Cytokines and the Skin Barrier


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
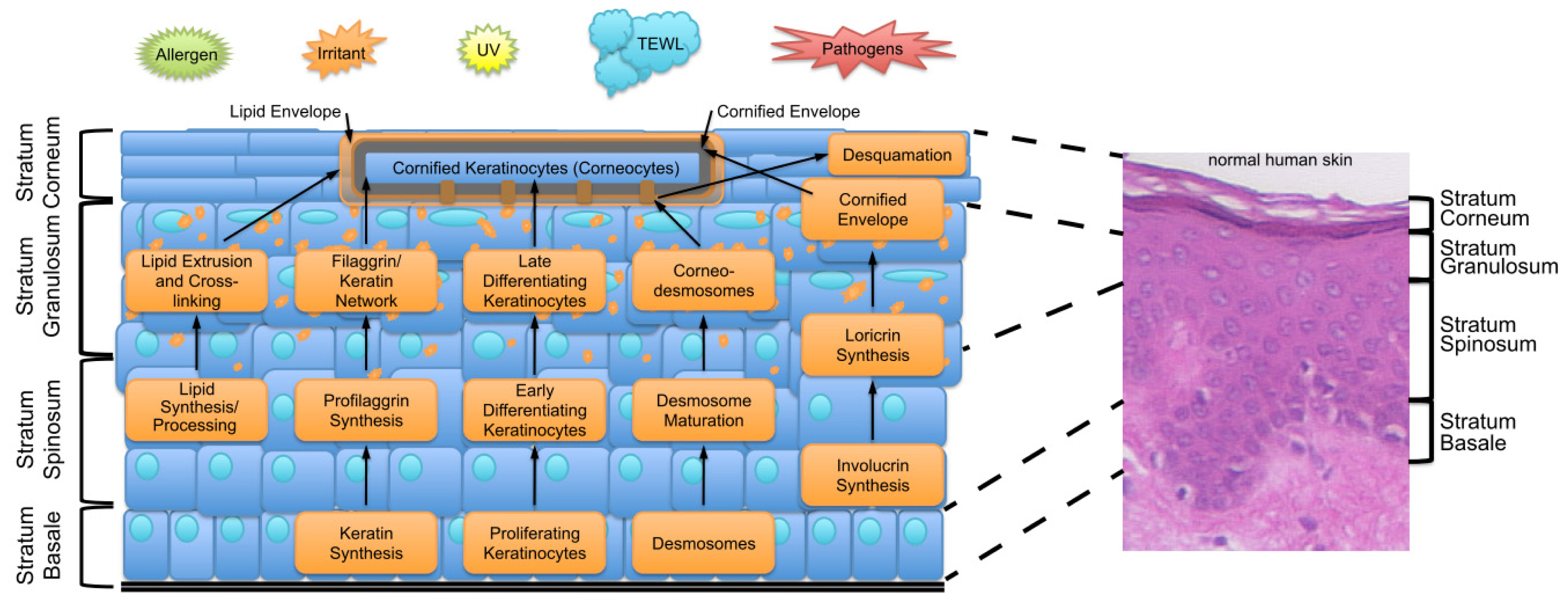
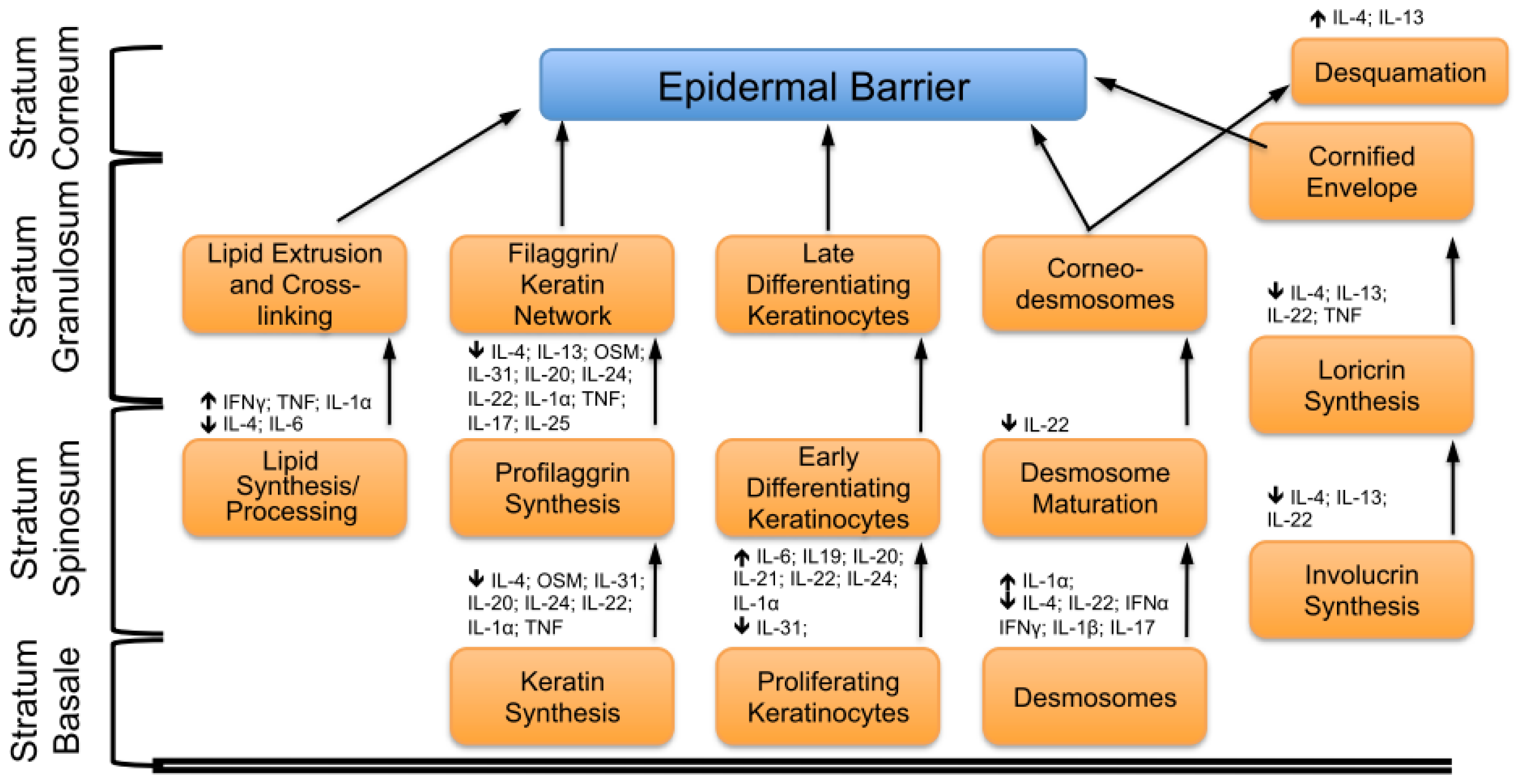
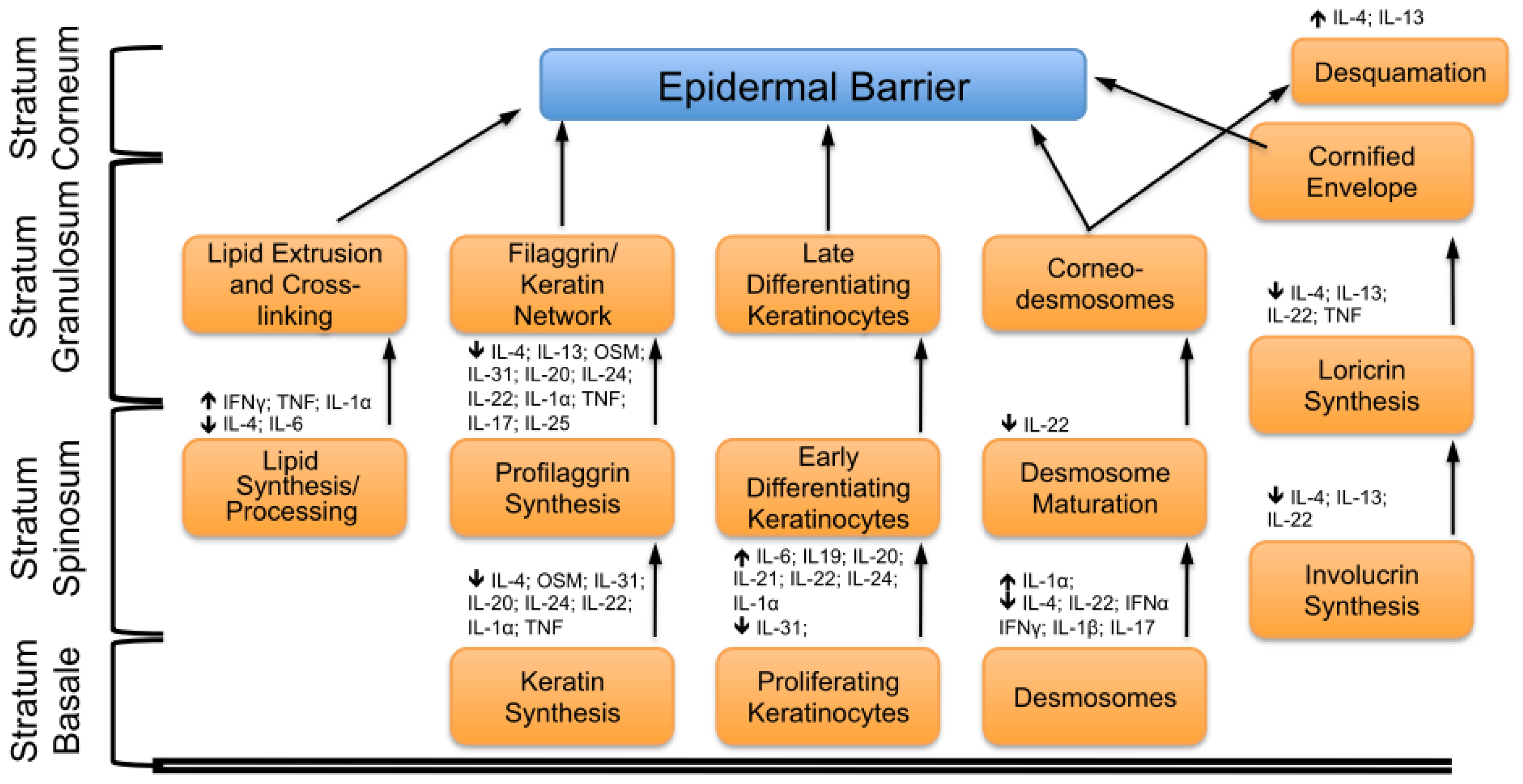
2. Key Features of the Epidermal Barrier
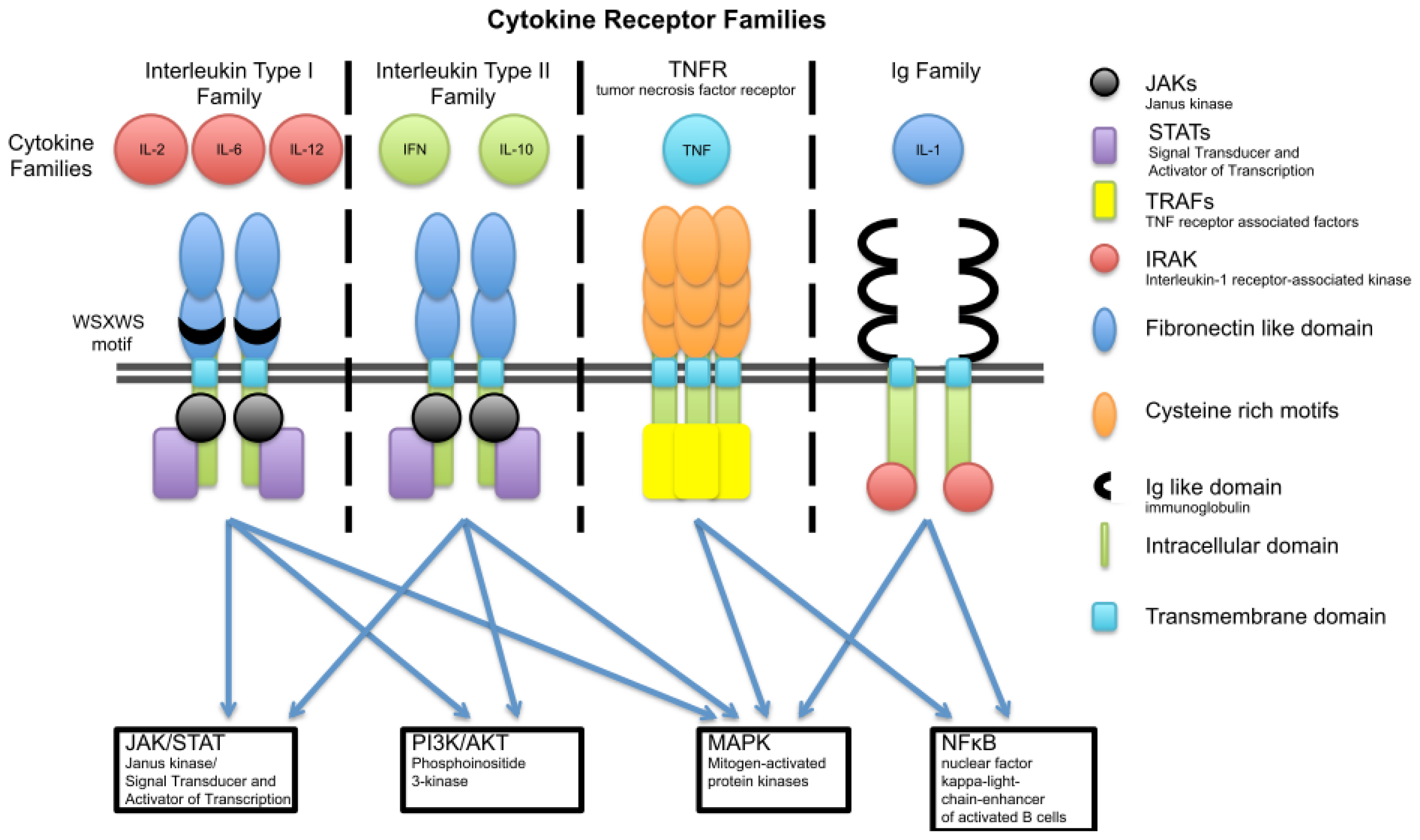
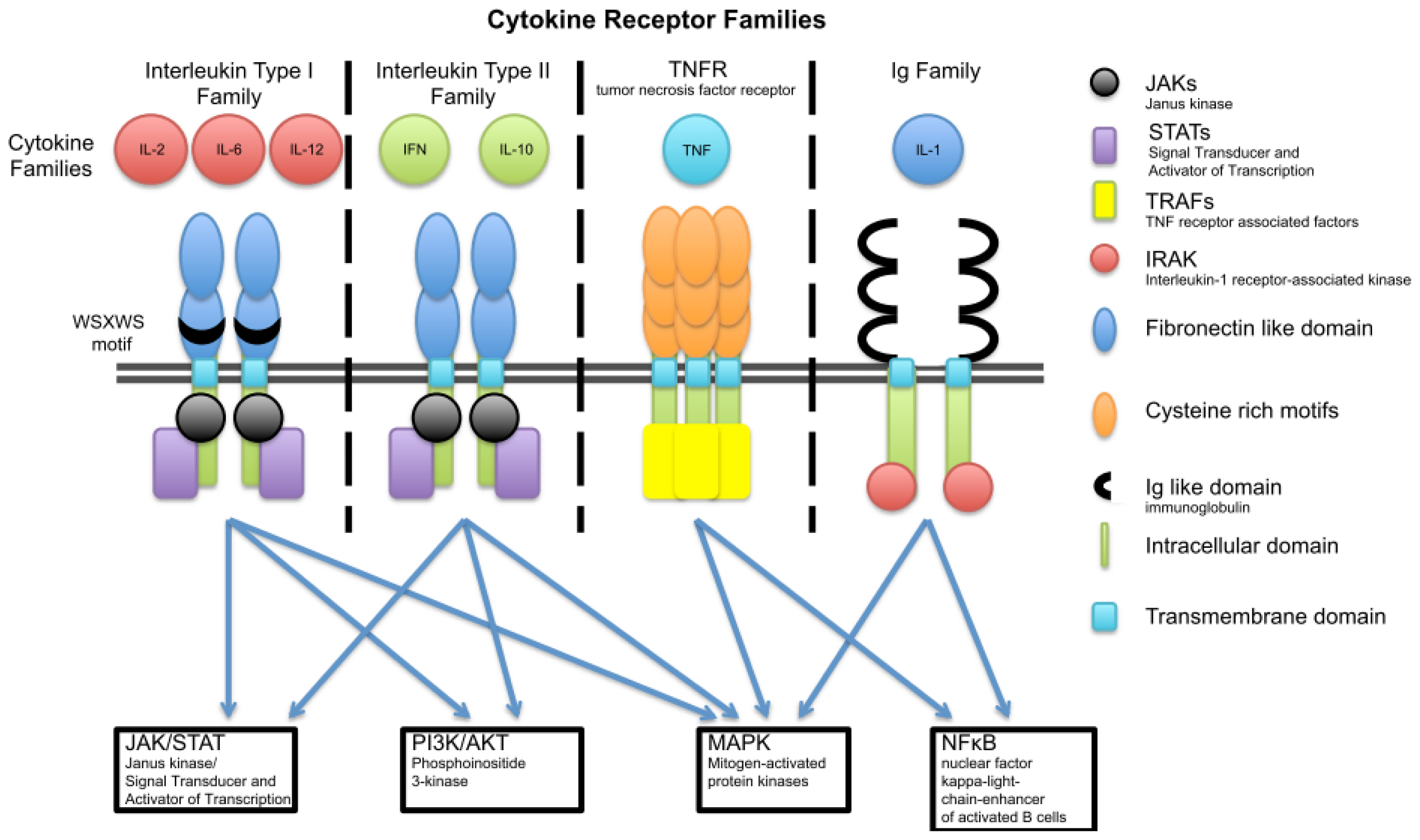
3. Cytokine Families
3.1. Interleukin Type I
3.1.1. γ-Chain (IL-2) Family
3.1.1.1. IL-4 and IL-13
3.1.1.2. IL-21
3.1.2. The IL-6 Family of Cytokines
3.1.2.1. IL-6
3.1.2.2. IL-31 and OSM
3.1.3. IL-12 Family
3.2. Interleukin Type II Cytokine Receptors
3.2.1. IL-10 Family
3.2.1.1. IL-19
3.2.1.2. IL-20
3.2.1.3. IL-22
3.2.1.4. IL-24
3.2.2. Interferons
IFNα and IFNγ
3.3. Ig Superfamily (Receptors with Extracellular Immunoglobulin (Ig)-Like Domains)
IL-1α and IL-1β
3.4. The TNF Family
TNFα
3.5. IL-17 Family
3.5.1. IL-17
3.5.2. IL-25 (IL-17E)
4. Concluding Remarks
Acknowledgments
Conflict of Interest
References
- Proksch, E.; Brandner, J.M.; Jensen, J.-M. The skin: An indispensable barrier. Exp. Dermatol 2008, 17, 1063–1072. [Google Scholar]
- Guttman-Yassky, E.; Nograles, K.E.; Krueger, J.G. Contrasting pathogenesis of atopic dermatitis and psoriasis--part I: Clinical and pathologic concepts. J. Allergy Clin. Immunol 2011, 127, 1110–1118. [Google Scholar]
- Bieber, T. Atopic dermatitis. Ann. Dermatol 2010, 22, 125. [Google Scholar]
- Parisi, R.; Symmons, D.P.M.; Griffiths, C.E.M.; Ashcroft, D.M. Global epidemiology of psoriasis: A systematic review of incidence and prevalence. J. Invest. Dermatol 2012, 133, 377–385. [Google Scholar]
- Schmuth, M.; Gruber, R.; Elias, P.M.; Williams, M.L. Ichthyosis update: Towards a function-driven model of pathogenesis of the disorders of cornification and the role of corneocyte proteins in these disorders. Adv. Dermatol 2007, 23, 231–256. [Google Scholar]
- Incorvaia, C.; Frati, F.; Verna, N.; D’Alò, S.; Motolese, A.; Pucci, S. Allergy and the skin. Clin. Exp. Immunol 2008, 153, 27–29. [Google Scholar]
- Candi, E.; Schmidt, R.; Melino, G. The cornified envelope: A model of cell death in the skin. Nat. Rev. Mol. Cell Biol 2005, 6, 328–340. [Google Scholar]
- Henry, J.; Toulza, E.; Hsu, C.-Y.; Pellerin, L.; Balica, S.; Mazereeuw-Hautier, J.; Paul, C.; Serre, G.; Jonca, N.; Simon, M. Update on the epidermal differentiation complex. Front. Biosci 2012, 17, 1517–1532. [Google Scholar]
- Kalinin, A.E.; Kajava, A.V.; Steinert, P.M. Epithelial barrier function: Assembly and structural features of the cornified cell envelope. Bioessays 2002, 24, 789–800. [Google Scholar]
- Harder, J.; Schröder, J.-M.; Gläser, R. The skin surface as antimicrobial barrier: Present concepts and future outlooks. Exp. Dermatol 2013, 22, 1–5. [Google Scholar]
- Gallo, R.L.; Hooper, L.V. Epithelial antimicrobial defence of the skin and intestine. Nat. Rev. Immunol 2012, 12, 503–516. [Google Scholar]
- Madison, K.C. Journal of investigative dermatology—Barrier function of the skin: [ldquo]La raison d’Etre[rdquo] of the epidermis. J. Invest. Dermatol 2003, 121, 231–241. [Google Scholar]
- Liao, W.; Lin, J.-X.; Leonard, W.J. IL-2 family cytokines: New insights into the complex roles of IL-2 as a broad regulator of T helper cell differentiation. Curr. Opin. Immunol 2011, 23, 598–604. [Google Scholar]
- Garbers, C.; Hermanns, H.M.; Schaper, F.; Müller-Newen, G.; Grötzinger, J.; Rose-John, S.; Scheller, J. Plasticity and cross-talk of interleukin 6-type cytokines. Cytokine Growth Factor Rev 2012, 23, 1–13. [Google Scholar]
- Jones, L.L.; Vignali, D.A.A. Molecular interactions within the IL-6/IL-12 cytokine/receptor superfamily. Immunol. Res 2011, 51, 5–14. [Google Scholar]
- Punnonen, J.; Yssel, H.; de Vries, J.E. The relative contribution of IL-4 and IL-13 to human IgE synthesis induced by activated CD4+ or CD8+ T cells. J. Allergy Clin. Immunol 1997, 100, 792–801. [Google Scholar]
- Neis, M.; Peters, B.; Dreuw, A.; Wenzel, J.; Bieber, T.; Mauch, C.; Krieg, T.; Stanzel, S.; HEINRICH, P.; MERK, H. Enhanced expression levels of IL-31 correlate with IL-4 and IL-13 in atopic and allergic contact dermatitis. J. Allergy Clin. Immunol 2006, 118, 930–937. [Google Scholar]
- Hamid, Q.; Boguniewicz, M.; Leung, D.Y. Differential in situ cytokine gene expression in acute versus chronic atopic dermatitis. J. Clin. Invest 1994, 94, 870–876. [Google Scholar]
- Howell, M.D.; Fairchild, H.R.; Kim, B.E.; Bin, L.; Boguniewicz, M.; Redzic, J.S.; Hansen, K.C.; Leung, D.Y.M. Th2 Cytokines act on S100/A11 to downregulate keratinocyte differentiation. J. Invest. Dermatol 2008, 128, 2248–2258. [Google Scholar]
- Kim, B.E.; Leung, D.Y.M.; Boguniewicz, M.; Howell, M.D. Loricrin and involucrin expression is down-regulated by Th2 cytokines through STAT-6. Clin. Immunol 2008, 126, 332–337. [Google Scholar]
- Kim, S.; Kim, H.-J.; Yang, H.S.; Kim, E.; Huh, I.-S.; Yang, J.-M. IL-31 serum protein and tissue mRNA levels in patients with atopic dermatitis. Ann. Dermatol 2011, 23, 468–473. [Google Scholar]
- Howell, M.D.; Kim, B.E.; Gao, P.; Grant, A.V.; Boguniewicz, M.; DeBenedetto, A.; Schneider, L.; Beck, L.A.; Barnes, K.C.; Leung, D.Y.M. Cytokine modulation of atopic dermatitis filaggrin skin expression. J. Allergy Clin. Immunol 2009, 124, R7–R12. [Google Scholar]
- Pellerin, L.; Henry, J.; Hsu, C.-Y.; Balica, S.; Jean-Decoster, C.; Méchin, M.-C.; Hansmann, B.; Rodríguez, E.; Weindinger, S.; Schmitt, A.-M.; et al. Defects of filaggrin-like proteins in both lesional and nonlesional atopic skin. J. Allergy Clin. Immunol. 2013. [Google Scholar] [CrossRef]
- Mischke, D.; Korge, B.P.; Marenholz, I.; Volz, A.; Ziegler, A. Genes encoding structural proteins of epidermal cornification and S100 calcium-binding proteins form a gene complex (“epidermal differentiation complex”) on human chromosome 1q21. J. Invest. Dermatol 1996, 106, 989–992. [Google Scholar]
- Marenholz, I. Identification of human epidermal differentiation complex (EDC)-encoded genes by subtractive hybridization of entire YACs to a gridded keratinocyte cDNA library. Genome Res 2001, 11, 341–355. [Google Scholar]
- Steinert, P.M.; Candi, E.; Kartasova, T.; Marekov, L.J. Small proline-rich proteins are cross-bridging proteins in the cornified cell envelopes of stratified squamous epithelia. J. Struct. Biol 1998, 122, 76–85. [Google Scholar]
- Hvid, M.; Johansen, C.; Deleuran, B.; Kemp, K.; Deleuran, M.; Vestergaard, C. Regulation of caspase 14 expression in keratinocytes by inflammatory cytokines—A possible link between reduced skin barrier function and inflammation? Exp. Dermatol 2011, 20, 633–636. [Google Scholar]
- Hoste, E.; Kemperman, P.; Devos, M.; Denecker, G.; Kezic, S.; Yau, N.; Gilbert, B.; Lippens, S.; de Groote, P.; Roelandt, R.; et al. Caspase-14 is required for filaggrin degradation to natural moisturizing factors in the skin. J. Invest. Dermatol 2011, 131, 2233–2241. [Google Scholar]
- Denecker, G.; Ovaere, P.; Vandenabeele, P.; Declercq, W. Caspase-14 reveals its secrets. J. Cell Biol 2008, 180, 451–458. [Google Scholar] [Green Version]
- Hatano, Y.; Adachi, Y.; Elias, P.M.; Crumrine, D.; Sakai, T.; Kurahashi, R.; Katagiri, K.; Fujiwara, S. The Th2 cytokine, interleukin-4, abrogates the cohesion of normal stratum corneum in mice: Implications for pathogenesis of atopic dermatitis. Exp. Dermatol 2013, 22, 30–35. [Google Scholar]
- Morizane, S.; Yamasaki, K.; Kajita, A.; Ikeda, K.; Zhan, M.; Aoyama, Y.; Gallo, R.L.; Iwatsuki, K. TH2 cytokines increase kallikrein 7 expression and function in patients with atopic dermatitis. J. Allergy Clin. Immunol 2012, 130, 259–261. [Google Scholar]
- Hatano, Y.; Terashi, H.; Arakawa, S.; Katagiri, K. Interleukin-4 suppresses the enhancement of ceramide synthesis and cutaneous permeability barrier functions induced by tumor necrosis factor-[alpha] and interferon-[gamma] in human epidermis. J. Invest. Dermatol 2005, 124, 786–792. [Google Scholar]
- Sawada, E.; Yoshida, N.; Sugiura, A.; Imokawa, G. Th1 cytokines accentuate but Th2 cytokines attenuate ceramide production in the stratum corneum of human epidermal equivalents: An implication for the disrupted barrier mechanism in atopic dermatitis. J. Dermatol. Sci 2012, 68, 25–35. [Google Scholar]
- Elbe-Bürger, A.; Egyed, A.; Olt, S.; Klubal, R.; Mann, U.; Rappersberger, K.; Rot, A.; Stingl, G. Overexpression of IL-4 alters the homeostasis in the skin. J. Invest. Dermatol 2002, 118, 767–778. [Google Scholar]
- Chan, L.S.; Robinson, N.; Xu, L. Expression of interleukin-4 in the epidermis of transgenic mice results in a pruritic inflammatory skin disease: An experimental animal model to study atopic dermatitis. J. Invest. Dermatol 2001, 117, 977–983. [Google Scholar]
- Sehra, S.; Yao, Y.; Howell, M.D.; Nguyen, E.T.; Kansas, G.S.; Leung, D.Y.M.; Travers, J.B.; Kaplan, M.H. IL-4 regulates skin homeostasis and the predisposition toward allergic skin inflammation. J. Immunol 2010, 184, 3186–3190. [Google Scholar]
- Zheng, T.; Oh, M.H.; Oh, S.Y.; Schroeder, J.T.; Glick, A.B.; Zhu, Z. Transgenic expression of interleukin-13 in the skin induces a pruritic dermatitis and skin remodeling. J. Invest. Dermatol 2009, 129, 742–751. [Google Scholar]
- Sivaprasad, U.; Warrier, M.R.; Gibson, A.M.; Chen, W.; Tabata, Y.; Bass, S.A.; Rothenberg, M.E.; Khurana Hershey, G.K. IL-13Rα2 has a protective role in a mouse model of cutaneous inflammation. J. Immunol 2010, 185, 6802–6808. [Google Scholar]
- Yagi, R.; Nagai, H.; Iigo, Y.; Akimoto, T.; Arai, T.; Kubo, M. Development of atopic dermatitis-like skin lesions in STAT6-deficient NC/Nga mice. J. Immunol 2002, 168, 2020–2027. [Google Scholar]
- Wills-Karp, M.; Finkelman, F.D. Untangling the complex web of IL-4- and IL-13-mediated signaling pathways. Sci. Signal. 2008, 1, pe55. [Google Scholar]
- Jin, H.; Oyoshi, M.K.; Le, Y.; Bianchi, T.; Koduru, S.; Mathias, C.B.; Kumar, L.; le Bras, S.; Young, D.; Collins, M.; et al. IL-21R is essential for epicutaneous sensitization and allergic skin inflammation in humans and mice. J. Clin. Invest 2009, 119, 47–60. [Google Scholar]
- Caruso, R.; Botti, E.; Sarra, M.; Esposito, M.; Stolfi, C.; Diluvio, L.; Giustizieri, M.L.; Pacciani, V.; Mazzotta, A.; Campione, E.; et al. Involvement of interleukin-21 in the epidermal hyperplasia of psoriasis. Nat. Med 2009, 15, 1013–1015. [Google Scholar]
- Sarra, M.; Caruso, R.; Cupi, M.L.; Monteleone, I.; Stolfi, C.; Campione, E.; Diluvio, L.; Mazzotta, A.; Botti, E.; Chimenti, S.; et al. IL-21 promotes skin recruitment of CD4(+) cells and drives IFN-γ-dependent epidermal hyperplasia. J. Immunol 2011, 186, 5435–5442. [Google Scholar]
- Coquet, J.M.; Kyparissoudis, K.; Pellicci, D.G.; Besra, G.; Berzins, S.P.; Smyth, M.J.; Godfrey, D. I. IL-21 is produced by NKT cells and modulates NKT cell activation and cytokine production. J. Immunol 2007, 178, 2827–2834. [Google Scholar]
- Monteleone, G.; Pallone, F.; MacDonald, T.T. Interleukin-21 (IL-21)-mediated pathways in T cell-mediated disease. Cytokine Growth Factor Rev 2009, 20, 185–191. [Google Scholar]
- Bonifati, C.; Carducci, M.; Cordiali Fei, P.; Trento, E.; Sacerdoti, G.; Fazio, M.; Ameglio, F. Correlated increases of tumour necrosis factor-alpha, interleukin-6 and granulocyte monocyte-colony stimulating factor levels in suction blister fluids and sera of psoriatic patients—Relationships with disease severity. Clin. Exp. Dermatol 1994, 19, 383–387. [Google Scholar]
- Sato, M.; Sawamura, D.; Ina, S.; Yaguchi, T.; Hanada, K.; Hashimoto, I. In vivo introduction of the interleukin 6 gene into human keratinocytes: Induction of epidermal proliferation by the fully spliced form of interleukin 6, but not by the alternatively spliced form. Arch. Dermatol. Res 1999, 291, 400–404. [Google Scholar]
- Sawamura, D.; Meng, X.; Ina, S.; Sato, M.; Tamai, K.; Hanada, K.; Hashimoto, I. Induction of keratinocyte proliferation and lymphocytic infiltration by in vivo introduction of the IL-6 gene into keratinocytes and possibility of keratinocyte gene therapy for inflammatory skin diseases using IL-6 mutant genes. J. Immunol 1998, 161, 5633–5639. [Google Scholar]
- Wang, X.-P.; Schunck, M.; Kallen, K.-J.; Neumann, C.; Trautwein, C.; Rose-John, S.; Proksch, E. The interleukin-6 cytokine system regulates epidermal permeability barrier homeostasis. J. Invest. Dermatol 2004, 123, 124–131. [Google Scholar]
- Cohen, T.; Nahari, D.; Cerem, L.W.; Neufeld, G.; Levi, B.Z. Interleukin 6 induces the expression of vascular endothelial growth factor. J. Biol. Chem 1996, 271, 736–741. [Google Scholar]
- Lin, Z.-Q.; Kondo, T.; Ishida, Y.; Takayasu, T.; Mukaida, N. Essential involvement of IL-6 in the skin wound-healing process as evidenced by delayed wound healing in IL-6-deficient mice. J. Leukoc. Biol 2003, 73, 713–721. [Google Scholar]
- Nobbe, S.; Dziunycz, P.; Muhleisen, B.; Bilsborough, J.; Dillon, S.R.; French, L.E.; Hofbauer, G.F. IL-31 expression by inflammatory cells is preferentially elevated in atopic dermatitis. Acta Derm. Venerol 2012, 92, 24–28. [Google Scholar]
- Raap, U.; Wichmann, K.; Bruder, M.; Ständer, S.; Wedi, B.; Kapp, A.; Werfel, T. Correlation of IL-31 serum levels with severity of atopic dermatitis. J. Allergy Clin. Immunol 2008, 122, 421–423. [Google Scholar]
- Dillon, S.R.; Sprecher, C.; Hammond, A.; Bilsborough, J.; Rosenfeld-Franklin, M.; Presnell, S.R.; Haugen, H.S.; Maurer, M.; Harder, B.; Johnston, J.; et al. Interleukin 31, a cytokine produced by activated T cells, induces dermatitis in mice. Nat. Immunol 2004, 5, 752–760. [Google Scholar]
- Cornelissen, C.; Marquardt, Y.; Czaja, K.; Wenzel, J.; Frank, J.; Lüscher-Firzlaff, J.; Lüscher, B.; Baron, J.M. IL-31 regulates differentiation and filaggrin expression in human organotypic skin models. J. Allergy Clin. Immunol 2012, 129, 426–433. [Google Scholar]
- Sonkoly, E.; Muller, A.; Lauerma, A.I.; Pivarcsi, A.; Soto, H.; Kemeny, L.; Alenius, H.; Dieu-Nosjean, M.-C.; Meller, S.; Rieker, J.; et al. IL-31: A new link between T cells and pruritus in atopic skin inflammation. J. Allergy Clin. Immunol 2006, 117, 411–417. [Google Scholar]
- Raap, U.; Wieczorek, D.; Gehring, M.; Pauls, I.; Ständer, S.; Kapp, A.; Wedi, B. Increased levels of serum IL-31 in chronic spontaneous urticaria. Exp. Dermatol 2010, 19, 464–466. [Google Scholar]
- Matsuda, H.; Watanabe, N.; Geba, G.P.; Sperl, J.; Tsudzuki, M.; Hiroi, J.; Matsumoto, M.; Ushio, H.; Saito, S.; Askenase, P.W.; et al. Development of atopic dermatitis-like skin lesion with IgE hyperproduction in NC/Nga mice. Int. Immunol 1997, 9, 461–466. [Google Scholar]
- Suto, H.; Matsuda, H.; Mitsuishi, K.; Hira, K.; Uchida, T.; Unno, T.; Ogawa, H.; Ra, C. NC/Nga mice: A mouse model for atopic dermatitis. Int. Arch. Allergy Immunol 1999, 120, 70–75. [Google Scholar]
- Cornelissen, C.; Lüscher-Firzlaff, J.; Baron, J.M.; Lüscher, B. Signaling by IL-31 and functional consequences. Eur. J. Cell Biol 2012, 91, 552–566. [Google Scholar]
- Boniface, K.; Diveu, C.; Morel, F.; Pedretti, N.; Froger, J.; Ravon, E.; Garcia, M.; Venereau, E.; Preisser, L.; Guignouard, E.; et al. Oncostatin M secreted by skin infiltrating T lymphocytes is a potent keratinocyte activator involved in skin inflammation. J. Immunol 2007, 178, 4615–4622. [Google Scholar]
- Gazel, A.; Rosdy, M.; Bertino, B.; Tornier, C.; Sahuc, F.; Blumenberg, M. A characteristic subset of psoriasis-associated genes is induced by oncostatin-M in reconstituted epidermis. J. Invest. Dermatol 2006, 126, 2647–2657. [Google Scholar]
- Chan, J.R.; Blumenschein, W.; Murphy, E.; Diveu, C.; Wiekowski, M.; Abbondanzo, S.; Lucian, L.; Geissler, R.; Brodie, S.; Kimball, A.B.; et al. IL-23 stimulates epidermal hyperplasia via TNF and IL-20R2-dependent mechanisms with implications for psoriasis pathogenesis. J. Exp. Med 2006, 203, 2577–2587. [Google Scholar]
- Zheng, Y.; Danilenko, D.M.; Valdez, P.; Kasman, I.; Eastham-Anderson, J.; Wu, J.; Ouyang, W. Interleukin-22, a TH17 cytokine, mediates IL-23-induced dermal inflammation and acanthosis. Nature 2006, 445, 648–651. [Google Scholar]
- Cua, D.J.; Sherlock, J.; Chen, Y.; Murphy, C.A.; Joyce, B.; Seymour, B.; Lucian, L.; To, W.; Kwan, S.; Churakova, T.; et al. Interleukin-23 rather than interleukin-12 is the critical cytokine for autoimmune inflammation of the brain. Nature 2003, 421, 744–748. [Google Scholar]
- Di Meglio, F.O.N.P. The role of IL-23 in the immunopathogenesis of psoriasis. F1000 Biol. Rep 2010, 2, 40. [Google Scholar]
- Zdanov, A. Structural analysis of cytokines comprising the IL-10 family. Cytokine Growth Factor Rev 2010, 21, 325–330. [Google Scholar]
- Kunz, S.; Wolk, K.; Witte, E.; Witte, K.; Doecke, W.-D.; Volk, H.D.; Sterry, W.; Asadullah, K.; Sabat, R. Interleukin (IL)-19, IL-20 and IL-24 are produced by and act on keratinocytes and are distinct from classical ILs. Exp. Dermatol 2006, 15, 991–1004. [Google Scholar]
- Otkjaer, K.; Kragballe, K.; Funding, A.T.; Clausen, J.T.; Noerby, P.L.; Steiniche, T.; Iversen, L. The dynamics of gene expression of interleukin-19 and interleukin-20 and their receptors in psoriasis. Br. J. Dermatol 2005, 153, 911–918. [Google Scholar]
- Sa, S.M.; Valdez, P.A.; Wu, J.; Jung, K.; Zhong, F.; Hall, L.; Kasman, I.; Winer, J.; Modrusan, Z.; Danilenko, D.M.; et al. The effects of IL-20 subfamily cytokines on reconstituted human epidermis suggest potential roles in cutaneous innate defense and pathogenic adaptive immunity in psoriasis. J. Immunol 2007, 178, 2229–2240. [Google Scholar]
- Parrish-Novak, J. Interleukins 19, 20, and 24 signal through two distinct receptor complexes. differences in receptor-ligand interactions mediate unique biological functions. J. Biol. Chem 2002, 277, 47517–47523. [Google Scholar]
- Blumberg, H.; Conklin, D.; Xu, W.F.; Grossmann, A.; Brender, T.; Carollo, S.; Eagan, M.; Foster, D.; Haldeman, B.A.; Hammond, A.; et al. Interleukin 20: Discovery, receptor identification, and role in epidermal function. Cell 2001, 104, 9–19. [Google Scholar]
- Rich, B.E.; Kupper, T.S. Cytokines: IL-20—A new effector in skin inflammation. Curr. Biol 2001, 11, R531–R534. [Google Scholar]
- Wolk, K.; Kunz, S.; Witte, E.; Friedrich, M.; Asadullah, K.; Sabat, R. IL-22 increases the innate immunity of tissues. Immunity 2004, 21, 241–254. [Google Scholar]
- Boniface, K.; Bernard, F.-X.; Garcia, M.; Gurney, A.L.; Lecron, J.-C.; Morel, F. IL-22 inhibits epidermal differentiation and induces proinflammatory gene expression and migration of human keratinocytes. J. Immunol 2005, 174, 3695–3702. [Google Scholar]
- Gutowska-Owsiak, D.; Schaupp, A.L.; Salimi, M.; Taylor, S.; Ogg, G.S. Interleukin-22 downregulates filaggrin expression and affects expression of profilaggrin processing enzymes. Br. J. Dermatol 2011, 165, 492–498. [Google Scholar]
- Egberts, F.; Heinrich, M.; Jensen, J.-M.; Winoto-Morbach, S.; Pfeiffer, S.; Wickel, M.; Schunck, M.; Steude, J.; Saftig, P.; Proksch, E.; et al. Cathepsin D is involved in the regulation of transglutaminase 1 and epidermal differentiation. J. Cell Sci 2004, 117, 2295–2307. [Google Scholar]
- Hsu, C.-Y.; Henry, J.; Raymond, A.-A.; Méchin, M.-C.; Pendaries, V.; Nassar, D.; Hansmann, B.; Balica, S.; Burlet-Schiltz, O.; Schmitt, A.-M.; et al. Deimination of human filaggrin-2 promotes its proteolysis by calpain 1. J. Biol. Chem 2011, 286, 23222–23233. [Google Scholar]
- Wolk, K.; Witte, E.; Wallace, E.; Döcke, W.D.; Kunz, S.; Asadullah, K.; Volk, H.D.; Sterry, W.; Sabat, R. IL-22 regulates the expression of genes responsible for antimicrobial defense, cellular differentiation, and mobility in keratinocytes: A potential role in psoriasis. Eur. J. Immunol 2006, 36, 1309–1323. [Google Scholar]
- Méhul, B.; Bernard, D.; Simonetti, L.; Bernard, M.A.; Schmidt, R. Identification and cloning of a new calmodulin-like protein from human epidermis. J. Biol. Chem 2000, 275, 12841–12847. [Google Scholar]
- Bergboer, J.G.M.; Tjabringa, G.S.; Kamsteeg, M.; van Vlijmen-Willems, I.M.J.J.; Rodijk-Olthuis, D.; Jansen, P.A.M.; Thuret, J.-Y.; Narita, M.; Ishida-Yamamoto, A.; Zeeuwen, P.L.J.M.; et al. Psoriasis risk genes of the late cornified envelope-3 group are distinctly expressed compared with genes of other LCE groups. Am. J. Pathol 2011, 178, 1470–1477. [Google Scholar]
- Sartor, R.B.; Muehlbauer, M. Microbial host interactions in IBD: Implications for pathogenesis and therapy. Curr. Gastroenterol. Rep 2007, 9, 497–507. [Google Scholar]
- Ma, H.-L.; Liang, S.; Li, J.; Napierata, L.; Brown, T.; Benoit, S.; Senices, M.; Gill, D.; Dunussi-Joannopoulos, K.; Collins, M.; et al. IL-22 is required for Th17 cell–mediated pathology in a mouse model of psoriasis-like skin inflammation. J. Clin. Invest 2008, 118, 597–607. [Google Scholar]
- Wolk, K.; Haugen, H.S.; Xu, W.; Witte, E.; Waggie, K.; Anderson, M.; Baur, E.; Witte, K.; Warszawska, K.; Philipp, S.; et al. IL-22 and IL-20 are key mediators of the epidermal alterations in psoriasis while IL-17 and IFN-γ are not. J. Mol. Med 2009, 87, 523–536. [Google Scholar]
- Bhawan, J.; Bansal, C.; Whren, K.; Schwertschlag, U. IL-11 psoriasis study group. K16 expression in uninvolved psoriatic skin: A possible marker of pre-clinical psoriasis. J. Cutan. Pathol. 2004, 31, 471–476. [Google Scholar]
- Poindexter, N.J.; Williams, R.R.; Powis, G.; Jen, E.; Caudle, A.S.; Chada, S.; Grimm, E.A. IL-24 is expressed during wound repair and inhibits TGFalpha-induced migration and proliferation of keratinocytes. Exp. Dermatol 2010, 19, 714–722. [Google Scholar]
- He, M.; Liang, P. IL-24 transgenic mice: In vivo evidence of overlapping functions for IL-20, IL-22, and IL-24 in the epidermis. J. Immunol 2010, 184, 1793–1798. [Google Scholar]
- Pestka, S.; Krause, C.D.; Sarkar, D.; Walter, M.R.; Shi, Y.; Fisher, P.B. Interleukin-10 and related cytokines and receptors. Annu. Rev. Immunol 2004, 22, 929–979. [Google Scholar]
- Feld, M.; Shpacovitch, V.M.; Fastrich, M.; Cevikbas, F.; Steinhoff, M. Interferon-γ induces upregulation and activation of the interleukin-31 receptor in human dermal microvascular endothelial cells. Exp. Dermatol 2010, 19, 921–923. [Google Scholar]
- Heise, R.; Neis, M.M.; Marquardt, Y.; Joussen, S.; Heinrich, P.C.; Merk, H.F.; Hermanns, H.M.; Baron, J.M. IL-31 receptor alpha expression in epidermal keratinocytes is modulated by cell differentiation and interferon gamma. J. Invest. Dermatol 2009, 129, 240–243. [Google Scholar]
- Dambacher, J.; Beigel, F.; Seiderer, J.; Haller, D.; Goke, B.; Auernhammer, C.J.; Brand, S. Interleukin 31 mediates MAP kinase and STAT1/3 activation in intestinal epithelial cells and its expression is upregulated in inflammatory bowel disease. Gut 2007, 56, 1257–1265. [Google Scholar] [Green Version]
- Albanesi, C.; Scarponi, C.; Cavani, A.; Federici, M.; Nasorri, F.; Girolomoni, G. Interleukin-17 is produced by both Th1 and Th2 lymphocytes, and modulates interferon-gamma- and interleukin-4-induced activation of human keratinocytes. J. Invest. Dermatol 2000, 115, 81–87. [Google Scholar]
- Tohyama, M.; Yang, L.; Hanakawa, Y.; Dai, X.; Shirakata, Y.; Sayama, K. IFN-α enhances IL-22 receptor expression in keratinocytes: A possible role in the development of psoriasis. J. Invest. Dermatol 2012, 132, 1933–1935. [Google Scholar]
- Tenaud, I.; Leroy, S.; Chebassier, N.; Dreno, B. Modulation in vitro of keratinocyte integrins by interferon-alpha and interferon-gamma. Int. J. Dermatol 2002, 41, 836–840. [Google Scholar]
- Litjens, S.H.M.; de Pereda, J.M.; Sonnenberg, A. Current insights into the formation and breakdown of hemidesmosomes. Trends Cell Biol 2006, 16, 376–383. [Google Scholar]
- Niculescu, C.; Ganguli-Indra, G.; Pfister, V.; Dupé, V.; Messaddeq, N.; de Arcangelis, A.; Georges-Labouesse, E. Conditional ablation of integrin alpha-6 in mouse epidermis leads to skin fragility and inflammation. Eur. J. Cell Biol 2011, 90, 270–277. [Google Scholar]
- Tsuji, T. Physiological and pathological roles of alpha3beta1 integrin. J. Membr. Biol 2004, 200, 115–132. [Google Scholar]
- Uchida, Y.; Hara, M.; Nishio, H.; Sidransky, E.; Inoue, S.; Otsuka, F.; Suzuki, A.; Elias, P.M.; Holleran, W.M.; Hamanaka, S. Epidermal sphingomyelins are precursors for selected stratum corneum ceramides. J. Lipid Res 2000, 41, 2071–2082. [Google Scholar]
- Hamanaka, S.; Hara, M.; Nishio, H.; Otsuka, F.; Suzuki, A.; Uchida, Y. Human epidermal glucosylceramides are major precursors of stratum corneum ceramides. J. Invest. Dermatol 2002, 119, 416–423. [Google Scholar]
- Barksby, H.E.; Lea, S.R.; Preshaw, P.M.; Taylor, J.J. The expanding family of interleukin-1 cytokines and their role in destructive inflammatory disorders. Clin. Exp. Immunol 2007, 149, 217–225. [Google Scholar]
- Portugal-Cohen, M.; Horev, L.; Ruffer, C.; Schlippe, G.; Voss, W.; Ma’or, Z.; Oron, M.; Soroka, Y.; Frušić-Zlotkin, M.; Milner, Y.; et al. Non-invasive skin biomarkers quantification of psoriasis and atopic dermatitis: Cytokines, antioxidants and psoriatic skin auto-fluorescence. Biomed. Pharmacother 2012, 66, 293–299. [Google Scholar]
- Wood, L.C.; Jackson, S.M.; Elias, P.M.; Grunfeld, C.; Feingold, K.R. Cutaneous barrier perturbation stimulates cytokine production in the epidermis of mice. J. Clin. Invest 1992, 90, 482–487. [Google Scholar]
- Barland, C.O.; Zettersten, E.; Brown, B.S.; Ye, J.; Elias, P.M.; Ghadially, R. Imiquimod-induced interleukin-1 alpha stimulation improves barrier homeostasis in aged murine epidermis. J. Invest. Dermatol 2004, 122, 330–336. [Google Scholar]
- O’Shaughnessy, R.F.L.; Choudhary, I.; Harper, J.I. Interleukin-1 alpha blockade prevents hyperkeratosis in an in vitro model of lamellar ichthyosis. Hum. Mol. Genet 2010, 19, 2594–2605. [Google Scholar]
- Jung, Y.-J.; Jung, M.; Kim, M.; Hong, S.-P.; Choi, E. H. IL-1[alpha] stimulation restores epidermal permeability and antimicrobial barriers compromised by topical tacrolimus. J. Invest. Dermatol 2010, 131, 698–705. [Google Scholar]
- Yano, S.; Banno, T.; Walsh, R.; Blumenberg, M. Transcriptional responses of human epidermal keratinocytes to cytokine interleukin-1. J. Cell. Physiol 2008, 214, 1–13. [Google Scholar]
- Groves, R.W.; Mizutani, H.; Kieffer, J.D.; Kupper, T.S. Inflammatory skin disease in transgenic mice that express high levels of interleukin 1 alpha in basal epidermis. Proc. Natl. Acad. Sci. USA 1995, 92, 11874. [Google Scholar]
- Dinarello, C.A. Immunological and inflammatory functions of the interleukin-1 family. Annu. Rev. Immunol 2009, 27, 519–550. [Google Scholar]
- Kirschner, N.; Poetzl, C.; von den Driesch, P.; Wladykowski, E.; Moll, I.; Behne, M.J.; Brandner, J.M. Alteration of tight junction proteins is an early event in psoriasis: Putative involvement of proinflammatory cytokines. Am. J. Pathol 2009, 175, 1095–1106. [Google Scholar]
- Sun, M.; Fink, P.J. A new class of reverse signaling costimulators belongs to the TNF family. J. Immunol 2007, 179, 4307–4312. [Google Scholar]
- Aggarwal, B.B. Signalling pathways of the TNF superfamily: A double-edged sword. Nat. Rev. Immunol 2003, 3, 745–756. [Google Scholar]
- Kim, B.E.; Howell, M.D.; Guttman-Yassky, E.; Guttman, E.; Gilleaudeau, P.M.; Cardinale, I.R.; Boguniewicz, M.; Krueger, J.G.; Leung, D.Y.M. TNF-α downregulates filaggrin and loricrin through c-Jun N-terminal kinase: Role for TNF-α antagonists to improve skin barrier. J. Invest. Dermatol 2011, 131, 1272–1279. [Google Scholar]
- Jensen, J.-M.; Schütze, S.; Förl, M.; Krönke, M.; Proksch, E. Roles for tumor necrosis factor receptor p55 and sphingomyelinase in repairing the cutaneous permeability barrier. J. Clin. Invest 1999, 104, 1761–1770. [Google Scholar]
- Lowes, M.A.; Bowcock, A.M.; Krueger, J.G. Pathogenesis and therapy of psoriasis. Nature 2007, 445, 866–873. [Google Scholar]
- Pasparakis, M. Role of NF-κB in epithelial biology. Immunol. Rev 2012, 246, 346–358. [Google Scholar]
- Reynolds, J.M.; Angkasekwinai, P.; Dong, C. IL-17 family member cytokines: Regulation and function in innate immunity. Cytokine Growth Factor Rev 2010, 21, 413–423. [Google Scholar]
- Chang, S.H.; Dong, C. Signaling of interleukin-17 family cytokines in immunity and inflammation. Cell. Signal 2011, 23, 1069–1075. [Google Scholar]
- Koga, C.; Kabashima, K.; Shiraishi, N.; Kobayashi, M.; Tokura, Y. Possible pathogenic role of Th17 cells for atopic dermatitis. J. Invest. Dermatol 2008, 128, 2625–2630. [Google Scholar]
- Gutowska-Owsiak, D.; Schaupp, A.L.; Salimi, M.; Selvakumar, T.A.; McPherson, T.; Taylor, S.; Ogg, G.S. IL-17 downregulates filaggrin and affects keratinocyte expression of genes associated with cellular adhesion. Exp. Dermatol 2012, 21, 104–10. [Google Scholar]
- Leonardi, C.; Matheson, R.; Zachariae, C.; Cameron, G.; Li, L.; Edson-Heredia, E.; Braun, D.; Banerjee, S. Anti-interleukin-17 monoclonal antibody ixekizumab in chronic plaque psoriasis. N. Engl. J. Med 2012, 366, 1190–1199. [Google Scholar]
- Papp, K.A.; Leonardi, C.; Menter, A.; Ortonne, J.-P.; Krueger, J.G.; Kricorian, G.; Aras, G.; Li, J.; Russell, C.B.; Thompson, E.H.Z.; et al. Brodalumab, an anti-interleukin-17-receptor antibody for psoriasis. N. Engl. J. Med 2012, 366, 1181–1189. [Google Scholar]
- Hvid, M.; Vestergaard, C.; Kemp, K.; Christensen, G.B.; Deleuran, B.; Deleuran, M. IL-25 in atopic dermatitis: A possible link between inflammation and skin barrier dysfunction? J. Invest. Dermatol 2010, 131, 150–157. [Google Scholar]
© 2013 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Hänel, K.H.; Cornelissen, C.; Lüscher, B.; Baron, J.M. Cytokines and the Skin Barrier. Int. J. Mol. Sci. 2013, 14, 6720-6745. https://doi.org/10.3390/ijms14046720
Hänel KH, Cornelissen C, Lüscher B, Baron JM. Cytokines and the Skin Barrier. International Journal of Molecular Sciences. 2013; 14(4):6720-6745. https://doi.org/10.3390/ijms14046720
Chicago/Turabian StyleHänel, Kai H., Christian Cornelissen, Bernhard Lüscher, and Jens Malte Baron. 2013. "Cytokines and the Skin Barrier" International Journal of Molecular Sciences 14, no. 4: 6720-6745. https://doi.org/10.3390/ijms14046720
APA StyleHänel, K. H., Cornelissen, C., Lüscher, B., & Baron, J. M. (2013). Cytokines and the Skin Barrier. International Journal of Molecular Sciences, 14(4), 6720-6745. https://doi.org/10.3390/ijms14046720