MicroRNA Transcriptomes Relate Intermuscular Adipose Tissue to Metabolic Risk

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
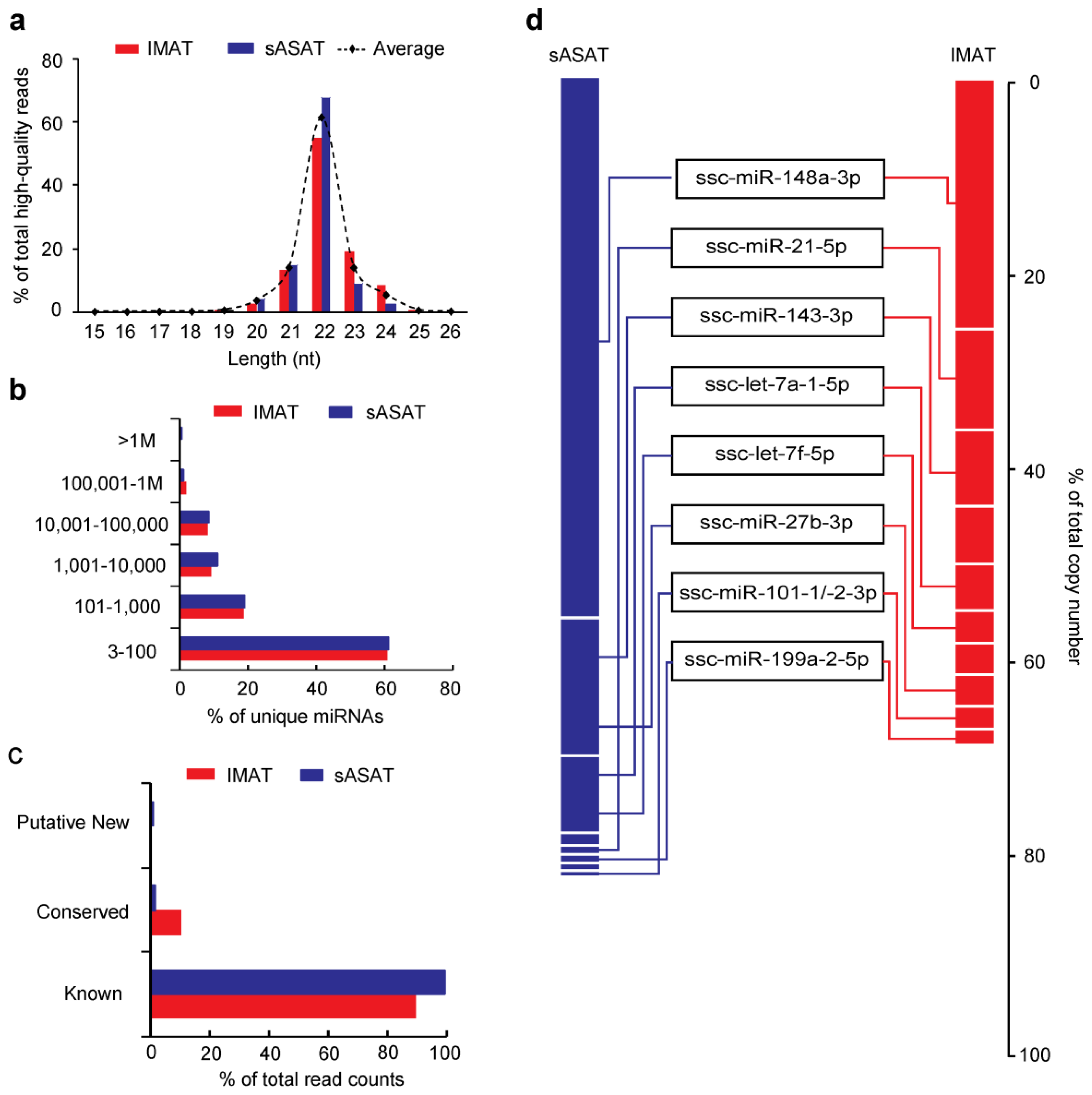
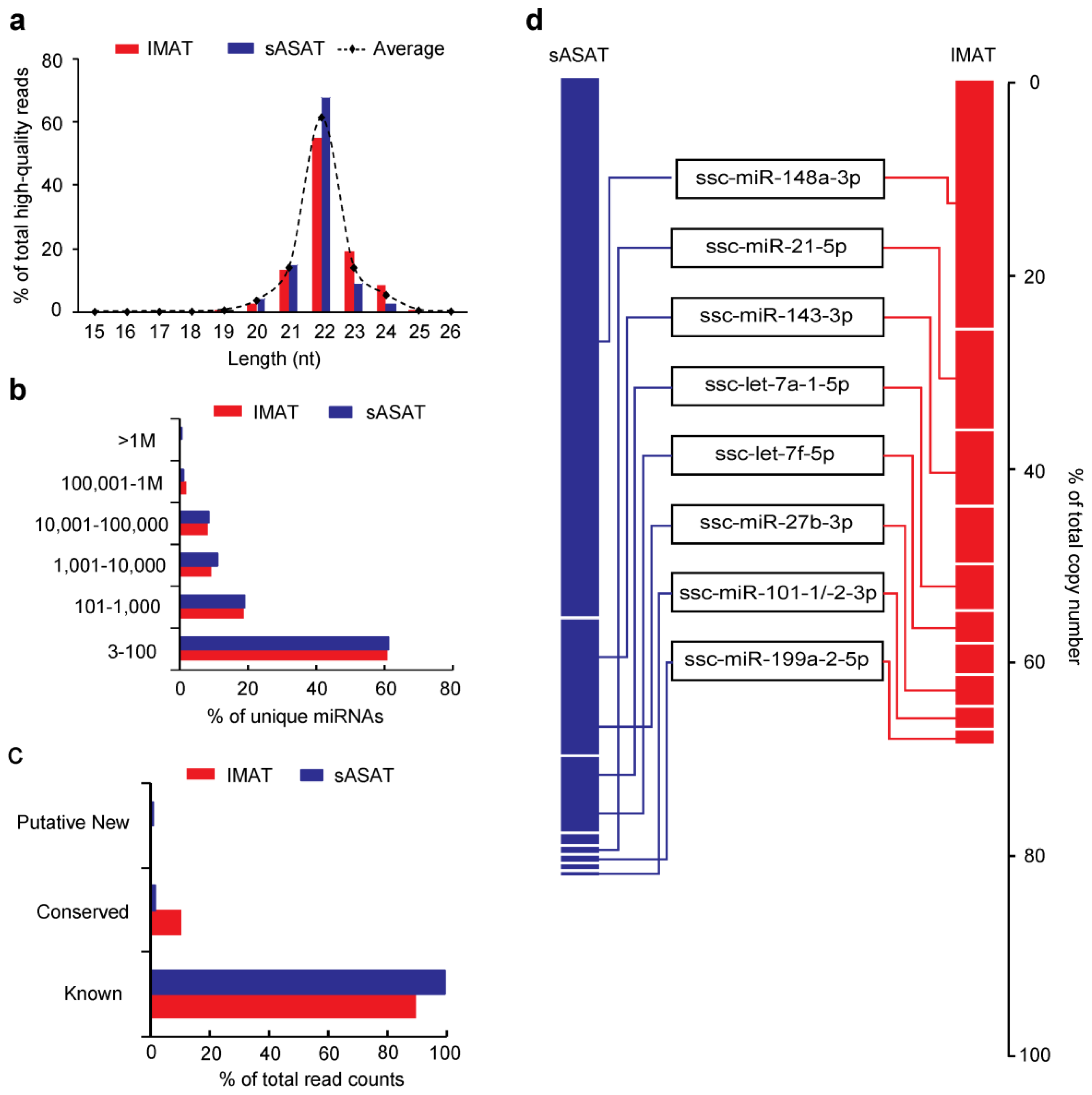
2.1. Transcriptome Sequencing Data
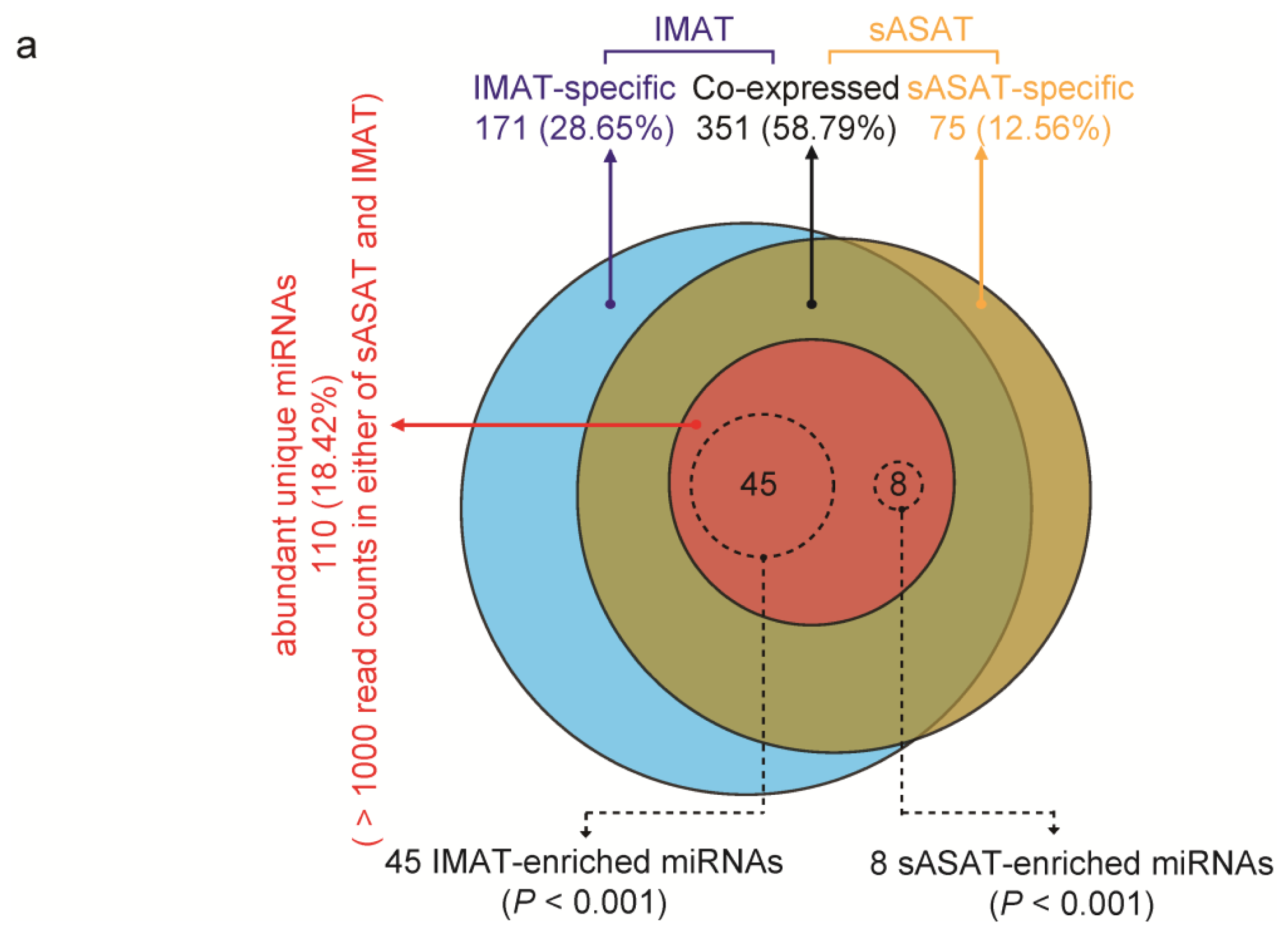
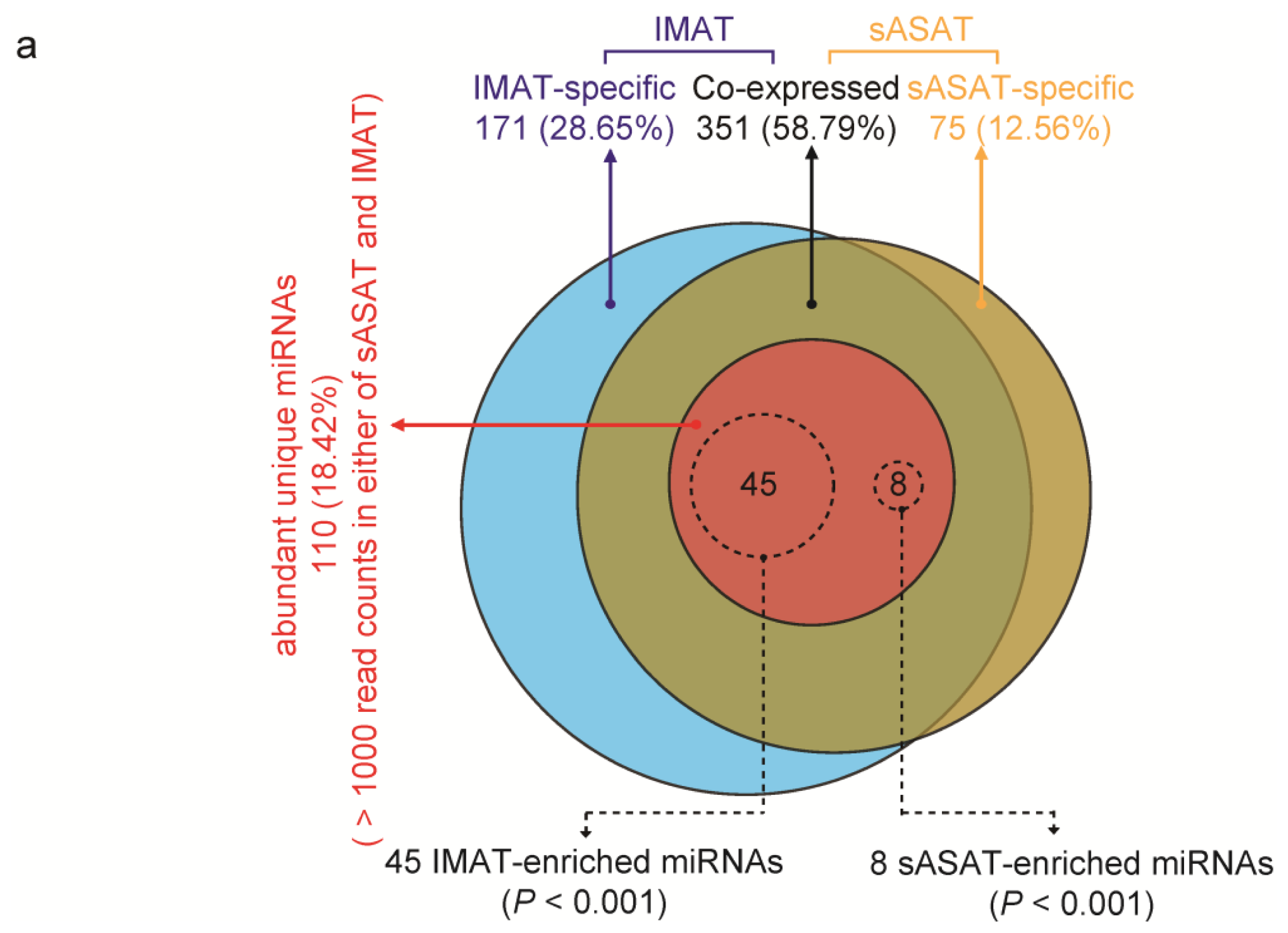
2.2. MiRNA Profiling of sASAT and IMAT
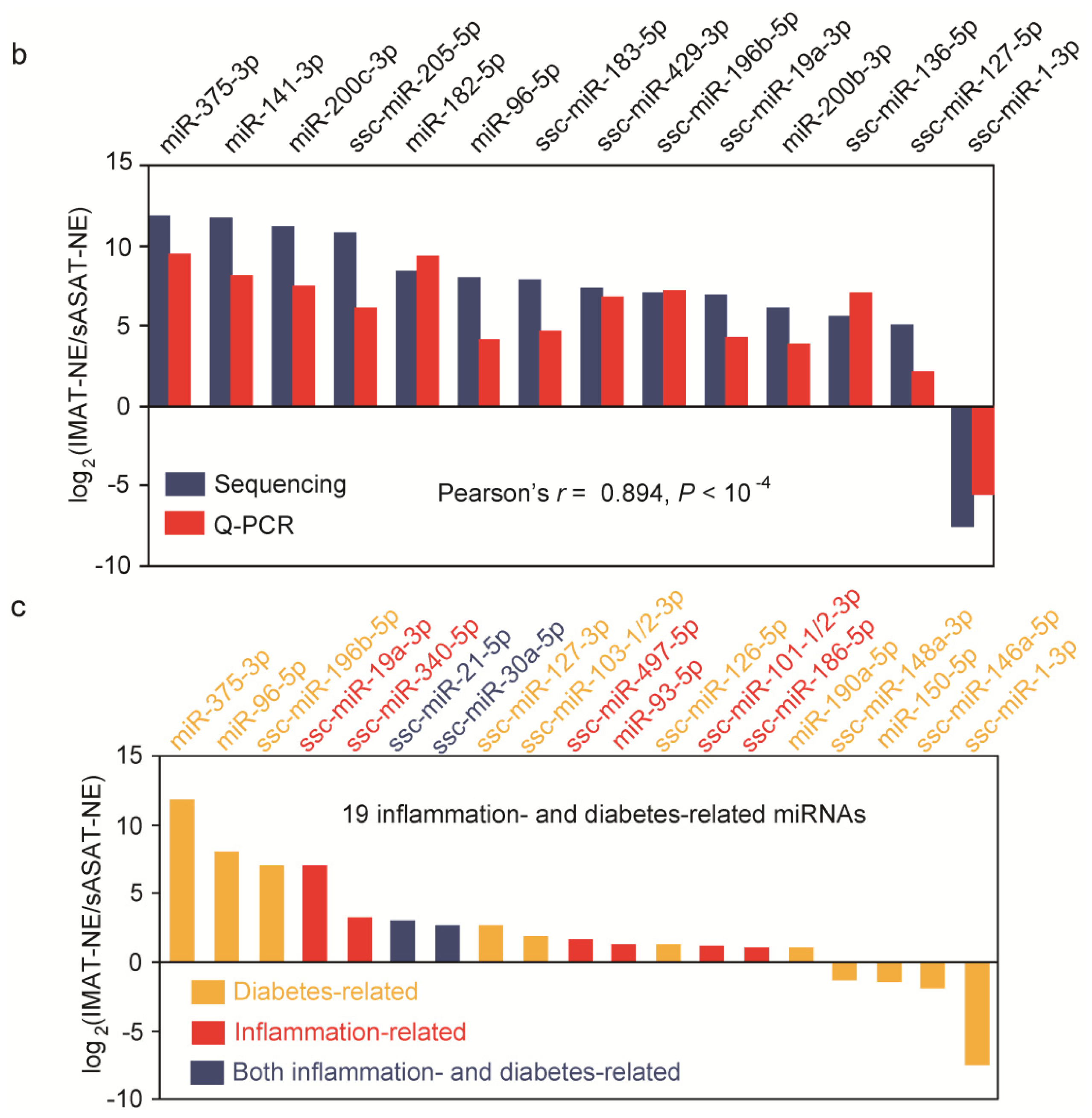
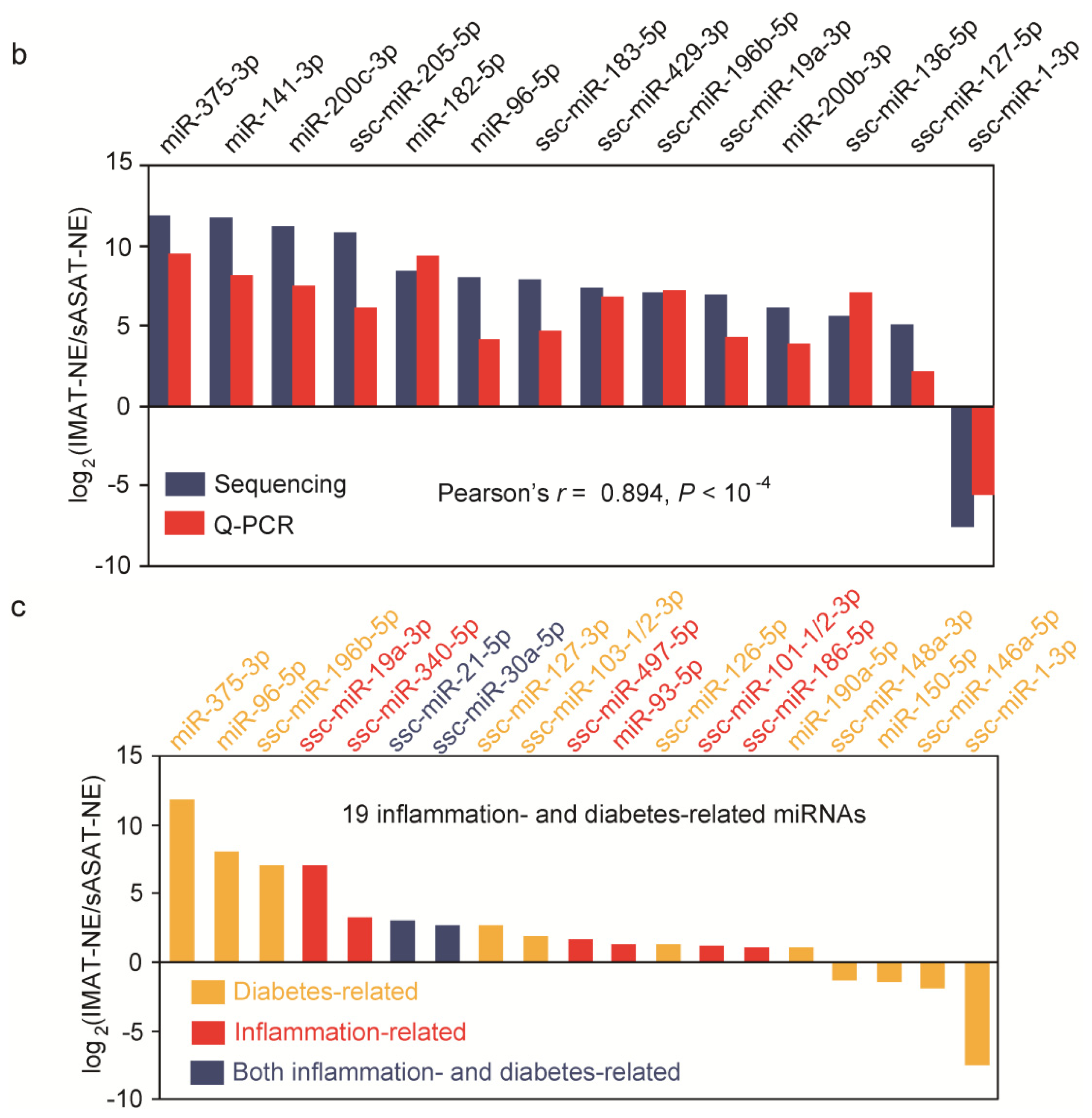
2.3. Inflammation- and Diabetes-Related miRNAs Enriched in IMAT
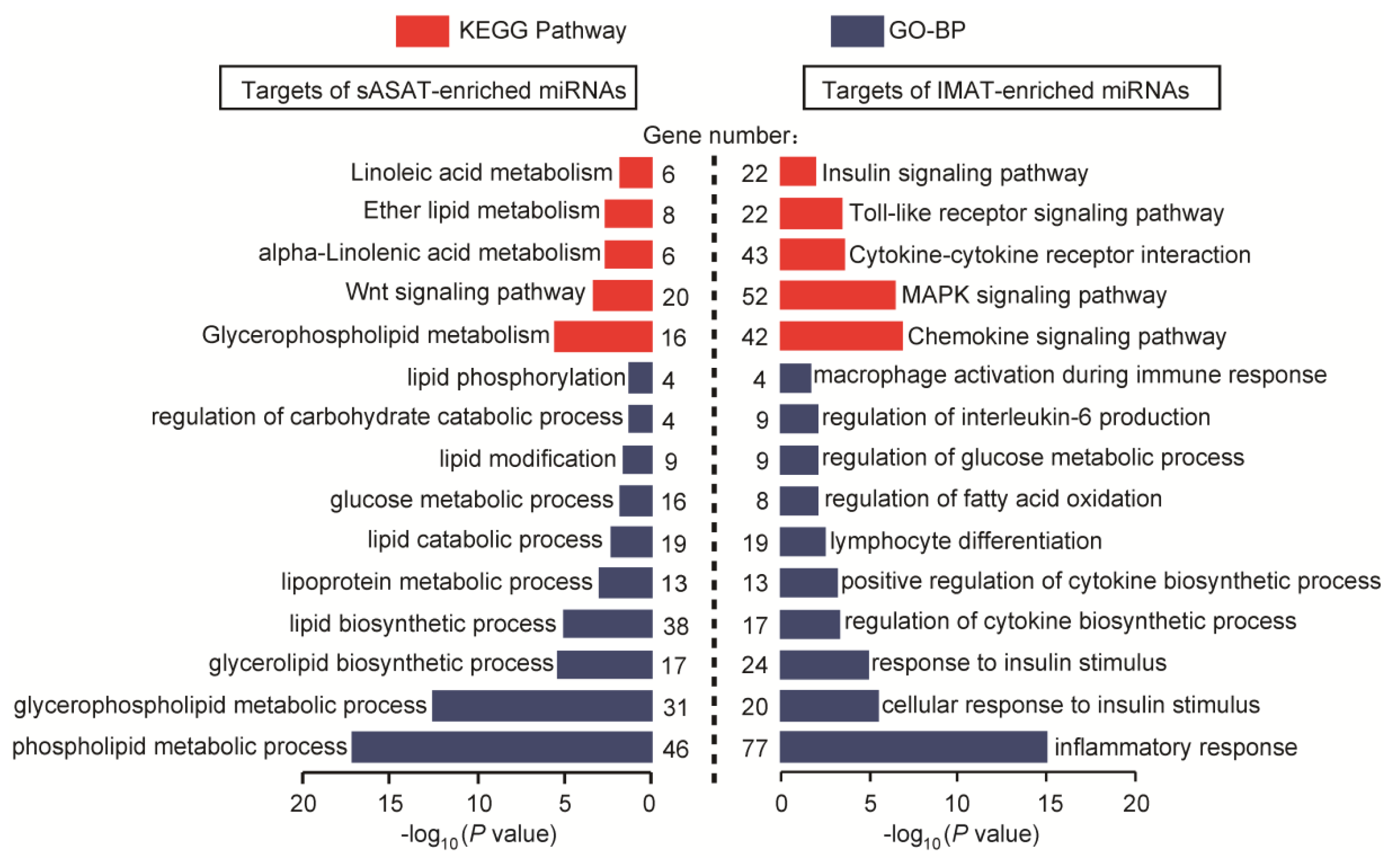
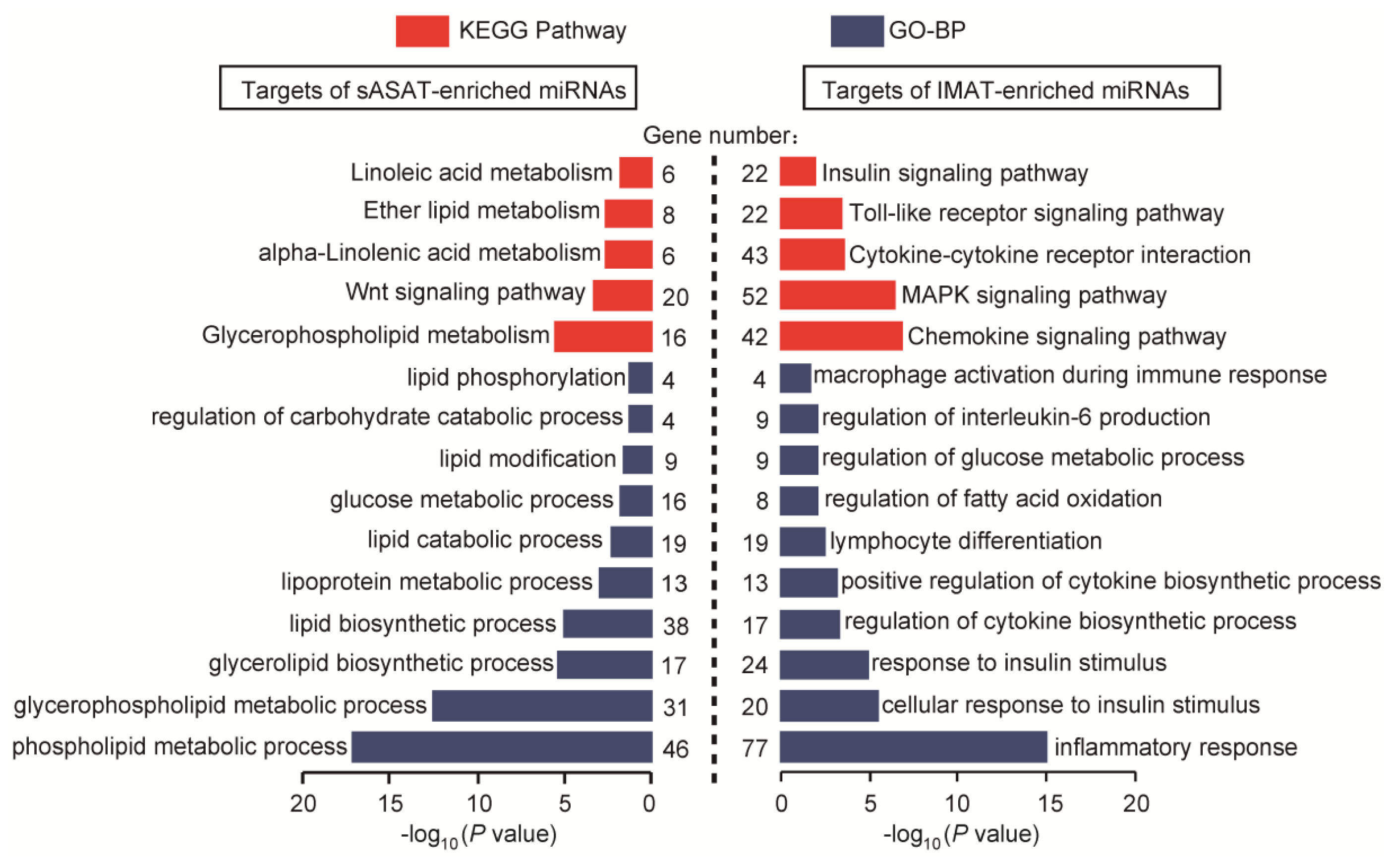
2.4. Functional Enrichment Analyses of miRNA Target Genes
3. Experimental Section
3.1. Animals and Sample Collection
3.2. Small RNA Libraries Construction and High-throughput Sequencing
3.3. Analysis of Small RNA-Sequencing Data
3.4. miRNA Differential Expression Analysis
3.5. Prediction and Functional Annotation of miRNA Target Genes
3.6. Q-PCR Validation
4. Conclusions
Acknowledgments
Conflict of Interest
References
- Zhang, Y.; Proenca, R.; Maffei, M.; Barone, M.; Leopold, L.; Friedman, J.M. Positional cloning of the mouse obese gene and its human homologue. Nature 1994, 372, 425–432. [Google Scholar]
- Kadowaki, T.; Yamauchi, T.; Kubota, N.; Hara, K.; Ueki, K.; Tobe, K. Adiponectin and adiponectin receptors in insulin resistance, diabetes, and the metabolic syndrome. J. Clin. Invest 2006, 116, 1784–1792. [Google Scholar]
- Lago, F.; Dieguez, C.; Gómez-Reino, J.; Gualillo, O. The emerging role of adipokines as mediators of inflammation and immune responses. Cytokine Growth Factor Rev 2007, 18, 313–325. [Google Scholar]
- Arner, P. Insulin resistance in type 2 diabetes-role of the adipokines. Curr. Mol. Med 2005, 5, 333–339. [Google Scholar]
- Dogru, T.; Sonmez, A.; Tasci, I.; Bozoglu, E.; Yilmaz, M.I.; Genc, H.; Erdem, G.; Gok, M.; Bingol, N.; Kilic, S. Plasma visfatin levels in patients with newly diagnosed and untreated type 2 diabetes mellitus and impaired glucose tolerance. Diabetes Res. Clin. Pract. Suppl 2007, 76, 24–29. [Google Scholar]
- Berg, A.H.; Scherer, P.E. Adipose tissue, inflammation, and cardiovascular disease. Circ. Res 2005, 96, 939–949. [Google Scholar]
- Gallagher, D.; Kuznia, P.; Heshka, S.; Albu, J.; Heymsfield, S.B.; Goodpaster, B.; Visser, M.; Harris, T.B. Adipose tissue in muscle: A novel depot similar in size to visceral adipose tissue. Am. J. Clin. Nutr 2005, 81, 903–910. [Google Scholar]
- Boettcher, M.; Machann, J.; Stefan, N.; Thamer, C.; Häring, H.U.; Claussen, C.D.; Fritsche, A.; Schick, F. Intermuscular adipose tissue (IMAT): Association with other adipose tissue compartments and insulin sensitivity. J. Magn. Reson. Imaging 2009, 29, 1340–1345. [Google Scholar]
- Freda, P.U.; Shen, W.; Heymsfield, S.B.; Reyes-Vidal, C.M.; Geer, E.B.; Bruce, J.N.; Gallagher, D. Lower visceral and subcutaneous but higher intermuscular adipose tissue depots in patients with growth hormone and insulin-like growth factor I excess due to acromegaly. J. Clin. Endocrinol. Metab 2008, 93, 2334–2343. [Google Scholar]
- Nelson, P.; Kiriakidou, M.; Sharma, A.; Maniataki, E.; Mourelatos, Z. The microRNA world: Small is mighty. Trends Biochem. Sci 2003, 28, 534–540. [Google Scholar]
- Xie, H.; Lim, B.; Lodish, H.F. MicroRNAs induced during adipogenesis that accelerate fat cell development are downregulated in obesity. Diabetes 2009, 58, 1050–1057. [Google Scholar]
- Kajimoto, K.; Naraba, H.; Iwai, N. MicroRNA and 3T3-L1 pre-adipocyte differentiation. RNA 2006, 12, 1626–1632. [Google Scholar]
- Ortega, F.J.; Moreno-Navarrete, J.M.; Pardo, G.; Sabater, M.; Hummel, M.; Ferrer, A.; Rodriguez-Hermosa, J.I.; Ruiz, B.; Ricart, W.; Peral, B. MiRNA expression profile of human subcutaneous adipose and during adipocyte differentiation. PLoS One 2010, 5, e9022. [Google Scholar]
- Wang, Q.; Li, Y.C.; Wang, J.; Kong, J.; Qi, Y.; Quigg, R.J.; Li, X. miR-17-92 cluster accelerates adipocyte differentiation by negatively regulating tumor-suppressor Rb2/p130. Proc. Natl. Acad. Sci. USA 2008, 105, 2889–2894. [Google Scholar]
- Lin, Q.; Gao, Z.; Alarcon, R.M.; Ye, J.; Yun, Z. A role of miR-27 in the regulation of adipogenesis. FEBS J 2009, 276, 2348–2358. [Google Scholar]
- Kim, S.Y.; Kim, A.Y.; Lee, H.W.; Son, Y.H.; Lee, G.Y.; Lee, J.-W.; Lee, Y.S.; Kim, J.B. miR-27a is a negative regulator of adipocyte differentiation via suppressing PPARγ expression. Biochem. Biophys. Res. Commun 2010, 392, 323–328. [Google Scholar]
- Kinoshita, M.; Ono, K.; Horie, T.; Nagao, K.; Nishi, H.; Kuwabara, Y.; Takanabe-Mori, R.; Hasegawa, K.; Kita, T.; Kimura, T. Regulation of adipocyte differentiation by activation of serotonin (5-HT) receptors 5-HT2AR and 5-HT2CR and involvement of microRNA-448-mediated repression of KLF5. Mol. Endocrinol 2010, 24, 1978–1987. [Google Scholar]
- Andersen, D.C.; Jensen, C.H.; Schneider, M.; Nossent, A.Y.; Eskildsen, T.; Hansen, J.L.; Teisner, B.; Sheikh, S.P. MicroRNA-15a fine-tunes the level of Delta-like 1 homolog (DLK1) in proliferating 3T3-L1 preadipocytes. Exp. Cell Res 2010, 316, 1681–1691. [Google Scholar]
- Martinelli, R.; Nardelli, C.; Pilone, V.; Buonomo, T.; Liguori, R.; Castanò, I.; Buono, P.; Masone, S.; Persico, G.; Forestieri, P. miR-519d overexpression is associated with human obesity. Obesity 2012, 18, 2170–2176. [Google Scholar]
- Ferland-McCollough, D.; Ozanne, S.; Siddle, K.; Willis, A.; Bushell, M. The involvement of microRNAs in Type 2 diabetes. Biochem. Soc. Trans 2010, 38, 1565. [Google Scholar]
- Wang, Q.; Wang, Y.; Minto, A.W.; Wang, J.; Shi, Q.; Li, X.; Quigg, R.J. MicroRNA-377 is up-regulated and can lead to increased fibronectin production in diabetic nephropathy. FASEB J 2008, 22, 4126–4135. [Google Scholar]
- Spurlock, M.E.; Gabler, N.K. The development of porcine models of obesity and the metabolic syndrome. J. Nutr 2008, 138, 397–402. [Google Scholar]
- Li, M.; Xia, Y.; Gu, Y.; Zhang, K.; Lang, Q.; Chen, L.; Guan, J.; Luo, Z.; Chen, H.; Li, Y. MicroRNAome of porcine pre-and postnatal development. PLoS One 2010, 5, e11541. [Google Scholar]
- Xie, S.-S.; Li, X.-Y.; Liu, T.; Cao, J.-H.; Zhong, Q.; Zhao, S.-H. Discovery of porcine microRNAs in multiple tissues by a Solexa deep sequencing approach. PLoS One 2011, 6, e16235. [Google Scholar]
- Kozomara, A.; Griffiths-Jones, S. miRBase: Integrating microRNA annotation and deep-sequencing data. Nucleic Acids Res 2011, 39, D152–D157. [Google Scholar]
- Qin, L.; Chen, Y.; Niu, Y.; Chen, W.; Wang, Q.; Xiao, S.; Li, A.; Xie, Y.; Li, J.; Zhao, X. A deep investigation into the adipogenesis mechanism: Profile of microRNAs regulating adipogenesis by modulating the canonical Wnt/β-catenin signaling pathway. BMC Genomics 2010, 11, 320. [Google Scholar]
- Kim, Y.J.; Hwang, S.J.; Bae, Y.C.; Jung, J.S. MiR-21 regulates adipogenic differentiation through the modulation of TGF-β signaling in mesenchymal stem cells derived from human adipose tissue. Stem Cells 2009, 27, 3093–3102. [Google Scholar]
- Hoekstra, M.; van der Lans, C.A.; Halvorsen, B.; Gullestad, L.; Kuiper, J.; Aukrust, P.; van Berkel, T.J.; Biessen, E.A. The peripheral blood mononuclear cell microRNA signature of coronary artery disease. Biochem. Biophys. Res. Commun 2010, 394, 792–797. [Google Scholar]
- Ro, S.; Park, C.; Young, D.; Sanders, K.M.; Yan, W. Tissue-dependent paired expression of miRNAs. Nucleic Acids Res 2007, 35, 5944–5953. [Google Scholar]
- Esau, C.; Kang, X.; Peralta, E.; Hanson, E.; Marcusson, E.G.; Ravichandran, L.V.; Sun, Y.; Koo, S.; Perera, R.J.; Jain, R. MicroRNA-143 regulates adipocyte differentiation. J. Biol. Chem 2004, 279, 52361–52365. [Google Scholar]
- Karbiener, M.; Fischer, C.; Nowitsch, S.; Opriessnig, P.; Papak, C.; Ailhaud, G.; Dani, C.; Amri, E.-Z.; Scheideler, M. microRNA miR-27b impairs human adipocyte differentiation and targets PPARγ. Biochem. Biophys. Res. Commun 2009, 390, 247–251. [Google Scholar]
- Carlsbecker, A.; Lee, J.-Y.; Roberts, C.J.; Dettmer, J.; Lehesranta, S.; Zhou, J.; Lindgren, O.; Moreno-Risueno, M.A.; Vatén, A.; Thitamadee, S. Cell signalling by microRNA165/6 directs gene dose-dependent root cell fate. Nature 2010, 465, 316–321. [Google Scholar]
- Romualdi, C.; Bortoluzzi, S.; d’Alessi, F.; Danieli, G.A. IDEG6: A web tool for detection of differentially expressed genes in multiple tag sampling experiments. Physiol. Genomics 2003, 12, 159–162. [Google Scholar]
- Okayama, H.; Schetter, A.; Harris, C. MicroRNAs and inflammation in the pathogenesis and progression of colon cancer. Dig. Dis 2012, 30, 9–15. [Google Scholar]
- Olivieri, F.; Spazzafumo, L.; Santini, G.; Lazzarini, R.; Albertini, M.C.; Rippo, M.R.; Galeazzi, R.; Abbatecola, A.M.; Marcheselli, F.; Monti, D. Age-related differences in the expression of circulating microRNAs: miR-21 as a new circulating marker of inflammaging. Mech. Ageing Dev 2012, 133, 675–685. [Google Scholar]
- Dai, L.; Zhang, X.; Hu, X.; Zhou, C.; Ao, Y. Silencing of microRNA-101 prevents IL-1b-induced extracellular matrix degradation in chondrocytes. Arthritis Res. Ther 2012, 14, R268. [Google Scholar]
- Trajkovski, M.; Hausser, J.; Soutschek, J.; Bhat, B.; Akin, A.; Zavolan, M.; Heim, M.H.; Stoffel, M. MicroRNAs 103 and 107 regulate insulin sensitivity. Nature 2011, 474, 649–653. [Google Scholar]
- Stittrich, A.-B.; Haftmann, C.; Sgouroudis, E.; Kühl, A.A.; Hegazy, A.N.; Panse, I.; Riedel, R.; Flossdorf, M.; Dong, J.; Fuhrmann, F. The microRNA miR-182 is induced by IL-2 and promotes clonal expansion of activated helper T lymphocytes. Nat. Immunol 2010, 11, 1057–1062. [Google Scholar]
- Sarver, A.L.; Li, L.; Subramanian, S. MicroRNA miR-183 functions as an oncogene by targeting the transcription factor EGR1 and promoting tumor cell migration. Cancer Res 2010, 70, 9570–9580. [Google Scholar]
- Wiklund, E.D.; Bramsen, J.B.; Hulf, T.; Dyrskjøt, L.; Ramanathan, R.; Hansen, T.B.; Villadsen, S.B.; Gao, S.; Ostenfeld, M.S.; Borre, M. Coordinated epigenetic repression of the miR-200 family and miR-205 in invasive bladder cancer. Int. J. Cancer 2011, 128, 1327–1334. [Google Scholar]
- Isacke, C. MicroRNA-200 family modulation in distinct breast cancer phenotypes. PLoS One 2012, 7, e47709. [Google Scholar]
- Garzon, R.; Volinia, S.; Liu, C.-G.; Fernandez-Cymering, C.; Palumbo, T.; Pichiorri, F.; Fabbri, M.; Coombes, K.; Alder, H.; Nakamura, T. MicroRNA signatures associated with cytogenetics and prognosis in acute myeloid leukemia. Blood 2008, 111, 3183–3189. [Google Scholar]
- Zaragosi, L.-E.; Wdziekonski, B.; Brigand, K.L.; Villageois, P.; Mari, B.; Waldmann, R.; Dani, C.; Barbry, P. Small RNA sequencing reveals miR-642a-3p as a novel adipocyte-specific microRNA and miR-30 as a key regulator of human adipogenesis. Genome Biol 2011, 12, R64. [Google Scholar]
- Gerin, I.; Bommer, G.T.; McCoin, C.S.; Sousa, K.M.; Krishnan, V.; MacDougald, O.A. Roles for miRNA-378/378* in adipocyte gene expression and lipogenesis. Am. J. Physiol. Endocrinol. Metab 2010, 299, E198–E206. [Google Scholar]
- Sun, L.; Xie, H.; Mori, M.A.; Alexander, R.; Yuan, B.; Hattangadi, S.M.; Liu, Q.; Kahn, C.R.; Lodish, H.F. Mir193b-365 is essential for brown fat differentiation. Nat. Cell Biol 2011, 13, 958–965. [Google Scholar]
- Yang, K.; He, Y.S.; Wang, X.Q.; Lu, L.; Chen, Q.J.; Liu, J.; Sun, Z.; Shen, W.F. MiR-146a inhibits oxidized low-density lipoprotein-induced lipid accumulation and inflammatory response via targeting toll-like receptor 4. FEBS Lett 2011, 585, 854–860. [Google Scholar]
- Krek, A.; Grün, D.; Poy, M.N.; Wolf, R.; Rosenberg, L.; Epstein, E.J.; MacMenamin, P.; da Piedade, I.; Gunsalus, K.C.; Stoffel, M. Combinatorial microRNA target predictions. Nat. Genet 2005, 37, 495–500. [Google Scholar]
- Lewis, B.P.; Burge, C.B.; Bartel, D.P. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell 2005, 120, 15–20. [Google Scholar]
- Griffiths-Jones, S.; Saini, H.K.; van Dongen, S.; Enright, A.J. miRBase: Tools for microRNA genomics. Nucleic Acids Res 2008, 36, D154–D158. [Google Scholar]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc 2008, 4, 44–57. [Google Scholar]
- Pruitt, K.D.; Tatusova, T.; Klimke, W.; Maglott, D.R. NCBI Reference Sequences: Current status, policy and new initiatives. Nucleic Acids Res 2009, 37, D32–D36. [Google Scholar]
- Gardner, P.P.; Daub, J.; Tate, J.G.; Nawrocki, E.P.; Kolbe, D.L.; Lindgreen, S.; Wilkinson, A.C.; Finn, R.D.; Griffiths-Jones, S.; Eddy, S.R.; et al. Rfam: Updates to the RNA families database. Nucleic Acids Res 2009, 37, D136–D140. [Google Scholar]
- Kohany, O.; Gentles, A.J.; Hankus, L.; Jurka, J. Annotation, submission and screening of repetitive elements in Repbase: RepbaseSubmitter and Censor. BMC Bioinforma 2006, 7, 474. [Google Scholar]
- Markham, N.R.; Zuker, M. UNAFold: Software for nucleic acid folding and hybridization. Methods Mol. Biol 2008, 453, 3–31. [Google Scholar]
- Hellemans, J.; Mortier, G.; de Paepe, A.; Speleman, F.; Vandesompele, J. qBase relative quantification framework and software for management and automated analysis of real-time quantitative PCR data. Genome Biol 2007, 8, R19. [Google Scholar]
© 2013 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Ma, J.; Yu, S.; Wang, F.; Bai, L.; Xiao, J.; Jiang, Y.; Chen, L.; Wang, J.; Jiang, A.; Li, M.; et al. MicroRNA Transcriptomes Relate Intermuscular Adipose Tissue to Metabolic Risk. Int. J. Mol. Sci. 2013, 14, 8611-8624. https://doi.org/10.3390/ijms14048611
Ma J, Yu S, Wang F, Bai L, Xiao J, Jiang Y, Chen L, Wang J, Jiang A, Li M, et al. MicroRNA Transcriptomes Relate Intermuscular Adipose Tissue to Metabolic Risk. International Journal of Molecular Sciences. 2013; 14(4):8611-8624. https://doi.org/10.3390/ijms14048611
Chicago/Turabian StyleMa, Jideng, Shuzhen Yu, Fengjiao Wang, Lin Bai, Jian Xiao, Yanzhi Jiang, Lei Chen, Jinyong Wang, Anan Jiang, Mingzhou Li, and et al. 2013. "MicroRNA Transcriptomes Relate Intermuscular Adipose Tissue to Metabolic Risk" International Journal of Molecular Sciences 14, no. 4: 8611-8624. https://doi.org/10.3390/ijms14048611
APA StyleMa, J., Yu, S., Wang, F., Bai, L., Xiao, J., Jiang, Y., Chen, L., Wang, J., Jiang, A., Li, M., & Li, X. (2013). MicroRNA Transcriptomes Relate Intermuscular Adipose Tissue to Metabolic Risk. International Journal of Molecular Sciences, 14(4), 8611-8624. https://doi.org/10.3390/ijms14048611