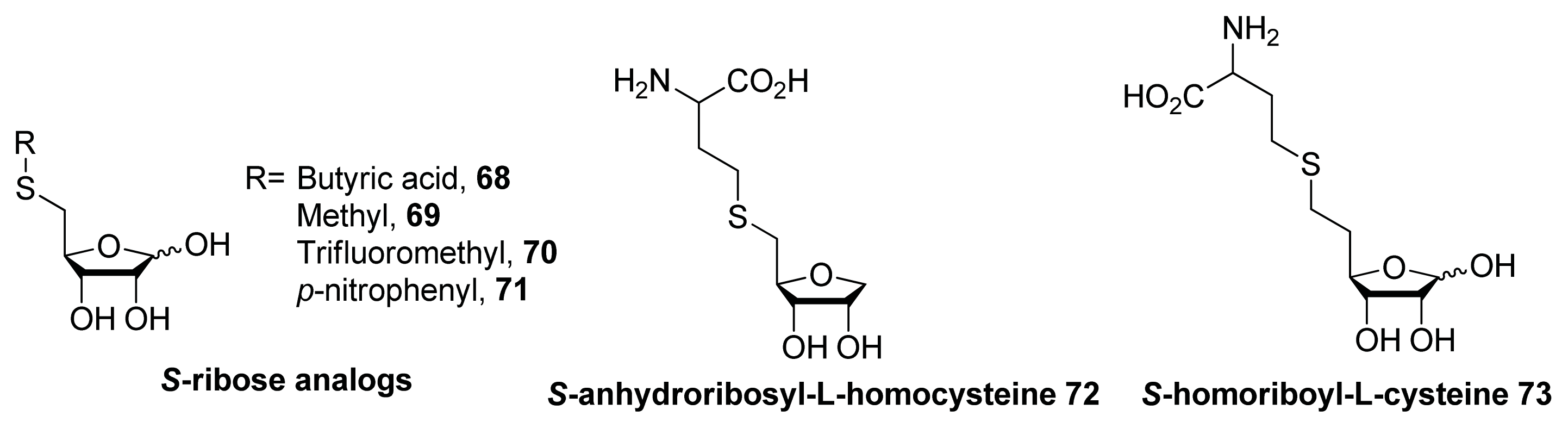
- Zhao, G.; Wan, W.; Mansouri, S.; Alfaro, J.F.; Bassler, B.L.; Cornell, K.A.; Zhou, Z.S. Chemical synthesis of S-ribosyl-l-homocysteine and activity assay as a LuxS substrate. Bioorg. Med. Chem. Lett 2003, 13, 3897–3900. [Google Scholar]
- Alfaro, J.F.; Zhang, T.; Wynn, D.P.; Karschner, E.L.; Zhou, Z.S. Synthesis of LuxS inhibitors targeting bacterial cell-cell communication. Org. Lett 2004, 6, 3043–3046. [Google Scholar]
- Zhu, J.; Dizin, E.; Hu, X.; Wavreille, A.-S.; Park, J.; Pei, D. S-Ribosylhomocysteinase (LuxS) is a mononuclear iron protein. Biochemistry 2003, 42, 4717–4726. [Google Scholar]
- Malladi, V.L.; Sobczak, A.J.; Meyer, T.M.; Pei, D.; Wnuk, S.F. Inhibition of LuxS by S-ribosylhomocysteine analogues containing a [4-aza]ribose ring. Bioorg. Med. Chem 2011, 19, 5507–5519. [Google Scholar]
- Shen, G.; Rajan, R.; Zhu, J.; Bell, C.E.; Pei, D. Design and synthesis of substrate and intermediate analogue inhibitors of S-ribosylhomocysteinase. J. Med. Chem 2006, 49, 3003–3011. [Google Scholar]
- Wnuk, S.F.; Robert, J.; Sobczak, A.J.; Meyers, B.P.; Malladi, V.L.; Zhu, J.; Gopishetty, B.; Pei, D. Inhibition of S-ribosylhomocysteinase (LuxS) by substrate analogues modified at the ribosyl C-3 position. Bioorg. Med. Chem 2009, 17, 6699–6706. [Google Scholar]
- Gopishetty, B.; Zhu, J.; Rajan, R.; Sobczak, A.J.; Wnuk, S.F.; Bell, C.E.; Pei, D. Probing the catalytic mechanism of S-ribosylhomocysteinase (LuxS) with catalytic intermediates and substrate analogues. J. Am. Chem. Soc 2009, 131, 1243–1250. [Google Scholar]
- Gram, L.; de Nys, R.; Maximilien, R.; Givskov, M.; Steinberg, P.; Kjelleberg, S. Inhibitory effects of secondary metabolites from the red alga delisea pulchra on swarming motility of Proteus mirabilis. Appl. Environ. Microbiol 1996, 62, 4284–4287. [Google Scholar]
- Givskov, M.; de Nys, R.; Manefield, M.; Gram, L.; Maximilien, R.; Eberl, L.; Molin, S.; Steinberg, P.D.; Kjelleberg, S. Eukaryotic interference with homoserine lactone-mediated prokaryotic signalling. J. Bacteriol 1996, 178, 6618–6622. [Google Scholar]
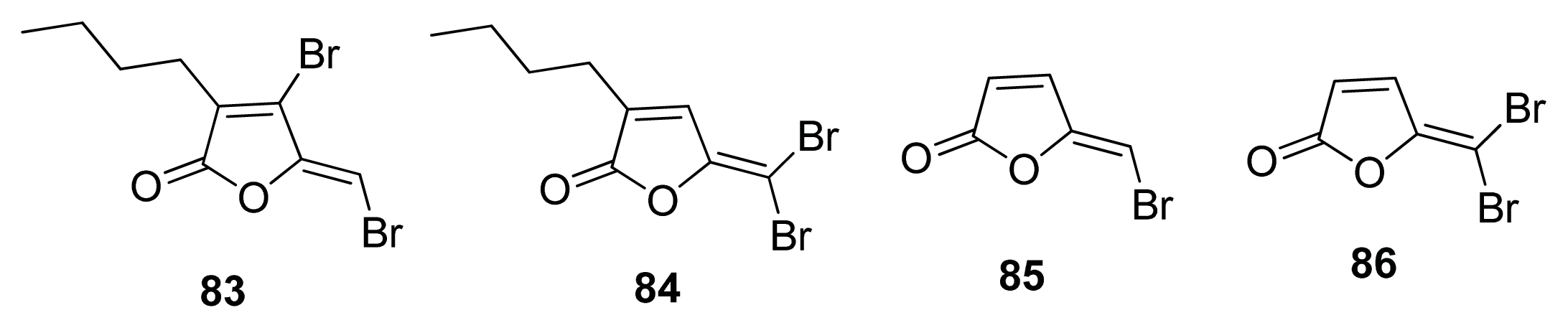
- Ren, D.; Sims, J.J.; Wood, T.K. Inhibition of biofilm formation and swarming of Escherichia coli by (5Z)-4-bromo-5-(bromomethylene)-3-butyl-2(5H)-furanone. Environ. Microbiol 2001, 3, 731–736. [Google Scholar]
- Defoirdt, T.; Crab, R.; Wood, T.K.; Sorgeloos, P.; Verstraete, W.; Bossier, P. Quorum sensing-disrupting brominated furanones protect the gnotobiotic brine shrimp Artemia franciscana from pathogenic Vibrio harveyi, Vibrio campbellii, and Vibrio parahaemolyticus isolates. Appl. Environ. Microbiol 2006, 72, 6419–6423. [Google Scholar]
- Janssens, J.C.; Steenackers, H.; Robijns, S.; Gellens, E.; Levin, J.; Zhao, H.; Hermans, K.; De Coster, D.; Verhoeven, T.L.; Marchal, K.; et al. Brominated furanones inhibit biofilm formation by Salmonella enterica serovar Typhimurium. Appl. Environ. Microbiol 2008, 74, 6639–6648. [Google Scholar]
- Shetye, G.; Singh, N.; Gao, X.; Bandyopadhyay, D.; Yan, A.; Luk, Y.-Y. Structures and biofilm inhibition activities of brominated furanones for Escherichia coli and Pseudomonas aeruginosa. Med. Chem. Comm 2013, 4, 1079–1084. [Google Scholar]
- Han, X.; Lu, C. Biological activity and identification of a peptide inhibitor of LuxS from Streptococcus suis serotype 2. FEMS Microbiol. Lett 2009, 294, 16–23. [Google Scholar]
- Roy, V.; Fernandes, R.; Tsao, C.-Y.; Bentley, W.E. cross species quorum quenching using a native ai-2 processing enzyme. ACS Chem. Biol 2009, 5, 223–232. [Google Scholar]
- Xue, X.; Pasparakis, G.; Halliday, N.; Winzer, K.; Howdle, S.M.; Cramphorn, C.J.; Cameron, N.R.; Gardner, P.M.; Davis, B.G.; Fernandez-Trillo, F.; et al. Synthetic polymers for simultaneous bacterial sequestration and quorum sense interference. Angew. Chem. Int. Ed. Engl 2011, 50, 9852–9856. [Google Scholar]
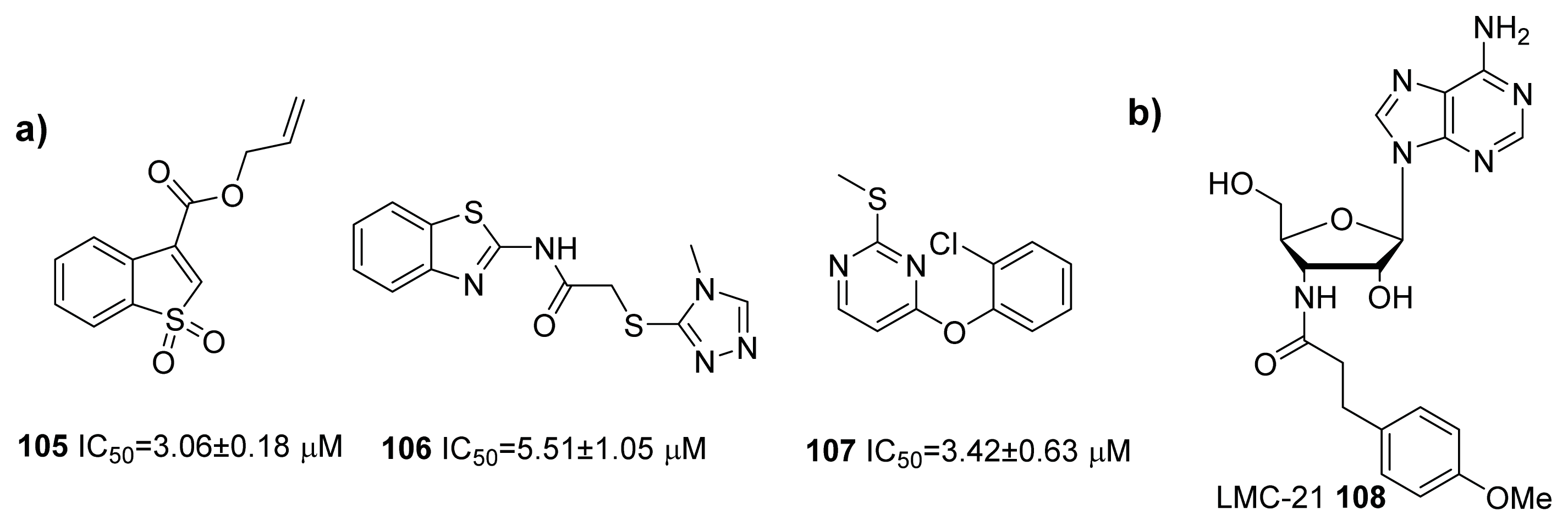
- Li, M.; Ni, N.; Chou, H.T.; Lu, C.D.; Tai, P.C.; Wang, B. Structure-based discovery and experimental verification of novel AI-2 quorum sensing inhibitors against Vibrio harveyi. Chem. Med. Chem 2008, 3, 1242–1249. [Google Scholar]
- Peng, H.; Cheng, Y.; Ni, N.; Li, M.; Choudhary, G.; Chou, H.T.; Lu, C.D.; Tai, P.C.; Wang, B. Synthesis and evaluation of new antagonists of bacterial quorum sensing in Vibrio harveyi. Chem. Med. Chem 2009, 4, 1457–1468. [Google Scholar]
- Ni, N.; Chou, H.T.; Wang, J.; Li, M.; Lu, C.D.; Tai, P.C.; Wang, B. Identification of boronic acids as antagonists of bacterial quorum sensing in Vibrio harveyi. Biochem. Biophys. Res. Commun 2008, 369, 590–594. [Google Scholar]
- Ni, N.; Choudhary, G.; Peng, H.; Li, M.; Chou, H.T.; Lu, C.D.; Gilbert, E.S.; Wang, B. Inhibition of quorum sensing in Vibrio harveyi by boronic acids. Chem. Biol. Drug. Des 2009, 74, 51–56. [Google Scholar]
- Ni, N.; Choudhary, G.; Li, M.; Wang, B. Pyrogallol and its analogs can antagonize bacterial quorum sensing in Vibrio harveyi. Bioorg. Med. Chem. Lett 2008, 18, 1567–1572. [Google Scholar]
- Ni, N.; Choudhary, G.; Li, M.; Wang, B. A new phenothiazine structural scaffold as inhibitors of bacterial quorum sensing in Vibrio harveyi. Biochem. Biophys. Res. Commun 2009, 382, 153–156. [Google Scholar]
- Zhu, P.; Peng, H.; Ni, N.; Wang, B.; Li, M. Novel AI-2 quorum sensing inhibitors in Vibrio harveyi identified through structure-based virtual screening. Bioorg. Med. Chem. Lett 2012, 22, 6413–6417. [Google Scholar]
- Brackman, G.; Celen, S.; Baruah, K.; Bossier, P.; Van Calenbergh, S.; Nelis, H.J.; Coenye, T. AI-2 quorum-sensing inhibitors affect the starvation response and reduce virulence in several Vibrio species, most likely by interfering with LuxPQ. Microbiology 2009, 155, 4114–4122. [Google Scholar]
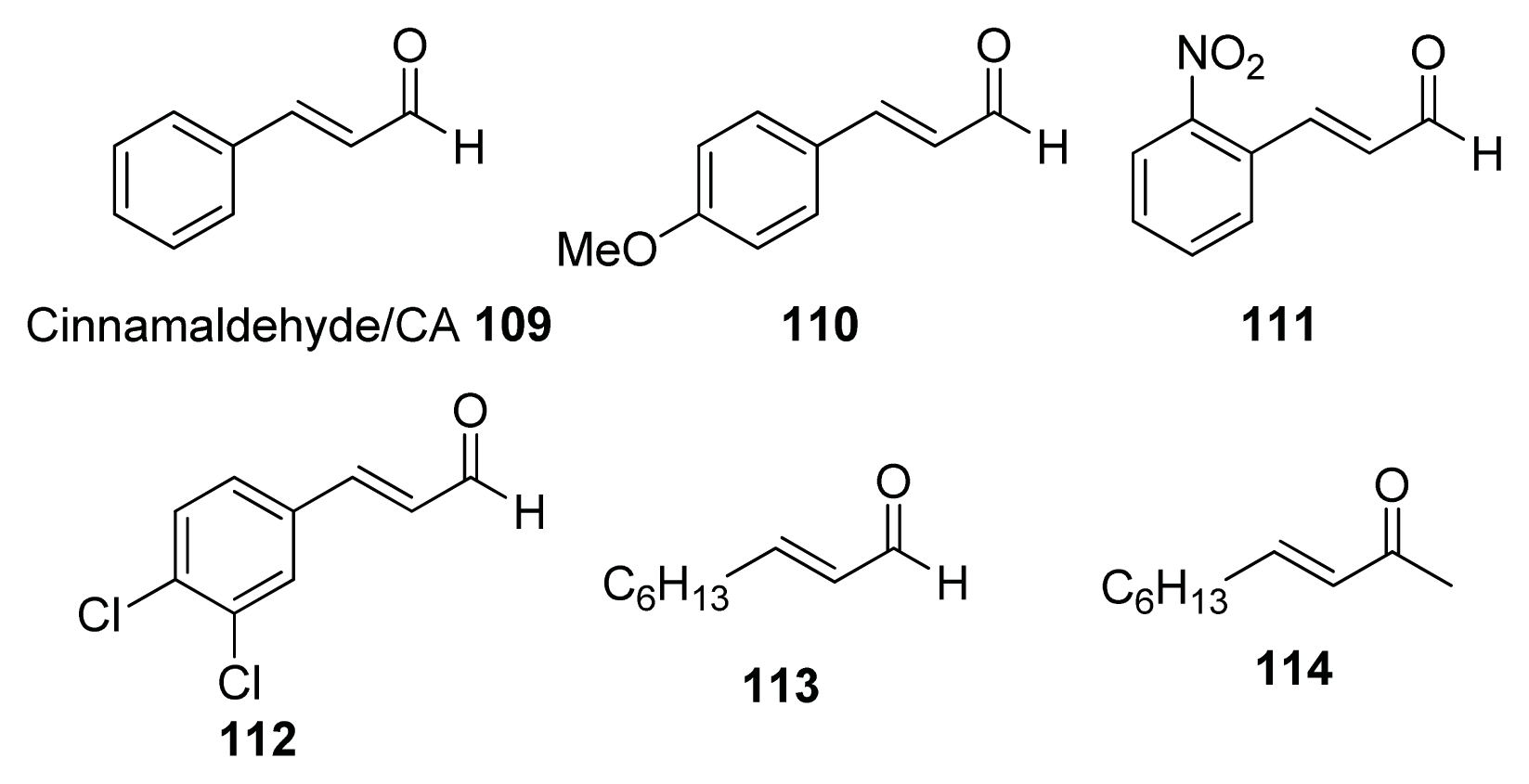
- Niu, C.; Afre, S.; Gilbert, E.S. Subinhibitory concentrations of cinnamaldehyde interfere with quorum sensing. Lett. Appl. Microbiol 2006, 43, 489–494. [Google Scholar]
- Brackman, G.; Defoirdt, T.; Miyamoto, C.; Bossier, P.; Van Calenbergh, S.; Nelis, H.; Coenye, T. Cinnamaldehyde and cinnamaldehyde derivatives reduce virulence in Vibrio spp. by decreasing the DNA-binding activity of the quorum sensing response regulator LuxR. BMC Microbiol 2008, 8, 149, :1–149:14.. [Google Scholar]
- Brackman, G.; Celen, S.; Hillaert, U.; Van Calenbergh, S.; Cos, P.; Maes, L.; Nelis, H.J.; Coenye, T. Structure-activity relationship of cinnamaldehyde analogs as inhibitors of AI-2 based quorum sensing and their effect on virulence of Vibrio spp. PLoS One 2011, 6, e16084. [Google Scholar]
- Brackman, G.; Al Quntar, A.A.; Enk, C.D.; Karalic, I.; Nelis, H.J.; Van Calenbergh, S.; Srebnik, M.; Coenye, T. Synthesis and evaluation of thiazolidinedione and dioxazaborocane analogues as inhibitors of AI-2 quorum sensing in Vibrio harveyi. Bioorg. Med. Chem 2013, 21, 660–667. [Google Scholar]
- Aharoni, R.; Bronstheyn, M.; Jabbour, A.; Zaks, B.; Srebnik, M.; Steinberg, D. Oxazaborolidine derivatives inducing autoinducer-2 signal transduction in Vibrio harveyi. Bioorg. Med. Chem 2008, 16, 1596–1604. [Google Scholar]
- Defoirdt, T.; Benneche, T.; Brackman, G.; Coenye, T.; Sorgeloos, P.; Scheie, A.A. A quorum sensing-disrupting brominated thiophenone with a promising therapeutic potential to treat luminescent vibriosis. PLoS One 2012, 7, e41788. [Google Scholar]
- Lowery, C.A.; McKenzie, K.M.; Qi, L.; Meijler, M.M.; Janda, K.D. Quorum sensing in Vibrio harveyi: Probing the specificity of the LuxP binding site. Bioorg. Med. Chem. Lett 2005, 15, 2395–2398. [Google Scholar]
- Kamaraju, K.; Smith, J.; Wang, J.; Roy, V.; Sintim, H.O.; Bentley, W.E.; Sukharev, S. Effects on membrane lateral pressure suggest permeation mechanisms for bacterial quorum signaling molecules. Biochemistry 2011, 50, 6983–6993. [Google Scholar]
- Zhu, J.; Hixon, M.S.; Globisch, D.; Kaufmann, G.F.; Janda, K.D. Mechanistic insights into the lsrk kinase required for autoinducer-2 quorum sensing activation. J. Am. Chem. Soc 2013, 135, 7827–7830. [Google Scholar]
- Furlani, R.E.; Yeagley, A.A.; Melander, C. A flexible approach to 1,4-di-substituted 2-aminoimidazoles that inhibit and disperse biofilms and potentiate the effects of beta-lactams against multi-drug resistant bacteria. Eur. J. Med. Chem 2013, 62, 59–70. [Google Scholar]
- Worthington, R.J.; Richards, J.J.; Melander, C. Small molecule control of bacterial biofilms. Org. Biomol. Chem 2012, 10, 7457–7474. [Google Scholar]
- Hoiby, N.; Bjarnsholt, T.; Givskov, M.; Molin, S.; Ciofu, O. Antibiotic resistance of bacterial biofilms. Int. J. Antimicrob. Agents 2010, 35, 322–332. [Google Scholar]
- Anderson, G.G.; O’Toole, G.A. Innate and induced resistance mechanisms of bacterial biofilms. Curr. Top. Microbiol. Immunol 2008, 322, 85–105. [Google Scholar]
- Stewart, P.S.; Costerton, J.W. Antibiotic resistance of bacteria in biofilms. Lancet 2001, 358, 135–138. [Google Scholar]
- Costerton, J.W.; Stewart, P.S.; Greenberg, E.P. Bacterial biofilms: A common cause of persistent infections. Science 1999, 284, 1318–1322. [Google Scholar]
- Mah, T.F.; O’Toole, G.A. Mechanisms of biofilm resistance to antimicrobial agents. Trends Microbiol 2001, 9, 34–39. [Google Scholar]
- Brackman, G.; Cos, P.; Maes, L.; Nelis, H.J.; Coenye, T. Quorum sensing inhibitors increase the susceptibility of bacterial biofilms to antibiotics in vitro and in vivo. Antimicrob. Agents Chemother 2011, 55, 2655–2661. [Google Scholar]
- Frezza, M.; Soulere, L.; Balestrino, D.; Gohar, M.; Deshayes, C.; Queneau, Y.; Forestier, C.; Doutheau, A. Ac2-DPD, the bis-(O)-acetylated derivative of 4,5-dihydroxy-2,3-pentanedione (DPD) is a convenient stable precursor of bacterial quorum sensing autoinducer AI-2. Bioorg. Med. Chem. Lett 2007, 17, 1428–1431. [Google Scholar]
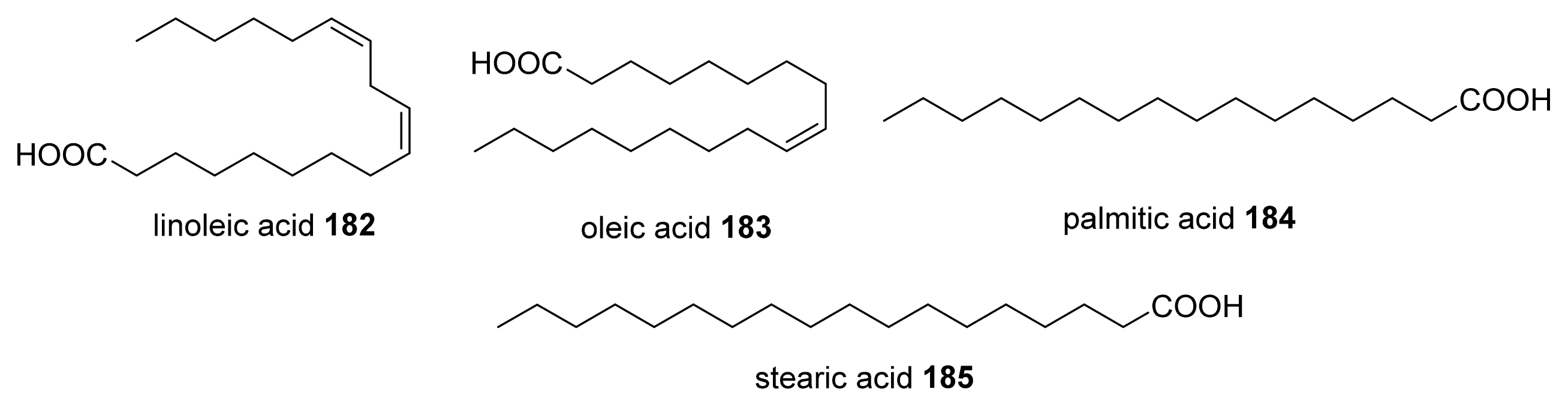
- Soni, K.A.; Jesudhasan, P.; Cepeda, M.; Widmer, K.; Jayaprakasha, G.K.; Patil, B.S.; Hume, M.E.; Pillai, S.D. Identification of ground beef-derived fatty acid inhibitors of autoinducer-2-based cell signaling. J. Food Prot 2008, 71, 134–138. [Google Scholar]
- Widmer, K.W.; Soni, K.A.; Hume, M.E.; Beier, R.C.; Jesudhasan, P.; Pillai, S.D. Identification of poultry meat-derived fatty acids functioning as quorum sensing signal inhibitors to autoinducer-2 (AI-2). J. Food Sci 2007, 72, M363–M368. [Google Scholar]
- Sun, J.; Daniel, R.; Wagner-Dobler, I.; Zeng, A.P. Is autoinducer-2 a universal signal for interspecies communication: A comparative genomic and phylogenetic analysis of the synthesis and signal transduction pathways. BMC Evol. Biol 2004, 4, 36, :1–36:11.. [Google Scholar]
- Winzer, K.; Hardie, K.R.; Williams, P. LuxS and autoinducer-2: Their contribution to quorum sensing and metabolism in bacteria. Adv. Appl. Microbiol 2003, 53, 291–396. [Google Scholar]
- Diggle, S.P.; Gardner, A.; West, S.A.; Griffin, A.S. Evolutionary theory of bacterial quorum sensing: When is a signal not a signal? Philos. Trans. R. Soc. Lond. B 2007, 362, 1241–1249. [Google Scholar]
- Hegde, M.; Englert, D.L.; Schrock, S.; Cohn, W.B.; Vogt, C.; Wood, T.K.; Manson, M.D.; Jayaraman, A. Chemotaxis to the quorum-sensing signal AI-2 requires the Tsr chemoreceptor and the periplasmic LsrB AI-2-binding protein. J. Bacteriol 2011, 193, 768–773. [Google Scholar]
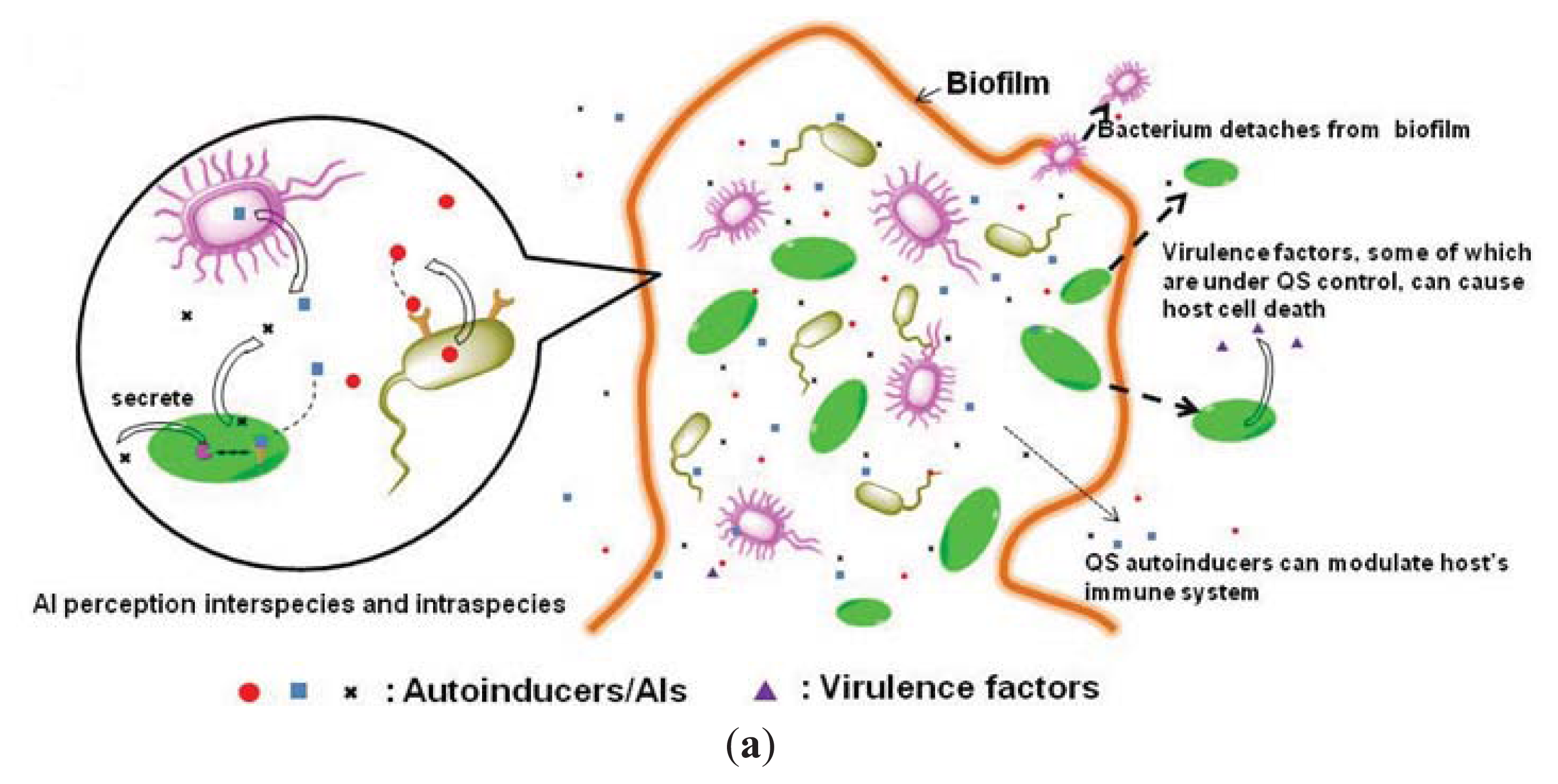
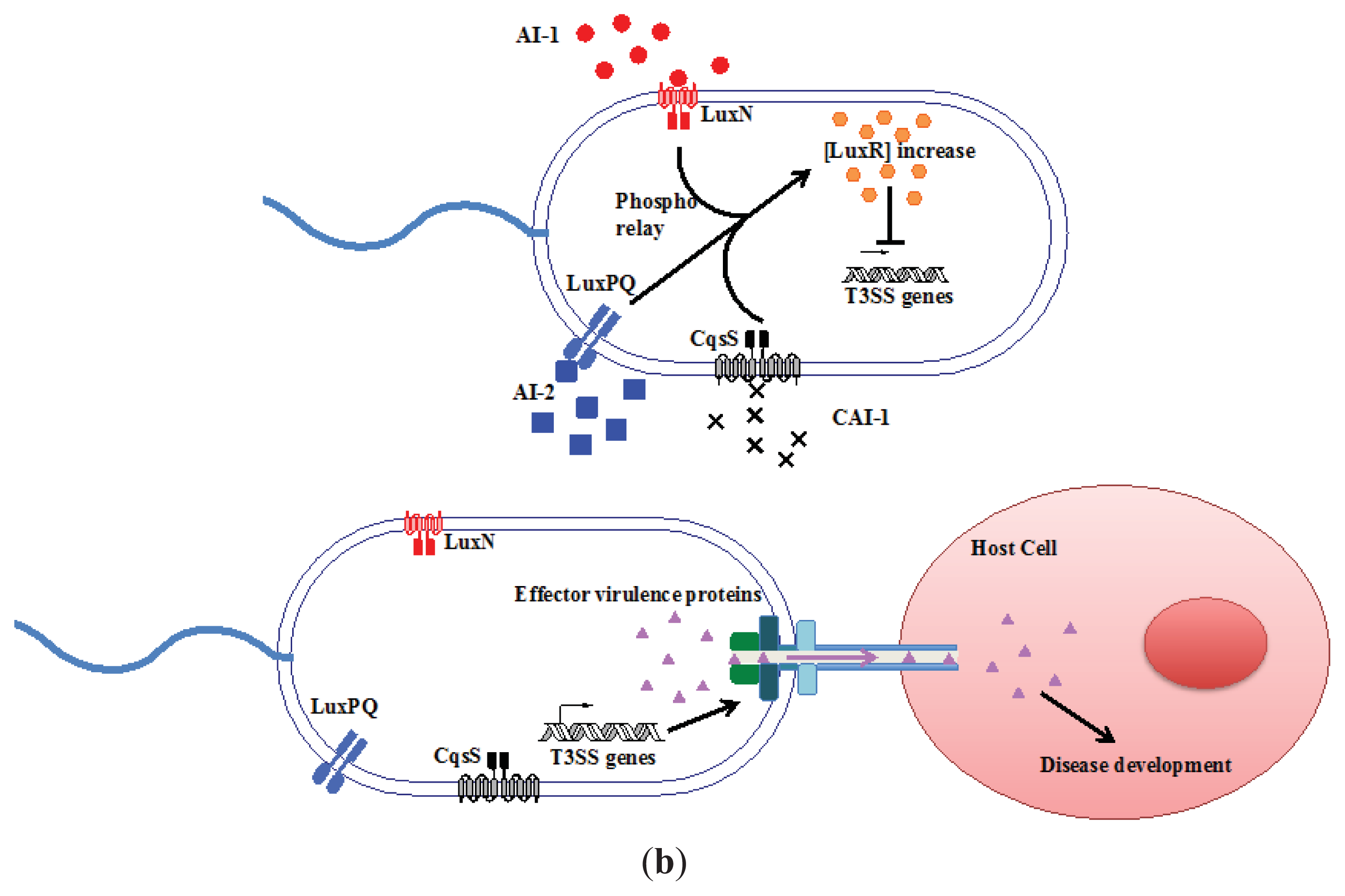





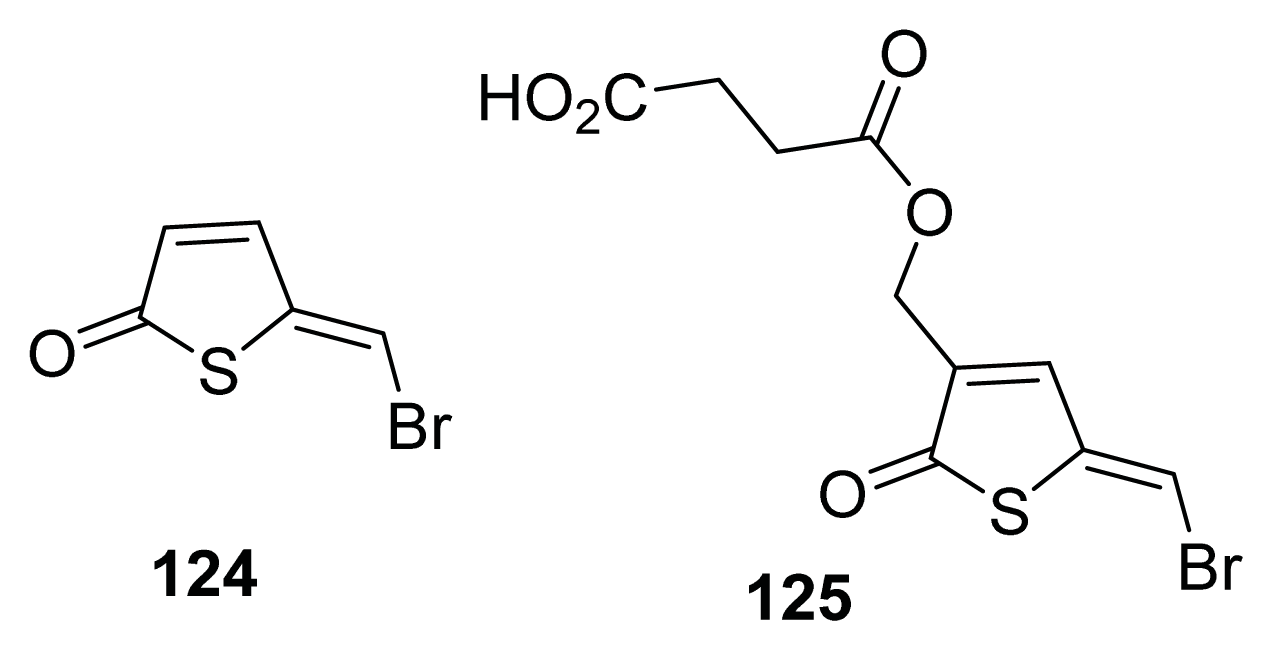
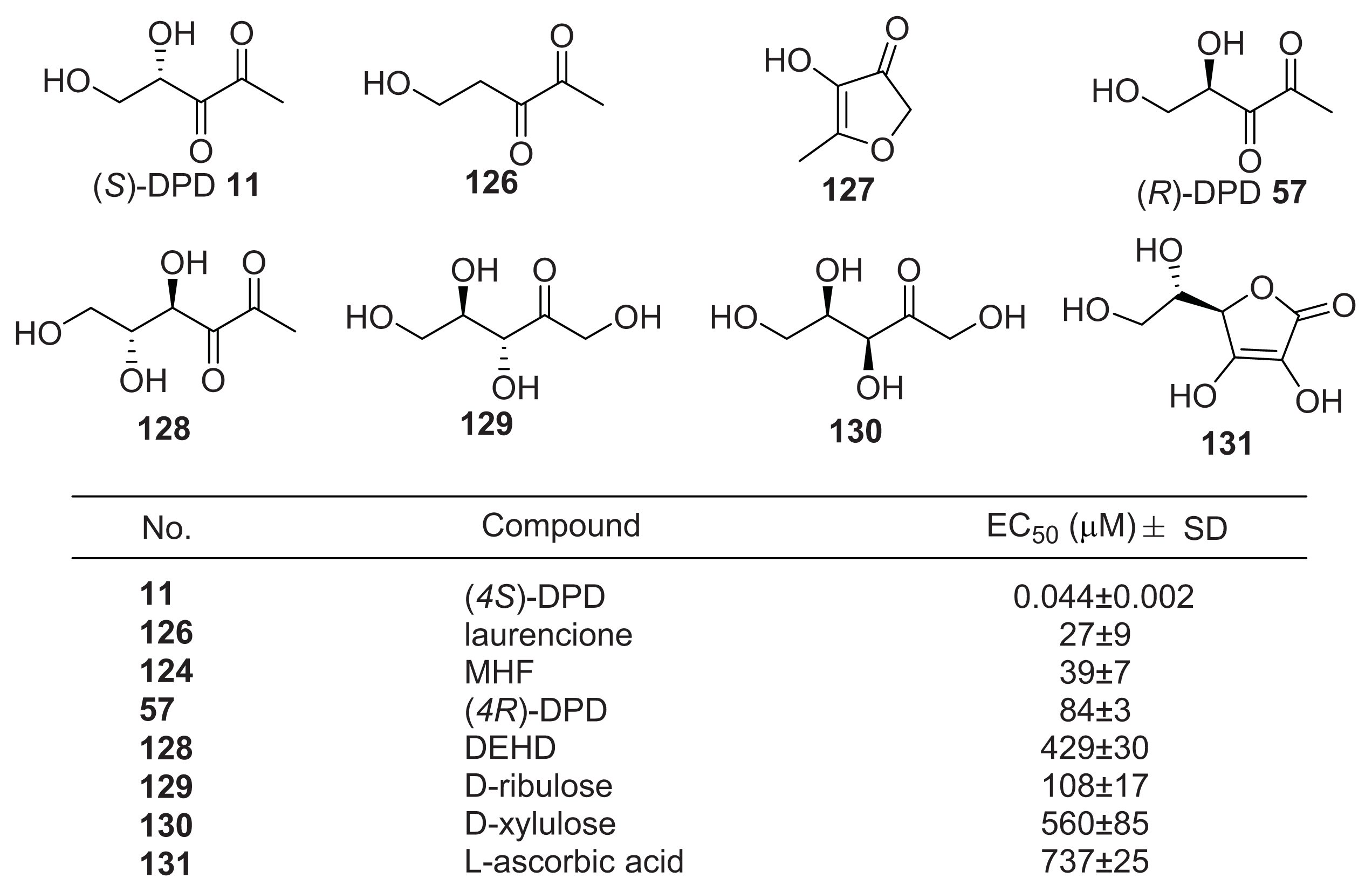

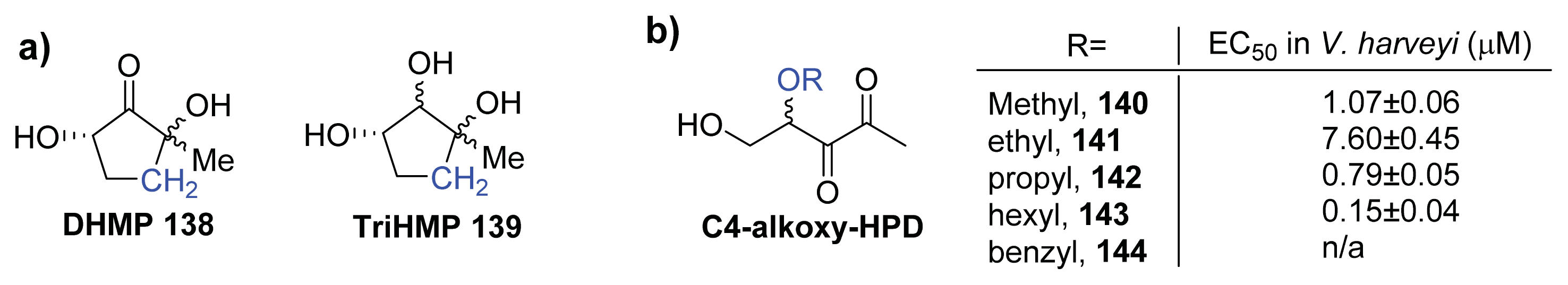
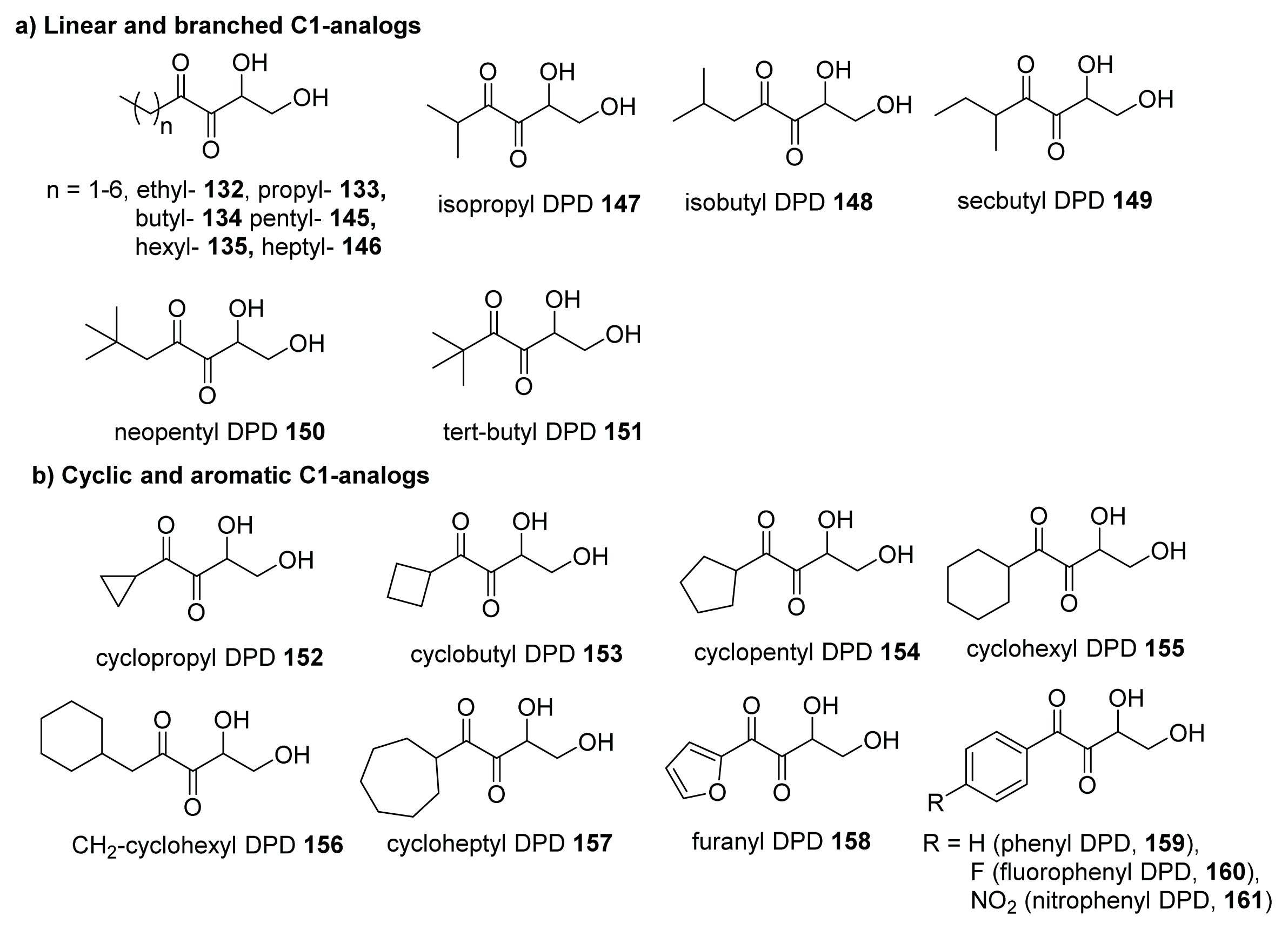
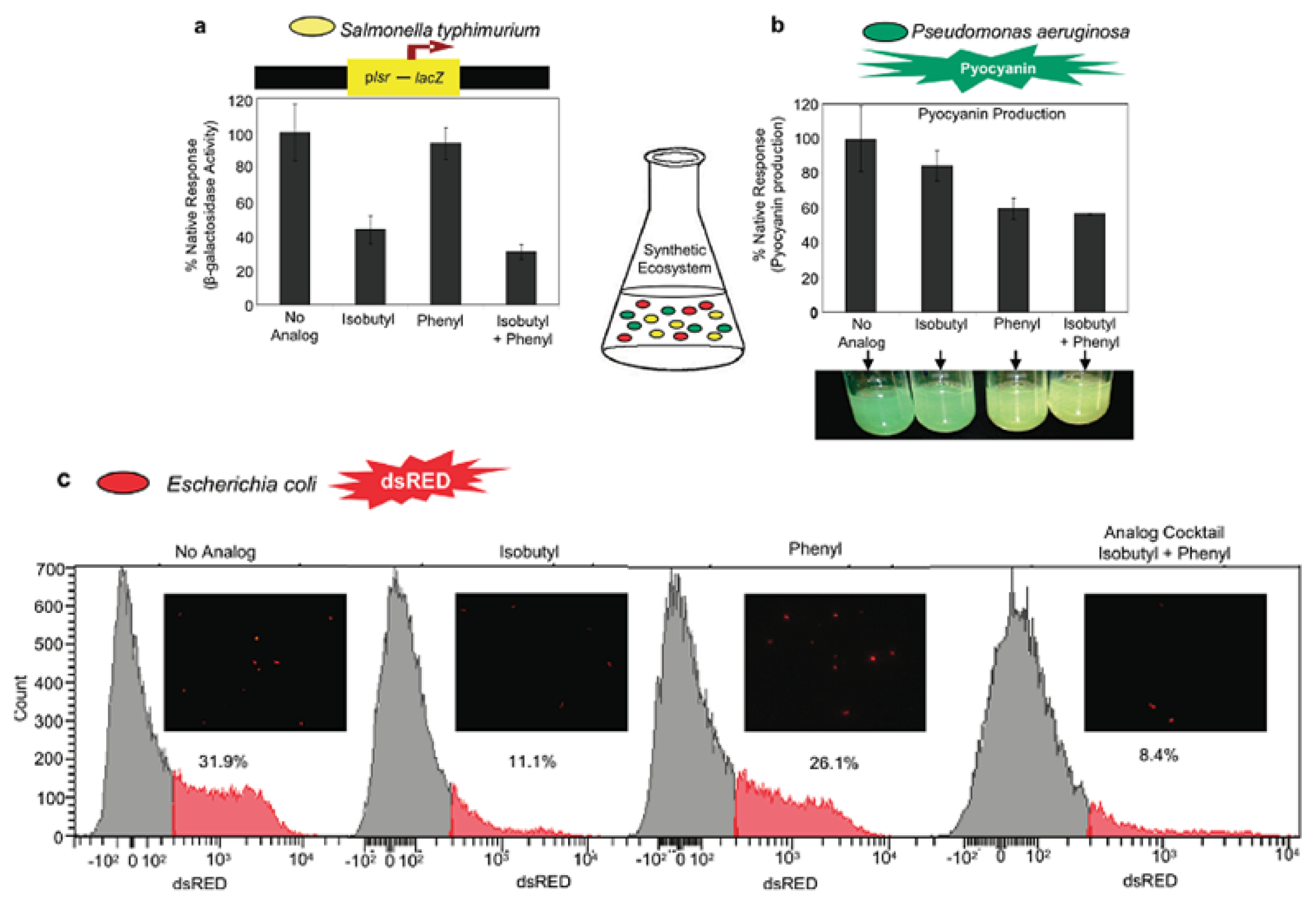

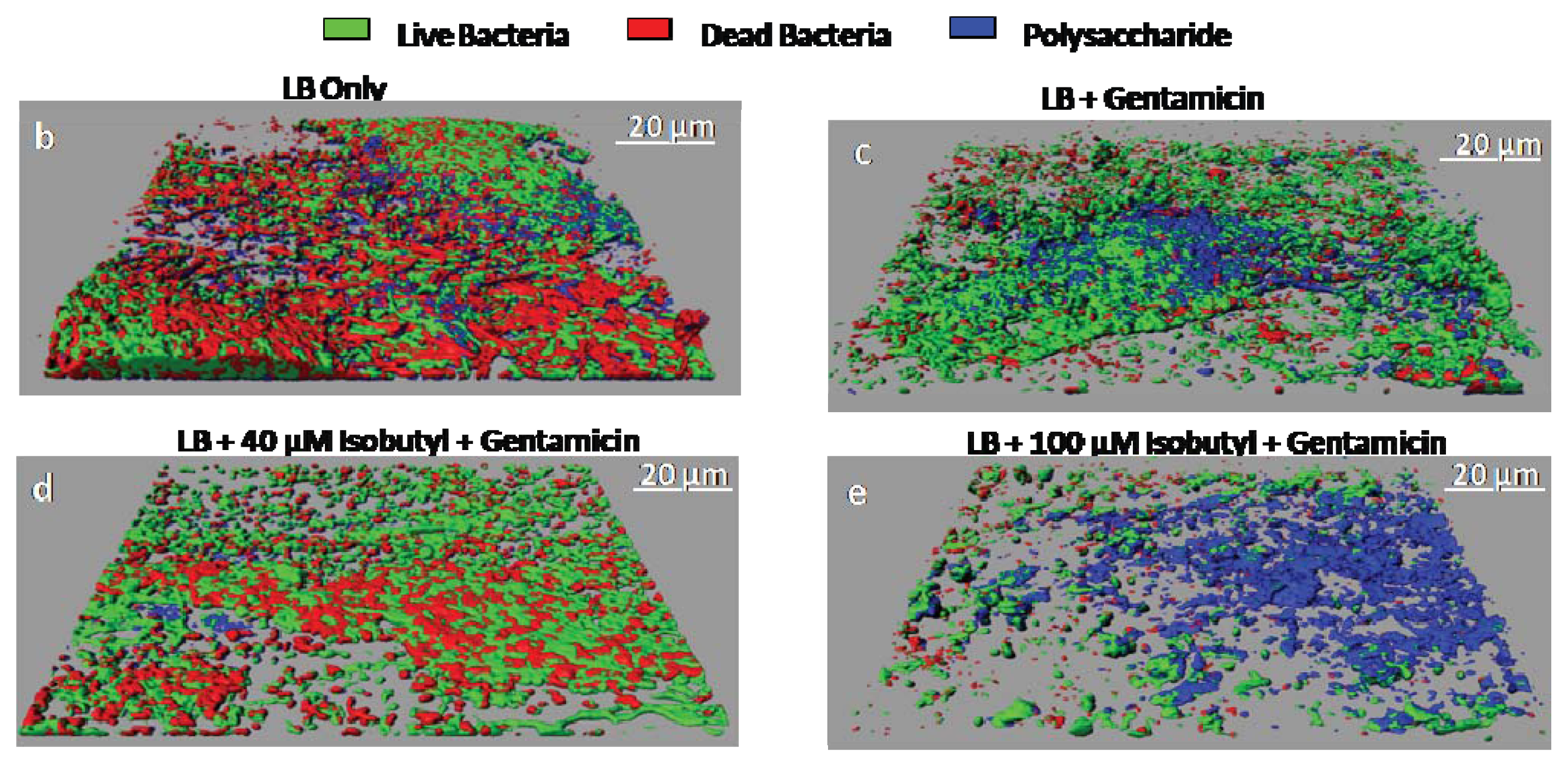
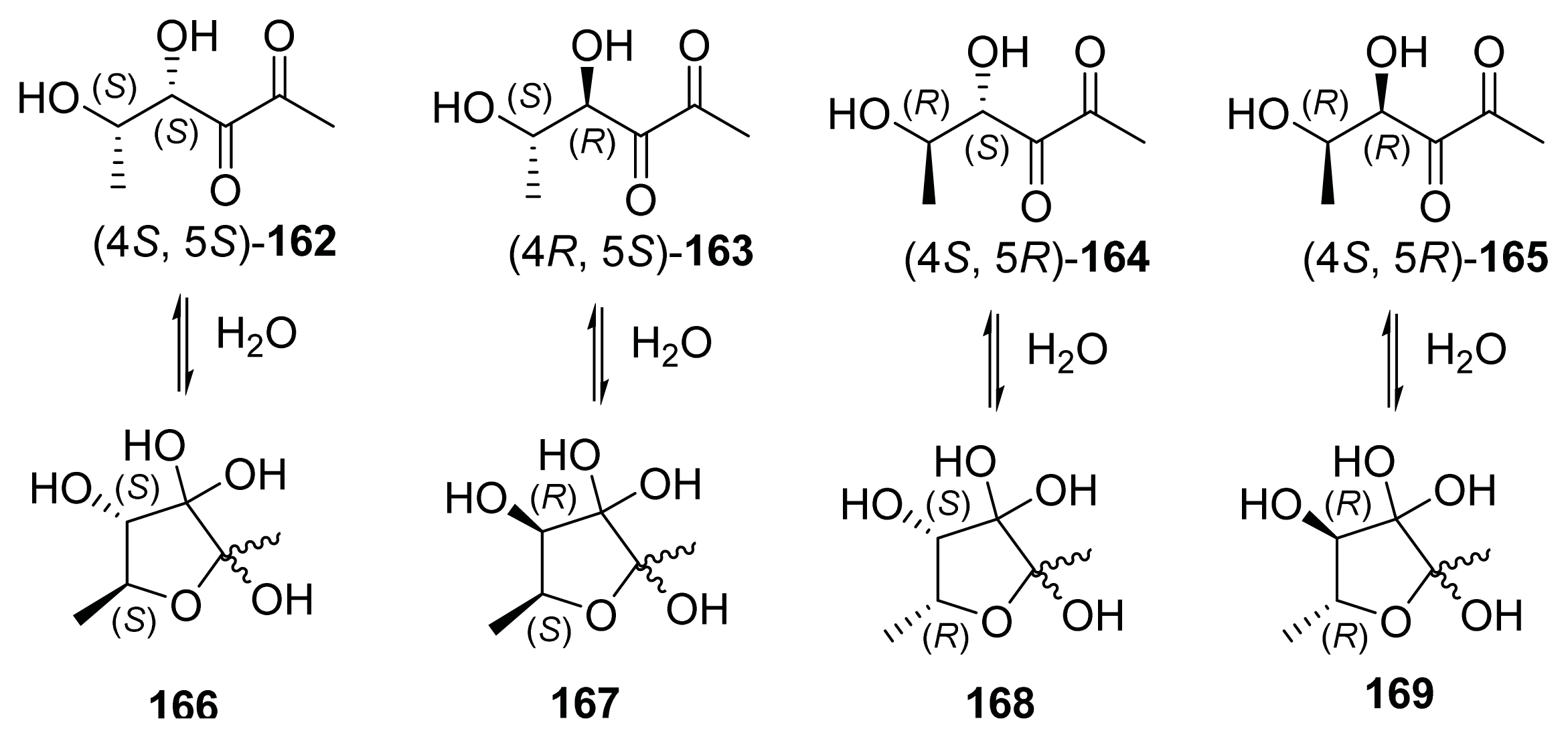
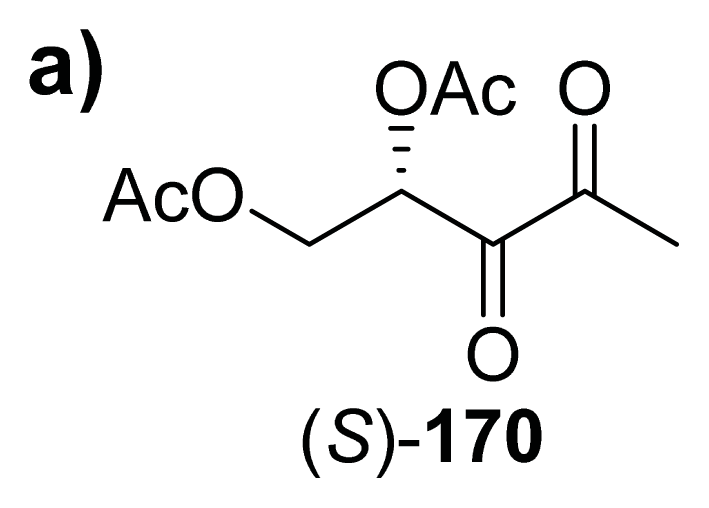
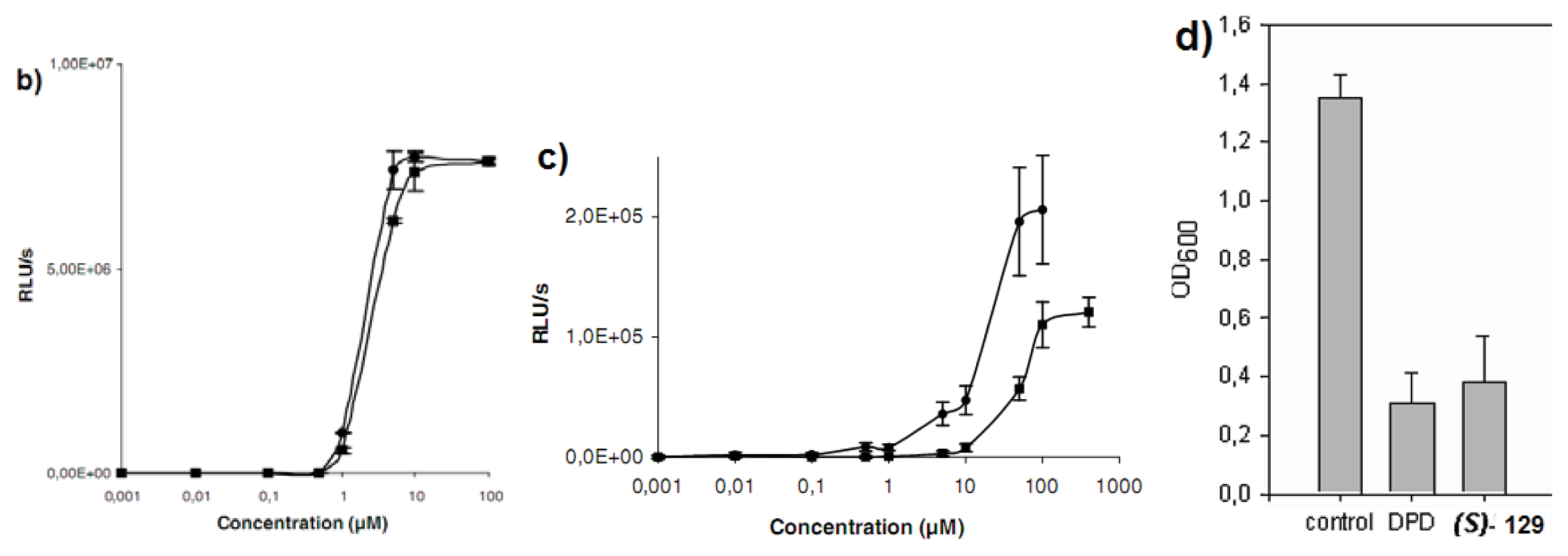
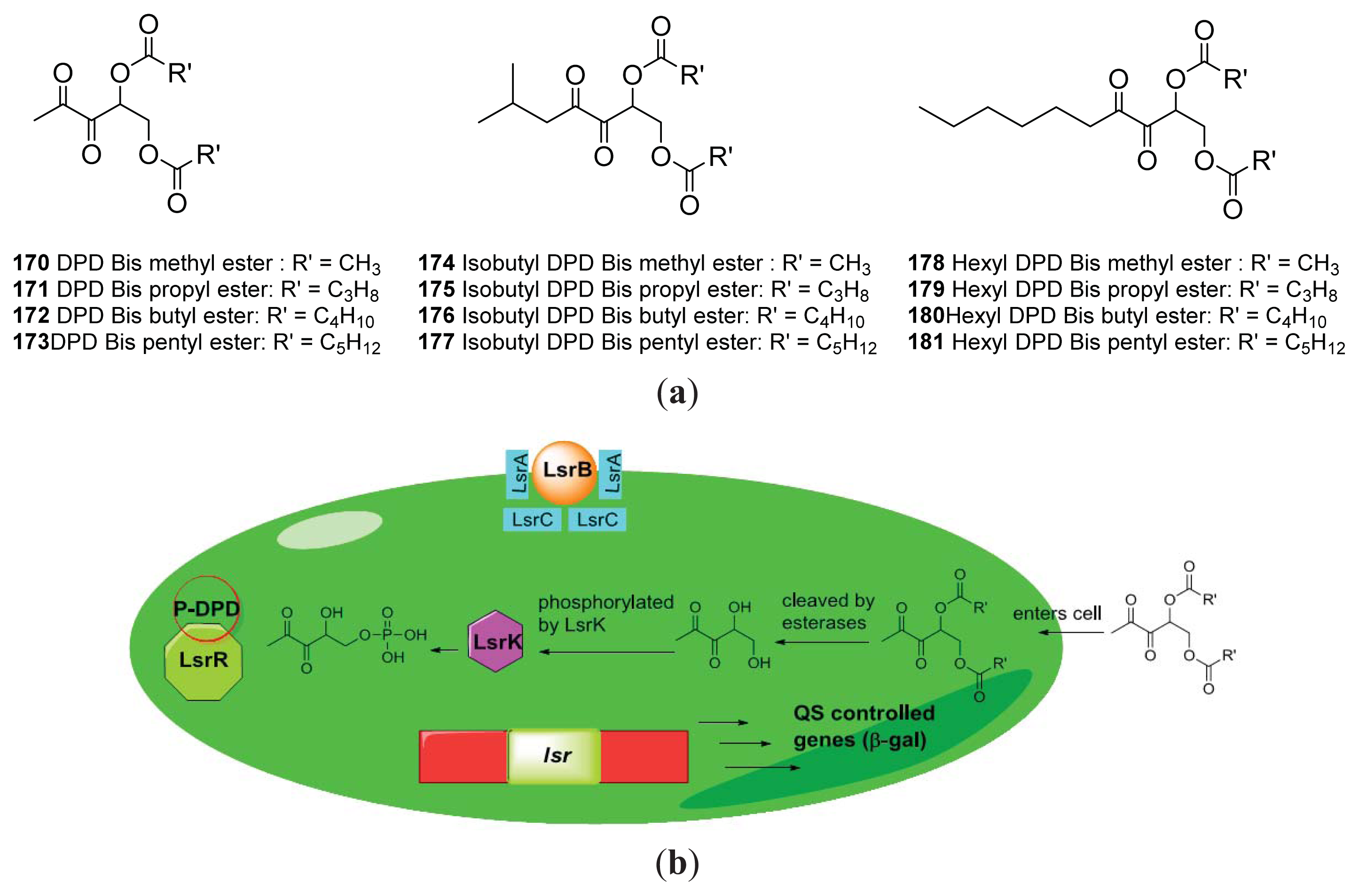



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Organism | Signaling molecules | Receptors | Phenotype |
|---|---|---|---|
| E. coli | AI-2 | LsrB, LsrR | Motility [33], biofilm formation [34] |
| S. typhimurium | AI-2 | LsrB, LsrR | InvF [35] |
| S. aeurus | AI-2, AIP | AgrC, AgrA | Biofilm formation [36] |
| S. anginosus | AI-2 | unknown | Susceptibility to antibiotics [37] |
| M. catarrhalis | AI-2 | unknown | Biofilm formation and antibiotic resistance [38] |
| H. pylori | AI-2 | TlpB | Motility [39] |
| V. cholerae | CAI-1, AI-2 | CqsS, LuxP | Biofilm formation, virulence factor production and protease [19,40] |
| V. harveyi | HAI-1, CAI-1, AI-2 | LuxN, CqsS, LuxP | Bioluminescence, biofilm formation, colony morphology, siderophore production, type III secretion and metalloprotease production [12,41,42] |
| V. fischeri | 3-oxo-C6-HSL, C8-HSL, AI-2 | AinR, LuxP, LuxR | Bioluminescence [43] |
| Y. pestis | 3-oxo-C8-HSL, 3-oxo-C6-HSL, AI-2 | LuxR homologue | Virulence factor expression [44] |
| A. actinomycetemcomitans | AI-2 | RbsB, LsrB | Optimal growth under iron starvation and biofilm development [45] |
© 2013 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Guo, M.; Gamby, S.; Zheng, Y.; Sintim, H.O. Small Molecule Inhibitors of AI-2 Signaling in Bacteria: State-of-the-Art and Future Perspectives for Anti-Quorum Sensing Agents. Int. J. Mol. Sci. 2013, 14, 17694-17728. https://doi.org/10.3390/ijms140917694
Guo M, Gamby S, Zheng Y, Sintim HO. Small Molecule Inhibitors of AI-2 Signaling in Bacteria: State-of-the-Art and Future Perspectives for Anti-Quorum Sensing Agents. International Journal of Molecular Sciences. 2013; 14(9):17694-17728. https://doi.org/10.3390/ijms140917694
Chicago/Turabian StyleGuo, Min, Sonja Gamby, Yue Zheng, and Herman O. Sintim. 2013. "Small Molecule Inhibitors of AI-2 Signaling in Bacteria: State-of-the-Art and Future Perspectives for Anti-Quorum Sensing Agents" International Journal of Molecular Sciences 14, no. 9: 17694-17728. https://doi.org/10.3390/ijms140917694
APA StyleGuo, M., Gamby, S., Zheng, Y., & Sintim, H. O. (2013). Small Molecule Inhibitors of AI-2 Signaling in Bacteria: State-of-the-Art and Future Perspectives for Anti-Quorum Sensing Agents. International Journal of Molecular Sciences, 14(9), 17694-17728. https://doi.org/10.3390/ijms140917694