The Role of Mitochondrial DNA Damage and Repair in the Resistance of BCR/ABL-Expressing Cells to Tyrosine Kinase Inhibitors


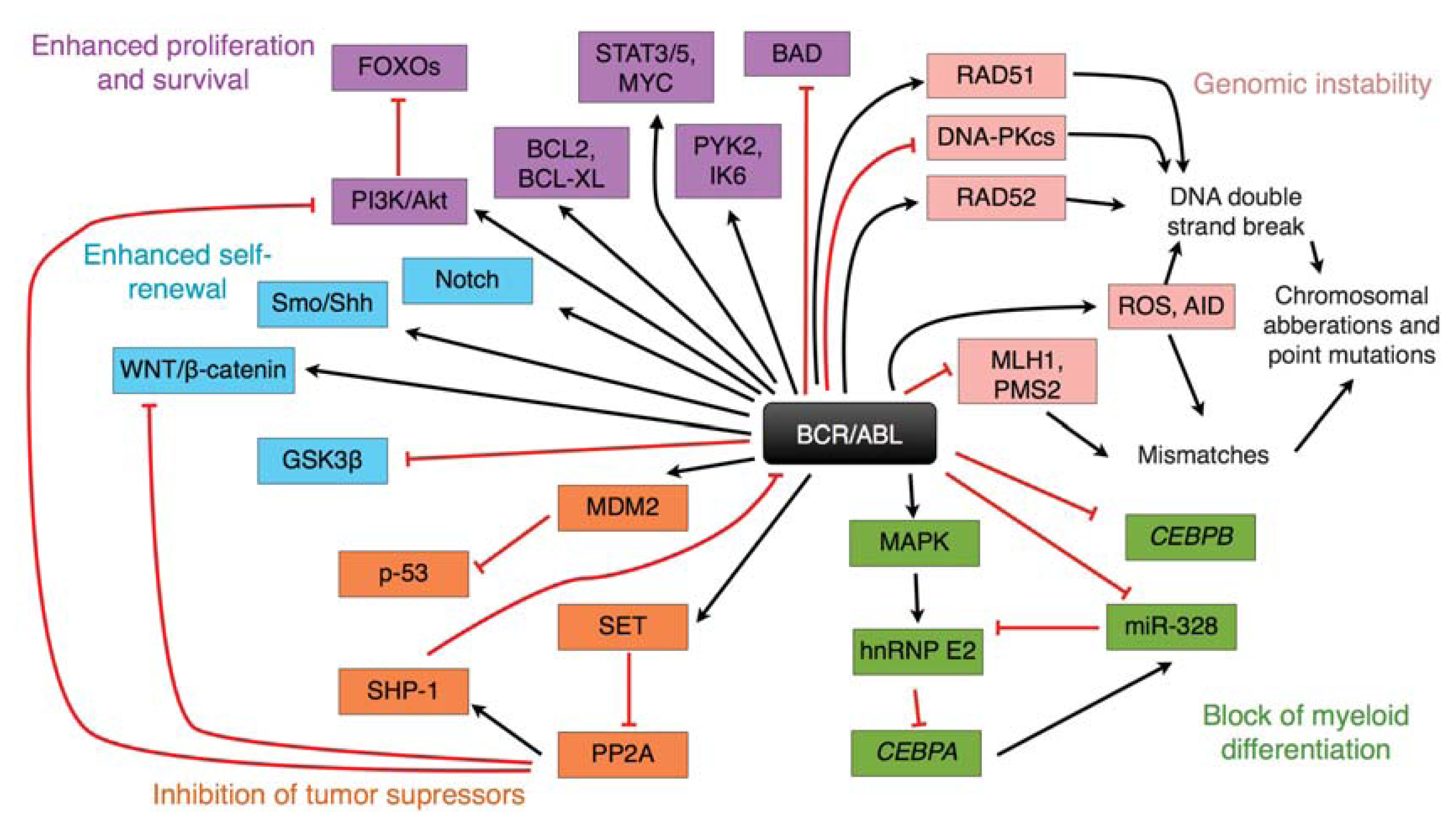{kind=link}
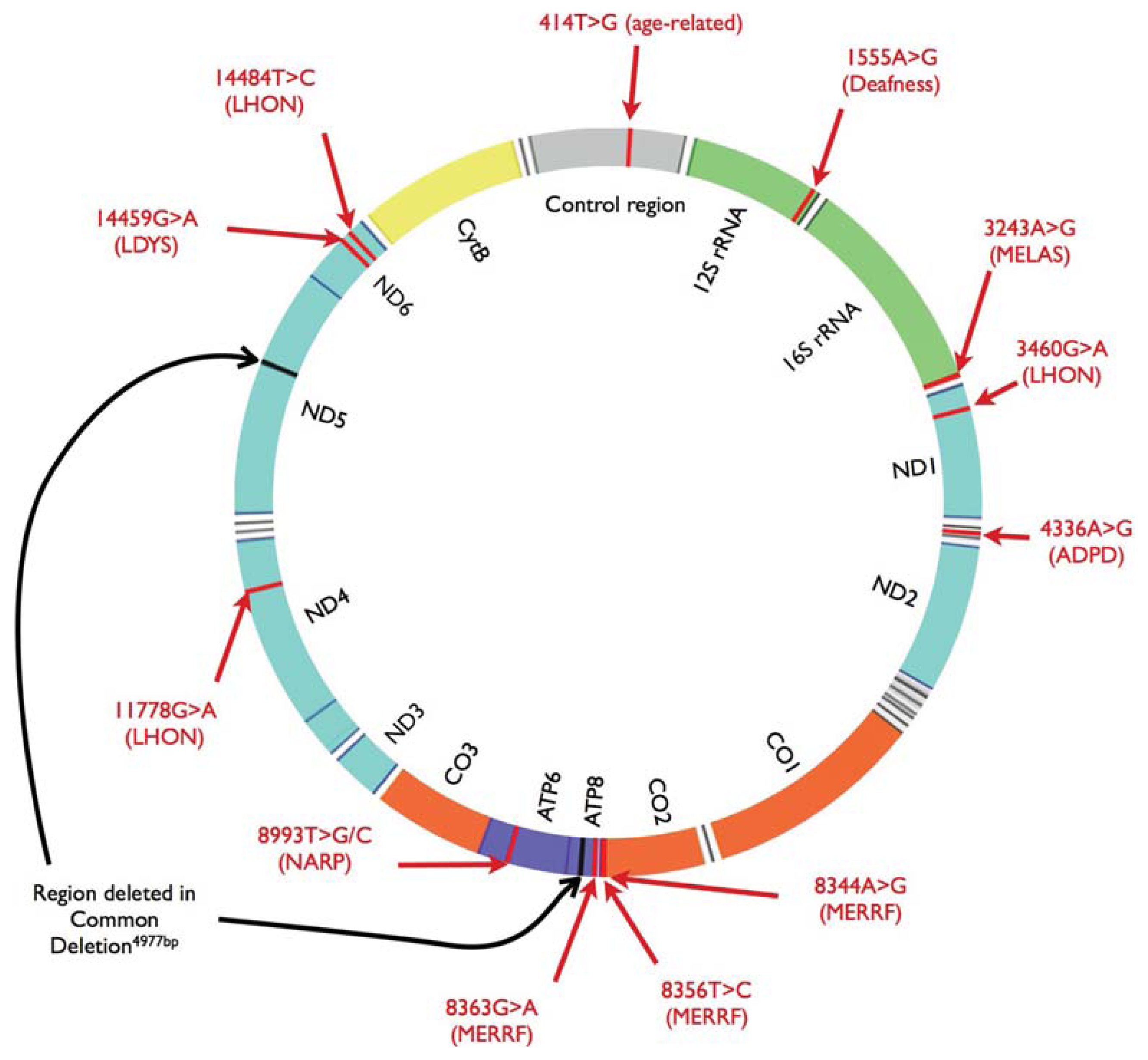{kind=link}
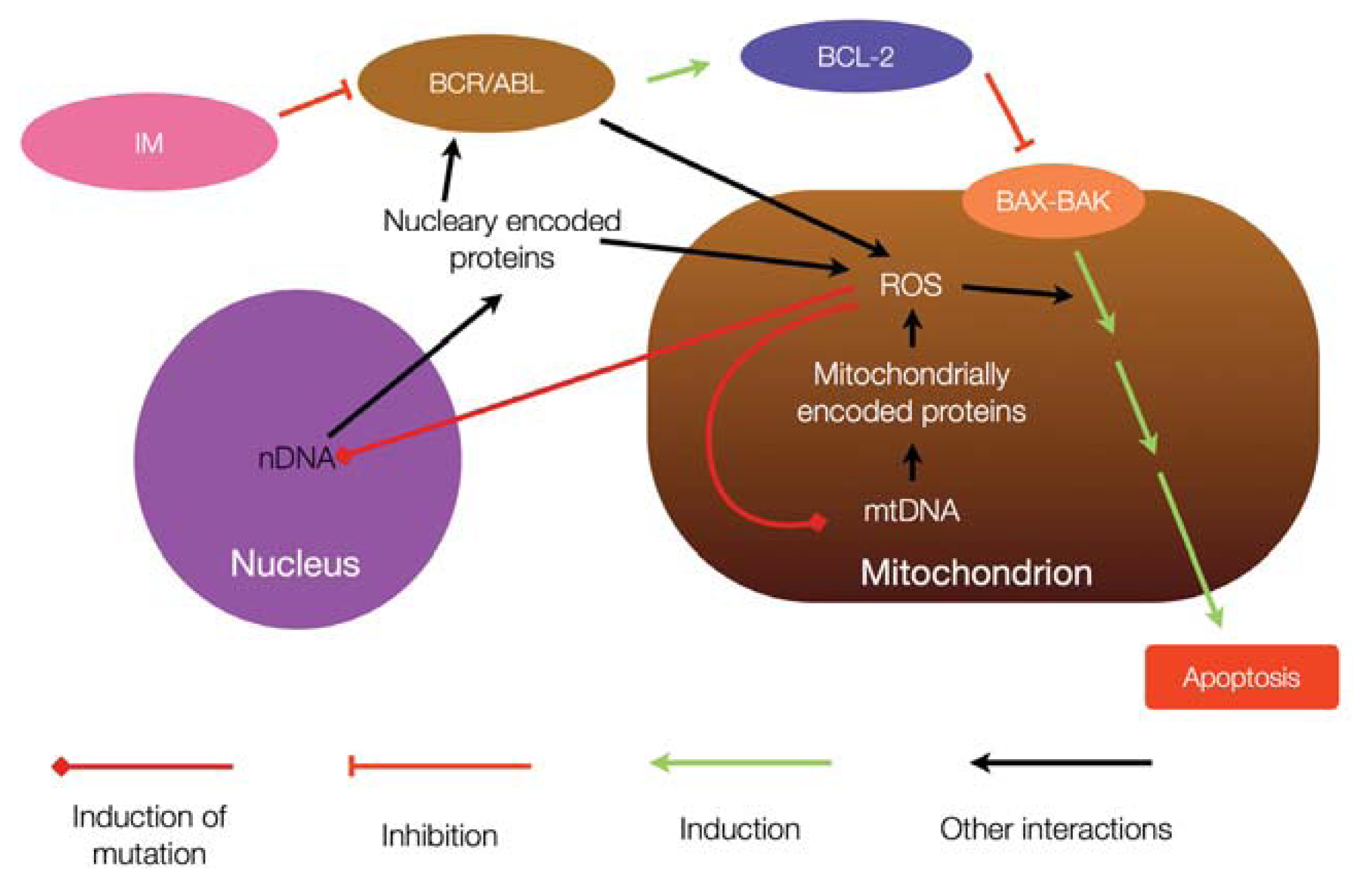
{kind=link}
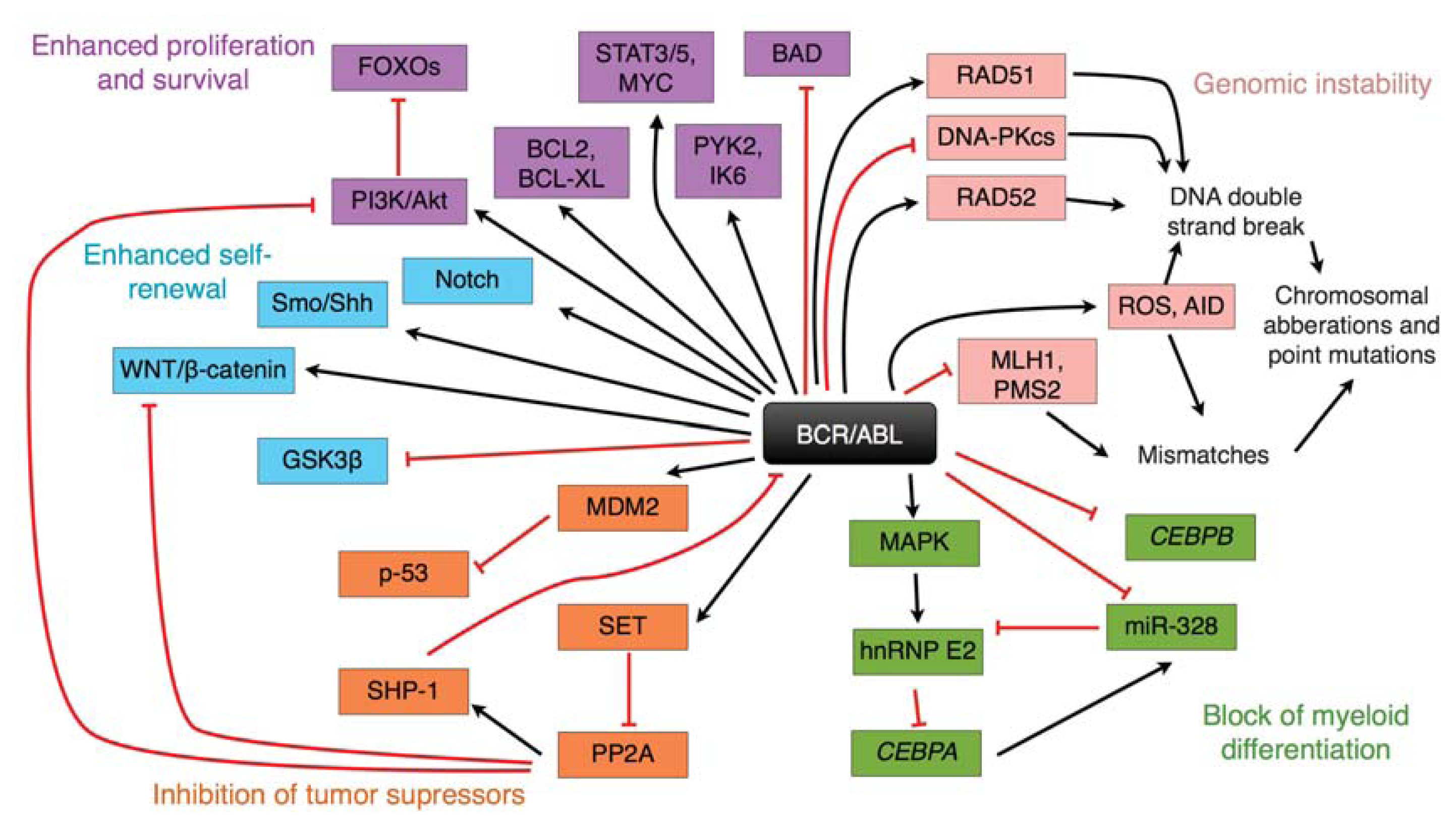
{kind=link}
Abstract
:1. Introduction
2. Imatinib Resistance
- mutations in the BCR/ABL1 kinase domain preventing the binding of imatinib
- clonal evolution
- pharmacokinetic variability
- amplification of the BCR/ABL gene
- overexpression of drug transporter genes
- overexpression of tyrosine kinases
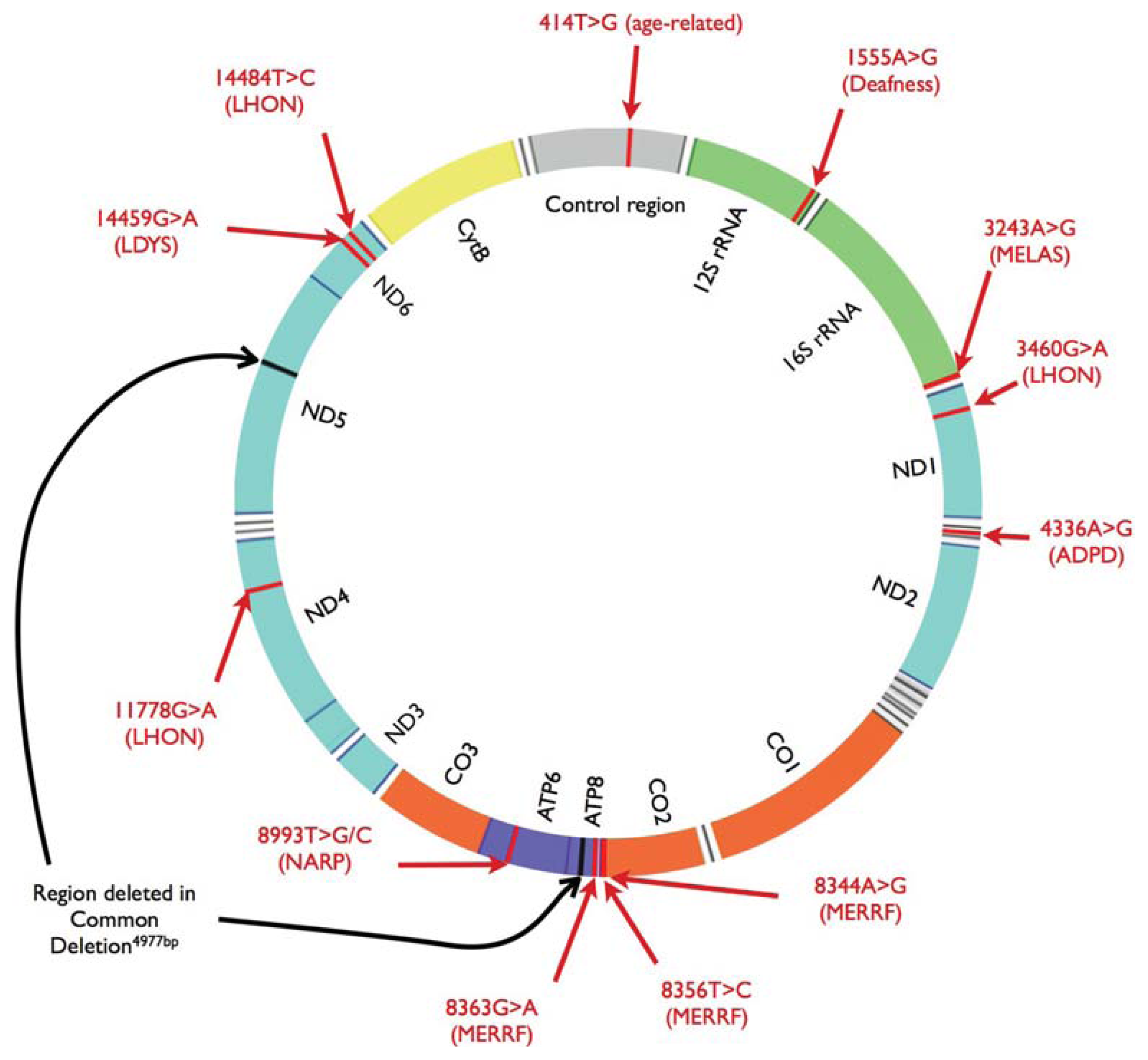
3. Mitochondrial DNA Damage and Repair
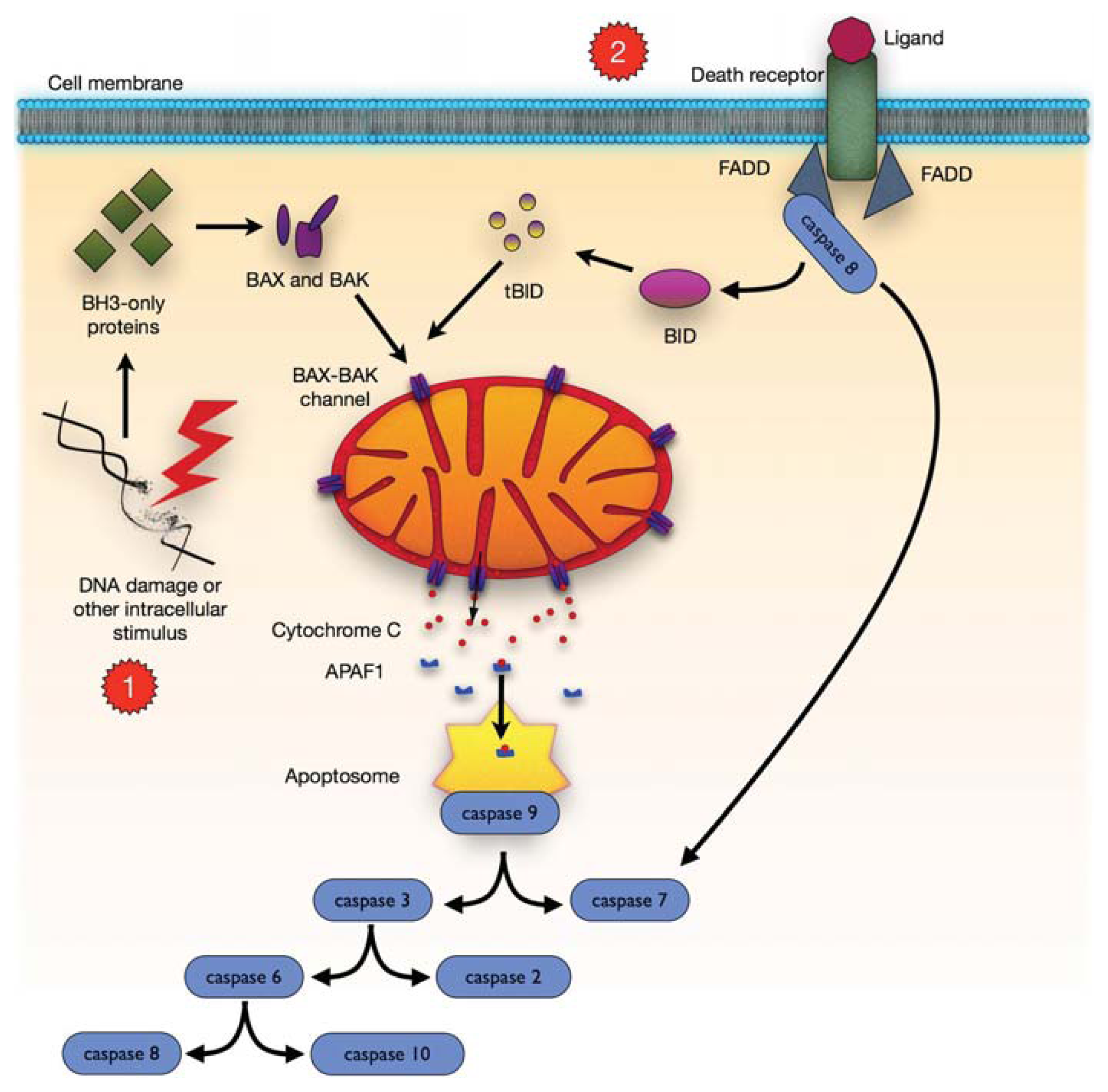
4. Mitochondrial-Dependent Apoptosis
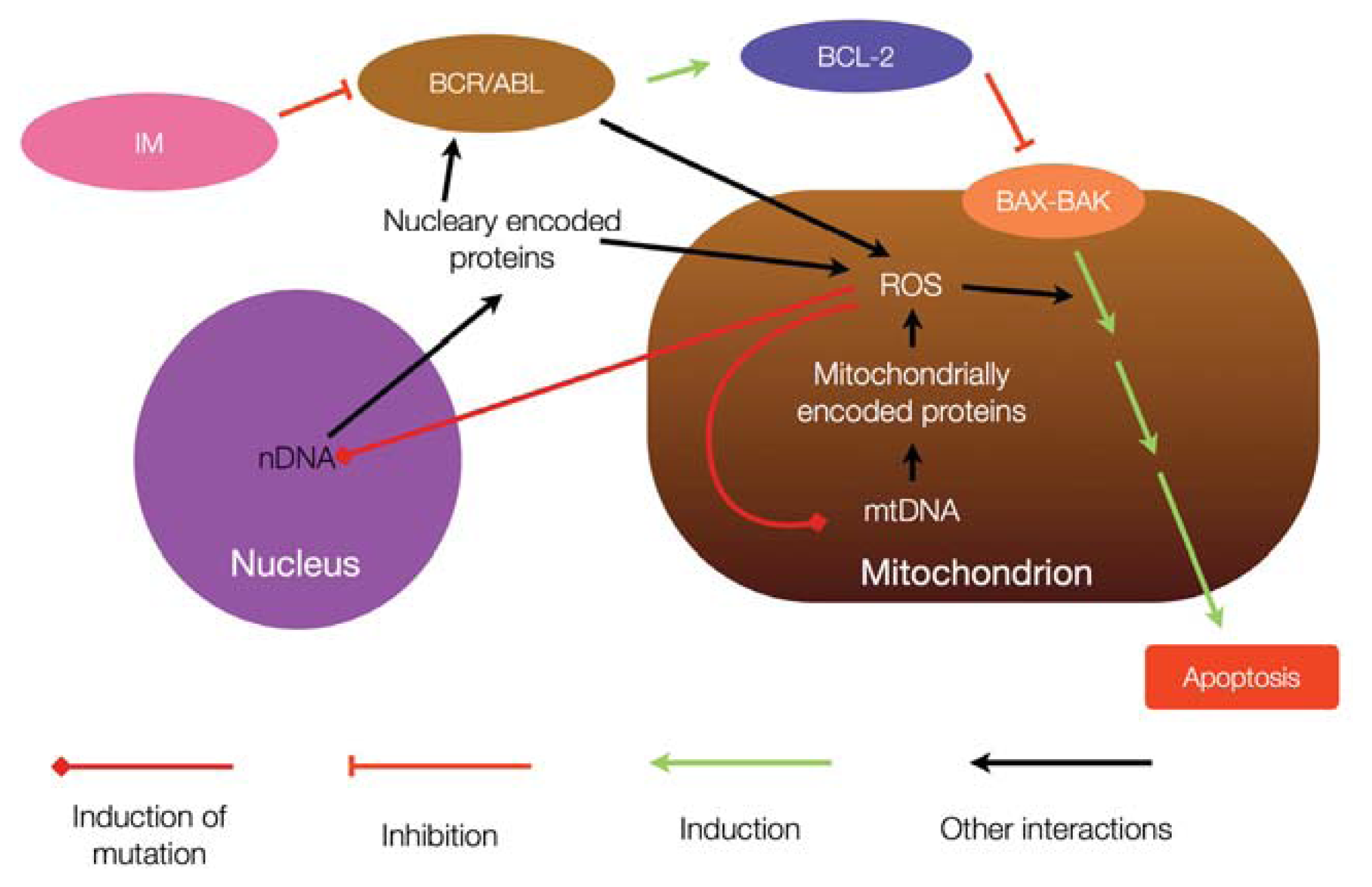
5. Relationship between Chronic Myeloid Leukemia and Mitochondrial Mutagenesis
6. Mitochondria and Resistance to Tyrosine Kinase Inhibitors
7. Conclusions
Acknowledgments
Conflict of Interest
References
- Druker, B.J. Translation of the Philadelphia chromosome into therapy for CML. Blood 2008, 112, 4808–4817. [Google Scholar]
- Shtivelman, E.; Lifshitz, B.; Gale, R.P.; Canaani, E. Fused transcript of abl and bcr genes in chronic myelogenous leukaemia. Nature 1985, 315, 550–554. [Google Scholar]
- Hughes, T.P.; Kaeda, J.; Branford, S.; Rudzki, Z.; Hochhaus, A.; Hensley, M.L.; Gathmann, I.; Bolton, A.E.; van Hoomissen, I.C.; Goldman, J.M.; et al. Frequency of major molecular responses to imatinib or interferon alfa plus cytarabine in newly diagnosed chronic myeloid leukemia. N. Engl. J. Med 2003, 349, 1423–1432. [Google Scholar]
- Rousselot, P.; Huguet, F.; Rea, D.; Legros, L.; Cayuela, J.M.; Maarek, O.; Blanchet, O.; Marit, G.; Gluckman, E.; Reiffers, J.; et al. Imatinib mesylate discontinuation in patients with chronic myelogenous leukemia in complete molecular remission for more than 2 years. Blood 2007, 109, 58–60. [Google Scholar]
- Melo, J.V.; Barnes, D.J. Chronic myeloid leukaemia as a model of disease evolution in human cancer. Nat. Rev. Cancer 2007, 7, 441–453. [Google Scholar]
- Perrotti, D.; Jamieson, C.; Goldman, J.; Skorski, T. Chronic myeloid leukemia: Mechanisms of blastic transformation. J. Clin. Investig 2010, 120, 2254–2264. [Google Scholar]
- Chomel, J.-C.; Turhan, A.G. Chronic myeloid leukemia stem cells in the era of targeted therapies: Resistance, persistence and long-term dormancy. Oncotarget 2011, 2, 713–727. [Google Scholar]
- Graham, S.M.; Jørgensen, H.G.; Allan, E.; Pearson, C.; Alcorn, M.J.; Richmond, L.; Holyoake, T.L. Primitive, quiescent, Philadelphia-positive stem cells from patients with chronic myeloid leukemia are insensitive to STI571 in vitro. Blood 2002, 99, 319–325. [Google Scholar]
- Branford, S.; Rudzki, Z.; Walsh, S.; Grigg, A.; Arthur, C.; Taylor, K.; Herrmann, R.; Lynch, K.P.; Hughes, T.P. High frequency of point mutations clustered within the adenosine triphosphate-binding region of BCR/ABL in patients with chronic myeloid leukemia or Ph-positive acute lymphoblastic leukemia who develop imatinib (STI571) resistance. Blood 2002, 99, 3472–3475. [Google Scholar]
- Miething, C.; Feihl, S.; Mugler, C.; Grundler, R.; von Bubnoff, N.; Lordick, F.; Peschel, C.; Duyster, J. The Bcr-Abl mutations T315I and Y253H do not confer a growth advantage in the absence of imatinib. Leukemia 2006, 20, 650–657. [Google Scholar]
- Soverini, S.; Colarossi, S.; Gnani, A.; Rosti, G.; Castagnetti, F.; Poerio, A.; Iacobucci, I.; Amabile, M.; Abruzzese, E.; Orlandi, E.; et al. GIMEMA working party on chronic myeloid leukemia contribution of ABL kinase domain mutations to imatinib resistance in different subsets of Philadelphia-positive patients: By the GIMEMA working party on chronic myeloid leukemia. Clin. Cancer Res 2006, 12, 7374–7379. [Google Scholar]
- Weisberg, E.; Manley, P.W.; Cowan-Jacob, S.W.; Hochhaus, A.; Griffin, J.D. Second generation inhibitors of BCR-ABL for the treatment of imatinib-resistant chronic myeloid leukaemia. Nat. Rev. Cancer 2007, 7, 345–356. [Google Scholar]
- Manley, P.W.; Cowan-Jacob, S.W.; Mestan, J. Advances in the structural biology, design and clinical development of Bcr-Abl kinase inhibitors for the treatment of chronic myeloid leukaemia. Biochim. Biophys. Acta 2005, 1754, 3–13. [Google Scholar]
- Weisberg, E.; Manley, P.; Mestan, J.; Cowan-Jacob, S.; Ray, A.; Griffin, J.D. AMN107 (nilotinib): A novel and selective inhibitor of BCR-ABL. Br. J. Cancer 2006, 94, 1765–1769. [Google Scholar]
- Amsberg, G.K.-V.; Schafhausen, P. Bosutinib in the management of chronic myelogenous leukemia. Biologics 2013, 7, 115–122. [Google Scholar]
- Press, R.D.; Kamel-Reid, S.; Ang, D. BCR-ABL1 RT-qPCR for monitoring the molecular response to tyrosine kinase inhibitors in chronic myeloid leukemia. J. Mol. Diagn. 2013. [Google Scholar] [CrossRef]
- Apperley, J.F. Part I: Mechanisms of resistance to imatinib in chronic myeloid leukaemia. Lancet Oncol 2007, 8, 1018–1029. [Google Scholar]
- Mahon, F.-X.; Belloc, F.; Lagarde, V.; Chollet, C.; Moreau-Gaudry, F.; Reiffers, J.; Goldman, J.M.; Melo, J.V. MDR1 gene overexpression confers resistance to imatinib mesylate in leukemia cell line models. Blood 2003, 101, 2368–2373. [Google Scholar]
- Jabbour, E.; Kantarjian, H.; Jones, D.; Talpaz, M.; Bekele, N.; O’Brien, S.; Zhou, X.; Luthra, R.; Garcia-Manero, G.; Giles, F.; et al. Frequency and clinical significance of BCR-ABL mutations in patients with chronic myeloid leukemia treated with imatinib mesylate. Leukemia 2006, 20, 1767–1773. [Google Scholar]
- Chandra, J.; Hackbarth, J.; Le, S.; Loegering, D.; Bone, N.; Bruzek, L.M.; Narayanan, V.L.; Adjei, A.A.; Kay, N.E.; Tefferi, A.; et al. Involvement of reactive oxygen species in adaphostin-induced cytotoxicity in human leukemia cells. Blood 2003, 102, 4512–4519. [Google Scholar]
- Chandra, J.; Tracy, J.; Loegering, D.; Flatten, K.; Verstovsek, S.; Beran, M.; Gorre, M.; Estrov, Z.; Donato, N.; Talpaz, M.; et al. Adaphostin-induced oxidative stress overcomes BCR/ABL mutation-dependent and -independent imatinib resistance. Blood 2006, 107, 2501–2506. [Google Scholar]
- Zhang, H.; Trachootham, D.; Lu, W.; Carew, J.; Giles, F.J.; Keating, M.J.; Arlinghaus, R.B.; Huang, P. Effective killing of Gleevec-resistant CML cells with T315I mutation by a natural compound PEITC through redox-mediated mechanism. Leukemia 2008, 22, 1191–1199. [Google Scholar]
- Sattler, M.; Verma, S.; Shrikhande, G.; Byrne, C.H.; Pride, Y.B.; Winkler, T.; Greenfield, E.A.; Salgia, R.; Griffin, J.D. The BCR/ABL tyrosine kinase induces production of reactive oxygen species in hematopoietic cells. J. Biol. Chem 2000, 275, 24273–24278. [Google Scholar]
- Kim, J.H.; Chu, S.C.; Gramlich, J.L.; Pride, Y.B.; Babendreier, E.; Chauhan, D.; Salgia, R.; Podar, K.; Griffin, J.D.; Sattler, M. Activation of the PI3K/mTOR pathway by BCR-ABL contributes to increased production of reactive oxygen species. Blood 2005, 105, 1717–1723. [Google Scholar]
- Koptyra, M.; Falinski, R.; Nowicki, M.O.; Stoklosa, T.; Majsterek, I.; Nieborowska-Skorska, M.; Blasiak, J.; Skorski, T. BCR/ABL kinase induces self-mutagenesis via reactive oxygen species to encode imatinib resistance. Blood 2006, 108, 319–327. [Google Scholar]
- Circu, M.L.; Aw, T.Y. Reactive oxygen species, cellular redox systems, and apoptosis. Free Radic. Biol. Med 2010, 48, 749–762. [Google Scholar]
- Kakkar, P.; Singh, B.K. Mitochondria: A hub of redox activities and cellular distress control. Mol. Cell. Biochem 2007, 305, 235–253. [Google Scholar]
- Richter, C. Oxidative damage to mitochondrial DNA and its relationship to ageing. Int. J. Biochem. Cell Biol 1995, 27, 647–653. [Google Scholar]
- McCord, J.M. Oxygen-derived free radicals in postischemic tissue injury. N. Engl. J. Med 1985, 312, 159–163. [Google Scholar]
- Trifunovic, A.; Wredenberg, A.; Falkenberg, M.; Spelbrink, J.N.; Rovio, A.T.; Bruder, C.E.; Bohlooly-Y, M.; Gidlöf, S.; Oldfors, A.; Wibom, R.; et al. Premature ageing in mice expressing defective mitochondrial DNA polymerase. Nature 2004, 429, 417–423. [Google Scholar]
- Wallace, D.C.; Brown, M.D.; Melov, S.; Graham, B.; Lott, M. Mitochondrial biology, degenerative diseases and aging. Biofactors 1998, 7, 187–190. [Google Scholar]
- Brown, M.D.; Wallace, D.C. Molecular basis of mitochondrial DNA disease. J. Bioenerg. Biomembr 1994, 26, 273–289. [Google Scholar]
- Green, D.R.; Reed, J.C. Mitochondria and apoptosis. Science 1998, 281, 1309–1312. [Google Scholar]
- Petit, P.X.; Susin, S.A.; Zamzami, N.; Mignotte, B.; Kroemer, G. Mitochondria and programmed cell death: Back to the future. FEBS Lett 1996, 396, 7–13. [Google Scholar]
- Hakem, R. DNA-damage repair; the good, the bad, and the ugly. EMBO J 2008, 27, 589–605. [Google Scholar]
- Manouvrier, S.; Rötig, A.; Hannebique, G.; Gheerbrandt, J.D.; Royer-Legrain, G.; Munnich, A.; Parent, M.; Grünfeld, J.P.; Largilliere, C.; Lombes, A. Point mutation of the mitochondrial tRNA(Leu) gene (A 3243 G) in maternally inherited hypertrophic cardiomyopathy, diabetes mellitus, renal failure, and sensorineural deafness. J. Med. Genet 1995, 32, 654–656. [Google Scholar]
- Van den Ouweland, J.M.; Lemkes, H.H.; Ruitenbeek, W.; Sandkuijl, L.A.; de Vijlder, M.F.; Struyvenberg, P.A.; van de Kamp, J.J.; Maassen, J.A. Mutation in mitochondrial tRNALeu(UUR) gene in a large pedigree with maternally transmitted type II diabetes mellitus and deafness. Nat. Genet 1992, 1, 368–371. [Google Scholar]
- Goto, Y.; Nonaka, I.; Horai, S. A mutation in the tRNALeu(UUR) gene associated with the MELAS subgroup of mitochondrial encephalomyopathies. Nature 1990, 348, 651–653. [Google Scholar]
- Fan, H.; Civalier, C.; Booker, J.K.; Gulley, M.L.; Prior, T.W.; Farber, R.A. Detection of common disease-causing mutations in mitochondrial DNA (mitochondrial encephalomyopathy, lactic acidosis with stroke-like episodes MTTL1 3243 A>G and myoclonic epilepsy associated with ragged-red fibers MTTK 8344 A>G) by real-time polymerase chain reaction. J. Mol. Diagn 2006, 8, 277–281. [Google Scholar]
- Gallardo, M.E.; Moreno-Loshuertos, R.; López, C.; Casqueiro, M.; Silva, J.; Bonilla, F.; Rodriguez de Cordoba, S.; Enríquez, J.A. m.6267G>A: A recurrent mutation in the human mitochondrial DNA that reduces cytochrome c oxidase activity and is associated with tumors. Hum. Mutat. 2006, 27, 575–582. [Google Scholar]
- Petros, J.A.; Baumann, A.K.; Ruiz-Pesini, E.; Amin, M.B.; Sun, C.Q.; Hall, J.; Lim, S.; Issa, M.M.; Flanders, W.D.; Hosseini, S.H.; et al. mtDNA mutations increase tumorigenicity in prostate cancer. Proc. Natl. Acad. Sci. USA 2005, 102, 719–724. [Google Scholar]
- Steffann, J.; Gigarel, N.; Corcos, J.; Bonnière, M.; Encha-Razavi, F.; Sinico, M.; Prevot, S.; Dumez, Y.; Yamgnane, A.; Frydman, R.; et al. Stability of the m.8993T→G mtDNA mutation load during human embryofetal development has implications for the feasibility of prenatal diagnosis in NARP syndrome. J. Med. Genet 2007, 44, 664–669. [Google Scholar]
- Marcelino, L.A.; Thilly, W.G. Mitochondrial mutagenesis in human cells and tissues. Mutat. Res 1999, 434, 177–203. [Google Scholar]
- Yu, M. Generation, function and diagnostic value of mitochondrial DNA copy number alterations in human cancers. Life Sci 2011, 89, 65–71. [Google Scholar]
- Lan, Q.; Lim, U.; Liu, C.-S.; Weinstein, S.J.; Chanock, S.; Bonner, M.R.; Virtamo, J.; Albanes, D.; Rothman, N. A prospective study of mitochondrial DNA copy number and risk of non-Hodgkin lymphoma. Blood 2008, 112, 4247–4249. [Google Scholar]
- Gredilla, R.; Bohr, V.A.; Stevnsner, T. Mitochondrial DNA repair and association with aging—An update. Exp. Gerontol 2010, 45, 478–488. [Google Scholar]
- Chen, Z.; Felsheim, R.; Wong, P.; Augustin, L.B.; Metz, R.; Kren, B.T.; Steer, C.J. Mitochondria isolated from liver contain the essential factors required for RNA/DNA oligonucleotide-targeted gene repair. Biochem. Biophys. Res. Commun. 2001, 285, 188–194. [Google Scholar]
- De Souza-Pinto, N.C.; Mason, P.A.; Hashiguchi, K.; Weissman, L.; Tian, J.; Guay, D.; Lebel, M.; Stevnsner, T.V.; Rasmussen, L.J.; Bohr, V.A. Novel DNA mismatch-repair activity involving YB-1 in human mitochondria. DNA Repair 2009, 8, 704–719. [Google Scholar]
- Blasiak, J.; Glowacki, S.; Kauppinen, A.; Kaarniranta, K. Mitochondrial and nuclear DNA damage and repair in age-related macular degeneration. Int. J. Mol. Sci 2013, 14, 2996–3010. [Google Scholar]
- Bohr, V.A. Repair of oxidative DNA damage in nuclear and mitochondrial DNA, and some changes with aging in mammalian cells. Free Radic. Biol. Med 2002, 32, 804–812. [Google Scholar]
- Liu, P.; Qian, L.; Sung, J.-S.; de Souza-Pinto, N.C.; Zheng, L.; Bogenhagen, D.F.; Bohr, V.A.; Wilson, D.M.; Shen, B.; Demple, B. Removal of oxidative DNA damage via FEN1-dependent long-patch base excision repair in human cell mitochondria. Mol. Cell. Biol 2008, 28, 4975–4987. [Google Scholar]
- Ballinger, S.W.; Patterson, C.; Yan, C.N.; Doan, R.; Burow, D.L.; Young, C.G.; Yakes, F.M.; van Houten, B.; Ballinger, C.A.; Freeman, B.A.; et al. Hydrogen peroxide- and peroxynitrite-induced mitochondrial DNA damage and dysfunction in vascular endothelial and smooth muscle cells. Circ. Res 2000, 86, 960–966. [Google Scholar]
- Taylor, R.C.; Cullen, S.P.; Martin, S.J. Apoptosis: Controlled demolition at the cellular level. Nat. Rev. Mol. Cell Biol 2008, 9, 231–241. [Google Scholar]
- Kroemer, G.; Galluzzi, L.; Brenner, C. Mitochondrial membrane permeabilization in cell death. Physiol. Rev 2007, 87, 99–163. [Google Scholar]
- Vaux, D.L. Apoptogenic factors released from mitochondria. Biochim. Biophys. Acta 2011, 1813, 546–550. [Google Scholar]
- Albeck, J.G.; Burke, J.M.; Aldridge, B.B.; Zhang, M.; Lauffenburger, D.A.; Sorger, P.K. Quantitative analysis of pathways controlling extrinsic apoptosis in single cells. Mol. Cell 2008, 30, 11–25. [Google Scholar]
- Shah, N.P.; Kasap, C.; Weier, C.; Balbas, M.; Nicoll, J.M.; Bleickardt, E.; Nicaise, C.; Sawyers, C.L. Transient potent BCR-ABL inhibition is sufficient to commit chronic myeloid leukemia cells irreversibly to apoptosis. Cancer Cell 2008, 14, 485–493. [Google Scholar]
- Czechowska, A.; Poplawski, T.; Drzewoski, J.; Blasiak, J. Imatinib (STI571) induces DNA damage in BCR/ABL-expressing leukemic cells but not in normal lymphocytes. Chem. Biol. Interact 2005, 152, 139–150. [Google Scholar]
- Leber, B.; Lin, J.; Andrews, D.W. Embedded together: The life and death consequences of interaction of the Bcl-2 family with membranes. Apoptosis 2007, 12, 897–911. [Google Scholar]
- Ralph, S.J.; Rodríguez-Enríquez, S.; Neuzil, J.; Saavedra, E.; Moreno-Sánchez, R. The causes of cancer revisited: “Mitochondrial malignancy” and ROS-induced oncogenic transformation—Why mitochondria are targets for cancer therapy. Mol. Aspects Med 2010, 31, 145–170. [Google Scholar]
- Pelicano, H.; Xu, R.-H.; Du, M.; Feng, L.; Sasaki, R.; Carew, J.S.; Hu, Y.; Ramdas, L.; Hu, L.; Keating, M.J.; et al. Mitochondrial respiration defects in cancer cells cause activation of Akt survival pathway through a redox-mediated mechanism. J. Cell Biol 2006, 175, 913–923. [Google Scholar]
- Gordan, J.D.; Thompson, C.B.; Simon, M.C. HIF and c-Myc: Sibling rivals for control of cancer cell metabolism and proliferation. Cancer Cell 2007, 12, 108–113. [Google Scholar]
- Horton, T.M.; Petros, J.A.; Heddi, A.; Shoffner, J.; Kaufman, A.E.; Graham, S.D.; Gramlich, T.; Wallace, D.C. Novel mitochondrial DNA deletion found in a renal cell carcinoma. Genes Chromosom. Cancer 1996, 15, 95–101. [Google Scholar]
- Máximo, V.; Soares, P.; Seruca, R.; Sobrinho-Simões, M. Comments on: Mutations in mitochondrial control region DNA in gastric tumours of Japanese patients, Tamura, et al. Eur. J. Cancer 1999, 35, 316–319. [Google Scholar]
- Ishikawa, K.; Koshikawa, N.; Takenaga, K.; Nakada, K.; Hayashi, J.-I. Reversible regulation of metastasis by ROS-generating mtDNA mutations. Mitochondrion 2008, 8, 339–344. [Google Scholar]
- Lee, H.C.; Yin, P.H.; Lu, C.Y.; Chi, C.W.; Wei, Y.H. Increase of mitochondria and mitochondrial DNA in response to oxidative stress in human cells. Biochem. J 2000, 348, 425–432. [Google Scholar]
- Todorov, I.N.; Todorov, G.I. Multifactorial nature of high frequency of mitochondrial DNA mutations in somatic mammalian cells. Biochemistry 2009, 74, 962–970. [Google Scholar]
- Carew, J.S.; Huang, P. Mitochondrial defects in cancer. Mol. Cancer 2002, 1. [Google Scholar] [CrossRef] [Green Version]
- Carew, J.S.; Zhou, Y.; Albitar, M.; Carew, J.D.; Keating, M.J.; Huang, P. Mitochondrial DNA mutations in primary leukemia cells after chemotherapy: Clinical significance and therapeutic implications. Leukemia 2003, 17, 1437–1447. [Google Scholar]
- Silkjaer, T.; Nørgaard, J.M.; Aggerholm, A.; Ebbesen, L.H.; Kjeldsen, E.; Hokland, P.; Nyvold, C.G. Characterization and prognostic significance of mitochondrial DNA variations in acute myeloid leukemia. Eur. J. Haematol 2013, 90, 385–396. [Google Scholar]
- Clayton, D.A.; Vinograd, J. Complex mitochondrial DNA in leukemic and normal human myeloid cells. Proc. Natl. Acad. Sci. USA 1969, 62, 1077–1084. [Google Scholar]
- Kujoth, G.C.; Hiona, A.; Pugh, T.D.; Someya, S.; Panzer, K.; Wohlgemuth, S.E.; Hofer, T.; Seo, A.Y.; Sullivan, R.; Jobling, W.A.; et al. Mitochondrial DNA mutations, oxidative stress, and apoptosis in mammalian aging. Science 2005, 309, 481–484. [Google Scholar]
- Wang, J.; Silva, J.P.; Gustafsson, C.M.; Rustin, P.; Larsson, N.G. Increased in vivo apoptosis in cells lacking mitochondrial DNA gene expression. Proc. Natl. Acad. Sci. USA 2001, 98, 4038–4043. [Google Scholar]
- Park, J.S.; Sharma, L.K.; Li, H.; Xiang, R.; Holstein, D.; Wu, J.; Lechleiter, J.; Naylor, S.L.; Deng, J.J.; Lu, J.; et al. A heteroplasmic, not homoplasmic, mitochondrial DNA mutation promotes tumorigenesis via alteration in reactive oxygen species generation and apoptosis. Hum. Mol. Genet 2009, 18, 1578–1589. [Google Scholar]
- Marin, J.J.G.; Hernandez, A.; Revuelta, I.E.; Gonzalez-Sanchez, E.; Gonzalez-Buitrago, J.M.; Perez, M.J. Mitochondrial genome depletion in human liver cells abolishes bile acid-induced apoptosis: Role of the Akt/mTOR survival pathway and Bcl-2 family proteins. Free Radic. Biol. Med 2013, 61, 218–228. [Google Scholar]
- Nieborowska-Skorska, M.; Kopinski, P.K.; Ray, R.; Hoser, G.; Ngaba, D.; Flis, S.; Cramer, K.; Reddy, M.M.; Koptyra, M.; Penserga, T.; et al. Rac2-MRC-cIII-generated ROS cause genomic instability in chronic myeloid leukemia stem cells and primitive progenitors. Blood 2012, 119, 4253–4263. [Google Scholar]
- Amarante-Mendes, G.P.; Naekyung Kim, C.; Liu, L.; Huang, Y.; Perkins, C.L.; Green, D.R.; Bhalla, K. Bcr-Abl exerts its antiapoptotic effect against diverse apoptotic stimuli through blockage of mitochondrial release of cytochrome C and activation of caspase-3. Blood 1998, 91, 1700–1705. [Google Scholar]
- Pellicano, F.; Copland, M.; Jørgensen, H.G.; Mountford, J.; Leber, B.; Holyoake, T.L. BMS-214662 induces mitochondrial apoptosis in chronic myeloid leukemia (CML) stem/progenitor cells, including CD34+38− cells, through activation of protein kinase Cbeta. Blood 2009, 114, 4186–4196. [Google Scholar]
- Kurosu, T.; Wu, N.; Oshikawa, G.; Kagechika, H.; Miura, O. Enhancement of imatinib-induced apoptosis of BCR/ABL-expressing cells by nutlin-3 through synergistic activation of the mitochondrial apoptotic pathway. Apoptosis 2010, 15, 608–620. [Google Scholar]
- Kurosu, T.; Ohki, M.; Wu, N.; Kagechika, H.; Miura, O. Sorafenib induces apoptosis specifically in cells expressing BCR/ABL by inhibiting its kinase activity to activate the intrinsic mitochondrial pathway. Cancer Res 2009, 69, 3927–3936. [Google Scholar]
- Kominsky, D.J.; Klawitter, J.; Brown, J.L.; Boros, L.G.; Melo, J.V.; Eckhardt, S.G.; Serkova, N.J. Abnormalities in glucose uptake and metabolism in imatinib-resistant human BCR-ABL-positive cells. Clin. Cancer Res 2009, 15, 3442–3450. [Google Scholar]
- Bentley, J.; Itchayanan, D.; Barnes, K.; McIntosh, E.; Tang, X.; Downes, C.P.; Holman, G.D.; Whetton, A.D.; Owen-Lynch, P.J.; Baldwin, S.A. Interleukin-3-mediated cell survival signals include phosphatidylinositol 3-kinase-dependent translocation of the glucose transporter GLUT1 to the cell surface. J. Biol. Chem 2003, 278, 39337–39348. [Google Scholar]
- Elstrom, R.L.; Bauer, D.E.; Buzzai, M.; Karnauskas, R.; Harris, M.H.; Plas, D.R.; Zhuang, H.; Cinalli, R.M.; Alavi, A.; Rudin, C.M.; et al. Akt stimulates aerobic glycolysis in cancer cells. Cancer Res 2004, 64, 3892–3899. [Google Scholar]
- Barnes, K.; McIntosh, E.; Whetton, A.D.; Daley, G.Q.; Bentley, J.; Baldwin, S.A. Chronic myeloid leukaemia: An investigation into the role of Bcr-Abl-induced abnormalities in glucose transport regulation. Oncogene 2005, 24, 3257–3267. [Google Scholar]
- Boren, J.; Cascante, M.; Marin, S.; Comín-Anduix, B.; Centelles, J.J.; Lim, S.; Bassilian, S.; Ahmed, S.; Lee, W.N.; Boros, L.G. Gleevec (STI571) influences metabolic enzyme activities and glucose carbon flow toward nucleic acid and fatty acid synthesis in myeloid tumor cells. J. Biol. Chem 2001, 276, 37747–37753. [Google Scholar]
- Gottschalk, S.; Anderson, N.; Hainz, C.; Eckhardt, S.G.; Serkova, N.J. Imatinib (STI571)-mediated changes in glucose metabolism in human leukemia BCR-ABL-positive cells. Clin. Cancer Res 2004, 10, 6661–6668. [Google Scholar]
- Klawitter, J.; Anderson, N.; Klawitter, J.; Christians, U.; Leibfritz, D.; Eckhardt, S.G.; Serkova, N.J. Time-dependent effects of imatinib in human leukaemia cells: A kinetic NMR-profiling study. Br. J. Cancer 2009, 100, 923–931. [Google Scholar]
- Kluza, J.; Jendoubi, M.; Ballot, C.; Dammak, A.; Jonneaux, A.; Idziorek, T.; Joha, S.; Dauphin, V.; Malet-Martino, M.; Balayssac, S.; et al. Exploiting mitochondrial dysfunction for effective elimination of imatinib-resistant leukemic cells. PLoS One 2011, 6, e21924. [Google Scholar]
- Zhao, F.; Mancuso, A.; Bui, T.V.; Tong, X.; Gruber, J.J.; Swider, C.R.; Sanchez, P.V.; Lum, J.J.; Sayed, N.; Melo, J.V.; et al. Imatinib resistance associated with BCR-ABL upregulation is dependent on HIF-1alpha-induced metabolic reprograming. Oncogene 2010, 29, 2962–2972. [Google Scholar]
- Warburg, O. On respiratory impairment in cancer cells. Science 1956, 124, 269–270. [Google Scholar]
- Mason, E.F.; Zhao, Y.; Goraksha-Hicks, P.; Coloff, J.L.; Gannon, H.; Jones, S.N.; Rathmell, J.C. Aerobic glycolysis suppresses p53 activity to provide selective protection from apoptosis upon loss of growth signals or inhibition of BCR-Abl. Cancer Res 2010, 70, 8066–8076. [Google Scholar]
© 2013 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Glowacki, S.; Synowiec, E.; Blasiak, J. The Role of Mitochondrial DNA Damage and Repair in the Resistance of BCR/ABL-Expressing Cells to Tyrosine Kinase Inhibitors. Int. J. Mol. Sci. 2013, 14, 16348-16364. https://doi.org/10.3390/ijms140816348
Glowacki S, Synowiec E, Blasiak J. The Role of Mitochondrial DNA Damage and Repair in the Resistance of BCR/ABL-Expressing Cells to Tyrosine Kinase Inhibitors. International Journal of Molecular Sciences. 2013; 14(8):16348-16364. https://doi.org/10.3390/ijms140816348
Chicago/Turabian StyleGlowacki, Sylwester, Ewelina Synowiec, and Janusz Blasiak. 2013. "The Role of Mitochondrial DNA Damage and Repair in the Resistance of BCR/ABL-Expressing Cells to Tyrosine Kinase Inhibitors" International Journal of Molecular Sciences 14, no. 8: 16348-16364. https://doi.org/10.3390/ijms140816348
APA StyleGlowacki, S., Synowiec, E., & Blasiak, J. (2013). The Role of Mitochondrial DNA Damage and Repair in the Resistance of BCR/ABL-Expressing Cells to Tyrosine Kinase Inhibitors. International Journal of Molecular Sciences, 14(8), 16348-16364. https://doi.org/10.3390/ijms140816348