Aptamers and Their Potential to Selectively Target Aspects of EGF, Wnt/β-Catenin and TGFβ–Smad Family Signaling

Abstract
:1. Aptamers: Emerging, but Re-Discovered Precious Tools in Diverse Studies
2. From Bench to Bedside: Aptamers Enter Diagnostics and Therapy
3. Aptamers and Their Use in Targeting of Signal Transduction Cascades
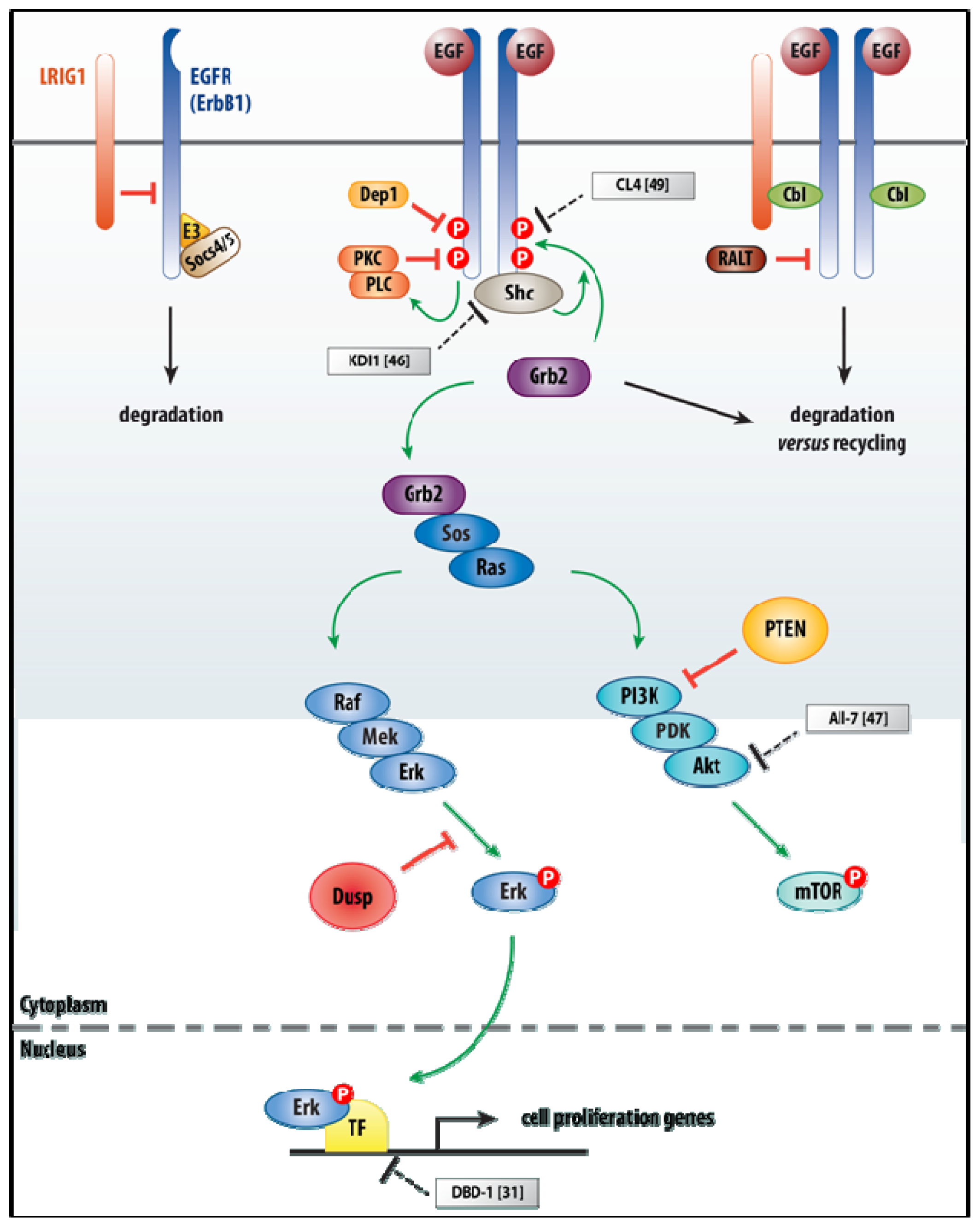
3.1. EGF Signaling
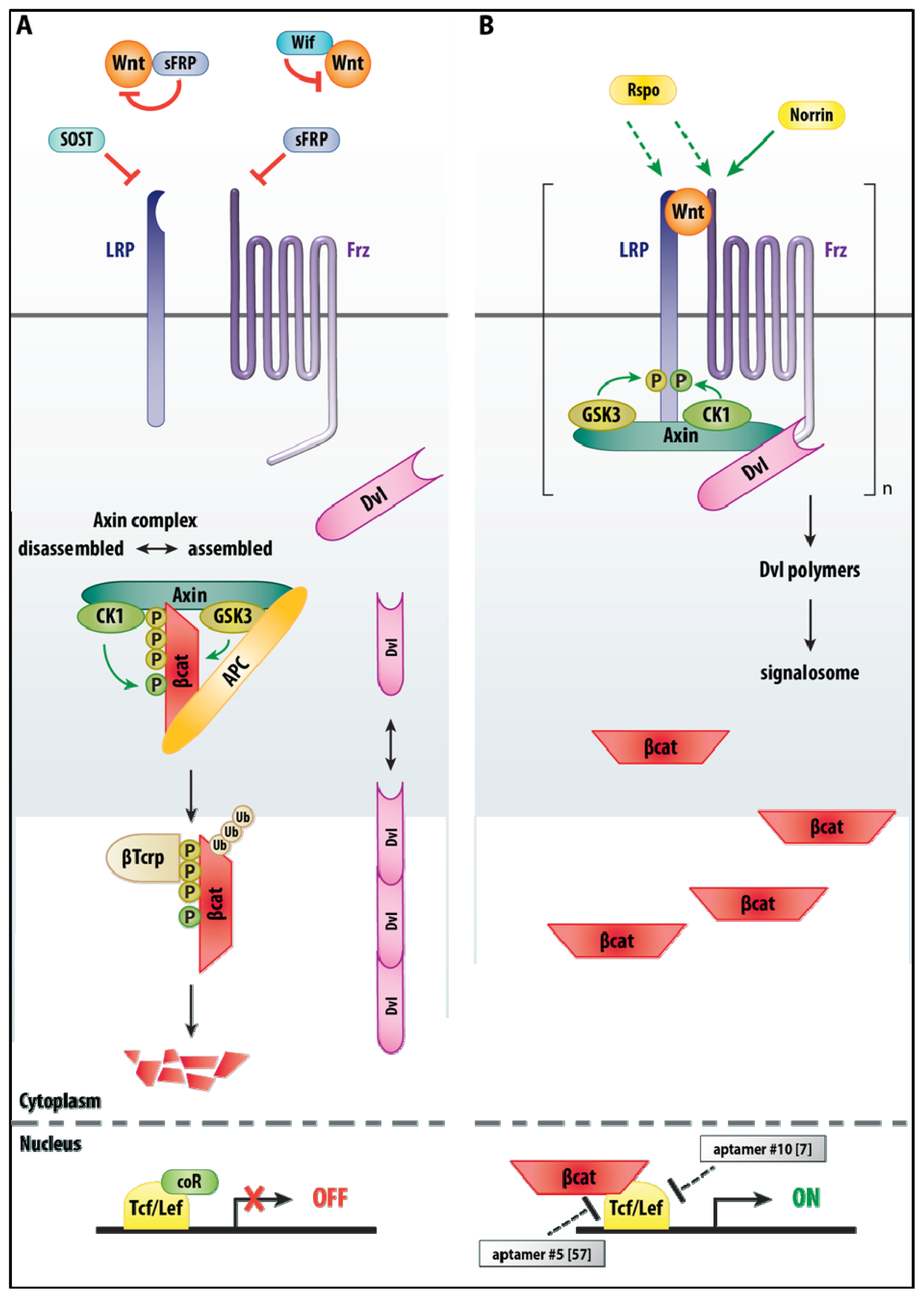
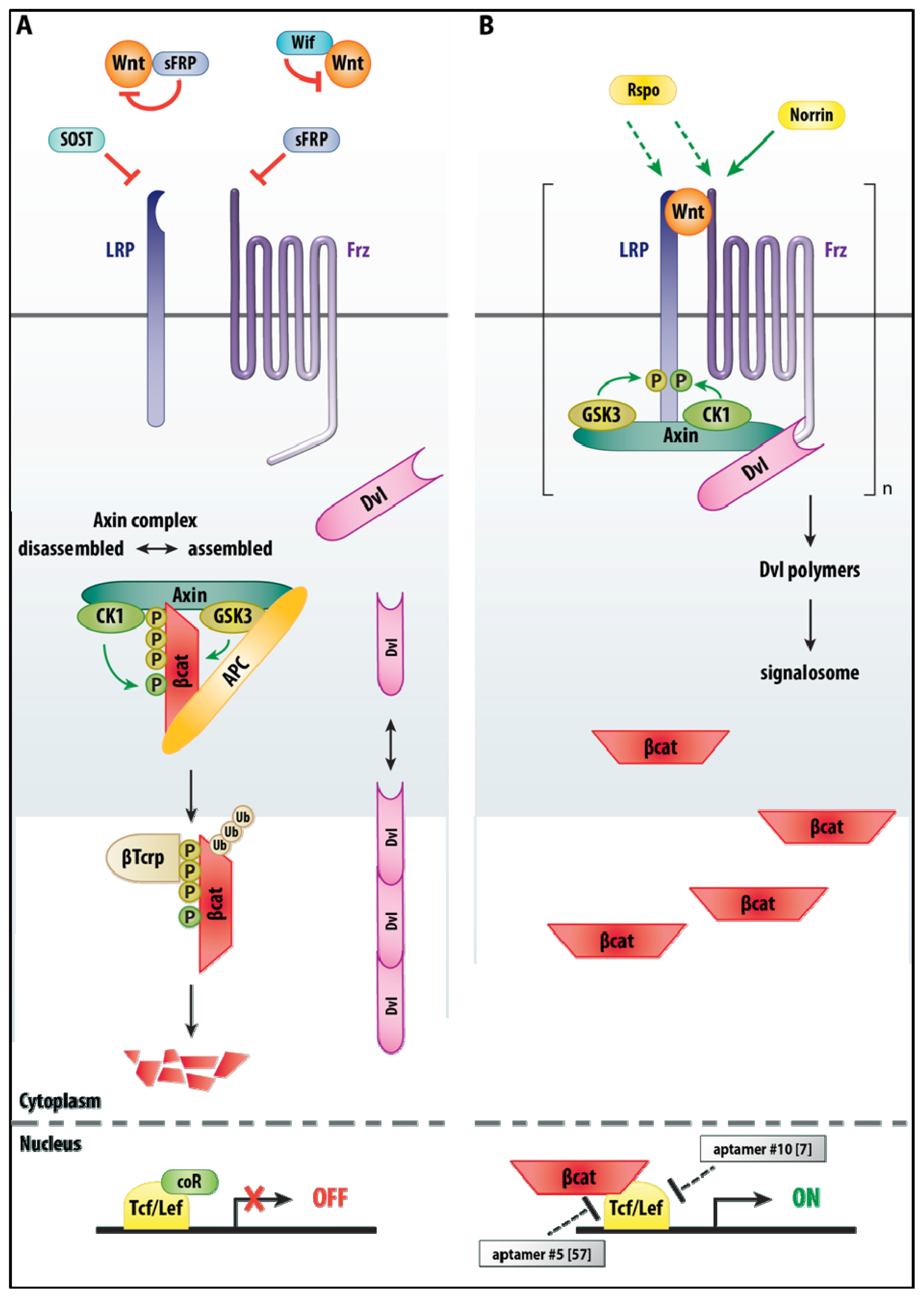
3.2. Wnt/β-Catenin Signaling
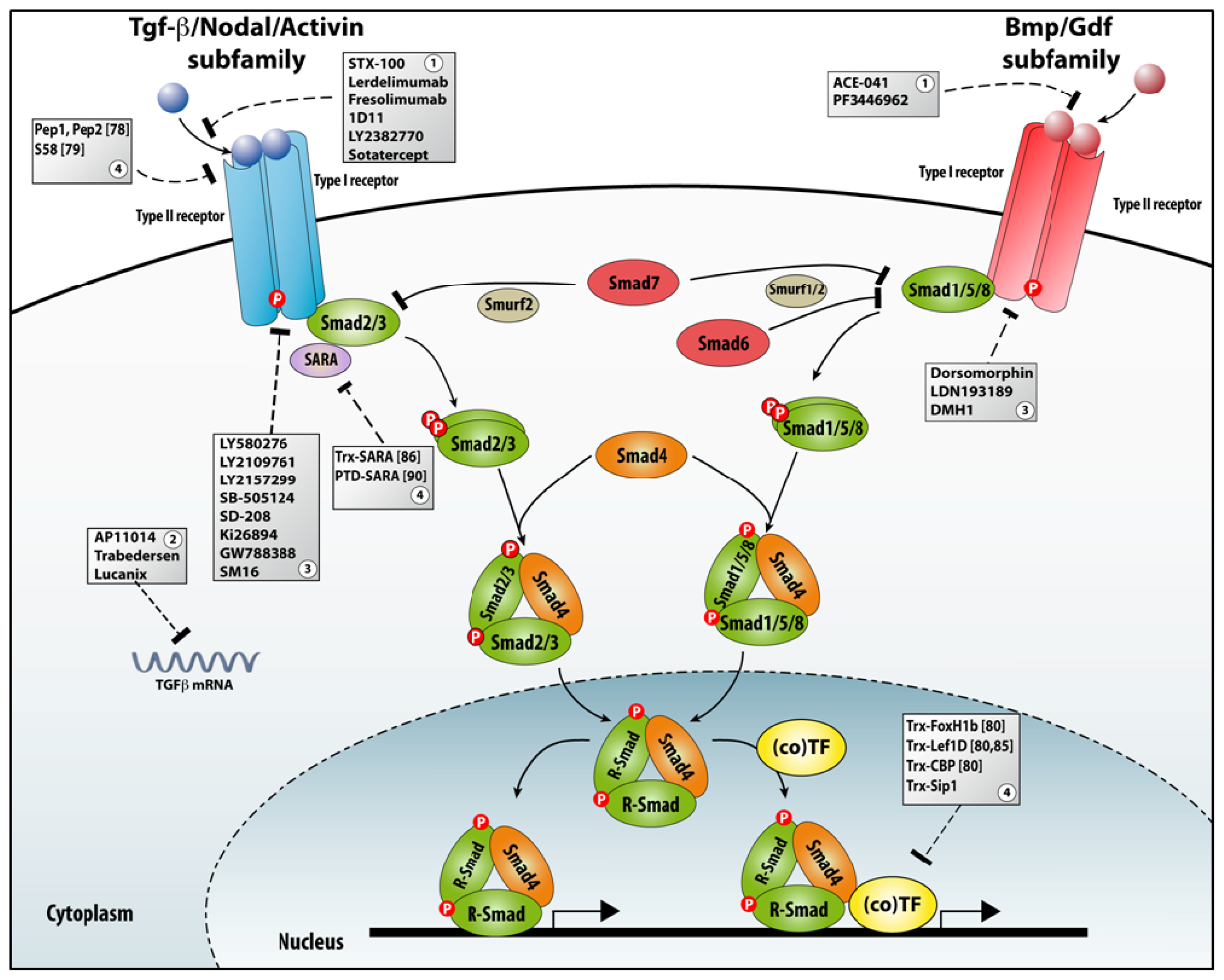
3.3. TGFβ Signaling
3.3.1. Different Aspects of TGFβ Signaling in Pathological Conditions
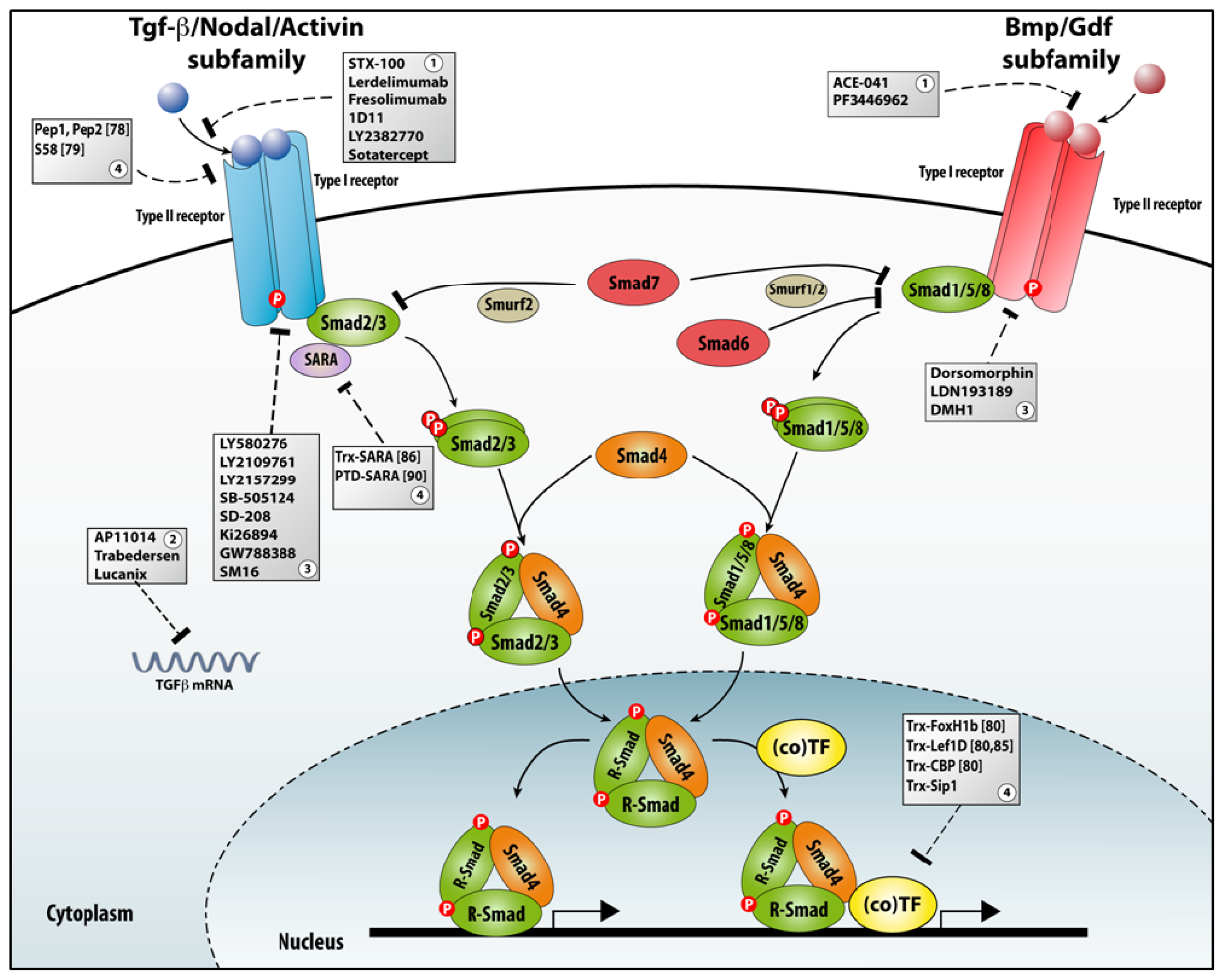
3.3.2. Many Roads to TGFβ Signaling Downregulation
3.3.3. Aptamers as a Tool for Studying TGFβ Signaling
4. A Growing Number of SIPs Provide New Potential Aptamers for Interfering with Smad Signaling
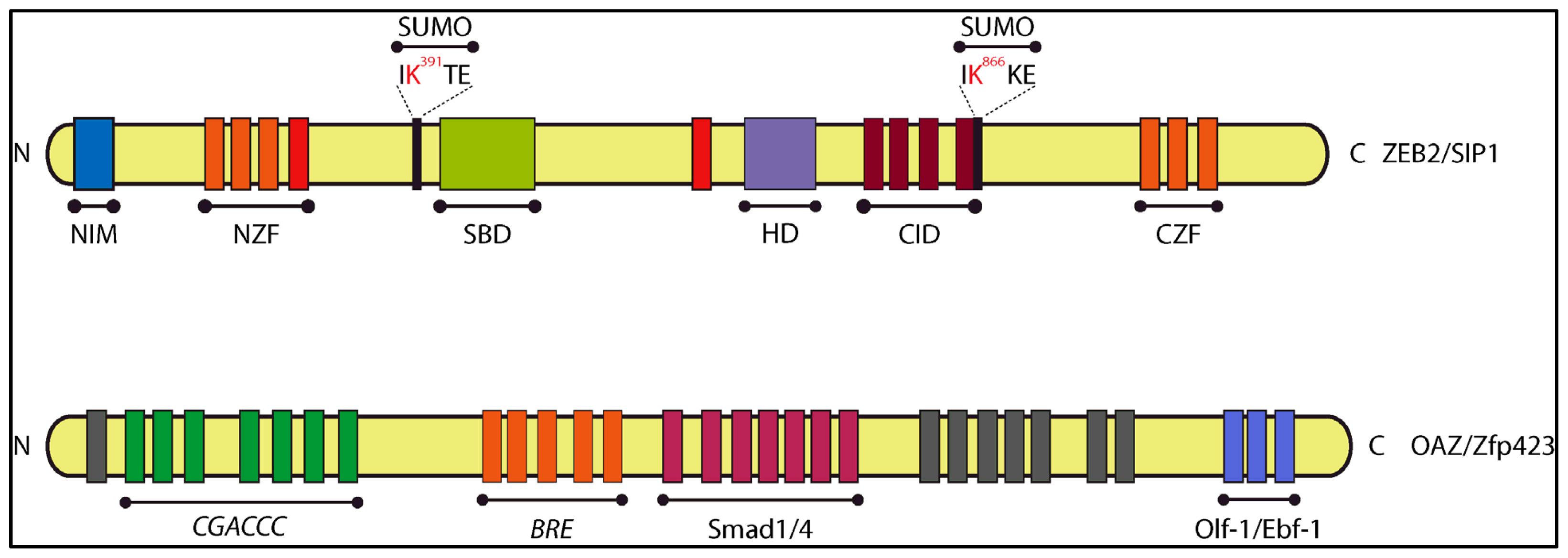
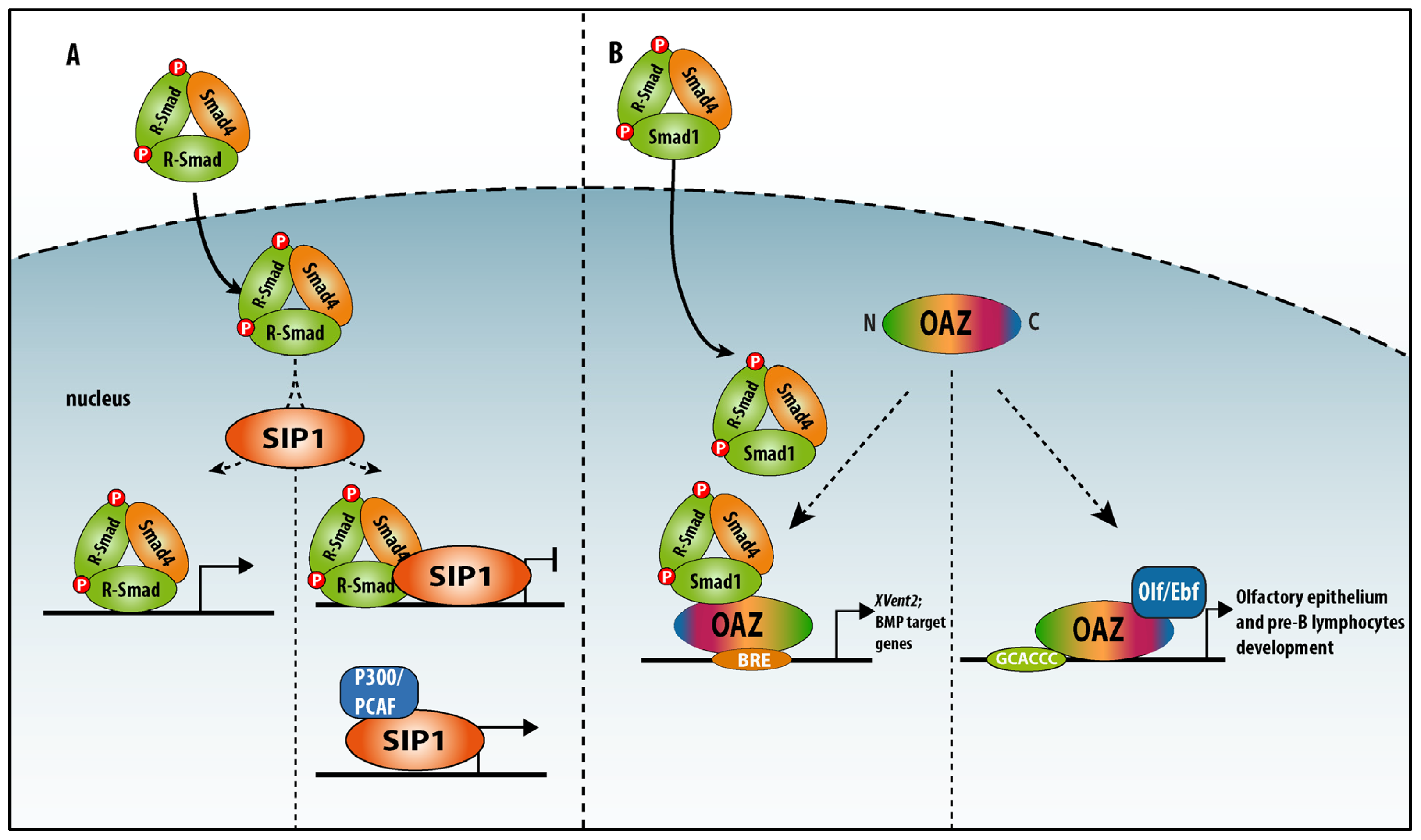
4.1. Zinc Finger TFs as SIPs: SIP1/Zeb2 and OAZ
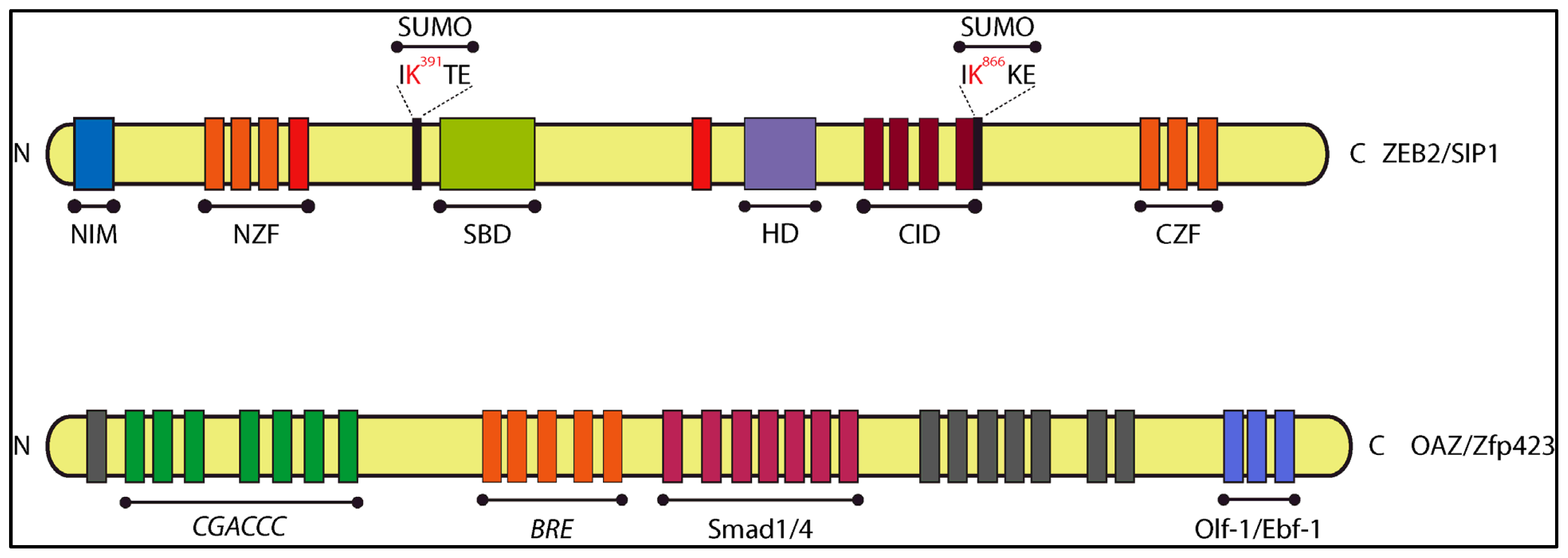
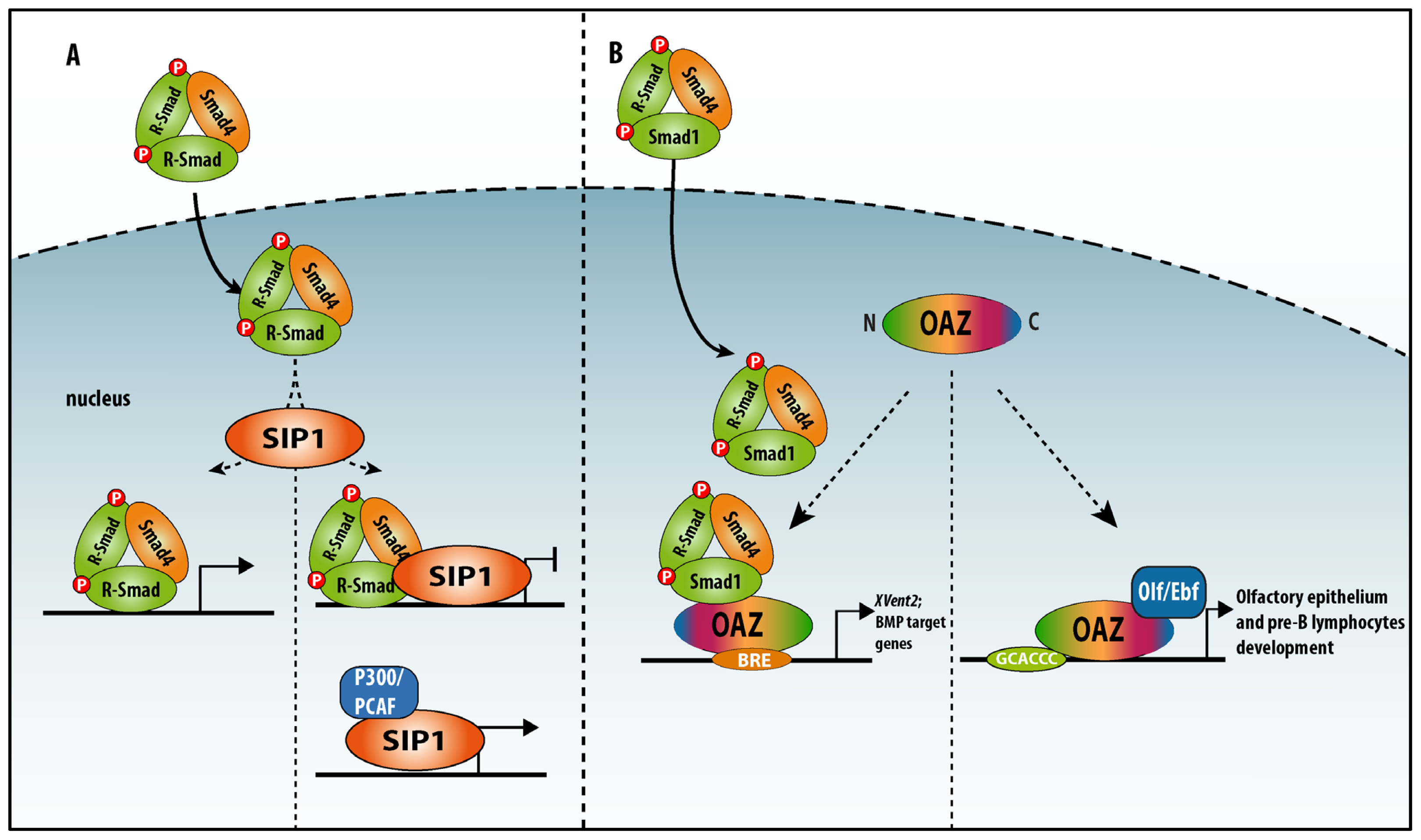
4.1.1. Smad-Interacting Protein-1 (Sip1, also Named Zeb2 and Zfhx1b)
4.1.2. OAZ (also named Zfp423)
4.2. Non-TF SIPs
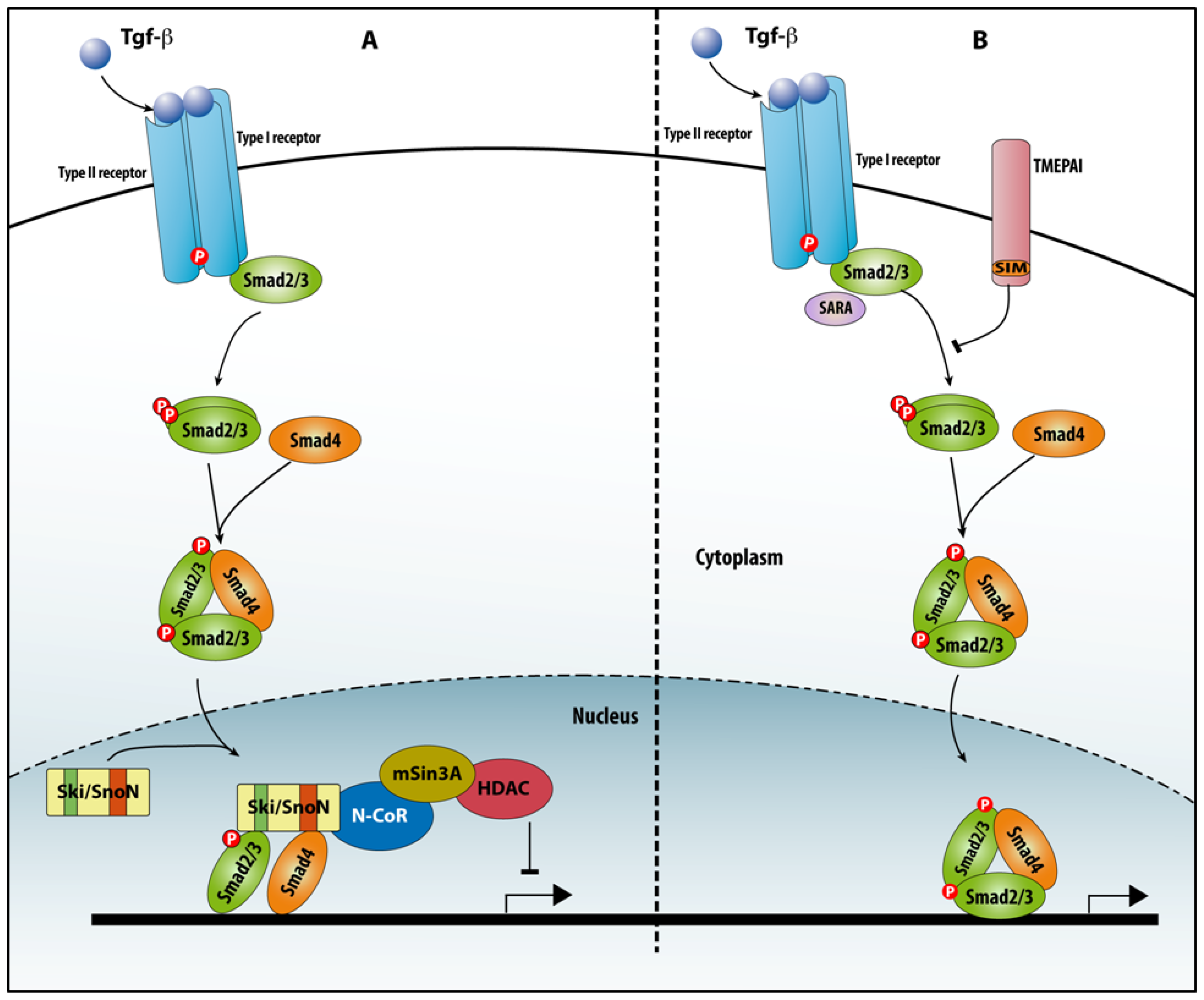
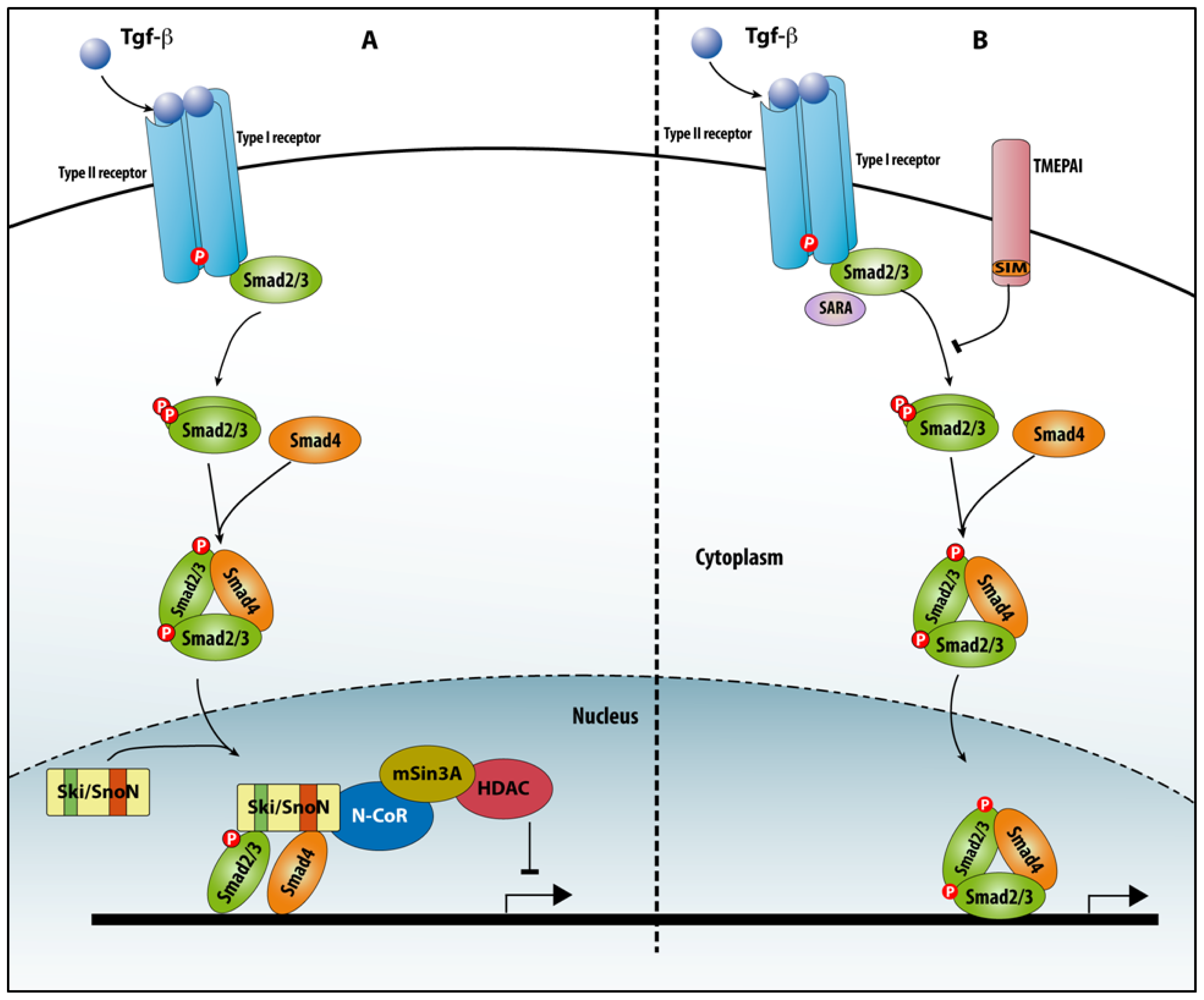
4.2.1. Ski/SnoN
4.2.2. TMEPAI
4.3. Mutated Smads in Cancer and Other Diseases
5. Conclusions and Future Perspectives
Acknowledgments
Conflict of Interest
References
- Ellington, A.D.; Szostak, J.W. In vitro selection of RNA molecules that bind specific ligands. Nature 1990, 346, 818–822. [Google Scholar]
- Ellington, A.D.; Szostak, J.W. Selection in vitro of single-stranded DNA molecules that fold into specific ligand-binding structures. Nature 1992, 355, 850–852. [Google Scholar]
- Stoltenburg, R.; Reinemann, C.; Strehlitz, B. SELEX—A (r)evolutionary method to generate high-affinity nucleic acid ligands. Biomol. Eng 2007, 24, 381–403. [Google Scholar]
- Cerchia, L.; Giangrande, P.H.; McNamara, J.O.; de Franciscis, V. Cell-specific aptamers for targeted therapies. Methods Mol. Biol 2009, 535, 59–78. [Google Scholar]
- Cerchia, L.; de Franciscis, V. Targeting cancer cells with nucleic acid aptamers. Trends Biotechnol 2010, 28, 517–525. [Google Scholar]
- Sefah, K.; Shangguan, D.; Xiong, X.; O’Donoghue, M.B.; Tan, W. Development of DNA aptamers using cell-SELEX. Nat. Protoc 2010, 5, 1169–1185. [Google Scholar]
- Park, M.W.; Choi, K.H.; Jeong, S. Inhibition of the DNA binding by the TCF-1 binding RNA aptamer. Biochem. Biophys. Res. Commun 2005, 330, 11–17. [Google Scholar]
- Zimmermann, G.R.; Wick, C.L.; Shields, T.P.; Jenison, R.D.; Pardi, A. Molecular interactions and metal binding in the theophylline-binding core of an RNA aptamer. RNA 2000, 6, 659–667. [Google Scholar]
- Ciesiolka, J.; Gorski, J.; Yarus, M. Selection of an RNA domain that binds Zn2+. RNA 1995, 1, 538–550. [Google Scholar]
- Famulok, M.; Hüttenhofer, A. In vitro selection analysis of neomycin binding RNAs with a mutagenized pool of variants of the 16S rRNA decoding region. Biochemistry 1996, 35, 4265–4270. [Google Scholar]
- Kraus, E.; James, W.; Barclay, A.N. Cutting edge: Novel RNA ligands able to bind CD4 antigen and inhibit CD4+ T lymphocyte function. J. Immunol 1998, 160, 5209–5212. [Google Scholar]
- Ohuchi, S.P.; Ohtsu, T.; Nakamura, Y. Selection of RNA aptamers against recombinant transforming growth factor-beta type III receptor displayed on cell surface. Biochimie 2006, 88, 897–904. [Google Scholar]
- Cerchia, L.; Ducongé, F.; Pestourie, C.; Boulay, J.; Aissouni, Y.; Gombert, K.; Tavitian, B.; de Franciscis, V.; Libri, D. Neutralizing aptamers from whole-cell SELEX inhibit the RET receptor tyrosine kinase. PLoS Biol 2005, 3, e123. [Google Scholar]
- Baines, I.C.; Colas, P. Peptide aptamers as guides for small-molecule drug discovery. Drug Discov. Today 2006, 11, 334–341. [Google Scholar]
- Cohen, B.A.; Colas, P.; Brent, R. An artificial cell-cycle inhibitor isolated from a combinatorial library. Proc. Natl. Acad. Sci. USA 1998, 95, 14272–14277. [Google Scholar]
- Geyer, C.R.; Brent, R. Selection of genetic agents from random peptide aptamer expression libraries. Methods Enzymol 2000, 328, 171–208. [Google Scholar]
- Ladner, R.C. Constrained peptides as binding entities. Trends Biotechnol 1995, 13, 426–430. [Google Scholar]
- Colas, P.; Cohen, B.; Jessen, T.; Grishina, I.; McCoy, J.; Brent, R. Genetic selection of peptide aptamers that recognize and inhibit cyclin-dependent kinase 2. Nature 1996, 380, 548–550. [Google Scholar]
- Abed, N.; Bickle, M.; Mari, B.; Schapira, M.; Sanjuan-España, R.; Sermesant, K.R.; Moncorgé, O.; Mouradian-Garcia, S.; Barbry, P.; Rudkin, B.B.; et al. A comparative analysis of perturbations caused by a gene knock-out, a dominant negative allele, and a set of peptide aptamers. Mole. Cell. Proteomics 2007, 6, 2110–2121. [Google Scholar]
- Abedi, M.R.; Caponigro, G.; Kamb, A. Green fluorescent protein as a scaffold for intracellular presentation of peptides. Nucleic Acids Res 1998, 26, 623–630. [Google Scholar]
- Norman, T.C.; Smith, D.L.; Sorger, P.K.; Drees, B.L.; O’Rourke, S.M.; Hughes, T.R.; Roberts, C.J.; Friend, S.H.; Fields, S.; Murray, A.W. Genetic selection of peptide inhibitors of biological pathways. Science 1999, 285, 591–595. [Google Scholar]
- Klevenz, B.; Butz, K.; Hoppe-Seyler, F. Peptide aptamers: Exchange of the thioredoxin-A scaffold by alternative platform proteins and its influence on target protein binding. Cell. Mol. Life Sci 2002, 59, 1993–1998. [Google Scholar]
- Johnson, S.; Evans, D.; Laurenson, S.; Paul, D.; Davies, A.G.; Ko Ferrigno, P.; Walti, C. Surface-immobilized peptide aptamers as probe molecules for protein detection. Anal. Chem 2008, 80, 978–983. [Google Scholar]
- Evans, D.; Johnson, S.; Laurenson, S.; Davies, G.; Ko Ferrigno, P.; Walti, C. Electrical protein detection in cell lysates using high-density peptide-aptamer microarrays. J. Biol 2008, 7, 3. [Google Scholar] [Green Version]
- Borghouts, C.; Kunz, C.; Groner, B. Current strategies for the development of peptide-based anti-cancer therapeutics. J. Pept. Sci 2005, 11, 713–726. [Google Scholar]
- Woodman, R.; Yeh, J.T.; Laurenson, S.; Ferrigno, P.K. Design and validation of a neutral protein scaffold for the presentation of peptide aptamers. J. Mol. Biol 2005, 352, 1118–1133. [Google Scholar]
- Miller, R.A.; Binkowski, B.F.; Belshaw, P.J. Ligand-regulated peptide aptamers that inhibit the 5′-AMP-activated protein kinase. J. Mol. Biol 2007, 365, 945–957. [Google Scholar]
- Geyer, C.R.; Colman-Lerner, A.; Brent, R. “Mutagenesis” by peptide aptamers identifies genetic network members and pathway connections. Proc. Natl. Acad. Sci. USA 1999, 96, 8567–8572. [Google Scholar]
- Xu, C.W.; Luo, Z. Inactivation of Ras function by allele-specific peptide aptamers. Oncogene 2002, 21, 5753–5757. [Google Scholar]
- Fabbrizio, E.; Le Cam, L.; Polanowska, J.; Kaczorek, M.; Lamb, N.; Brent, R.; Sardet, C. Inhibition of mammalian cell proliferation by genetically selected peptide aptamers that functionally antagonize E2F activity. Oncogene 1999, 18, 4357–4363. [Google Scholar]
- Nagel-Wolfrum, K.; Buerger, C.; Wittig, I.; Butz, K.; Hoppe-Seyler, F.; Groner, B. The interaction of specific peptide aptamers with the DNA binding domain and the dimerization domain of the transcription factor Stat3 inhibits transactivation and induces apoptosis in tumor cells. Mol. Cancer Res 2004, 2, 170–182. [Google Scholar]
- Tomai, E.; Butz, K.; Lohrey, C.; von Weizsäcker, F.; Zentgraf, H.; Hoppe-Seyler, F. Peptide aptamer-mediated inhibition of target proteins by sequestration into aggresomes. J. Boil. Chem 2006, 281, 21345–21352. [Google Scholar]
- Nouvion, A.; Thibaut, J.; Lohez, O.D.; Venet, S.; Colas, P.; Gillet, G.; Lalle, P. Modulation of Nr-13 antideath activity by peptide aptamers. Oncogene 2007, 26, 701–710. [Google Scholar]
- De Chassey, B.; Mikaelian, I.; Mathieu, A.; Bickle, M.; Olivier, D.; Nègre, D.; Cosset, F.; Rudkin, B.B.; Colas, P. An antiproliferative genetic screening identifies a peptide aptamer that targets calcineurin and up-regulates its activity. Mol. Cell. Proteomics 2007, 6, 451–459. [Google Scholar]
- Bardou, C.; Borie, C.; Bickle, M.; Rudkin, B.B.; Colas, P. Peptide aptamers for small molecule drug discovery. Methods Mol. Boil 2009, 535, 373–388. [Google Scholar]
- Bunka, D.H.; Platonova, O.; Stockley, P.G. Development of aptamer therapeutics. Curr. Opin. Pharmacol 2010, 10, 557–562. [Google Scholar]
- Cunningham, E.T.; Adamis, A.P.; Altaweel, M.; Aiello, L.P.; Bressler, N.M.; D’Amico, D.J.; Goldbaum, M.; Guyer, D.R.; Katz, B.; Patel, M.; et al. A phase II randomized double-masked trial of pegaptanib, an anti-vascular endothelial growth factor aptamer, for diabetic macular edema. Ophthalmology 2005, 112, 1747–1757. [Google Scholar]
- Sundaram, P.; Kurniawan, H.; Byrne, M.E.; Wower, J. Therapeutic RNA aptamers in clinical trials. Eur. J. Pharm. Sci 2013, 48, 259–271. [Google Scholar]
- Bouchard, P.R.; Hutabarat, R.M.; Thompson, K.M. Discovery and development of therapeutic aptamers. Annu. Rev. Pharmacol. Toxicol 2010, 50, 237–257. [Google Scholar]
- Keefe, A.D.; Cload, S.T. SELEX with modified nucleotides. Curr. Opin. Chem. Biol 2008, 12, 448–456. [Google Scholar]
- Warren, C.M.; Landgraf, R. Signalling through ERBB receptors: Multiple layers of diversity and control. Cell. Signal 2006, 18, 923–933. [Google Scholar]
- Yarden, Y.; Sliwkowski, M.X. Untangling the ErbB signalling network. Nat. Rev. Mol. Cell Biol 2001, 2, 127–137. [Google Scholar]
- Casalini, P.; Iorio, M.V.; Galmozzi, E.; Ménard, S. Role of HER receptors family in development and differentiation. J. Cell. Physiol 2004, 200, 343–350. [Google Scholar]
- Di Modugno, F.; Mottolese, M.; DeMonte, L.; Trono, P.; Balsamo, M.; Conidi, A.; Melucci, E.; Terrenato, I.; Belleudi, F.; Torrisi, M.R.; et al. The cooperation between hMena overexpression and HER2 signalling in breast cancer. PLoS One 2010, 5, e15852. [Google Scholar]
- Avraham, R.; Yarden, Y. Feedback regulation of EGFR signalling: Decision making by early and delayed loops. Nat. Rev. Mol. Cell Biol 2011, 12, 104–117. [Google Scholar]
- Buerger, C.; Nagel-Wolfrum, K.; Kunz, C.; Wittig, I.; Butz, K.; Hoppe-Seyler, F.; Groner, B. Sequence-specific peptide aptamers, interacting with the intracellular domain of the epidermal growth factor receptor, interfere with Stat3 activation and inhibit the growth of tumor cells. J. Coil. Chem 2003, 278, 37610–37621. [Google Scholar]
- Kunz, C.; Borghouts, C.; Buerger, C.; Groner, B. Peptide aptamers with binding specificity for the intracellular domain of the ErbB2 receptor interfere with AKT signalling and sensitize breast cancer cells to Taxol. Mol. Cancer Res 2006, 4, 983–998. [Google Scholar]
- Schwarze, S.R.; Hruska, K.A.; Dowdy, S.F. Protein transduction: unrestricted delivery into all cells? Trends Cell Biol 2000, 10, 290–295. [Google Scholar]
- Esposito, C.L.; Passaro, D.; Longobardo, I.; Condorelli, G.; Marotta, P.; Affuso, A.; de Franciscis, V.; Cerchia, L. A neutralizing RNA aptamer against EGFR causes selective apoptotic cell death. PLoS One 2011, 6, e24071. [Google Scholar]
- Clevers, H.; Nusse, R. Wnt/β-catenin signalling and disease. Cell 2012, 149, 1192–1205. [Google Scholar]
- MacDonald, B.T.; Tamai, K.; He, X. Wnt/beta-catenin signalling: Components, mechanisms, and diseases. Dev. Cell 2009, 17, 9–26. [Google Scholar]
- Angers, S.; Moon, R.T. Proximal events in Wnt signal transduction. Nat. Rev. Mol. Cell Biol 2009, 10, 468–477. [Google Scholar]
- Roberts, D.M.; Slep, K.C.; Peifer, M. It takes more than two to tango: Dishevelled polymerization and Wnt signalling. Nat. Struct. Mol. Biol. 2007, 14, 463–465. [Google Scholar]
- Huang, S.A.; Mishina, Y.M.; Liu, S.; Cheung, A.; Stegmeier, F.; Michaud, G.A.; Charlat, O.; Wiellette, E.; Zhang, Y.; Wiessner, S.; et al. Tankyrase inhibition stabilizes axin and antagonizes Wnt signalling. Nature 2009, 461, 614–620. [Google Scholar]
- Huber, A.H.; Nelson, W.J.; Weis, W.I. Three-dimensional structure of the armadillo repeat region of beta-catenin. Cell 1997, 90, 871–882. [Google Scholar]
- Lepourcelet, M.; Chen, Y.P.; France, D.S.; Wang, H.; Crews, P.; Petersen, F.; Bruseo, C.; Wood, A.W.; Shivdasani, R.A. Small-molecule antagonists of the oncogenic Tcf/beta-catenin protein complex. Cancer Cell 2004, 5, 91–102. [Google Scholar]
- Lee, S.K.; Park, M.W.; Yang, E.G.; Yu, J.; Jeong, S. An RNA aptamer that binds to the beta-catenin interaction domain of TCF-1 protein. Biochem. Biophys. Res. Commun 2005, 327, 294–299. [Google Scholar]
- Jeong, S.; Lee, H.K.; Kim, M.Y. Use of RNA aptamers for the modulation of cancer cell signalling. Methods Mol. Boil 2009, 542, 363–377. [Google Scholar]
- Drabsch, Y.; Dijke, P. TGF-β signalling and its role in cancer progression and metastasis. Cancer Metastasis Rev 2012, 31, 553–568. [Google Scholar]
- Dooley, S.; Dijke, P. TGF-β in progression of liver disease. Cell Tissue Res 2011, 347, 245–256. [Google Scholar]
- Seuntjens, E.; Umans, L.; Zwijsen, A.; Sampaolesi, M.; Verfaillie, C.M.; Huylebroeck, D. Transforming growth factor type beta and Smad family signalling in stem cell function. Cytokine Growth Factor Rev 2009, 20, 449–458. [Google Scholar]
- Massagué, J. TGFβ signalling in context. Nat. Rev. Mol. Cell Biol 2012, 13, 616–630. [Google Scholar]
- Allendorph, G.P.; Vale, W.W.; Choe, S. Structure of the ternary signalling complex of a TGF-beta superfamily member. Proc. Natl. Acad. Sci. USA 2006, 103, 7643–7648. [Google Scholar]
- Groppe, J.; Hinck, C.S.; Samavarchi-Tehrani, P.; Zubieta, C.; Schuermann, J.P.; Taylor, A.B.; Schwarz, P.M.; Wrana, J.L.; Hinck, A.P. Cooperative assembly of TGF-beta superfamily signalling complexes is mediated by two disparate mechanisms and distinct modes of receptor binding. Mol. Cell 2008, 29, 157–168. [Google Scholar]
- Nickel, J.; Sebald, W.; Groppe, J.C.; Mueller, T.D. Intricacies of BMP receptor assembly. Cytokine Growth Factor Rev 2009, 20, 367–377. [Google Scholar]
- Mu, Y.; Gudey, S.K.; Landström, M. Non-Smad signalling pathways. Cell Tissue Res 2011, 347, 11–20. [Google Scholar]
- Kang, J. S.; Liu, C.; Derynck, R. New regulatory mechanisms of TGF-beta receptor function. Trends Cell Biol 2009, 19, 385–394. [Google Scholar]
- McDonald, J.; Bayrak-Toydemir, P.; Pyeritz, R.E. Hereditary hemorrhagic telangiectasia: An overview of diagnosis, management, and pathogenesis. Genet. Med 2011, 13, 607–616. [Google Scholar]
- Castonguay, R.; Werner, E.D.; Matthews, R.G.; Presman, E.; Mulivor, A.W.; Solban, N.; Sako, D.; Pearsall, R.S.; Underwood, K.W.; Seehra, J.; et al. Soluble endoglin specifically binds bone morphogenetic proteins 9 and 10 via its orphan domain, inhibits blood vessel formation, and suppresses tumor growth. J. Biol. Chem 2011, 286, 30034–30046. [Google Scholar]
- Cai, J.; Pardali, E.; Sánchez-Duffhues, G.; ten Dijke, P. BMP signalling in vascular diseases. FEBS Lett 2012, 586, 1993–2002. [Google Scholar]
- López-Hernández, F.J.; Lopez-Novoa, J.M. Role of TGF-β in chronic kidney disease: An integration of tubular, glomerular and vascular effects. Cell Tissue Res 2011, 347, 141–154. [Google Scholar]
- Summers, K.M.; West, J.A.; Hattam, A.; Stark, D.; McGill, J.J.; West, M.J. Recent developments in the diagnosis of marfan syndrome and related disorders. Med. J. Aust 2012, 197, 494–497. [Google Scholar]
- Hoffjan, S. Genetic dissection of marfan syndrome and related connective tissue disorders: An update 2012. Mol. Syndromol 2012, 3, 47–58. [Google Scholar]
- Akhurst, R.J.; Hata, A. Targeting the TGFβ signalling pathway in disease. Nat. Rev. Drug Discov 2012, 11, 790–811. [Google Scholar]
- Raje, N.; Vallet, S. Sotatercept, a soluble activin receptor type 2A IgG-Fc fusion protein for the treatment of anemia and bone loss. Curr. Opin. Mol. Ther 2010, 12, 586–597. [Google Scholar]
- Bennett, C.F.; Swayze, E.E. RNA targeting therapeutics: Molecular mechanisms of antisense oligonucleotides as a therapeutic platform. Annu. Rev. Pharmacol. Toxicol 2010, 50, 259–293. [Google Scholar]
- Hong, C.C.; Yu, P.B. Applications of small molecule BMP inhibitors in physiology and disease. Cytokine Growth Factor Rev 2009, 20, 409–418. [Google Scholar]
- Li, L.; Orner, B.P.; Huang, T.; Hinck, A.P.; Kiessling, L.L. Peptide ligands that use a novel binding site to target both TGF-β receptors. Mol. BioSyst 2010, 6, 2392. [Google Scholar]
- Zhu, X.; Li, L.; Zou, L; Zhu, X.; Xian, G.; Li, H.; Tan, Y.; Xie, L. A novel aptamer targeting TGF-β receptor II inhibits transdifferentiation of human tenon’s fibroblasts into myofibroblast. Invest. Ophthalmol. Vis. Sci. 2012, 5, 6897–6903. [Google Scholar]
- Cui, Q.; Lim, S.K.; Zhao, B.; Hoffmann, F.M. Selective inhibition of TGF-beta responsive genes by Smad-interacting peptide aptamers from FoxH1, Lef1 and CBP. Oncogene 2005, 24, 3864–3874. [Google Scholar]
- Labbé, E.; Letamendia, A.; Attisano, L. Association of Smads with lymphoid enhancer binding factor 1/T cell-specific factor mediates cooperative signalling by the transforming growth factor-beta and wnt pathways. Proc. Natl. Acad. Sci. USA 2000, 97, 8358–8363. [Google Scholar]
- Goodman, R.H.; Smolik, S. CBP/p300 in cell growth, transformation, and development. Genes Dev 2000, 14, 1553–1577. [Google Scholar]
- Feng, X.H.; Zhang, Y.; Wu, R.Y.; Derynck, R. The tumor suppressor Smad4/DPC4 and transcriptional adaptor CBP/p300 are coactivators for smad3 in TGF-beta-induced transcriptional activation. Genes Dev 1998, 12, 2153–2163. [Google Scholar]
- Topper, J.N.; DiChiara, M.R.; Brown, J.D.; Williams, A.J.; Falb, D.; Collins, T.; Gimbrone, M.A. CREB binding protein is a required coactivator for Smad-dependent, transforming growth factor beta transcriptional responses in endothelial cells. Proc. Natl. Acad. Sci. USA 1998, 95, 9506–9511. [Google Scholar]
- Lim, S.K.; Hoffmann, F.M. Smad4 cooperates with lymphoid enhancer-binding factor 1/T cell-specific factor to increase c-myc expression in the absence of TGF-beta signalling. Proc. Natl. Acad. Sci. USA 2006, 103, 18580–18585. [Google Scholar]
- Zhao, B.M.; Hoffmann, F.M. Inhibition of transforming growth factor-beta1-induced signalling and epithelial-to-mesenchymal transition by the Smad-binding peptide aptamer Trx-SARA. Mol. Boil. Cell 2006, 17, 3819–3831. [Google Scholar]
- Tsukazaki, T.; Chiang, T.A.; Davison, A.F.; Attisano, L.; Wrana, J.L. SARA, a FYVE domain protein that recruits Smad2 to the TGFbeta receptor. Cell 1998, 95, 779–791. [Google Scholar]
- Shi, W.; Chang, C.; Nie, S.; Xie, S.; Wan, M.; Cao, X. Endofin acts as a Smad anchor for receptor activation in BMP signalling. J. Cell Sci 2007, 120, 1216–1224. [Google Scholar]
- Wu, G.; Chen, Y.G.; Ozdamar, B.; Gyuricza, C.A.; Chong, P.A.; Wrana, J.L.; Massague, J.; Shi, Y. Structural basis of Smad2 recognition by the Smad anchor for receptor activation. Science 2000, 287, 92–97. [Google Scholar]
- Huang, C.; Du, R.; Zhang, P.; Meng, H.; Jia, H.; Song, Y.; Li, M.; Zhang, Y.; Sun, S. Expression, purification, and functional characterization of recombinant PTD-SARA. Acta Biochim. Biophys. Sin 2011, 43, 110–177. [Google Scholar]
- Verschueren, K.; Remacle, J.E.; Collart, C.; Kraft, H.; Baker, B.S.; Tylzanowski, P.; Nelles, L.; Wuytens, G.; Su, M.T.; Bodmer, R.; et al. SIP1, a novel zinc finger/homeodomain repressor, interacts with Smad proteins and binds to 5′-CACCT sequences in candidate target genes. J. Boil. Chem 1999, 274, 20489–20498. [Google Scholar]
- Weng, Q.; Chen, Y.; Wang, H.; Xu, X.; Yang, B.; He, Q.; Shou, W.; Chen, Y.; Higashi, Y.; van den Berghe, V.; et al. Dual-mode modulation of Smad signalling by Smad-interacting protein Sip1 is required for myelination in the central nervous system. Neuron 2012, 73, 713–728. [Google Scholar]
- Conidi, A.; Cazzola, S.; Beets, K.; Coddens, K.; Collart, C.; Cornelis, F.; Cox, L.; Joke, D.; Dobreva, M.P.; Dries, R.; et al. Few Smad proteins and many Smad-interacting proteins yield multiple functions and action modes in TGFβ/BMP signalling in vivo. Cytokine Growth Factor Rev 2011, 22, 287–300. [Google Scholar]
- Remacle, J.E.; Kraft, H.; Lerchner, W.; Wuytens, G.; Collart, C.; Verschueren, K.; Smith, J.C.; Huylebroeck, D. New mode of DNA binding of multi-zinc finger transcription factors: DeltaEF1 family members bind with two hands to two target sites. EMBO J 1999, 18, 5073–5084. [Google Scholar]
- Van Grunsven, L.A.; Taelman, V.; Michiels, C.; Verstappen, G.; Souopgui, J.; Nichane, M.; Moens, E.; Opdecamp, K.; Vanhomwegen, J.; Kricha, S.; et al. XSip1 neuralizing activity involves the co-repressor CtBP and occurs through BMP dependent and independent mechanisms. Dev. Boil 2007, 306, 34–49. [Google Scholar]
- Verstappen, G.; van Grunsven, L.A.; Michiels, C.; van de Putte, T.; Souopgui, J.; van Damme, J.; Bellefroid, E.; Vandekerckhove, J.; Huylebroeck, D. Atypical Mowat-Wilson patient confirms the importance of the novel association between ZFHX1B/SIP1 and NuRD corepressor complex. Hum. Mol. Genet 2008, 17, 1175–1183. [Google Scholar]
- Van Grunsven, L.A.; Taelman, V.; Michiels, C.; Opdecamp, K.; Huylebroeck, D.; Bellefroid, E.J. deltaEF1 and SIP1 are differentially expressed and have overlapping activities during Xenopus embryogenesis. Dev. Dyn 2006, 235, 1491–1500. [Google Scholar]
- Bracken, C.P.; Gregory, P.A.; Kolesnikoff, N.; Bert, A.G.; Wang, J.; Shannon, M.F.; Goodall, G.J. A double-negative feedback loop between ZEB1-SIP1 and the microRNA-200 family regulates epithelial-mesenchymal transition. Cancer Res 2008, 68, 7846–7854. [Google Scholar]
- Park, S.; Gaur, A.B.; Lengyel, E.; Peter, M.E. The miR-200 family determines the epithelial phenotype of cancer cells by targeting the E-cadherin repressors ZEB1 and ZEB2. Genes Dev 2008, 22, 894–907. [Google Scholar]
- Gregory, P.A.; Bert, A.G.; Paterson, E.L.; Barry, S.C.; Tsykin, A.; Farshid, G.; Vadas, M.A.; Khew-Goodall, Y.; Goodall, G.J. The miR-200 family and miR-205 regulate epithelial to mesenchymal transition by targeting ZEB1 and SIP1. Nat. Cell Boil 2008, 10, 593–601. [Google Scholar]
- Christoffersen, N.R.; Silahtaroglu, A.; Orom, U.A.; Kauppinen, S.; Lund, A.H. MiR-200b mediates post-transcriptional repression of ZFHX1B. RNA 2007, 13, 1172–1178. [Google Scholar]
- Polytarchou, C.; Iliopoulos, D.; Struhl, K. An integrated transcriptional regulatory circuit that reinforces the breast cancer stem cell state. Proc. Natl. Acad. Sci. USA 2012, 109, 14470–14475. [Google Scholar]
- Lai, Z.C.; Rushton, E.; Bate, M.; Rubin, G.M. Loss of function of the Drosophila zfh-1 gene results in abnormal development of mesodermally derived tissues. Proc. Natl. Acad. Sci. USA 1993, 90, 4122–4126. [Google Scholar]
- Mowat, D.R.; Croaker, G.D.; Cass, D.T.; Kerr, B.A.; Chaitow, J.; Adès, L.C.; Chia, N.L.; Wilson, M.J. Hirschsprung disease, microcephaly, mental retardation, and characteristic facial features: Delineation of a new syndrome and identification of a locus at chromosome 2q22-q23. J. Med. Genet 1998, 35, 617–623. [Google Scholar]
- Wakamatsu, N.; Yamada, Y.; Yamada, K.; Ono, T.; Nomura, N.; Taniguchi, H.; Kitoh, H.; Mutoh, N.; Yamanaka, T.; Mushiake, K.; et al. Mutations in SIP1, encoding Smad interacting protein-1, cause a form of Hirschsprung disease. Nat. Genet 2001, 27, 369–370. [Google Scholar]
- Cacheux, V.; Dastot-Le Moal, F.; Kääriäinen, H.; Bondurand, N.; Rintala, R.; Boissier, B.; Wilson, M.; Mowat, D.; Goossens, M. Loss-of-function mutations in SIP1 Smad interacting protein 1 result in a syndromic Hirschsprung disease. Hum. Mol. Genet 2001, 10, 1503–1510. [Google Scholar]
- Garavelli, L.; Zollino, M.; Mainardi, P.C.; Gurrieri, F.; Rivieri, F.; Soli, F.; Verri, R.; Albertini, E.; Favaron, E.; Zignani, M.; et al. Mowat-Wilson syndrome: Facial phenotype changing with age: Study of 19 Italian patients and review of the literature. Am. J. Med. Genet. A 2009, 149A, 417–426. [Google Scholar]
- Conidi, A.; van den Berghe, V.; Stryjewska, A.; Xue, H.; Chen, Y.G.; Seuntjens, E.; Huylebroeck, D. Four amino acids within a tandem QxVx repeat in a predicted extended alpha-helix of the Smad-binding domain of Sip1 are necessary for binding to activated Smad proteins. PLoS One submitted for revision. 2013. [Google Scholar]
- Hata, A.; Seoane, J.; Lagna, G.; Montalvo, E.; Hemmati-Brivanlou, A.; Massague, J. OAZ uses distinct DNA- and protein-binding zinc fingers in separate BMP-Smad and Olf signalling pathways. Cell 2000, 100, 229–240. [Google Scholar]
- Roby, Y.A.; Bushey, M.A.; Cheng, L.E.; Kulaga, H.M.; Lee, S.J.; Reed, R.R. Zfp423/OAZ mutation reveals the importance of Olf/EBF transcription activity in olfactory neuronal maturation. J. Neurosci 2012, 32, 13679a–13688a. [Google Scholar]
- Lin, H.; Grosschedl, R. Failure of B-cell differentiation in mice lacking the transcription factor EBF. Nature 1995, 376, 263–267. [Google Scholar]
- Tsai, R.Y.; Reed, R.R. Cloning and functional characterization of Roaz, a zinc finger protein that interacts with O/E-1 to regulate gene expression: implications for olfactory neuronal development. J. Neurosci 1997, 17, 4159–4169. [Google Scholar]
- Tsai, R.Y.; Reed, R.R. Identification of DNA recognition sequences and protein interaction domains of the multiple-Zn-finger protein Roaz. Mol. Cell.Biol 1998, 18, 6447–6456. [Google Scholar]
- Doyle, A.J.; Doyle, J.J.; Bessling, S.L.; Maragh, S.; Lindsay, M.E.; Schepers, D.; Gillis, E.; Mortier, G.; Homfray, T.; Sauls, K.; et al. Mutations in the TGF-β repressor SKI cause Shprintzen-Goldberg syndrome with aortic aneurysm. Nat. Genet 2012, 44, 1249–1254. [Google Scholar]
- Luo, K.; Stroschein, S.L.; Wang, W.; Chen, D.; Martens, E.; Zhou, S.; Zhou, Q. The Ski oncoprotein interacts with the Smad proteins to repress TGFbeta signalling. Genes Dev 1999, 13, 2196–2206. [Google Scholar]
- Watanabe, Y.; Itoh, S.; Goto, T.; Ohnishi, E.; Inamitsu, M.; Itoh, F.; Satoh, K.; Wiercinska, E.; Yang, W.; Shi, L.; et al. TMEPAI, a transmembrane TGF-beta-inducible protein, sequesters Smad proteins from active participation in TGF-beta signalling. Mol. Cell 2010, 37, 123–134. [Google Scholar]
- Fleming, N.I.; Jorissen, R.N.; Mouradov, D.; Christie, M.; Sakthianandeswaren, A.; Palmieri, M.; Day, F.; Li, S.; Tsui, C.; Lipton, L.; et al. SMAD2, SMAD3 and SMAD4 mutations in colorectal cancer. Cancer Res 2013, 73, 725–735. [Google Scholar]
- Van de Laar, I.M.B.H.; Oldenburg, R.A.; Pals, G.; Roos-Hesselink, J.W.; de Graaf, B.M.; Verhagen, J.M.A.; Hoedemaekers, Y.M.; Willemsen, R.; Severijnen, L.; Venselaar, H.; et al. Mutations in SMAD3 cause a syndromic form of aortic aneurysms and dissections with early-onset osteoarthritis. Nat. Genet 2011, 43, 121–126. [Google Scholar]
- Jones, J.A.; Spinale, F.G.; Ikonomidis, J.S. Transforming growth factor-beta signalling in thoracic aortic aneurysm development: A paradox in pathogenesis. J. Vasc. Res 2009, 46, 119–137. [Google Scholar]
- Akhurst, R.J. The paradoxical TGF-β vasculopathies. Nat. Genet 2012, 44, 838–839. [Google Scholar]
- Guida, E.; Bisso, A.; Fenollar-Ferrer, C.; Napoli, M.; Anselmi, C.; Girardini, J.E.; Carloni, P.; del Sal, G. Peptide aptamers targeting mutant p53 induce apoptosis in tumor cells. Cancer Res 2008, 68, 6550–6558. [Google Scholar]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Aptamers | Antibodies | |
|---|---|---|
| Production | Chemical process carried out in vitro and at low cost standardized process | Requires, in most cases, an animal, increasing the cost activity; stability may vary from batch to batch |
| Targets | Any protein and any site, including toxin-specific targeting of mutant proteins, as well as of post-transcriptionally modified proteins or differentially expressed isoforms | Some epitopes difficult to target; toxins excluded as these are not tolerated by the animal; can target protein modifications, albeit often with low specificity and/or affinity |
| Selection/screening | Iterative in vitro selection procedure allows the obtaining of highly specific aptamers | Screening of large panels of (monoclonal) antibodies is fairly time-consuming and expensive |
| Modifications | Many chemical modifications available to increase stability or cellular uptake | Relatively few chemical modifications available |
| Immunogenicity | None reported that exceeds other antisense oligonucleotides or macromolecules | Proven immunogenicity, especially relevant to non-humanized antibodies |
| Scaffold | Structural element | Application | References | Remark |
|---|---|---|---|---|
| Thioredoxin A (TrxA) | 1 loop | yeast two-hybrid phage display mammalian cells | [18,25] | |
| Staphylococcal nuclease | 1 loop | functional screening | [21] | |
| Human Stefin A | 3 sites | yeast two-hybrid | [26] | |
| Green Fluorescent Protein (GFP) | loop randomization | visual screening | [20] | |
| FKBP12-(peptide)-FRB-GST | fusion of three domains | screen for kinase inhibitors | [27] | ligand-regulated peptide aptamers (LiRPs; rapamycin) |
| NCT number | Target (aptamer + adjuvant) | Conditions | Phase/s | Funded by |
|---|---|---|---|---|
| NCT00950638 | C5 complement (ARC1905 aptamer) | AMD | 1 | Industry |
| NCT00709527 | C5 complement (ARC1905 aptamer + Lucentis) | AMD | 1 | Industry |
| NCT01089517 | PDGF (E10030 aptamer + Lucentis) | AMD | 2 | Industry |
| NCT00569140 | PDGF (E10030 aptamer) | AMD | 1 | Industry |
| NCT00312351 | VEGF (Macugen) | Macular Degeneration | 4 | Industry |
| NCT00021736 | VEGF | AMD and Choroidal Neovascularization | 2/3 | Industry |
| NCT00215670 | VEGF (Macugen) | AMD | 2/3 | Industry |
| NCT00321997 | VEGF (Macugen) | AMD | 2/3 | Industry |
| NCT00040313 | VEGF (Macugen) | DME | 2 | Industry |
| NCT01487070 | VEGF (Macugen) | Proliferative Diabetic Retinopathy | 1 | Other |
| NCT01487044 | VEGF (Macugen) | DME | Other/Industry | |
| NCT00632242 | vWF (ARC1779) | Purpura Thrombotic, Thrombocytopenic von Willebrand Disease Type-2b | 2 | Industry |
| NCT01034410 | Nucleolin (AS1411) | Acute Myeloid Leukemia | 2 | Industry |
| NCT01191372 | BAX499 (ARC19499) | Hemophilia | 1/2 | Industry |
| NCT01194934 | CXCL12/SDF-1 (NOX-A12) | Hematopoietic Stem Cell Transplantation | 1 | Industry |
| NCT00976378 | CXCL12/SDF-1 (NOX-A12) | Autologous Stem Cell Transplantation | 1 | Industry/Other |
| NCT00976729 | CCL2/MCP-1 (NOX-E36) | Chronic Inflammatory Diseases Type 2 Diabetes Mellitus Systemic Lupus Erythematosus | 1 | Industry |
| NCT00113997 | REG1 | Healthy | 1 | NIH |
| NCT00056199 | von Hippel-Lindau Disease | 1 | NIH |
© 2013 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Conidi, A.; Van den Berghe, V.; Huylebroeck, D. Aptamers and Their Potential to Selectively Target Aspects of EGF, Wnt/β-Catenin and TGFβ–Smad Family Signaling. Int. J. Mol. Sci. 2013, 14, 6690-6719. https://doi.org/10.3390/ijms14046690
Conidi A, Van den Berghe V, Huylebroeck D. Aptamers and Their Potential to Selectively Target Aspects of EGF, Wnt/β-Catenin and TGFβ–Smad Family Signaling. International Journal of Molecular Sciences. 2013; 14(4):6690-6719. https://doi.org/10.3390/ijms14046690
Chicago/Turabian StyleConidi, Andrea, Veronique Van den Berghe, and Danny Huylebroeck. 2013. "Aptamers and Their Potential to Selectively Target Aspects of EGF, Wnt/β-Catenin and TGFβ–Smad Family Signaling" International Journal of Molecular Sciences 14, no. 4: 6690-6719. https://doi.org/10.3390/ijms14046690
APA StyleConidi, A., Van den Berghe, V., & Huylebroeck, D. (2013). Aptamers and Their Potential to Selectively Target Aspects of EGF, Wnt/β-Catenin and TGFβ–Smad Family Signaling. International Journal of Molecular Sciences, 14(4), 6690-6719. https://doi.org/10.3390/ijms14046690