Integration of Multiple Signaling Pathways Determines Differences in the Osteogenic Potential and Tissue Regeneration of Neural Crest-Derived and Mesoderm-Derived Calvarial Bones


{kind=link}
{kind=link}
Abstract
:1. Introduction
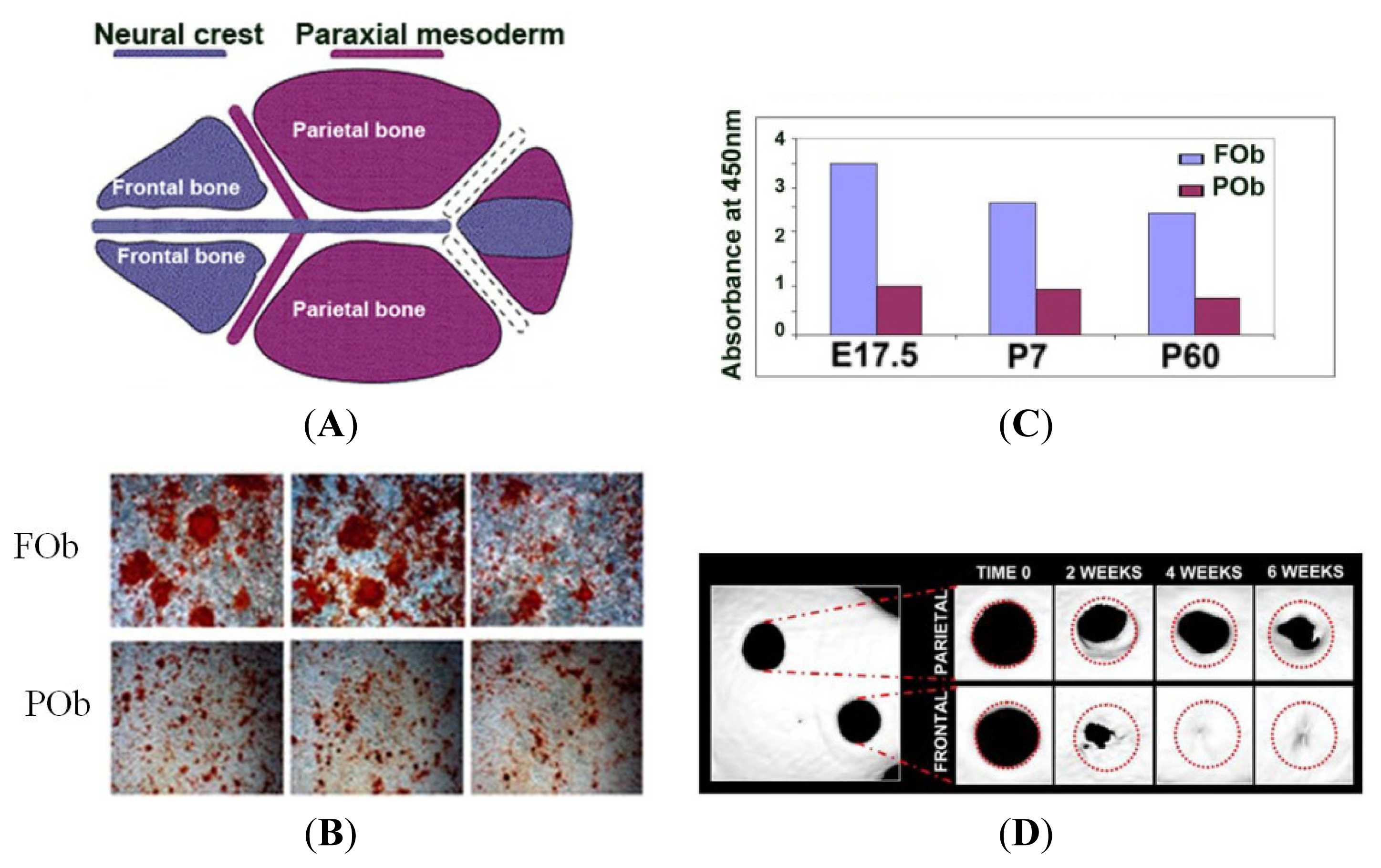
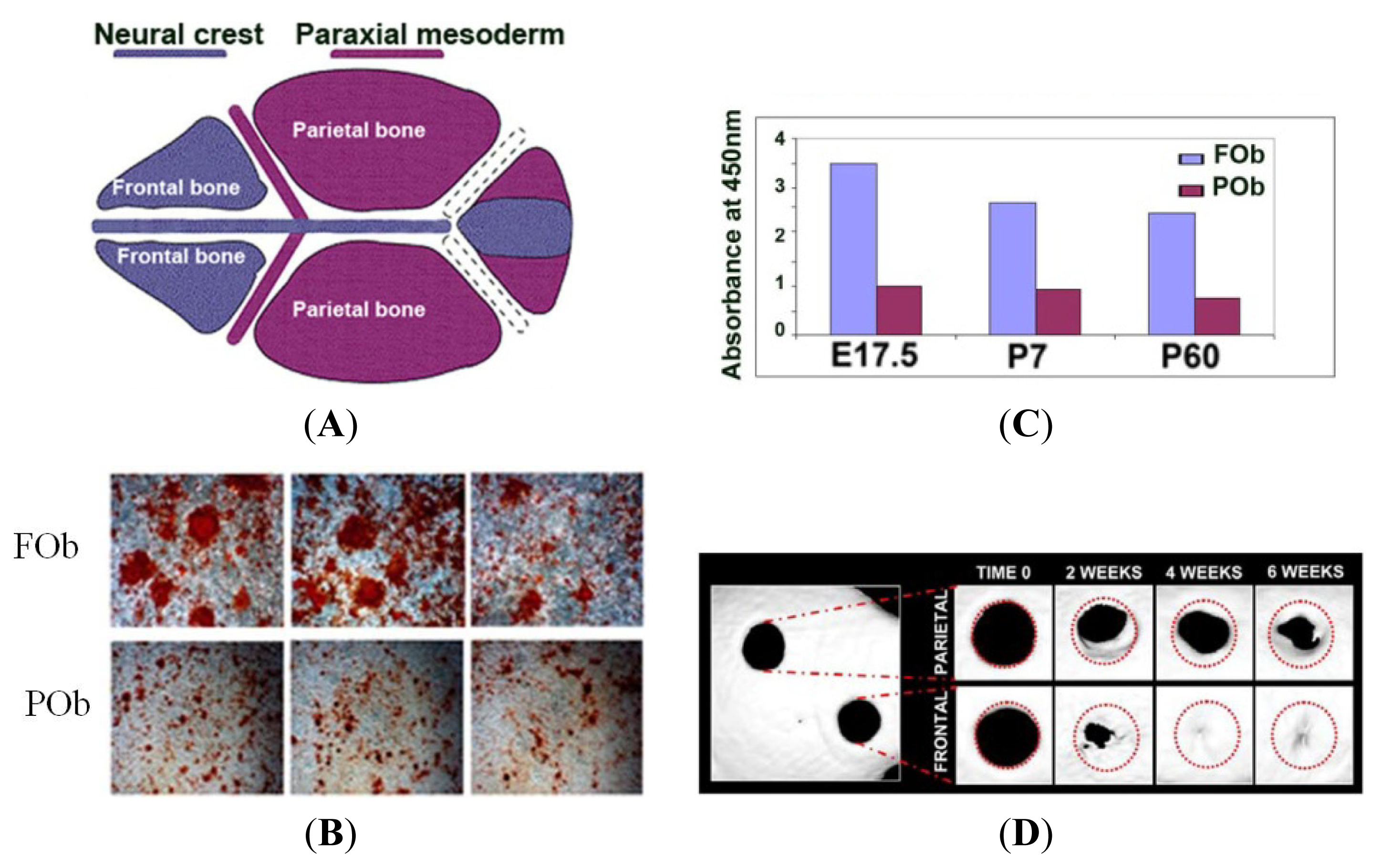
2. Development of the Mammalian Calvarium: A Model to Study the Integration of Multiple Signaling Pathways
3. The Neural Crest
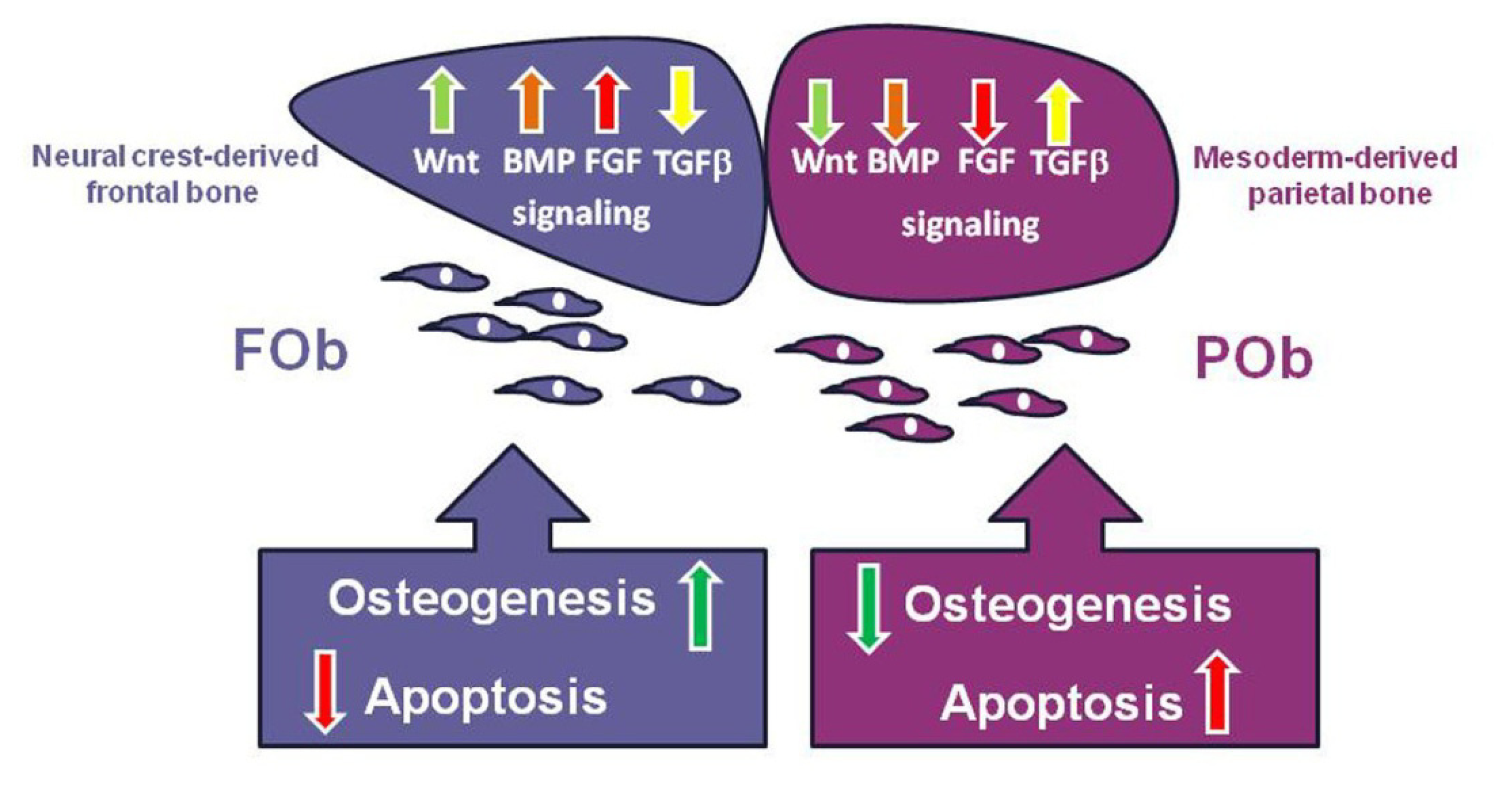
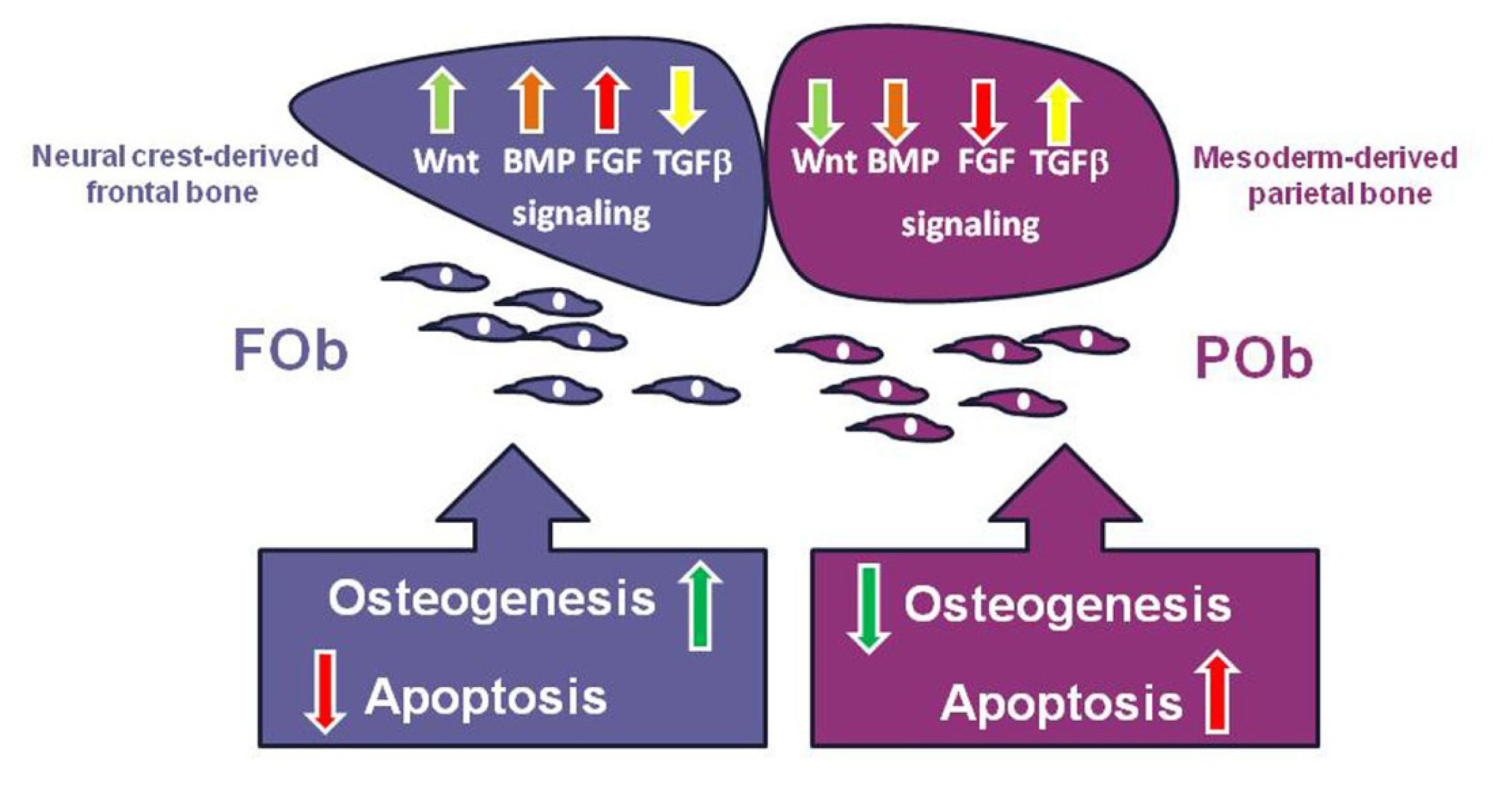
4. The Key Signaling Pathways and How They Achieve Coordination through Cross-Talk
4.1. The FGF Signaling Pathway
4.2. The Wnt Signaling Pathway
4.3. The BMP Signaling Pathway
4.4. The TGF-β Signaling Pathway
5. Integration of Multiple Pathways Controlling Apoptotic Activity in Osteoblasts
6. Conclusions
Acknowledgments
Conflict of Interest
References
- Morriss-Kay, G.M. Derivation of the mammalian skull vault. J. Anat 2001, 199, 143–151. [Google Scholar]
- Lian, J.B.; Stein, G.S.; Javed, A.; van Wijnen, A.J.; Stein, J.L.; Montecino, M.; Hassan, M.Q.; Gaur, T.; Lengner, C.J.; Young, D.W. Networks and hubs for the transcriptional control of osteoblastogenesis. Rev. Endocr. Metab. Disord 2006, 7, 1–16. [Google Scholar]
- Long, F. Building strong bones: Molecular regulation of the osteoblast lineage. Nat. Rev. Mol. Cell Biol 2012, 13, 27–38. [Google Scholar]
- Abzhanov, A.; Rodda, S.J.; McMahon, A.P.; Tabin, C.J. Regulation of skeletogenic differentiation in cranial dermal bone. Development 2007, 134, 3133–3144. [Google Scholar]
- Crane, J.F.; Trainor, P.A. Neural crest stem and progenitor cells. Annu. Rev. Cell Dev. Biol 2006, 22, 267–286. [Google Scholar]
- Noden, D.M. Interactions and fates of avian craniofacial mesenchyme. Development 1988, 103, 121–140. [Google Scholar]
- Le Lievre, C.S. Participation of neural crest-derived cells in the genesis of the skull in birds. J. Embryol. Exp. Morphol 1978, 47, 17–37. [Google Scholar]
- Couly, G.F.; Coltey, P.M.; Le Douarin, N.M. The triple origin of skull in higher vertebrates: A study in quail-chick chimeras. Development 1993, 117, 409–429. [Google Scholar]
- Jiang, X.; Iseki, S.; Maxson, R.E.; Sucov, H.M.; Morriss-Kay, G.M. Tissue origins and interactions in the mammalian skull vault. Dev. Biol 2002, 241, 106–116. [Google Scholar]
- Yoshida, T.; Vivatbutsiri, P.; Morriss-Kay, G.; Saga, Y.; Iseki, S. Cell lineage in mammalian craniofacial mesenchyme. Mech. Dev 2008, 125, 797–808. [Google Scholar]
- Quarto, N.; Wan, D.C.; Kwan, M.D.; Panetta, N.J.; Li, S.; Longaker, M.T. Origin matters: Differences in embryonic tissue origin and Wnt signaling determine the osteogenic potential and healing capacity of frontal and parietal calvarial bones. J. Bone Miner Res 2010, 25, 1680–1694. [Google Scholar]
- Le Douarin, N.M.; Dupin, E. The neural crest in vertebrate evolution. Curr. Opin. Genet. Dev 2012, 22, 381–389. [Google Scholar]
- Bronner, M.E.; LeDouarin, N.M. Development and evolution of the neural crest: An overview. Dev. Biol 2012, 366, 2–9. [Google Scholar]
- Stuhlmiller, T.J.; Garcia-Castro, M.I. Current perspectives of the signaling pathways directing neural crest induction. Cell Mol. Life Sci 2012, 69, 3715–3737. [Google Scholar]
- Donoghue, P.C.; Graham, A.; Kelsh, R.N. The origin and evolution of the neural crest. Bioessays 2008, 30, 530–541. [Google Scholar]
- Ishii, M.; Arias, A.C.; Liu, L.; Chen, Y.B.; Bronner, M.E.; Maxson, R.E. A stable cranial neural crest cell line from mouse. Stem Cells Dev 2012, 21, 3069–3080. [Google Scholar]
- Karsenty, G. Minireview: Transcriptional control of osteoblast differentiation. Endocrinology 2001, 142, 2731–2733. [Google Scholar]
- Karsenty, G.; Wagner, E.F. Reaching a genetic and molecular understanding of skeletal development. Dev. Cell 2002, 2, 389–406. [Google Scholar]
- Karsenty, G. Transcriptional control of skeletogenesis. Annu. Rev. Genomics Hum. Genet 2008, 9, 183–196. [Google Scholar]
- Muenke, M.; Schell, U. Fibroblast-growth-factor receptor mutations in human skeletal disorders. Trends Genet 1995, 11, 308–313. [Google Scholar]
- Naski, M.C.; Ornitz, D.M. FGF signaling in skeletal development. Front. Biosci 1998, 3, d781–d794. [Google Scholar]
- Ornitz, D.M.; Itoh, N. Fibroblast growth factors. Genome Biol. 2001. [Google Scholar] [CrossRef]
- Marie, P.J. Fibroblast growth factor signaling controlling bone formation: An update. Gene 2012, 498, 1–4. [Google Scholar]
- Mansukhani, A.; Bellosta, P.; Sahni, M.; Basilico, C. Signaling by fibroblast growth factors (FGF) and fibroblast growth factor receptor 2 (FGFR2)-activating mutations blocks mineralization and induces apoptosis in osteoblasts. J. Cell Biol 2000, 149, 1297–1308. [Google Scholar]
- Cowan, C.M.; Quarto, N.; Warren, S.M.; Salim, A.; Longaker, M.T. Age-related changes in the biomolecular mechanisms of calvarial osteoblast biology affect fibroblast growth factor-2 signaling and osteogenesis. J. Biol. Chem 2003, 278, 32005–32013. [Google Scholar]
- Fakhry, A.; Ratisoontorn, C.; Vedhachalam, C.; Salhab, I.; Koyama, E.; Leboy, P.; Pacifici, M.; Kirschner, R.E.; Nah, H.D. Effects of FGF-2/−9 in calvarial bone cell cultures: Differentiation stage-dependent mitogenic effect, inverse regulation of BMP-2 and noggin, and enhancement of osteogenic potential. Bone 2005, 36, 254–266. [Google Scholar]
- Quarto, N.; Longaker, M.T. FGF-2 inhibits osteogenesis in mouse adipose tissue-derived stromal cells and sustains their proliferative and osteogenic potential state. Tissue Eng 2006, 12, 1405–1418. [Google Scholar]
- Beenken, A.; Mohammadi, M. The FGF family: Biology, pathophysiology and therapy. Nat. Rev. Drug Discov 2009, 8, 235–253. [Google Scholar]
- Chaudhary, L.R.; Avioli, L.V. Extracellular-signal regulated kinase signaling pathway mediates downregulation of type I procollagen gene expression by FGF-2, PDGF-BB, and okadaic acid in osteoblastic cells. J. Cell Biochem 2000, 76, 354–359. [Google Scholar]
- Kim, H.J.; Kim, J.H.; Bae, S.C.; Choi, J.Y.; Kim, H.J.; Ryoo, H.M. The protein kinase C pathway plays a central role in the fibroblast growth factor-stimulated expression and transactivation activity of Runx2. J. Biol. Chem 2003, 278, 319–326. [Google Scholar]
- Deng, Z.L.; Sharff, K.A.; Tang, N.; Song, W.X.; Luo, J.; Luo, X.; Chen, J.; Bennett, E.; Reid, R.; Manning, D.; et al. Regulation of osteogenic differentiation during skeletal development. Front. Biosci 2008, 13, 2001–2021. [Google Scholar]
- Dailey, L.; Ambrosetti, D.; Mansukhani, A.; Basilico, C. Mechanisms underlying differential responses to FGF signaling. Cytokine Growth Factor Rev 2005, 16, 233–247. [Google Scholar]
- Iseki, S.; Wilkie, A.O.; Morriss-Kay, G.M. Fgfr1 and Fgfr2 have distinct differentiation- and proliferation-related roles in the developing mouse skull vault. Development 1999, 126, 5611–5620. [Google Scholar]
- Bonaventure, J.; El Ghouzzi, V. Molecular and cellular bases of syndromic craniosynostoses. Expert Rev. Mol. Med 2003, 5, 1–17. [Google Scholar]
- Marie, P.J.; Coffin, J.D.; Hurley, M.M. FGF and FGFR signaling in chondrodysplasias and craniosynostosis. J. Cell Biochem 2005, 96, 888–896. [Google Scholar]
- Tanimoto, Y.; Yokozeki, M.; Hiura, K.; Matsumoto, K.; Nakanishi, H.; Matsumoto, T.; Marie, P.J.; Moriyama, K. A soluble form of fibroblast growth factor receptor 2 (FGFR2) with S252W mutation acts as an efficient inhibitor for the enhanced osteoblastic differentiation caused by FGFR2 activation in Apert syndrome. J. Biol. Chem 2004, 279, 45926–45934. [Google Scholar]
- Park, J.; Park, O.J.; Yoon, W.J.; Kim, H.J.; Choi, K.Y.; Cho, T.J.; Ryoo, H.M. Functional characterization of a novel FGFR2 mutation, E731K, in craniosynostosis. J. Cell Biochem 2012, 113, 457–464. [Google Scholar]
- Ohbayashi, N.; Shibayama, M.; Kurotaki, Y.; Imanishi, M.; Fujimori, T.; Itoh, N.; Takada, S. FGF18 is required for normal cell proliferation and differentiation during osteogenesis and chondrogenesis. Genes Dev 2002, 16, 870–879. [Google Scholar]
- Liu, Z.; Xu, J.; Colvin, J.S.; Ornitz, D.M. Coordination of chondrogenesis and osteogenesis by fibroblast growth factor 18. Genes Dev 2002, 16, 859–869. [Google Scholar]
- Hung, I.H.; Yu, K.; Lavine, K.J.; Ornitz, D.M. FGF9 regulates early hypertrophic chondrocyte differentiation and skeletal vascularization in the developing stylopod. Dev. Biol 2007, 307, 300–313. [Google Scholar]
- Montero, A.; Okada, Y.; Tomita, M.; Ito, M.; Tsurukami, H.; Nakamura, T.; Doetschman, T.; Coffin, J.D.; Hurley, M.M. Disruption of the fibroblast growth factor-2 gene results in decreased bone mass and bone formation. J. Clin. Invest 2000, 105, 1085–1093. [Google Scholar]
- Moore, R.; Ferretti, P.; Copp, A.; Thorogood, P. Blocking endogenous FGF-2 activity prevents cranial osteogenesis. Dev. Biol 2002, 243, 99–114. [Google Scholar]
- Xiao, L.; Sobue, T.; Esliger, A.; Kronenberg, M.S.; Coffin, J.D.; Doetschman, T.; Hurley, M.M. Disruption of the Fgf2 gene activates the adipogenic and suppresses the osteogenic program in mesenchymal marrow stromal stem cells. Bone 2010, 47, 360–370. [Google Scholar]
- Liu, Z.; Lavine, K.J.; Hung, I.H.; Ornitz, D.M. FGF18 is required for early chondrocyte proliferation, hypertrophy and vascular invasion of the growth plate. Dev. Biol 2007, 302, 80–91. [Google Scholar]
- Quarto, N.; Behr, B.; Li, S.; Longaker, M.T. Differential FGF ligands and FGF receptors expression pattern in frontal and parietal calvarial bones. Cells Tissues Organs 2009, 190, 158–169. [Google Scholar]
- Behr, B.; Panetta, N.J.; Longaker, M.T.; Quarto, N. Different endogenous threshold levels of Fibroblast Growth Factor-ligands determine the healing potential of frontal and parietal bones. Bone 2010, 47, 281–294. [Google Scholar]
- Li, S.; Quarto, N.; Longaker, M.T. Activation of FGF signaling mediates proliferative and osteogenic differences between neural crest derived frontal and mesoderm parietal derived bone. PLoS One 2010, 5, e14033. [Google Scholar]
- Baroni, T.; Carinci, P.; Lilli, C.; Bellucci, C.; Aisa, M.C.; Scapoli, L.; Volinia, S.; Carinci, F.; Pezzetti, F.; Calvitti, M.; et al. P253R fibroblast growth factor receptor-2 mutation induces RUNX2 transcript variants and calvarial osteoblast differentiation. J. Cell Physiol 2005, 202, 524–535. [Google Scholar]
- Teplyuk, N.M.; Haupt, L.M.; Ling, L.; Dombrowski, C.; Mun, F.K.; Nathan, S.S.; Lian, J.B.; Stein, J.L.; Stein, G.S.; Cool, S.M.; van Wijnen, A.J. The osteogenic transcription factor Runx2 regulates components of the fibroblast growth factor/proteoglycan signaling axis in osteoblasts. J. Cell Biochem 2009, 107, 144–154. [Google Scholar]
- Marie, P.J.; Kaabeche, K.; Guenou, H. Roles of FGFR2 and twist in human craniosynostosis: Insights from genetic mutations in cranial osteoblasts. Front. Oral Biol 2008, 12, 144–159. [Google Scholar]
- Miraoui, H.; Marie, P.J. Pivotal role of Twist in skeletal biology and pathology. Gene 2010, 468, 1–7. [Google Scholar]
- Rice, D.P.; Aberg, T.; Chan, Y.; Tang, Z.; Kettunen, P.J.; Pakarinen, L.; Maxson, R.E.; Thesleff, I. Integration of FGF and TWIST in calvarial bone and suture development. Development 2000, 127, 1845–1855. [Google Scholar]
- Rice, R.; Rice, D.P.; Thesleff, I. Foxc1 integrates Fgf and Bmp signalling independently of twist or noggin during calvarial bone development. Dev. Dyn 2005, 233, 847–852. [Google Scholar]
- Kim, H.J.; Rice, D.P.; Kettunen, P.J.; Thesleff, I. FGF-, BMP- and Shh-mediated signalling pathways in the regulation of cranial suture morphogenesis and calvarial bone development. Development 1998, 125, 1241–1251. [Google Scholar]
- Logan, C.Y.; Nusse, R. The Wnt signaling pathway in development and disease. Annu. Rev. Cell Dev. Biol 2004, 20, 781–810. [Google Scholar]
- Clevers, H.; Nusse, R. Wnt/beta-catenin signaling and disease. Cell 2012, 149, 1192–1205. [Google Scholar]
- Hartmann, C. A Wnt canon orchestrating osteoblastogenesis. Trends Cell Biol 2006, 16, 151–158. [Google Scholar]
- Krishnan, V.; Bryant, H.U.; Macdougald, O.A. Regulation of bone mass by Wnt signaling. J. Clin. Invest 2006, 116, 1202–1209. [Google Scholar]
- Monroe, D.G.; McGee-Lawrence, M.E.; Oursler, M.J.; Westendorf, J.J. Update on Wnt signaling in bone cell biology and bone disease. Gene 2012, 492, 1–18. [Google Scholar]
- Davis, L.A.; Zur Nieden, N.I. Mesodermal fate decisions of a stem cell: The Wnt switch. Cell Mol. Life Sci 2008, 65, 2658–2674. [Google Scholar]
- Leupin, O.; Piters, E.; Halleux, C.; Hu, S.; Kramer, I.; Morvan, F.; Bouwmeester, T.; Schirle, M.; Bueno-Lozano, M.; et al. Bone overgrowth-associated mutations in the LRP4 gene impair sclerostin facilitator function. J. Biol. Chem 2011, 286, 19489–19500. [Google Scholar]
- Lee, J.S.; Ishimoto, A.; Yanagawa, S. Characterization of mouse dishevelled (Dvl) proteins in Wnt/Wingless signaling pathway. J. Biol. Chem 1999, 274, 21464–21470. [Google Scholar]
- Seto, E.S.; Bellen, H.J. The ins and outs of Wingless signaling. Trends Cell Biol 2004, 14, 45–53. [Google Scholar]
- Ikeda, S.; Kishida, S.; Yamamoto, H.; Murai, H.; Koyama, S.; Kikuchi, A. Axin, a negative regulator of the Wnt signaling pathway, forms a complex with GSK-3beta and beta-catenin and promotes GSK-3beta-dependent phosphorylation of beta-catenin. EMBO J 1998, 17, 1371–1384. [Google Scholar]
- He, T.C.; Sparks, A.B.; Rago, C.; Hermeking, H.; Zawel, L.; da Costa, L.T.; Morin, P.J.; Vogelstein, B.; Kinzler, K.W. Identification of c-MYC as a target of the APC pathway. Science 1998, 281, 1509–1512. [Google Scholar]
- Tetsu, O.; McCormick, F. Beta-catenin regulates expression of cyclin D1 in colon carcinoma cells. Nature 1999, 398, 422–426. [Google Scholar]
- Jho, E.H.; Zhang, T.; Domon, C.; Joo, C.K.; Freund, J.N.; Costantini, F. Wnt/beta-catenin/Tcf signaling induces the transcription of Axin2, a negative regulator of the signaling pathway. Mol. Cell Biol 2002, 22, 1172–1183. [Google Scholar]
- Hill, T.P.; Spater, D.; Taketo, M.M.; Birchmeier, W.; Hartmann, C. Canonical Wnt/beta-catenin signaling prevents osteoblasts from differentiating into chondrocytes. Dev. Cell 2005, 8, 727–738. [Google Scholar]
- Day, T.F.; Guo, X.; Garrett-Beal, L.; Yang, Y. Wnt/beta-catenin signaling in mesenchymal progenitors controls osteoblast and chondrocyte differentiation during vertebrate skeletogenesis. Dev. Cell 2005, 8, 739–750. [Google Scholar]
- Westendorf, J.J.; Kahler, R.A.; Schroeder, T.M. Wnt signaling in osteoblasts and bone diseases. Gene 2004, 341, 19–39. [Google Scholar]
- Glass, D.A., 2nd; Bialek, P.; Ahn, J.D.; Starbuck, M.; Patel, M.S.; Clevers, H.; Taketo, M.M.; Long, F.; McMahon, A.P.; Lang, R.A.; Karsenty, G. Canonical Wnt signaling in differentiated osteoblasts controls osteoclast differentiation. Dev. Cell 2005, 8, 751–764. [Google Scholar]
- Holmen, S.L.; Zylstra, C.R.; Mukherjee, A.; Sigler, R.E.; Faugere, M.C.; Bouxsein, M.L.; Deng, L.; Clemens, T.L.; Williams, B.O. Essential role of beta-catenin in postnatal bone acquisition. J. Biol. Chem 2005, 280, 21162–21168. [Google Scholar]
- Boyden, L.M.; Mao, J.; Belsky, J.; Mitzner, L.; Farhi, A.; Mitnick, M.A.; Wu, D.; Insogna, K.; Lifton, R.P. High bone density due to a mutation in LDL-receptor-related protein 5. N. Engl. J. Med 2002, 346, 1513–1521. [Google Scholar]
- Little, R.D.; Carulli, J.P.; del Mastro, R.G.; Dupuis, J.; Osborne, M.; Folz, C.; Manning, S.P.; Swain, P.M.; Zhao, S.C.; Eustace, B.; et al. A mutation in the LDL receptor-related protein 5 gene results in the autosomal dominant high-bone-mass trait. Am. J. Hum. Genet 2002, 70, 11–19. [Google Scholar]
- Ai, M.; Holmen, S.L.; van Hul, W.; Williams, B.O.; Warman, M.L. Reduced affinity to and inhibition by DKK1 form a common mechanism by which high bone mass-associated missense mutations in LRP5 affect canonical Wnt signaling. Mol. Cell Biol 2005, 25, 4946–4955. [Google Scholar]
- Semenov, M.V.; He, X. LRP5 mutations linked to high bone mass diseases cause reduced LRP5 binding and inhibition by SOST. J. Biol. Chem 2006, 281, 38276–38284. [Google Scholar]
- Qiu, W.; Andersen, T.E.; Bollerslev, J.; Mandrup, S.; Abdallah, B.M.; Kassem, M. Patients with high bone mass phenotype exhibit enhanced osteoblast differentiation and inhibition of adipogenesis of human mesenchymal stem cells. J. Bone Miner. Res 2007, 22, 1720–1731. [Google Scholar]
- Gong, Y.; Slee, R.B.; Fukai, N.; Rawadi, G.; Roman-Roman, S.; Reginato, A.M.; Wang, H.; Cundy, T.; Glorieux, F.H.; Lev, D.; et al. LDL receptor-related protein 5 (LRP5) affects bone accrual and eye development. Cell 2001, 107, 513–523. [Google Scholar]
- Fei, Y.; Xiao, L.; Doetschman, T.; Coffin, D.J.; Hurley, M.M. Fibroblast growth factor 2 stimulation of osteoblast differentiation and bone formation is mediated by modulation of the Wnt signaling pathway. J. Biol. Chem 2011, 286, 40575–40583. [Google Scholar]
- Reinhold, M.I.; Naski, M.C. Direct interactions of Runx2 and canonical Wnt signaling induce FGF18. J. Biol. Chem 2007, 282, 3653–3663. [Google Scholar]
- Sampath, T.K.; Muthukumaran, N.; Reddi, A.H. Isolation of osteogenin, an extracellular matrix-associated, bone-inductive protein, by heparin affinity chromatography. Proc. Natl. Acad. Sci. USA 1987, 84, 7109–7113. [Google Scholar]
- Wang, E.A.; Rosen, V.; Cordes, P.; Hewick, R.M.; Kriz, M.J.; Luxenberg, D.P.; Sibley, B.S.; Wozney, J.M. Purification and characterization of other distinct bone-inducing factors. Proc. Natl. Acad. Sci. USA 1988, 85, 9484–9488. [Google Scholar]
- Celeste, A.J.; Iannazzi, J.A.; Taylor, R.C.; Hewick, R.M.; Rosen, V.; Wang, E.A.; Wozney, J.M. Identification of transforming growth factor beta family members present in bone-inductive protein purified from bovine bone. Proc. Natl. Acad. Sci. USA 1990, 87, 9843–9847. [Google Scholar]
- Feng, X.H.; Derynck, R. Specificity and versatility in tgf-beta signaling through Smads. Annu. Rev. Cell Dev. Biol 2005, 21, 659–693. [Google Scholar]
- Nishimura, R.; Hata, K.; Matsubara, T.; Wakabayashi, M.; Yoneda, T. Regulation of bone and cartilage development by network between BMP signalling and transcription factors. J. Biochem 2012, 151, 247–254. [Google Scholar]
- Fujii, M.; Takeda, K.; Imamura, T.; Aoki, H.; Sampath, T.K.; Enomoto, S.; Kawabata, M.; Kato, M.; Ichijo, H.; Miyazono, K. Roles of bone morphogenetic protein type I receptors and Smad proteins in osteoblast and chondroblast differentiation. Mol. Biol. Cell 1999, 10, 3801–3813. [Google Scholar]
- Gori, F.; Thomas, T.; Hicok, K.C.; Spelsberg, T.C.; Riggs, B.L. Differentiation of human marrow stromal precursor cells: Bone morphogenetic protein-2 increases OSF2/CBFA1, enhances osteoblast commitment, and inhibits late adipocyte maturation. J. Bone Miner. Res 1999, 14, 1522–1535. [Google Scholar]
- Javed, A.; Bae, J.S.; Afzal, F.; Gutierrez, S.; Pratap, J.; Zaidi, S.K.; Lou, Y.; van Wijnen, A.J.; Stein, J.L.; Stein, G.S.; Lian, J.B. Structural coupling of Smad and Runx2 for execution of the BMP2 osteogenic signal. J. Biol. Chem 2008, 283, 8412–8422. [Google Scholar]
- Javed, A.; Afzal, F.; Bae, J.S.; Gutierrez, S.; Zaidi, K.; Pratap, J.; van Wijnen, A.J.; Stein, J.L.; Stein, G.S.; Lian, J.B. Specific residues of RUNX2 are obligatory for formation of BMP2-induced RUNX2-SMAD complex to promote osteoblast differentiation. Cells Tissues Organs 2009, 189, 133–137. [Google Scholar]
- Kokabu, S.; Gamer, L.; Cox, K.; Lowery, J.; Tsuji, K.; Raz, R.; Economides, A.; Katagiri, T.; Rosen, V. BMP3 suppresses osteoblast differentiation of bone marrow stromal cells via interaction with Acvr2b. Mol. Endocrinol 2012, 26, 87–94. [Google Scholar]
- Warren, S.M.; Brunet, L.J.; Harland, R.M.; Economides, A.N.; Longaker, M.T. The BMP antagonist noggin regulates cranial suture fusion. Nature 2003, 422, 625–629. [Google Scholar]
- Reinhold, M.I.; Abe, M.; Kapadia, R.M.; Liao, Z.; Naski, M.C. FGF18 represses noggin expression and is induced by calcineurin. J. Biol. Chem 2004, 279, 38209–38219. [Google Scholar]
- Choi, K.Y.; Kim, H.J.; Lee, M.H.; Kwon, T.G.; Nah, H.D.; Furuichi, T.; Komori, T.; Nam, S.H.; Kim, Y.J.; Kim, H.J.; Ryoo, H.M. Runx2 regulates FGF2-induced Bmp2 expression during cranial bone development. Dev. Dyn 2005, 233, 115–121. [Google Scholar]
- Chen, Y.; Whetstone, H.C.; Youn, A.; Nadesan, P.; Chow, E.C.; Lin, A.C.; Alman, B.A. Beta-catenin signaling pathway is crucial for bone morphogenetic protein 2 to induce new bone formation. J. Biol. Chem 2007, 282, 526–533. [Google Scholar]
- Lin, G.L.; Hankenson, K.D. Integration of BMP, Wnt, and notch signaling pathways in osteoblast differentiation. J. Cell Biochem 2011, 112, 3491–3501. [Google Scholar]
- Mundlos, S.; Otto, F.; Mundlos, C.; Mulliken, J.B.; Aylsworth, A.S.; Albright, S.; Lindhout, D.; Cole, W.G.; Henn, W.; Knoll, J.H.; et al. Mutations involving the transcription factor CBFA1 cause cleidocranial dysplasia. Cell 1997, 89, 773–779. [Google Scholar]
- Lee, B.; Thirunavukkarasu, K.; Zhou, L.; Pastore, L.; Baldini, A.; Hecht, J.; Geoffroy, V.; Ducy, P.; Karsenty, G. Missense mutations abolishing DNA binding of the osteoblast-specific transcription factor OSF2/CBFA1 in cleidocranial dysplasia. Nat. Genet 1997, 16, 307–310. [Google Scholar]
- Derynck, R.; Akhurst, R.J. Differentiation plasticity regulated by TGF-beta family proteins in development and disease. Nat. Cell Biol 2007, 9, 1000–1004. [Google Scholar]
- Roelen, B.A.; Dijke, P. Controlling mesenchymal stem cell differentiation by TGFBeta family members. J. Orthop. Sci 2003, 8, 740–748. [Google Scholar]
- Maeda, S.; Hayashi, M.; Komiya, S.; Imamura, T.; Miyazono, K. Endogenous TGF-beta signaling suppresses maturation of osteoblastic mesenchymal cells. EMBO J 2004, 23, 552–563. [Google Scholar]
- Tang, Y.; Wu, X.; Lei, W.; Pang, L.; Wan, C.; Shi, Z.; Zhao, L.; Nagy, T.R.; Peng, X.; Hu, J.; et al. TGF-beta1-induced migration of bone mesenchymal stem cells couples bone resorption with formation. Nat. Med 2009, 15, 757–765. [Google Scholar]
- Wrana, J.L.; Maeno, M.; Hawrylyshyn, B.; Yao, K.L.; Domenicucci, C.; Sodek, J. Differential effects of transforming growth factor-beta on the synthesis of extracellular matrix proteins by normal fetal rat calvarial bone cell populations. J. Cell Biol 1988, 106, 915–924. [Google Scholar]
- Chen, D.; Zhao, M.; Mundy, G.R. Bone morphogenetic proteins. Growth Factors 2004, 22, 233–241. [Google Scholar]
- Quarto, N.; Leonard, B.; Li, S.; Marchand, M.; Anderson, E.; Behr, B.; Francke, U.; Reijo-Pera, R.; Chiao, E.; Longaker, M.T. Skeletogenic phenotype of human Marfan embryonic stem cells faithfully phenocopied by patient-specific induced-pluripotent stem cells. Proc. Natl. Acad. Sci. USA 2012, 109, 215–220. [Google Scholar]
- Quarto, N.; Li, S.; Renda, A.; Longaker, M.T. Exogenous activation of BMP-2 signaling overcomes TGFbeta-mediated inhibition of osteogenesis in Marfan embryonic stem cells and Marfan patient-specific induced pluripotent stem cells. Stem Cells 2012, 30, 2709–2719. [Google Scholar]
- Ten Dijke, P.; Arthur, H.M. Extracellular control of TGFbeta signalling in vascular development and disease. Nat. Rev. Mol. Cell Biol 2007, 8, 857–869. [Google Scholar]
- Ramirez, F.; Sakai, L.Y.; Dietz, H.C.; Rifkin, D.B. Fibrillin microfibrils: Multipurpose extracellular networks in organismal physiology. Physiol. Genomics 2004, 19, 151–154. [Google Scholar]
- Bolar, N.; van Laer, L.; Loeys, B.L. Marfan syndrome: From gene to therapy. Curr. Opin. Pediatr 2012, 24, 498–504. [Google Scholar]
- Manolagas, S.C. Cell number versus cell vigor—What really matters to a regenerating skeleton? Endocrinology 1999, 140, 4377–4381. [Google Scholar]
- Palumbo, C.; Ferretti, M.; de Pol, A. Apoptosis during intramembranous ossification. J. Anat 2003, 203, 589–598. [Google Scholar]
- Xing, L.; Boyce, B.F. Regulation of apoptosis in osteoclasts and osteoblastic cells. Biochem. Biophys. Res. Commun 2005, 328, 709–720. [Google Scholar]
- Hunziker, A.; Jensen, M.H.; Krishna, S. Stress-specific response of the p53-Mdm2 feedback loop. BMC Syst. Biol 2010, 4, 94. [Google Scholar]
- Kim, D.J.; Koh, J.M.; Lee, O.; Kim, N.J.; Lee, Y.S.; Kim, Y.S.; Park, J.Y.; Lee, K.U.; Kim, G.S. Homocysteine enhances apoptosis in human bone marrow stromal cells. Bone 2006, 39, 582–590. [Google Scholar]
- Sheng, M.H.; Lau, K.H.; Mohan, S.; Baylink, D.J.; Wergedal, J.E. High osteoblastic activity in C3H/HeJ mice compared to C57BL/6J mice is associated with low apoptosis in C3H/HeJ osteoblasts. Calcif. Tissue Int 2006, 78, 293–301. [Google Scholar]
- Rice, D.P.; Kim, H.J.; Thesleff, I. Apoptosis in murine calvarial bone and suture development. Eur. J. Oral Sci 1999, 107, 265–275. [Google Scholar]
- Alejandre Alcazar, M.A.; Morty, R.E.; Lendzian, L.; Vohlen, C.; Oestreicher, I.; Plank, C.; Schneider, H.; Dotsch, J. Inhibition of TGF-beta signaling and decreased apoptosis in IUGR-associated lung disease in rats. PLoS One 2011, 6, e26371. [Google Scholar]
- Heger, J.; Warga, B.; Meyering, B.; Abdallah, Y.; Schluter, K.D.; Piper, H.M.; Euler, G. TGFbeta receptor activation enhances cardiac apoptosis via SMAD activation and concomitant NO release. J. Cell Physiol 2011, 226, 2683–2690. [Google Scholar]
- Kimura, N.; Matsuo, R.; Shibuya, H.; Nakashima, K.; Taga, T. BMP2-induced apoptosis is mediated by activation of the TAK1-p38 kinase pathway that is negatively regulated by Smad6. J. Biol. Chem 2000, 275, 17647–17652. [Google Scholar]
- Chen, S.; Guttridge, D.C.; You, Z.; Zhang, Z.; Fribley, A.; Mayo, M.W.; Kitajewski, J.; Wang, C.Y. Wnt-1 signaling inhibits apoptosis by activating beta-catenin/T cell factor-mediated transcription. J. Cell Biol 2001, 152, 87–96. [Google Scholar]
- Liu, Z.; Shen, J.; Pu, K.; Katus, H.A.; Ploger, F.; Tiefenbacher, C.P.; Chen, X.; Braun, T. GDF5 and BMP2 inhibit apoptosis via activation of BMPR2 and subsequent stabilization of XIAP. Biochim. Biophys. Acta 2009, 1793, 1819–1827. [Google Scholar]
- Shimizu, T.; Kayamori, T.; Murayama, C.; Miyamoto, A. Bone morphogenetic protein (BMP)-4 and BMP-7 suppress granulosa cell apoptosis via different pathways: BMP-4 via PI3K/PDK-1/Akt and BMP-7 via PI3K/PDK-1/PKC. Biochem. Biophys. Res. Commun 2012, 417, 869–873. [Google Scholar]
- Macias, D.; Ganan, Y.; Sampath, T.K.; Piedra, M.E.; Ros, M.A.; Hurle, J.M. Role of BMP-2 and OP-1 (BMP-7) in programmed cell death and skeletogenesis during chick limb development. Development 1997, 124, 1109–1117. [Google Scholar]
- Bodine, P.V. Wnt signaling control of bone cell apoptosis. Cell Res 2008, 18, 248–253. [Google Scholar]
- Satoh, Y.; Ishiguro, Y.; Sakuraba, H.; Kawaguchi, S.; Hiraga, H.; Fukuda, S.; Nakane, A. Cyclosporine regulates intestinal epithelial apoptosis via TGF-beta-related signaling. Am. J. Physiol. Gastrointest Liver Physiol 2009, 297, G514–G519. [Google Scholar]
- Almeida, M.; Han, L.; Bellido, T.; Manolagas, S.C.; Kousteni, S. Wnt proteins prevent apoptosis of both uncommitted osteoblast progenitors and differentiated osteoblasts by beta-catenin-dependent and -independent signaling cascades involving Src/ERK and phosphatidylinositol 3-kinase/AKT. J. Biol. Chem 2005, 280, 41342–41351. [Google Scholar]
- Li, S.; Meyer, N.P.; Quarto, N.; Longaker, M.T. Integration of multiple signaling regulates through apoptosis the differential osteogenic potential of neural crest-derived and mesoderm derived osteoblasts. PLoS One 2012, in press. [Google Scholar]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Senarath-Yapa, K.; Li, S.; Meyer, N.P.; Longaker, M.T.; Quarto, N. Integration of Multiple Signaling Pathways Determines Differences in the Osteogenic Potential and Tissue Regeneration of Neural Crest-Derived and Mesoderm-Derived Calvarial Bones. Int. J. Mol. Sci. 2013, 14, 5978-5997. https://doi.org/10.3390/ijms14035978
Senarath-Yapa K, Li S, Meyer NP, Longaker MT, Quarto N. Integration of Multiple Signaling Pathways Determines Differences in the Osteogenic Potential and Tissue Regeneration of Neural Crest-Derived and Mesoderm-Derived Calvarial Bones. International Journal of Molecular Sciences. 2013; 14(3):5978-5997. https://doi.org/10.3390/ijms14035978
Chicago/Turabian StyleSenarath-Yapa, Kshemendra, Shuli Li, Nathaniel P. Meyer, Michael T. Longaker, and Natalina Quarto. 2013. "Integration of Multiple Signaling Pathways Determines Differences in the Osteogenic Potential and Tissue Regeneration of Neural Crest-Derived and Mesoderm-Derived Calvarial Bones" International Journal of Molecular Sciences 14, no. 3: 5978-5997. https://doi.org/10.3390/ijms14035978
APA StyleSenarath-Yapa, K., Li, S., Meyer, N. P., Longaker, M. T., & Quarto, N. (2013). Integration of Multiple Signaling Pathways Determines Differences in the Osteogenic Potential and Tissue Regeneration of Neural Crest-Derived and Mesoderm-Derived Calvarial Bones. International Journal of Molecular Sciences, 14(3), 5978-5997. https://doi.org/10.3390/ijms14035978