Chemical Inhibitors and microRNAs (miRNA) Targeting the Mammalian Target of Rapamycin (mTOR) Pathway: Potential for Novel Anticancer Therapeutics

Abstract
:
1. Introduction
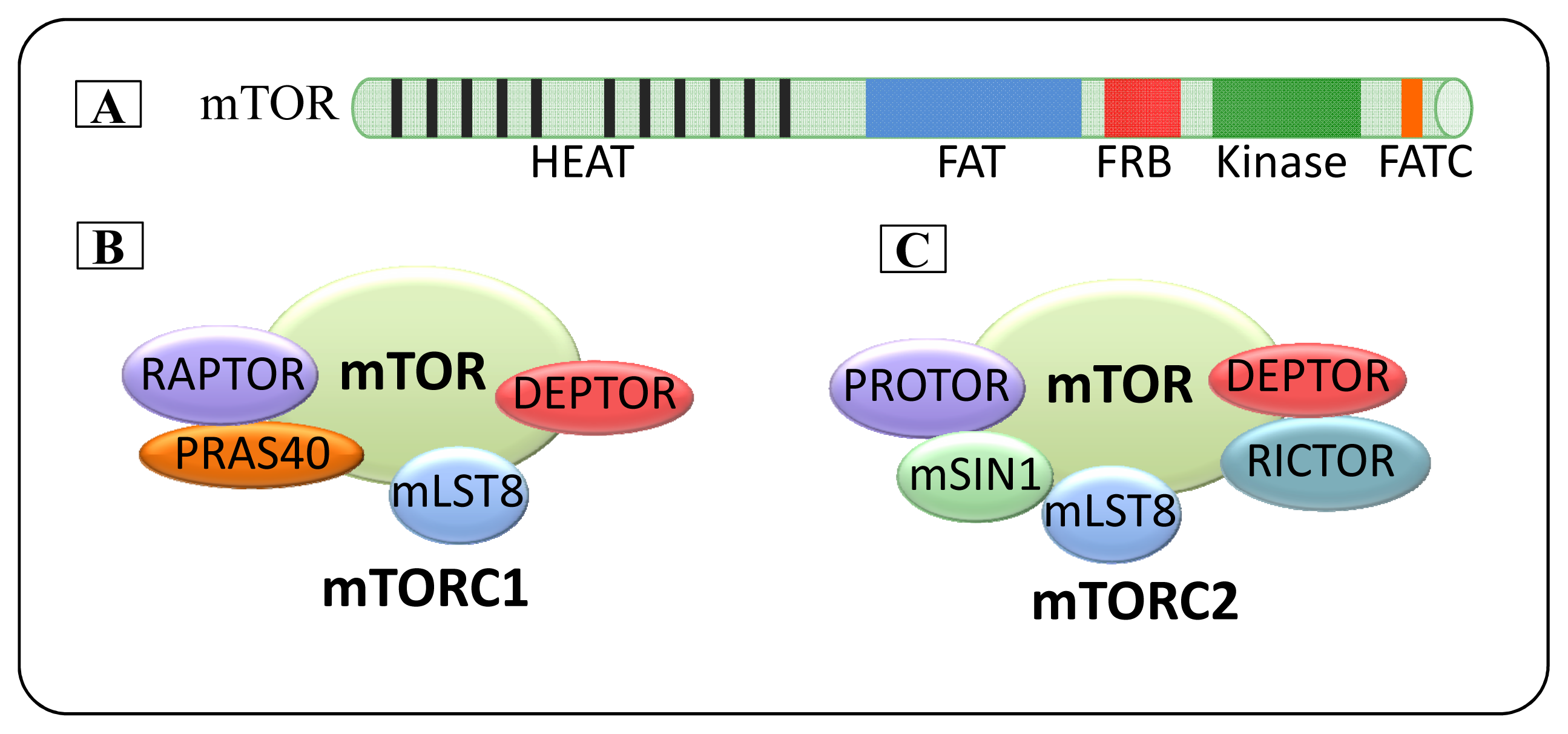
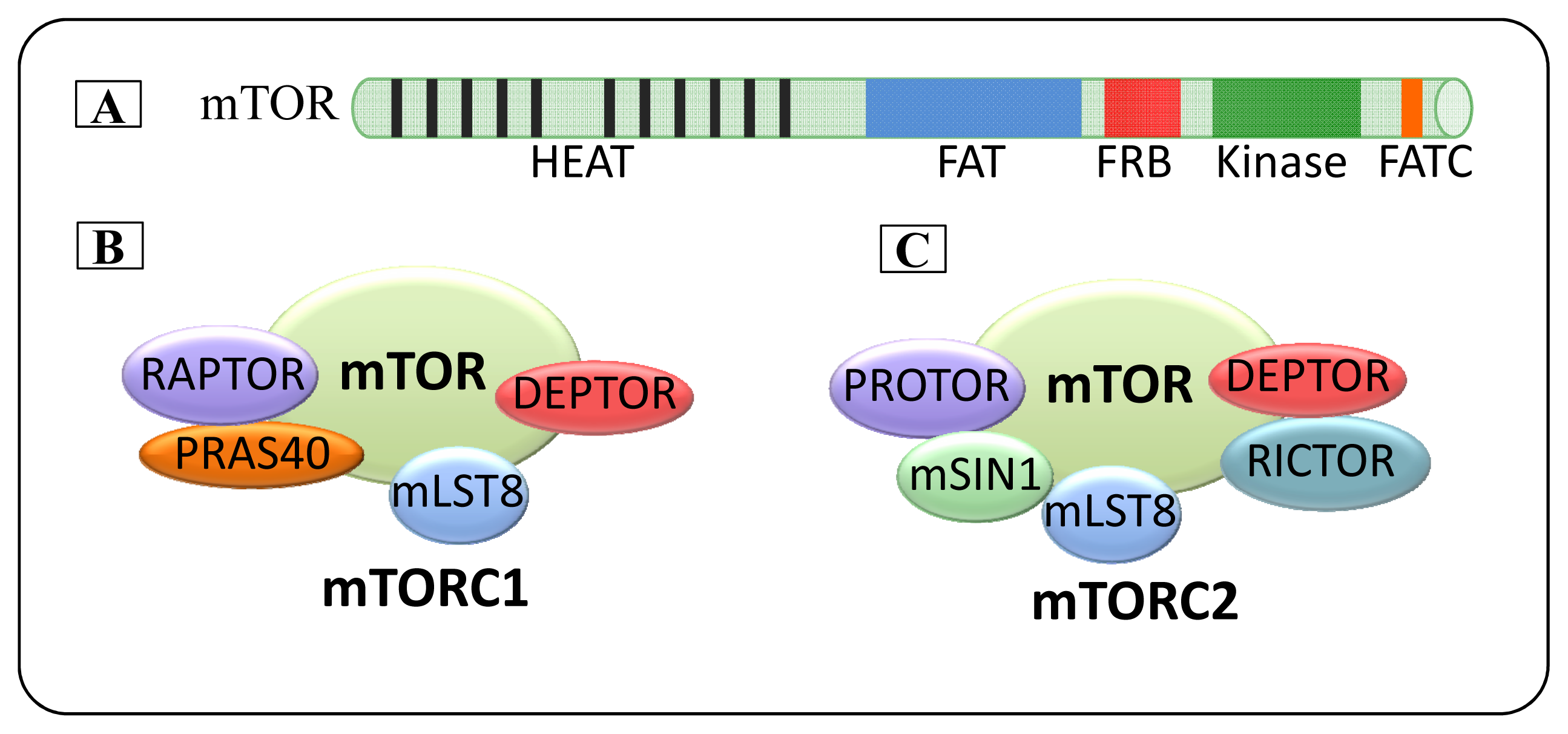
2. mTOR
2.1. mTOR Complexes
2.1.1. mTORC1
2.1.2. mTORC2
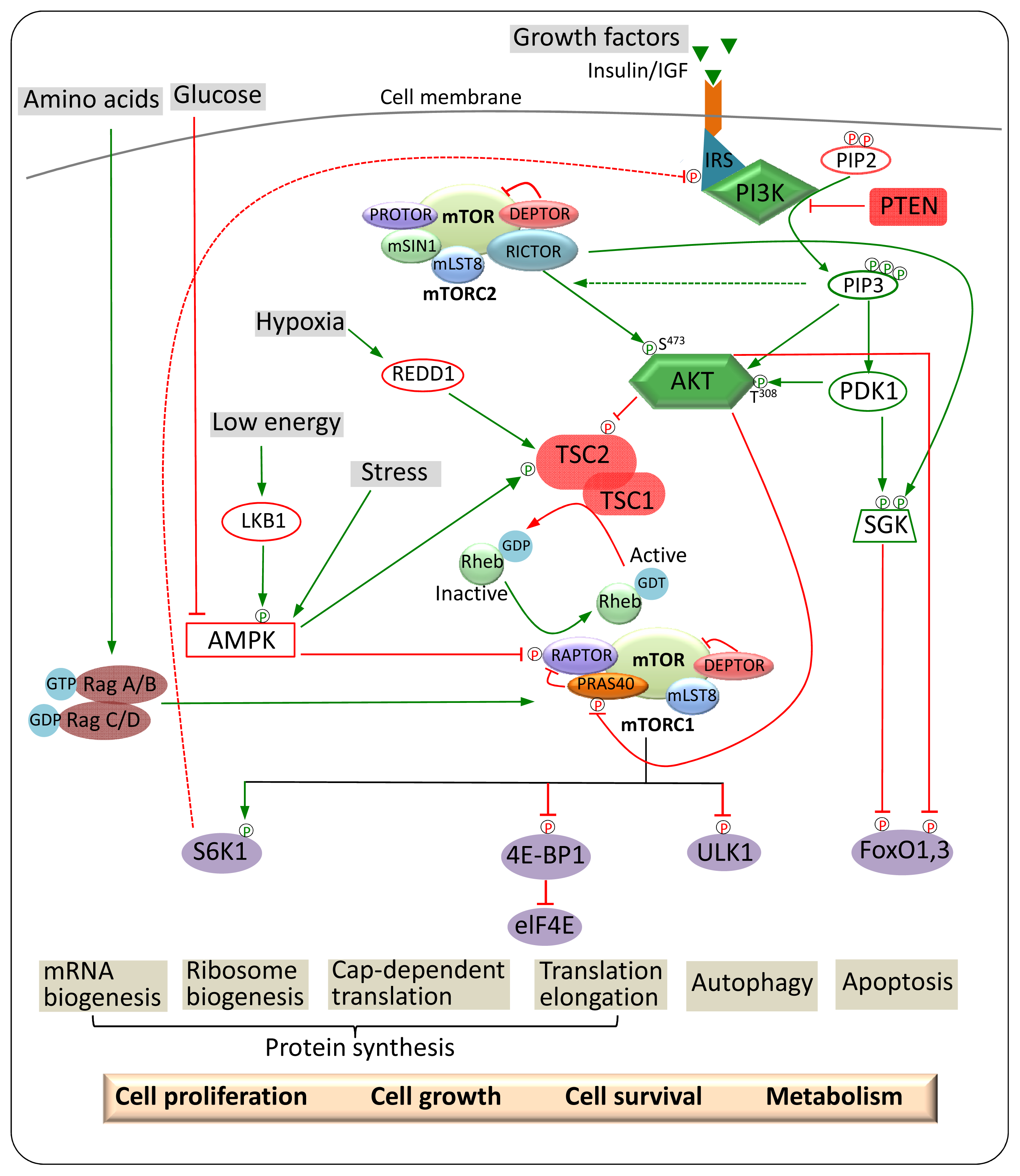
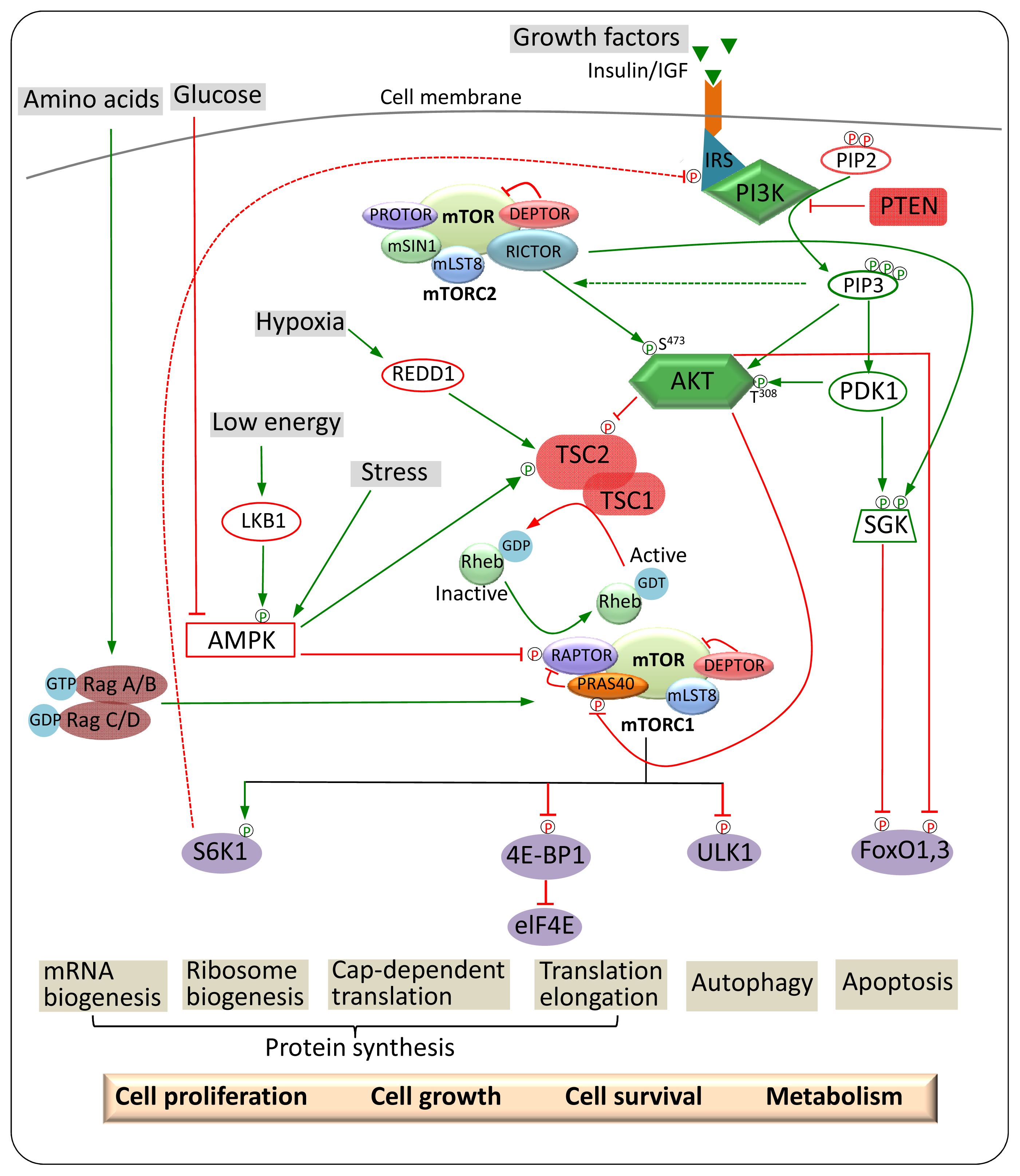
3. mTOR Regulation and Signalling
3.1. Upstream Regulators of mTORC1
3.1.1. Nutrients
3.1.2. Growth Factors
3.1.3. Cellular Energy Status and Stress
3.2. Upstream and Downstream Regulators of mTORC2
4. mTOR Signalling Pathways in Cancer
4.1. Downstream Targets of mTORC1 in Cancer
4.1.1. 4E-BPs
4.1.2. S6 Kinases
4.2. Downstream Targets of mTORC2 in Cancer
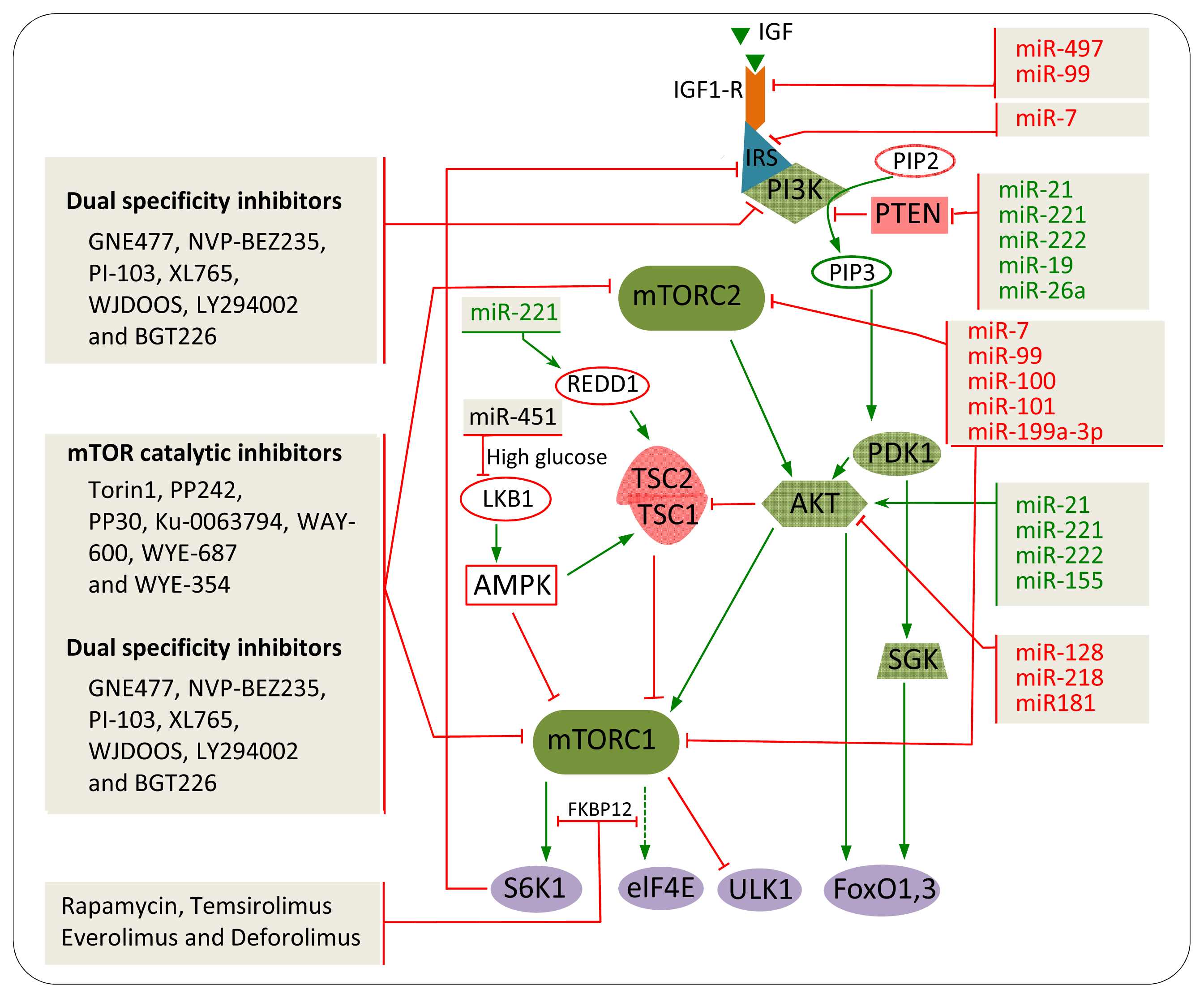
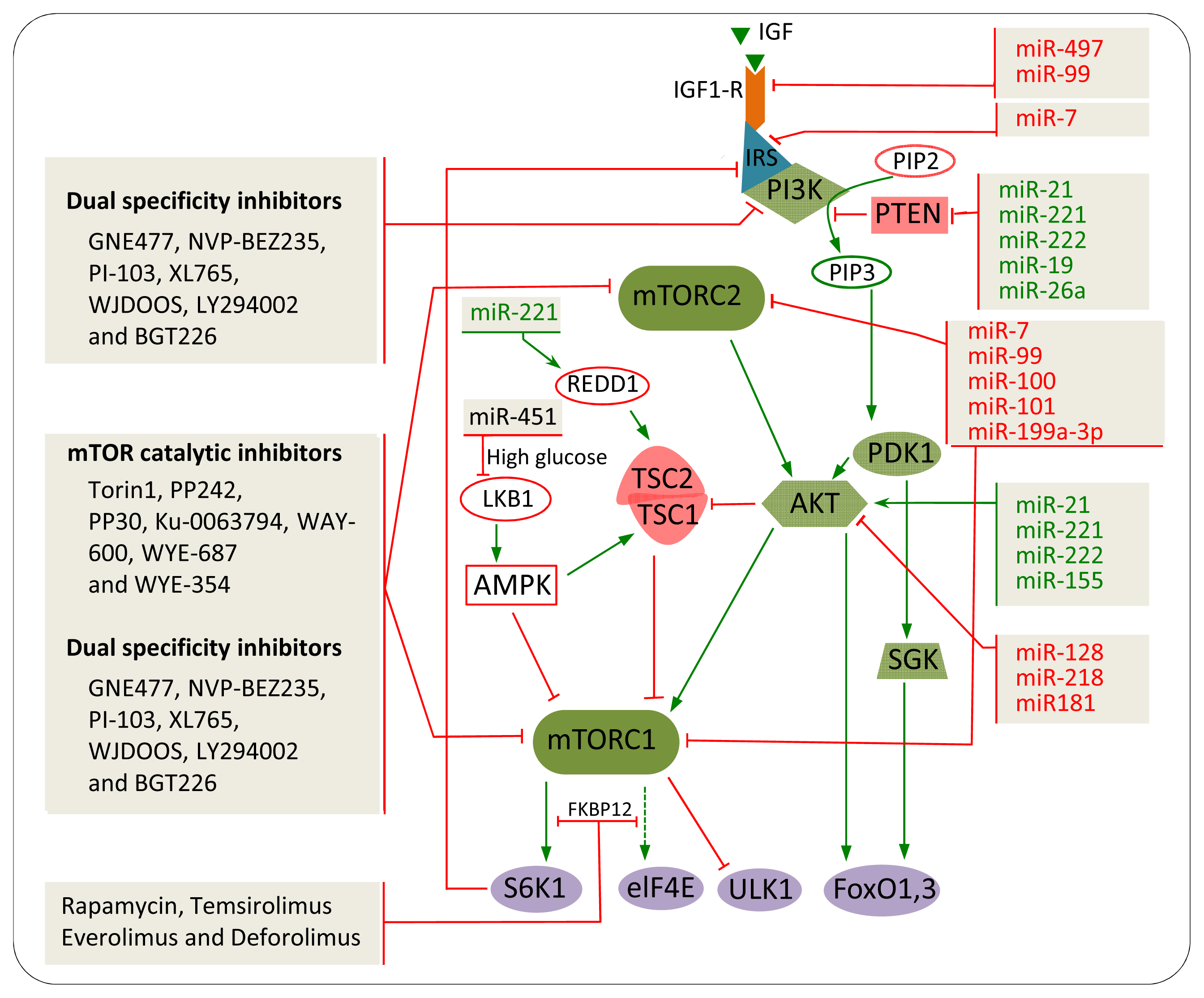
5. The Impact of mTOR Inhibitors in Cancer
5.1. Rapamycin and Rapalogs
5.2. mTOR Catalytic Inhibitors
5.3. Dual Specificity Inhibitors
6. miRNAs
7. miRNAs Involved in mTOR Regulation in Cancer
7.1. IGF-R
7.2. PTEN
7.3. AKT
7.4. mTOR
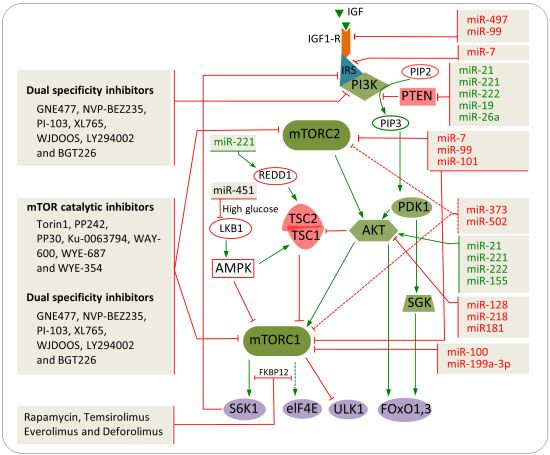
8. Combination of miRNAs and Chemical Inhibitors to Target the mTOR Pathway
9. Conclusion
Acknowledgements
Conflict of Interest
References
- Brown, E.J.; Albers, M.W.; Shin, T.B.; Ichikawa, K.; Keith, C.T.; Lane, W.S.; Schreiber, S.L. A mammalian protein targeted by G1-arresting rapamycin-receptor complex. Nature 1994, 369, 756–758. [Google Scholar]
- Chiu, M.I.; Katz, H.; Berlin, V. RAPT1, a mammalian homolog of yeast Tor, interacts with the FKBP12/rapamycin complex. Proc. Natl. Acad. Sci. USA 1994, 91, 12574–12578. [Google Scholar]
- Heitman, J.; Movva, N.R.; Hall, M.N. Targets for cell cycle arrest by the immunosuppressant rapamycin in yeast. Science 1991, 253, 905–909. [Google Scholar]
- Sabatini, D.M.; Erdjument-Bromage, H.; Lui, M.; Tempst, P.; Snyder, S.H. RAFT1: A mammalian protein that binds to FKBP12 in a rapamycin-dependent fashion and is homologous to yeast TORs. Cell 1994, 78, 35–43. [Google Scholar]
- Vezina, C.; Kudelski, A.; Sehgal, S.N. Rapamycin (AY-22,989), a new antifungal antibiotic. I. Taxonomy of the producing streptomycete and isolation of the active principle. J. Antibiot. (Tokyo) 1975, 28, 721–726. [Google Scholar]
- Choi, J.; Chen, J.; Schreiber, S.L.; Clardy, J. Structure of the FKBP12-rapamycin complex interacting with the binding domain of human FRAP. Science 1996, 273, 239–242. [Google Scholar]
- Abraham, R.T. PI 3-kinase related kinases: ‘Big’ players in stress-induced signaling pathways. DNA Repair (Amst) 2004, 3, 883–887. [Google Scholar]
- Hay, N.; Sonenberg, N. Upstream and downstream of mTOR. Genes Dev 2004, 18, 1926–1945. [Google Scholar]
- Sabers, C.J.; Martin, M.M.; Brunn, G.J.; Williams, J.M.; Dumont, F.J.; Wiederrecht, G.; Abraham, R.T. Isolation of a protein target of the FKBP12-rapamycin complex in mammalian cells. J. Biol. Chem 1995, 270, 815–822. [Google Scholar]
- Yonezawa, K.; Yoshino, K.I.; Tokunaga, C.; Hara, K. Kinase activities associated with mTOR. Curr. Top Microbiol. Immunol 2004, 279, 271–282. [Google Scholar]
- Brunn, G.J.; Fadden, P.; Haystead, T.A.; Lawrence, J.C., Jr. The mammalian target of rapamycin phosphorylates sites having a (Ser/Thr)-Pro motif and is activated by antibodies to a region near its COOH terminus. J. Biol. Chem. 1997, 272, 32547–32550. [Google Scholar]
- Burnett, P.E.; Barrow, R.K.; Cohen, N.A.; Snyder, S.H.; Sabatini, D.M. RAFT1 phosphorylation of the translational regulators p70 S6 kinase and 4E-BP1. Proc. Natl. Acad. Sci. USA 1998, 95, 1432–1437. [Google Scholar]
- Isotani, S.; Hara, K.; Tokunaga, C.; Inoue, H.; Avruch, J.; Yonezawa, K. Immunopurified mammalian target of rapamycin phosphorylates and activates p70 S6 kinase alpha in vitro. J. Biol. Chem 1999, 274, 34493–34498. [Google Scholar]
- Andrade, M.A.; Bork, P. HEAT repeats in the Huntington’s disease protein. Nat. Genet 1995, 11, 115–116. [Google Scholar]
- Bosotti, R.; Isacchi, A.; Sonnhammer, E.L. FAT: A novel domain in PIK-related kinases. Trends Biochem. Sci 2000, 25, 225–227. [Google Scholar]
- Jacinto, E.; Hall, M.N. Tor signalling in bugs, brain and brawn. Nat. Rev. Mol. Cell Biol 2003, 4, 117–126. [Google Scholar]
- Sekulic, A.; Hudson, C.C.; Homme, J.L.; Yin, P.; Otterness, D.M.; Karnitz, L.M.; Abraham, R.T. A direct linkage between the phosphoinositide 3-kinase-AKT signaling pathway and the mammalian target of rapamycin in mitogen-stimulated and transformed cells. Cancer Res 2000, 60, 3504–3513. [Google Scholar]
- Guertin, D.A.; Sabatini, D.M. Defining the role of mTOR in cancer. Cancer Cell 2007, 12, 9–22. [Google Scholar]
- Sabatini, D.M. mTOR and cancer: Insights into a complex relationship. Nat. Rev. Cancer 2006, 6, 729–734. [Google Scholar]
- Hara, K.; Maruki, Y.; Long, X.; Yoshino, K.; Oshiro, N.; Hidayat, S.; Tokunaga, C.; Avruch, J.; Yonezawa, K. Raptor, a binding partner of target of rapamycin (TOR), mediates TOR action. Cell 2002, 110, 177–189. [Google Scholar]
- Kim, D.H.; Sarbassov, D.D.; Ali, S.M.; King, J.E.; Latek, R.R.; Erdjument-Bromage, H.; Tempst, P.; Sabatini, D.M. mTOR interacts with raptor to form a nutrient-sensitive complex that signals to the cell growth machinery. Cell 2002, 110, 163–175. [Google Scholar]
- Nojima, H.; Tokunaga, C.; Eguchi, S.; Oshiro, N.; Hidayat, S.; Yoshino, K.; Hara, K.; Tanaka, N.; Avruch, J.; Yonezawa, K. The mammalian target of rapamycin (mTOR) partner, raptor, binds the mTOR substrates p70 S6 kinase and 4E-BP1 through their TOR signaling (TOS) motif. J. Biol. Chem 2003, 278, 15461–15464. [Google Scholar]
- Schalm, S.S.; Fingar, D.C.; Sabatini, D.M.; Blenis, J. TOS motif-mediated raptor binding regulates 4E-BP1 multisite phosphorylation and function. Curr. Biol 2003, 13, 797–806. [Google Scholar]
- Kim, D.H.; Sarbassov, D.D.; Ali, S.M.; Latek, R.R.; Guntur, K.V.; Erdjument-Bromage, H.; Tempst, P.; Sabatini, D.M. GbetaL, a positive regulator of the rapamycin-sensitive pathway required for the nutrient-sensitive interaction between raptor and mTOR. Mol. Cell 2003, 11, 895–904. [Google Scholar]
- Guertin, D.A.; Stevens, D.M.; Thoreen, C.C.; Burds, A.A.; Kalaany, N.Y.; Moffat, J.; Brown, M.; Fitzgerald, K.J.; Sabatini, D.M. Ablation in mice of the mTORC components raptor, rictor, or mLST8 reveals that mTORC2 is required for signaling to Akt-FOXO and PKCalpha but not S6K1. Dev. Cell 2006, 11, 859–871. [Google Scholar]
- Sancak, Y.; Thoreen, C.C.; Peterson, T.R.; Lindquist, R.A.; Kang, S.A.; Spooner, E.; Carr, S.A.; Sabatini, D.M. PRAS40 is an insulin-regulated inhibitor of the mTORC1 protein kinase. Mol. Cell 2007, 25, 903–915. [Google Scholar]
- Vander Haar, E.; Lee, S.I.; Bandhakavi, S.; Griffin, T.J.; Kim, D.H. Insulin signalling to mTOR mediated by the Akt/PKB substrate PRAS40. Nat. Cell Biol 2007, 9, 316–323. [Google Scholar]
- Wang, L.; Harris, T.E.; Roth, R.A.; Lawrence, J.C., Jr. PRAS40 regulates mTORC1 kinase activity by functioning as a direct inhibitor of substrate binding. J. Biol. Chem. 2007, 282, 20036–20044. [Google Scholar]
- Peterson, T.R.; Laplante, M.; Thoreen, C.C.; Sancak, Y.; Kang, S.A.; Kuehl, W.M.; Gray, N.S.; Sabatini, D.M. DEPTOR is an mTOR inhibitor frequently overexpressed in multiple myeloma cells and required for their survival. Cell 2009, 137, 873–886. [Google Scholar]
- Sarbassov, D.D.; Ali, S.M.; Kim, D.H.; Guertin, D.A.; Latek, R.R.; Erdjument-Bromage, H.; Tempst, P.; Sabatini, D.M. Rictor, a novel binding partner of mTOR, defines a rapamycin-insensitive and raptor-independent pathway that regulates the cytoskeleton. Curr. Biol 2004, 14, 1296–1302. [Google Scholar]
- Frias, M.A.; Thoreen, C.C.; Jaffe, J.D.; Schroder, W.; Sculley, T.; Carr, S.A.; Sabatini, D.M. mSin1 is necessary for Akt/PKB phosphorylation and its isoforms define three distinct mTORC2s. Curr. Biol 2006, 16, 1865–1870. [Google Scholar]
- Yang, Q.; Inoki, K.; Ikenoue, T.; Guan, K.L. Identification of Sin1 as an essential TORC2 component required for complex formation and kinase activity. Genes Dev 2006, 20, 2820–2832. [Google Scholar]
- Ikenoue, T.; Inoki, K.; Yang, Q.; Zhou, X.; Guan, K.L. Essential function of TORC2 in PKC and Akt turn motif phosphorylation, maturation and signalling. EMBO J 2008, 27, 1919–1931. [Google Scholar]
- Pearce, L.R.; Huang, X.; Boudeau, J.; Pawlowski, R.; Wullschleger, S.; Deak, M.; Ibrahim, A.F.; Gourlay, R.; Magnuson, M.A.; Alessi, D.R. Identification of Protor as a novel Rictor-binding component of mTOR complex-2. Biochem. J 2007, 405, 513–522. [Google Scholar]
- Zoncu, R.; Efeyan, A.; Sabatini, D.M. mTOR: From growth signal integration to cancer, diabetes and ageing. Nat. Rev. Mol. Cell Biol 2011, 12, 21–35. [Google Scholar]
- Hara, K.; Yonezawa, K.; Weng, Q.P.; Kozlowski, M.T.; Belham, C.; Avruch, J. Amino acid sufficiency and mTOR regulate p70 S6 kinase and eIF-4E BP1 through a common effector mechanism. J. Biol. Chem 1998, 273, 14484–14494. [Google Scholar]
- Nicklin, P.; Bergman, P.; Zhang, B.; Triantafellow, E.; Wang, H.; Nyfeler, B.; Yang, H.; Hild, M.; Kung, C.; Wilson, C.; et al. Bidirectional transport of amino acids regulates mTOR and autophagy. Cell 2009, 136, 521–534. [Google Scholar]
- Wang, X.; Campbell, L.E.; Miller, C.M.; Proud, C.G. Amino acid availability regulates p70 S6 kinase and multiple translation factors. Biochem. J 1998, 334, 261–267. [Google Scholar]
- Shigemitsu, K.; Tsujishita, Y.; Hara, K.; Nanahoshi, M.; Avruch, J.; Yonezawa, K. Regulation of translational effectors by amino acid and mammalian target of rapamycin signaling pathways. Possible involvement of autophagy in cultured hepatoma cells. J. Biol. Chem 1999, 274, 1058–1065. [Google Scholar]
- Shigemitsu, K.; Tsujishita, Y.; Miyake, H.; Hidayat, S.; Tanaka, N.; Hara, K.; Yonezawa, K. Structural requirement of leucine for activation of p70 S6 kinase. FEBS Lett 1999, 447, 303–306. [Google Scholar]
- Findlay, G.M.; Yan, L.; Procter, J.; Mieulet, V.; Lamb, R.F. A MAP4 kinase related to Ste20 is a nutrient-sensitive regulator of mTOR signalling. Biochem. J 2007, 403, 13–20. [Google Scholar]
- Gulati, P.; Gaspers, L.D.; Dann, S.G.; Joaquin, M.; Nobukuni, T.; Natt, F.; Kozma, S.C.; Thomas, A.P.; Thomas, G. Amino acids activate mTOR complex 1 via Ca2+/CaM signaling to hVps34. Cell Metab 2008, 7, 456–465. [Google Scholar]
- Nobukuni, T.; Joaquin, M.; Roccio, M.; Dann, S.G.; Kim, S.Y.; Gulati, P.; Byfield, M.P.; Backer, J.M.; Natt, F.; Bos, J.L.; et al. Amino acids mediate mTOR/raptor signaling through activation of class 3 phosphatidylinositol 3OH-kinase. Proc. Natl. Acad. Sci. USA 2005, 102, 14238–14243. [Google Scholar]
- Yan, L.; Mieulet, V.; Burgess, D.; Findlay, G.M.; Sully, K.; Procter, J.; Goris, J.; Janssens, V.; Morrice, N.A.; Lamb, R.F. PP2A T61 epsilon is an inhibitor of MAP4K3 in nutrient signaling to mTOR. Mol. Cell 2010, 37, 633–642. [Google Scholar]
- Tee, A.R.; Fingar, D.C.; Manning, B.D.; Kwiatkowski, D.J.; Cantley, L.C.; Blenis, J. Tuberous sclerosis complex-1 and -2 gene products function together to inhibit mammalian target of rapamycin (mTOR)-mediated downstream signaling. Proc. Natl. Acad. Sci. USA 2002, 99, 13571–13576. [Google Scholar]
- Smith, E.M.; Finn, S.G.; Tee, A.R.; Browne, G.J.; Proud, C.G. The tuberous sclerosis protein TSC2 is not required for the regulation of the mammalian target of rapamycin by amino acids and certain cellular stresses. J. Biol. Chem 2005, 280, 18717–18727. [Google Scholar]
- Kim, E.; Goraksha-Hicks, P.; Li, L.; Neufeld, T.P.; Guan, K.L. Regulation of TORC1 by Rag GTPases in nutrient response. Nat. Cell Biol 2008, 10, 935–945. [Google Scholar]
- Sancak, Y.; Peterson, T.R.; Shaul, Y.D.; Lindquist, R.A.; Thoreen, C.C.; Bar-Peled, L.; Sabatini, D.M. The Rag GTPases bind raptor and mediate amino acid signaling to mTORC1. Science 2008, 320, 1496–1501. [Google Scholar]
- Shaw, R.J. mTOR signaling: RAG GTPases transmit the amino acid signal. Trends Biochem. Sci 2008, 33, 565–568. [Google Scholar]
- Sancak, Y.; Bar-Peled, L.; Zoncu, R.; Markhard, A.L.; Nada, S.; Sabatini, D.M. Ragulator-Rag complex targets mTORC1 to the lysosomal surface and is necessary for its activation by amino acids. Cell 2010, 141, 290–303. [Google Scholar]
- Fingar, D.C.; Blenis, J. Target of rapamycin (TOR): An integrator of nutrient and growth factor signals and coordinator of cell growth and cell cycle progression. Oncogene 2004, 23, 3151–3171. [Google Scholar]
- Inoki, K.; Li, Y.; Zhu, T.; Wu, J.; Guan, K.L. TSC2 is phosphorylated and inhibited by Akt and suppresses mTOR signalling. Nat. Cell Biol 2002, 4, 648–657. [Google Scholar]
- Potter, C.J.; Pedraza, L.G.; Xu, T. Akt regulates growth by directly phosphorylating Tsc2. Nat. Cell Biol 2002, 4, 658–665. [Google Scholar]
- Garami, A.; Zwartkruis, F.J.; Nobukuni, T.; Joaquin, M.; Roccio, M.; Stocker, H.; Kozma, S.C.; Hafen, E.; Bos, J.L.; Thomas, G. Insulin activation of Rheb, a mediator of mTOR/S6K/4E-BP signaling, is inhibited by TSC1 and 2. Mol. Cell 2003, 11, 1457–1466. [Google Scholar]
- Zhang, Y.; Gao, X.; Saucedo, L.J.; Ru, B.; Edgar, B.A.; Pan, D. Rheb is a direct target of the tuberous sclerosis tumour suppressor proteins. Nat. Cell Biol 2003, 5, 578–581. [Google Scholar]
- Long, X.; Lin, Y.; Ortiz-Vega, S.; Yonezawa, K.; Avruch, J. Rheb binds and regulates the mTOR kinase. Curr. Biol 2005, 15, 702–713. [Google Scholar]
- Ma, L.; Chen, Z.; Erdjument-Bromage, H.; Tempst, P.; Pandolfi, P.P. Phosphorylation and functional inactivation of TSC2 by Erk implications for tuberous sclerosis and cancer pathogenesis. Cell 2005, 121, 179–193. [Google Scholar]
- Murphy, L.O.; Blenis, J. MAPK signal specificity: The right place at the right time. Trends Biochem. Sci 2006, 31, 268–275. [Google Scholar]
- Roux, P.P.; Ballif, B.A.; Anjum, R.; Gygi, S.P.; Blenis, J. Tumor-promoting phorbol esters and activated Ras inactivate the tuberous sclerosis tumor suppressor complex via p90 ribosomal S6 kinase. Proc. Natl. Acad. Sci. USA 2004, 101, 13489–13494. [Google Scholar]
- Dennis, P.B.; Jaeschke, A.; Saitoh, M.; Fowler, B.; Kozma, S.C.; Thomas, G. Mammalian TOR: A homeostatic ATP sensor. Science 2001, 294, 1102–1105. [Google Scholar]
- Hardie, D.G. AMP-activated/SNF1 protein kinases: Conserved guardians of cellular energy. Nat. Rev. Mol. Cell Biol 2007, 8, 774–785. [Google Scholar]
- Corradetti, M.N.; Inoki, K.; Bardeesy, N.; DePinho, R.A.; Guan, K.L. Regulation of the TSC pathway by LKB1: Evidence of a molecular link between tuberous sclerosis complex and Peutz-Jeghers syndrome. Genes Dev 2004, 18, 1533–1538. [Google Scholar]
- Inoki, K.; Zhu, T.; Guan, K.L. TSC2 mediates cellular energy response to control cell growth and survival. Cell 2003, 115, 577–590. [Google Scholar]
- Gwinn, D.M.; Shackelford, D.B.; Egan, D.F.; Mihaylova, M.M.; Mery, A.; Vasquez, D.S.; Turk, B.E.; Shaw, R.J. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol. Cell 2008, 30, 214–226. [Google Scholar]
- Brugarolas, J.; Lei, K.; Hurley, R.L.; Manning, B.D.; Reiling, J.H.; Hafen, E.; Witters, L.A.; Ellisen, L.W.; Kaelin, W.G., Jr. Regulation of mTOR function in response to hypoxia by REDD1 and the TSC1/TSC2 tumor suppressor complex. Genes Dev. 2004, 18, 2893–2904. [Google Scholar] [Green Version]
- DeYoung, M.P.; Horak, P.; Sofer, A.; Sgroi, D.; Ellisen, L.W. Hypoxia regulates TSC1/2-mTOR signaling and tumor suppression through REDD1-mediated 14-3-3 shuttling. Genes Dev 2008, 22, 239–251. [Google Scholar]
- Feng, Z.; Hu, W.; de Stanchina, E.; Teresky, A.K.; Jin, S.; Lowe, S.; Levine, A.J. The regulation of AMPK beta1, TSC2 and PTEN expression by p53: Stress, cell and tissue specificity and the role of these gene products in modulating the IGF-1-AKT-mTOR pathways. Cancer Res 2007, 67, 3043–3053. [Google Scholar]
- Budanov, A.V.; Karin, M. p53 target genes sestrin1 and sestrin2 connect genotoxic stress and mTOR signaling. Cell 2008, 134, 451–460. [Google Scholar]
- Sarbassov, D.D.; Guertin, D.A.; Ali, S.M.; Sabatini, D.M. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science 2005, 307, 1098–1101. [Google Scholar]
- Manning, B.D.; Cantley, L.C. AKT/PKB signaling: Navigating downstream. Cell 2007, 129, 1261–1274. [Google Scholar]
- Guertin, D.A.; Sabatini, D.M. An expanding role for mTOR in cancer. Trends Mol. Med 2005, 11, 353–361. [Google Scholar]
- Laplante, M.; Sabatini, D.M. An emerging role of mTOR in lipid biosynthesis. Curr. Biol 2009, 19, R1046–R1052. [Google Scholar]
- Calnan, D.R.; Brunet, A. The FoxO code. Oncogene 2008, 27, 2276–2288. [Google Scholar]
- Garcia-Martinez, J.M.; Alessi, D.R. mTOR complex 2 (mTORC2) controls hydrophobic motif phosphorylation and activation of serum- and glucocorticoid-induced protein kinase 1 (SGK1). Biochem. J 2008, 416, 375–385. [Google Scholar]
- Jacinto, E.; Loewith, R.; Schmidt, A.; Lin, S.; Ruegg, M.A.; Hall, A.; Hall, M.N. Mammalian TOR complex 2 controls the actin cytoskeleton and is rapamycin insensitive. Nat. Cell Biol 2004, 6, 1122–1128. [Google Scholar]
- Menon, S.; Manning, B.D. Common corruption of the mTOR signaling network in human tumors. Oncogene 2008, 27, S43–S51. [Google Scholar]
- Yuan, T.L.; Cantley, L.C. PI3K pathway alterations in cancer: Variations on a theme. Oncogene 2008, 27, 5497–5510. [Google Scholar]
- Roberts, P.J.; Der, C.J. Targeting the Raf-MEK-ERK mitogen-activated protein kinase cascade for the treatment of cancer. Oncogene 2007, 26, 3291–3310. [Google Scholar]
- Shaw, R.J.; Cantley, L.C. Ras, PI(3)K and mTOR signalling controls tumour cell growth. Nature 2006, 441, 424–430. [Google Scholar]
- Engelman, J.A.; Luo, J.; Cantley, L.C. The evolution of phosphatidylinositol 3-kinases as regulators of growth and metabolism. Nat. Rev. Genet 2006, 7, 606–619. [Google Scholar]
- Johannessen, C.M.; Reczek, E.E.; James, M.F.; Brems, H.; Legius, E.; Cichowski, K. The NF1 tumor suppressor critically regulates TSC2 and mTOR. Proc. Natl. Acad. Sci. USA 2005, 102, 8573–8578. [Google Scholar]
- Rousseau, D.; Gingras, A.C.; Pause, A.; Sonenberg, N. The eIF4E-binding proteins 1 and 2 are negative regulators of cell growth. Oncogene 1996, 13, 2415–2420. [Google Scholar]
- Akcakanat, A.; Sahin, A.; Shaye, A.N.; Velasco, M.A.; Meric-Bernstam, F. Comparison of Akt/mTOR signaling in primary breast tumors and matched distant metastases. Cancer 2008, 112, 2352–2358. [Google Scholar]
- Armengol, G.; Rojo, F.; Castellvi, J.; Iglesias, C.; Cuatrecasas, M.; Pons, B.; Baselga, J.; Ramon y Cajal, S. 4E-binding protein 1: A key molecular “funnel factor” in human cancer with clinical implications. Cancer Res 2007, 67, 7551–7555. [Google Scholar]
- Castellvi, J.; Garcia, A.; Rojo, F.; Ruiz-Marcellan, C.; Gil, A.; Baselga, J.; Ramon y Cajal, S. Phosphorylated 4E binding protein 1: A hallmark of cell signaling that correlates with survival in ovarian cancer. Cancer 2006, 107, 1801–1811. [Google Scholar]
- Wendel, H.G.; De Stanchina, E.; Fridman, J.S.; Malina, A.; Ray, S.; Kogan, S.; Cordon-Cardo, C.; Pelletier, J.; Lowe, S.W. Survival signalling by Akt and eIF4E in oncogenesis and cancer therapy. Nature 2004, 428, 332–337. [Google Scholar]
- Wendel, H.G.; Silva, R.L.; Malina, A.; Mills, J.R.; Zhu, H.; Ueda, T.; Watanabe-Fukunaga, R.; Fukunaga, R.; Teruya-Feldstein, J.; Pelletier, J.; Lowe, S.W. Dissecting eIF4E action in tumorigenesis. Genes Dev 2007, 21, 3232–3237. [Google Scholar]
- Ruggero, D.; Montanaro, L.; Ma, L.; Xu, W.; Londei, P.; Cordon-Cardo, C.; Pandolfi, P.P. The translation factor eIF-4E promotes tumor formation and cooperates with c-Myc in lymphomagenesis. Nat. Med 2004, 10, 484–486. [Google Scholar]
- Petroulakis, E.; Parsyan, A.; Dowling, R.J.; LeBacquer, O.; Martineau, Y.; Bidinosti, M.; Larsson, O.; Alain, T.; Rong, L.; Mamane, Y.; et al. p53-dependent translational control of senescence and transformation via 4E-BPs. Cancer Cell 2009, 16, 439–446. [Google Scholar]
- Clemens, M.J.; Bommer, U.A. Translational control: The cancer connection. Int. J. Biochem. Cell Biol 1999, 31, 1–23. [Google Scholar]
- De Benedetti, A.; Harris, A.L. eIF4E expression in tumors: Its possible role in progression of malignancies. Int. J. Biochem. Cell Biol 1999, 31, 59–72. [Google Scholar]
- Mamane, Y.; Petroulakis, E.; Rong, L.; Yoshida, K.; Ler, L.W.; Sonenberg, N. eIF4E—from translation to transformation. Oncogene 2004, 23, 3172–3179. [Google Scholar]
- Graff, J.R.; Konicek, B.W.; Vincent, T.M.; Lynch, R.L.; Monteith, D.; Weir, S.N.; Schwier, P.; Capen, A.; Goode, R.L.; Dowless, M.S.; et al. Therapeutic suppression of translation initiation factor eIF4E expression reduces tumor growth without toxicity. J. Clin. Invest 2007, 117, 2638–2648. [Google Scholar]
- Fumagalli, S.; Thomas, G. S6 Phosphorylation and Signal Transduction. In Translational Control of Gene Expression; Sonenberg, N., Hershey, J.W.B., Mathews, M., Eds.; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 2000; pp. 695–717. [Google Scholar]
- Fingar, D.C.; Salama, S.; Tsou, C.; Harlow, E.; Blenis, J. Mammalian cell size is controlled by mTOR and its downstream targets S6K1 and 4EBP1/eIF4E. Genes Dev 2002, 16, 1472–1487. [Google Scholar]
- Zhou, H.Y.; Wong, A.S. Activation of p70S6K induces expression of matrix metalloproteinase 9 associated with hepatocyte growth factor-mediated invasion in human ovarian cancer cells. Endocrinology 2006, 147, 2557–2566. [Google Scholar]
- Couch, F.J.; Wang, X.Y.; Wu, G.J.; Qian, J.; Jenkins, R.B.; James, C.D. Localization of PS6K to chromosomal region 17q23 and determination of its amplification in breast cancer. Cancer Res 1999, 59, 1408–1411. [Google Scholar]
- Jastrzebski, K.; Hannan, K.M.; Tchoubrieva, E.B.; Hannan, R.D.; Pearson, R.B. Coordinate regulation of ribosome biogenesis and function by the ribosomal protein S6 kinase, a key mediator of mTOR function. Growth Factors 2007, 25, 209–226. [Google Scholar]
- Kwon, H.K.; Bae, G.U.; Yoon, J.W.; Kim, Y.K.; Lee, H.Y.; Lee, H.W.; Han, J.W. Constitutive activation of p70S6k in cancer cells. Arch. Pharm. Res 2002, 25, 685–690. [Google Scholar]
- Barlund, M.; Monni, O.; Kononen, J.; Cornelison, R.; Torhorst, J.; Sauter, G.; Kallioniemi, O.-P.; Kallioniemi, A. Multiple genes at 17q23 undergo amplification and overexpression in breast cancer. Cancer Res 2000, 60, 5340–5344. [Google Scholar]
- Heinonen, H.; Nieminen, A.; Saarela, M.; Kallioniemi, A.; Klefstrom, J.; Hautaniemi, S.; Monni, O. Deciphering downstream gene targets of PI3K/mTOR/p70S6K pathway in breast cancer. BMC Genomics 2008, 9. [Google Scholar] [CrossRef]
- Pon, Y.L.; Zhou, H.Y.; Cheung, A.N.; Ngan, H.Y.; Wong, A.S. p70 S6 kinase promotes epithelial to mesenchymal transition through snail induction in ovarian cancer cells. Cancer Res 2008, 68, 6524–6532. [Google Scholar]
- Hsu, P.P.; Sabatini, D.M. Cancer cell metabolism: Warburg and beyond. Cell 2008, 134, 703–707. [Google Scholar]
- Guertin, D.A.; Stevens, D.M.; Saitoh, M.; Kinkel, S.; Crosby, K.; Sheen, J.H.; Mullholland, D.J.; Magnuson, M.A.; Wu, H.; Sabatini, D.M. mTOR complex 2 is required for the development of prostate cancer induced by Pten loss in mice. Cancer Cell 2009, 15, 148–159. [Google Scholar]
- Hoang, B.; Frost, P.; Shi, Y.; Belanger, E.; Benavides, A.; Pezeshkpour, G.; Cappia, S.; Guglielmelli, T.; Gera, J.; Lichtenstein, A. Targeting TORC2 in multiple myeloma with a new mTOR kinase inhibitor. Blood 2010, 116, 4560–4568. [Google Scholar]
- Masri, J.; Bernath, A.; Martin, J.; Jo, O.D.; Vartanian, R.; Funk, A.; Gera, J. mTORC2 activity is elevated in gliomas and promotes growth and cell motility via overexpression of rictor. Cancer Res 2007, 67, 11712–11720. [Google Scholar]
- Chen, J.; Zheng, X.F.; Brown, E.J.; Schreiber, S.L. Identification of an 11-kDa FKBP12-rapamycin-binding domain within the 289-kDa FKBP12-rapamycin-associated protein and characterization of a critical serine residue. Proc. Natl. Acad. Sci. USA 1995, 92, 4947–4951. [Google Scholar]
- Oshiro, N.; Yoshino, K.; Hidayat, S.; Tokunaga, C.; Hara, K.; Eguchi, S.; Avruch, J.; Yonezawa, K. Dissociation of raptor from mTOR is a mechanism of rapamycin-induced inhibition of mTOR function. Genes Cells 2004, 9, 359–366. [Google Scholar]
- Sarbassov, D.D.; Ali, S.M.; Sengupta, S.; Sheen, J.H.; Hsu, P.P.; Bagley, A.F.; Markhard, A.L.; Sabatini, D.M. Prolonged rapamycin treatment inhibits mTORC2 assembly and Akt/PKB. Mol. Cell 2006, 22, 159–168. [Google Scholar]
- Faivre, S.; Kroemer, G.; Raymond, E. Current development of mTOR inhibitors as anticancer agents. Nat. Rev. Drug Discov 2006, 5, 671–688. [Google Scholar]
- Petroulakis, E.; Mamane, Y.; Le Bacquer, O.; Shahbazian, D.; Sonenberg, N. mTOR signaling: Implications for cancer and anticancer therapy. Br. J. Cancer 2006, 94, 195–199. [Google Scholar]
- Geoerger, B.; Kerr, K.; Tang, C.B.; Fung, K.M.; Powell, B.; Sutton, L.N.; Phillips, P.C.; Janss, A.J. Antitumor activity of the rapamycin analog CCI-779 in human primitive neuroectodermal tumor/medulloblastoma models as single agent and in combination chemotherapy. Cancer Res 2001, 61, 1527–1532. [Google Scholar]
- Grewe, M.; Gansauge, F.; Schmid, R.M.; Adler, G.; Seufferlein, T. Regulation of cell growth and cyclin D1 expression by the constitutively active FRAP-p70s6K pathway in human pancreatic cancer cells. Cancer Res 1999, 59, 3581–3587. [Google Scholar]
- Pang, H.; Faber, L.E. Estrogen and rapamycin effects on cell cycle progression in T47D breast cancer cells. Breast Cancer Res. Treat 2001, 70, 21–26. [Google Scholar]
- Seufferlein, T.; Rozengurt, E. Rapamycin inhibits constitutive p70s6k phosphorylation, cell proliferation and colony formation in small cell lung cancer cells. Cancer Res 1996, 56, 3895–3897. [Google Scholar]
- van der Poel, H.G.; Hanrahan, C.; Zhong, H.; Simons, J.W. Rapamycin induces Smad activity in prostate cancer cell lines. Urol. Res 2003, 30, 380–386. [Google Scholar]
- Guba, M.; von Breitenbuch, P.; Steinbauer, M.; Koehl, G.; Flegel, S.; Hornung, M.; Bruns, C.J.; Zuelke, C.; Farkas, S.; Anthuber, M.; et al. Rapamycin inhibits primary and metastatic tumor growth by antiangiogenesis: Involvement of vascular endothelial growth factor. Nat. Med 2002, 8, 128–135. [Google Scholar]
- Ballou, L.M.; Lin, R.Z. Rapamycin and mTOR kinase inhibitors. J. Chem. Biol 2008, 1, 27–36. [Google Scholar]
- Rizzieri, D.A.; Feldman, E.; Dipersio, J.F.; Gabrail, N.; Stock, W.; Strair, R.; Rivera, V.M.; Albitar, M.; Bedrosian, C.L.; Giles, F.J. A phase 2 clinical trial of deforolimus (AP23573, MK-8669), a novel mammalian target of rapamycin inhibitor, in patients with relapsed or refractory hematologic malignancies. Clin. Cancer Res 2008, 14, 2756–2762. [Google Scholar]
- Dowling, R.J.; Pollak, M.; Sonenberg, N. Current status and challenges associated with targeting mTOR for cancer therapy. Biodrugs 2009, 23, 77–91. [Google Scholar]
- Kapoor, A.; Figlin, R.A. Targeted inhibition of mammalian target of rapamycin for the treatment of advanced renal cell carcinoma. Cancer 2009, 115, 3618–3630. [Google Scholar]
- O’Reilly, K.E.; Rojo, F.; She, Q.B.; Solit, D.; Mills, G.B.; Smith, D.; Lane, H.; Hofmann, F.; Hicklin, D.J.; Ludwig, D.L.; et al. mTOR inhibition induces upstream receptor tyrosine kinase signaling and activates Akt. Cancer Res 2006, 66, 1500–1508. [Google Scholar]
- Choo, A.Y.; Yoon, S.O.; Kim, S.G.; Roux, P.P.; Blenis, J. Rapamycin differentially inhibits S6Ks and 4E-BP1 to mediate cell-type-specific repression of mRNA translation. Proc. Natl. Acad. Sci. USA 2008, 105, 17414–17419. [Google Scholar]
- Thoreen, C.C.; Kang, S.A.; Chang, J.W.; Liu, Q.; Zhang, J.; Gao, Y.; Reichling, L.J.; Sim, T.; Sabatini, D.M.; Gray, N.S. An ATP-competitive mammalian target of rapamycin inhibitor reveals rapamycin-resistant functions of mTORC1. J. Biol. Chem 2009, 284, 8023–8032. [Google Scholar]
- Feldman, M.E.; Apsel, B.; Uotila, A.; Loewith, R.; Knight, Z.A.; Ruggero, D.; Shokat, K.M. Active-site inhibitors of mTOR target rapamycin-resistant outputs of mTORC1 and mTORC2. PLoS Biol. 2009, 7. [Google Scholar] [CrossRef]
- Yu, K.; Toral-Barza, L.; Shi, C.; Zhang, W.G.; Lucas, J.; Shor, B.; Kim, J.; Verheijen, J.; Curran, K.; Malwitz, D.J.; et al. Biochemical, cellular and in vivo activity of novel ATP-competitive and selective inhibitors of the mammalian target of rapamycin. Cancer Res 2009, 69, 6232–6240. [Google Scholar]
- Garcia-Martinez, J.M.; Moran, J.; Clarke, R.G.; Gray, A.; Cosulich, S.C.; Chresta, C.M.; Alessi, D.R. Ku-0063794 is a specific inhibitor of the mammalian target of rapamycin (mTOR). Biochem. J 2009, 421, 29–42. [Google Scholar]
- Shor, B.; Gibbons, J.J.; Abraham, R.T.; Yu, K. Targeting mTOR globally in cancer: Thinking beyond rapamycin. Cell Cycle 2009, 8, 3831–3837. [Google Scholar]
- Janes, M.R.; Limon, J.J.; So, L.; Chen, J.; Lim, R.J.; Chavez, M.A.; Vu, C.; Lilly, M.B.; Mallya, S.; Ong, S.T.; et al. Effective and selective targeting of leukemia cells using a TORC1/2 kinase inhibitor. Nat. Med 2010, 16, 205–213. [Google Scholar]
- Liu, P.; Cheng, H.; Roberts, T.M.; Zhao, J.J. Targeting the phosphoinositide 3-kinase pathway in cancer. Nat. Rev. Drug Discov 2009, 8, 627–644. [Google Scholar]
- Heffron, T.P.; Berry, M.; Castanedo, G.; Chang, C.; Chuckowree, I.; Dotson, J.; Folkes, A.; Gunzner, J.; Lesnick, J.D.; Lewis, C.; et al. Identification of GNE-477, a potent and efficacious dual PI3K/mTOR inhibitor. Bioorg. Med. Chem. Lett 2010, 20, 2408–2411. [Google Scholar]
- Li, T.; Wang, J.; Wang, X.; Yang, N.; Chen, S.M.; Tong, L.J.; Yang, C.H.; Meng, L.H.; Ding, J. WJD008, a dual phosphatidylinositol 3-kinase (PI3K)/mammalian target of rapamycin inhibitor, prevents PI3K signaling and inhibits the proliferation of transformed cells with oncogenic PI3K mutant. J. Pharmacol. Exp. Ther 2010, 334, 830–838. [Google Scholar]
- Brachmann, S.; Fritsch, C.; Maira, S.M.; Garcia-Echeverria, C. PI3K and mTOR inhibitors: A new generation of targeted anticancer agents. Curr. Opin. Cell Biol 2009, 21, 194–198. [Google Scholar]
- Cao, P.; Maira, S.M.; Garcia-Echeverria, C.; Hedley, D.W. Activity of a novel, dual PI3-kinase/mTor inhibitor NVP-BEZ235 against primary human pancreatic cancers grown as orthotopic xenografts. Br. J. Cancer 2009, 100, 1267–1276. [Google Scholar]
- Serra, V.; Markman, B.; Scaltriti, M.; Eichhorn, P.J.; Valero, V.; Guzman, M.; Botero, M.L.; Llonch, E.; Atzori, F.; Di Cosimo, S.; et al. NVP-BEZ235, a dual PI3K/mTOR inhibitor, prevents PI3K signaling and inhibits the growth of cancer cells with activating PI3K mutations. Cancer Res 2008, 68, 8022–8030. [Google Scholar]
- Manara, M.C.; Nicoletti, G.; Zambelli, D.; Ventura, S.; Guerzoni, C.; Landuzzi, L.; Lollini, P.L.; Maira, S.M.; Garcia-Echeverria, C.; Mercuri, M.; et al. NVP-BEZ235 as a new therapeutic option for sarcomas. Clin. Cancer Res 2010, 16, 530–540. [Google Scholar]
- Raynaud, F.I.; Eccles, S.; Clarke, P.A.; Hayes, A.; Nutley, B.; Alix, S.; Henley, A.; Di-Stefano, F.; Ahmad, Z.; Guillard, S.; et al. Pharmacologic characterization of a potent inhibitor of class I phosphatidylinositide 3-kinases. Cancer Res 2007, 67, 5840–5850. [Google Scholar]
- Zhang, C.Z.; Han, L.; Zhang, A.-L.; Fu, Y.-C.; Yue, X.; Wang, G.-X.; Jia, Z.-F.; Pu, P.-Y.; Zhang, Q.-Y.; Kang, C.-S. MicroRNA-221 and microRNA-222 regulate gastric carcinoma cell proliferation and radioresistance by targeting PTEN. BMC Cancer 2010, 10, 367:1–367:10. [Google Scholar]
- Garofalo, M.; di Leva, G.; Romano, G.; Nuovo, G.; Suh, S.S.; Ngankeu, A.; Taccioli, C.; Pichiorri, F.; Alder, H.; Secchiero, P.; et al. miR-221&222 regulate TRAIL resistance and enhance tumorigenicity through PTEN and TIMP3 downregulation. Cancer Cell 2009, 16, 498–509. [Google Scholar]
- Kefas, B.; Godlewski, J.; Comeau, L.; Li, Y.; Abounader, R.; Hawkinson, M.; Lee, J.; Fine, H.; Chiocca, E.A.; Lawler, S.; Purow, B. microRNA-7 inhibits the epidermal growth factor receptor and the Akt pathway and is down-regulated in glioblastoma. Cancer Res 2008, 68, 3566–3572. [Google Scholar]
- Zhang, J.; Han, L.; Ge, Y.; Zhou, X.; Zhang, A.; Zhang, C.; Zhong, Y.; You, Y.; Pu, P.; Kang, C. miR-221/222 promote malignant progression of glioma through activation of the Akt pathway. Int. J. Oncol 2010, 36, 913–920. [Google Scholar]
- Zhou, X.; Ren, Y.; Moore, L.; Mei, M.; You, Y.; Xu, P.; Wang, B.; Wang, G.; Jia, Z.; Pu, P.; Zhang, W.; Kang, C. Downregulation of miR-21 inhibits EGFR pathway and suppresses the growth of human glioblastoma cells independent of PTEN status. Lab. Invest 2010, 90, 144–155. [Google Scholar]
- Doghman, M.; El Wakil, A.; Cardinaud, B.; Thomas, E.; Wang, J.; Zhao, W.; Peralta-Del Valle, M.H.; Figueiredo, B.C.; Zambetti, G.P.; Lalli, E. Regulation of insulin-like growth factor-mammalian target of rapamycin signaling by microRNA in childhood adrenocortical tumors. Cancer Res 2010, 70, 4666–4675. [Google Scholar]
- Guo, S.T.; Jiang, C.C.; Wang, G.P.; Li, Y.P.; Wang, C.Y.; Guo, X.Y.; Yang, R.H.; Feng, Y.; Wang, F.H.; Tseng, H.Y.; et al. MicroRNA-497 targets insulin-like growth factor 1 receptor and has a tumour suppressive role in human colorectal cancer. Oncogene 2012. [Google Scholar] [CrossRef]
- Jiang, L.; Liu, X.; Chen, Z.; Jin, Y.; Heidbreder, C.E.; Kolokythas, A.; Wang, A.; Dai, Y.; Zhou, X. MicroRNA-7 targets IGF1R (insulin-like growth factor 1 receptor) in tongue squamous cell carcinoma cells. Biochem. J 2010, 432, 199–205. [Google Scholar]
- Kong, K.L.; Kwong, D.L.; Chan, T.H.; Law, S.Y.; Chen, L.; Li, Y.; Qin, Y.R.; Guan, X.Y. MicroRNA-375 inhibits tumour growth and metastasis in oesophageal squamous cell carcinoma through repressing insulin-like growth factor 1 receptor. Gut 2012, 61, 33–42. [Google Scholar]
- Li, D.; Liu, X.; Lin, L.; Hou, J.; Li, N.; Wang, C.; Wang, P.; Zhang, Q.; Zhang, P.; Zhou, W.; et al. MicroRNA-99a inhibits hepatocellular carcinoma growth and correlates with prognosis of patients with hepatocellular carcinoma. J. Biol. Chem 2011, 286, 36677–36685. [Google Scholar]
- Darido, C.; Georgy, S.R.; Wilanowski, T.; Dworkin, S.; Auden, A.; Zhao, Q.; Rank, G.; Srivastava, S.; Finlay, M.J.; Papenfuss, A.T.; et al. Targeting of the tumor suppressor GRHL3 by a miR-21-dependent proto-oncogenic network results in PTEN loss and tumorigenesis. Cancer Cell 2011, 20, 635–648. [Google Scholar]
- Meng, F.; Henson, R.; Wehbe-Janek, H.; Ghoshal, K.; Jacob, S.T.; Patel, T. MicroRNA-21 regulates expression of the PTEN tumor suppressor gene in human hepatocellular cancer. Gastroenterology 2007, 133, 647–658. [Google Scholar]
- Ng, R.; Song, G.; Roll, G.R.; Frandsen, N.M.; Willenbring, H. A microRNA-21 surge facilitates rapid cyclin D1 translation and cell cycle progression in mouse liver regeneration. J. Clin. Invest 2012, 122, 1097–1108. [Google Scholar]
- Fang, Y.; Xue, J.L.; Shen, Q.; Chen, J.; Tian, L. MicroRNA-7 inhibits tumor growth and metastasis by targeting the phosphoinositide 3-kinase/Akt pathway in hepatocellular carcinoma. Hepatology 2012, 55, 1852–1862. [Google Scholar]
- Sun, D.; Lee, Y.S.; Malhotra, A.; Kim, H.K.; Matecic, M.; Evans, C.; Jensen, R.V.; Moskaluk, C.A.; Dutta, A. miR-99 family of MicroRNAs suppresses the expression of prostate-specific antigen and prostate cancer cell proliferation. Cancer Res 2011, 71, 1313–1324. [Google Scholar]
- Pineau, P.; Volinia, S.; McJunkin, K.; Marchio, A.; Battiston, C.; Terris, B.; Mazzaferro, V.; Lowe, S.W.; Croce, C.M.; Dejean, A. miR-221 overexpression contributes to liver tumorigenesis. Proc. Natl. Acad. Sci. USA 2010, 107, 264–269. [Google Scholar]
- Nagaraja, A.K.; Creighton, C.J.; Yu, Z.; Zhu, H.; Gunaratne, P.H.; Reid, J.G.; Olokpa, E.; Itamochi, H.; Ueno, N.T.; Hawkins, S.M.; et al. A link between mir-100 and FRAP1/mTOR in clear cell ovarian cancer. Mol. Endocrinol 2010, 24, 447–463. [Google Scholar]
- Merkel, O.; Hamacher, F.; Laimer, D.; Sifft, E.; Trajanoski, Z.; Scheideler, M.; Egger, G.; Hassler, M.R.; Thallinger, C.; Schmatz, A.; et al. Identification of differential and functionally active miRNAs in both anaplastic lymphoma kinase (ALK)+ and ALK− anaplastic large-cell lymphoma. Proc. Natl. Acad. Sci. USA 2010, 107, 16228–16233. [Google Scholar]
- Fornari, F.; Milazzo, M.; Chieco, P.; Negrini, M.; Calin, G.A.; Grazi, G.L.; Pollutri, D.; Croce, C.M.; Bolondi, L.; Gramantieri, L. MiR-199a-3p regulates mTOR and c-Met to influence the doxorubicin sensitivity of human hepatocarcinoma cells. Cancer Res 2010, 70, 5184–5193. [Google Scholar]
- Duan, Z.; Choy, E.; Harmon, D.; Liu, X.; Susa, M.; Mankin, H.; Hornicek, F. MicroRNA-199a-3p is downregulated in human osteosarcoma and regulates cell proliferation and migration. Mol. Cancer Ther 2011, 10, 1337–1345. [Google Scholar]
- Godlewski, J.; Nowicki, M.O.; Bronisz, A.; Nuovo, G.; Palatini, J.; de Lay, M.; Van Brocklyn, J.; Ostrowski, M.C.; Chiocca, E.A.; Lawler, S.E. MicroRNA-451 regulates LKB1/AMPK signaling and allows adaptation to metabolic stress in glioma cells. Mol. Cell 2010, 37, 620–632. [Google Scholar]
- Nan, Y.; Han, L.; Zhang, A.; Wang, G.; Jia, Z.; Yang, Y.; Yue, X.; Pu, P.; Zhong, Y.; Kang, C. MiRNA-451 plays a role as tumor suppressor in human glioma cells. Brain Res 2010, 1359, 14–21. [Google Scholar]
- Godlewski, J.; Bronisz, A.; Nowicki, M.O.; Chiocca, E.A.; Lawler, S. microRNA-451: A conditional switch controlling glioma cell proliferation and migration. Cell Cycle 2010, 9, 2742–2748. [Google Scholar]
- Huse, J.T.; Brennan, C.; Hambardzumyan, D.; Wee, B.; Pena, J.; Rouhanifard, S.H.; Sohn-Lee, C.; le Sage, C.; Agami, R.; Tuschl, T.; et al. The PTEN-regulating microRNA miR-26a is amplified in high-grade glioma and facilitates gliomagenesis in vivo. Genes Dev 2009, 23, 1327–1337. [Google Scholar]
- Pekarsky, Y.; Santanam, U.; Cimmino, A.; Palamarchuk, A.; Efanov, A.; Maximov, V.; Volinia, S.; Alder, H.; Liu, C.G.; Rassenti, L.; et al. Tcl1 expression in chronic lymphocytic leukemia is regulated by miR-29 and miR-181. Cancer Res 2006, 66, 11590–11593. [Google Scholar]
- Laine, J.; Kunstle, G.; Obata, T.; Sha, M.; Noguchi, M. The protooncogene TCL1 is an Akt kinase coactivator. Mol. Cell 2000, 6, 395–407. [Google Scholar]
- Olive, V.; Bennett, M.J.; Walker, J.C.; Ma, C.; Jiang, I.; Cordon-Cardo, C.; Li, Q.J.; Lowe, S.W.; Hannon, G.J.; He, L. miR-19 is a key oncogenic component of mir-17-92. Genes Dev 2009, 23, 2839–2849. [Google Scholar]
- Godlewski, J.; Nowicki, M.O.; Bronisz, A.; Williams, S.; Otsuki, A.; Nuovo, G.; Raychaudhury, A.; Newton, H.B.; Chiocca, E.A.; Lawler, S. Targeting of the Bmi-1 oncogene/stem cell renewal factor by microRNA-128 inhibits glioma proliferation and self-renewal. Cancer Res 2008, 68, 9125–9130. [Google Scholar]
- Uesugi, A.; Kozaki, K.; Tsuruta, T.; Furuta, M.; Morita, K.; Imoto, I.; Omura, K.; Inazawa, J. The tumor suppressive microRNA miR-218 targets the mTOR component Rictor and inhibits AKT phosphorylation in oral cancer. Cancer Res 2011, 71, 5765–5778. [Google Scholar]
- Rajewsky, N. microRNA target predictions in animals. Nat. Genet 2006, 38, S8–S13. [Google Scholar]
- Roccaro, A.M.; Sacco, A.; Chen, C.; Runnels, J.; Leleu, X.; Azab, F.; Azab, A.K.; Jia, X.; Ngo, H.T.; Melhem, M.R.; et al. microRNA expression in the biology, prognosis and therapy of Waldenstrom macroglobulinemia. Blood 2009, 113, 4391–4402. [Google Scholar]
- Bousquet, E.; Mazieres, J.; Privat, M.; Rizzati, V.; Casanova, A.; Ledoux, A.; Mery, E.; Couderc, B.; Favre, G.; Pradines, A. Loss of RhoB expression promotes migration and invasion of human bronchial cells via activation of AKT1. Cancer Res 2009, 69, 6092–6099. [Google Scholar]
- Roccaro, A.M.; Sacco, A.; Jia, X.; Banwait, R.; Maiso, P.; Azab, F.; Flores, L.; Manier, S.; Azab, A.K.; Ghobrial, I.M. Mechanisms of activity of the TORC1 inhibitor everolimus in waldenstrom macroglobulinemia. Clin. Cancer Res 2012, 18, 6609–6622. [Google Scholar]
- Wang, F.Z.; Weber, F.; Croce, C.; Liu, C.G.; Liao, X.; Pellett, P.E. Human cytomegalovirus infection alters the expression of cellular microRNA species that affect its replication. J. Virol 2008, 82, 9065–9074. [Google Scholar]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| miRNA | Cancer Cells | Mechanism | Expression | Activity | Biological effects | Reference |
|---|---|---|---|---|---|---|
| miR-21 | Human squamous cell carcinoma; human hepatocellular cancer (HCC); human glioblastoma (GBM) xenograft model; hepa1, 6 mouse hepatoma | Induced loss of GRHL3 and PTEN expression leading to increased activity of mTOR signalling; targeting PTEN gene through the 3′-UTR binding site and repressing its expression leading to increase activity of AKT and mTOR kinase pathways; downregulation of miR-21 inhibited Akt and affected mTOR activity; activates Akt1/mTORC1-mediated cyclin D1 translation by inhibiting Rhob | Overexpressed | Oncomir | Increased tumour cell proliferation; Promotes cell survival and proliferation; Promotes cell growth; Accelerates hepatocyte proliferation | [142, 148–150] |
| miR-7 | Human glioblastoma; human hepatocellular carcinoma (HCC) | Targets IRS; upstream regulator of Akt/mTOR pathway; regulates the expression of mTOR by directly binding to target sites within the 3′-UTR region | Downregulated | Tumour suppressor | Regulates cell invasion | [140,151] |
| miR-99 | Human prostate cancer; hepatocellular carcinoma; adrenocortical tumour | Suppresses gene expression by directly binding to the 3′-UTR of IGF-1R and mTOR | Downregulated | Tumour suppressor | Regulates cell growth Induces cell cycle arrest | [143,147, 152] |
| miR-221 miR-222 | Human glioblastomas; Human gastric cancer; Non-small cell lung cancer; Hepatocellular carcinoma | Activates Akt by regulation of common gene expression in gliomagenesis; Regulate PTEN by targeting PTEN; 3′-UTR binding sequences; Enhance cellular migration through blocking PTEN expression leading to activation of the AKT/mTOR pathway; miR-221 targets REDD-1, a regulator of the mTOR kinase signalling | Overexpressed | Oncomir | Induces cell proliferation and cell invasion | [138,139, 141,153] |
| miR-100 | Adrenocortical tumour Clear cell ovarian cancer | Interacts with the mTOR 3′-UTR and Raptor genes | Downregulated | Tumour suppressor | Regulation of mitosis and cytokinesis | [143,154] |
| miR-101 | Engrafted anaplastic large-cell lymphoma mouse models | Targets mTOR 3′-UTR and suppress mTOR | Downregulated | Tumour suppressor | Reduces tumour growth | [155] |
| miR-199 | Hepatocellular carcinoma Osteosarcoma | miR-199a-3p suppresses gene expression by directly binding to the 3′-UTR of mTOR | Downregulated | Tumour suppressor | G1-phase cell cycle arrest Reduces invasive capability Inhibition of cell migration and cell growth | [156,157] |
| miR-451 | Glioblastoma | Affects mTOR pathway through AMPK by targeting LKB1, 14-3-3ζ(zeta) and TSC1 in response to glucose | Downregulated | Gene regulator | Regulates cell survival and responsiveness to glucose deprivation | [158–160] |
| miR-26 | Murine glioma model | Directly binds to the B2 and B3 sites in the 3′-UTR of PTEN | Overexpressed | Oncomir | Mediates translation and reduces steady-state levels of the protein | [161] |
| miR-181 | Lymphocytic Leukaemia | Targets TCL-1 a co-activator of Akt, which enhances Akt kinase activity | Downregulated | Tumour suppressor | Mediates Akt translation | [162,163] |
| miR-19 | Em-myc model of mouse B-cell lymphoma | Mediates the oncogenic activity miR-17-92 cluster by inhibiting PTEN | Overexpressed | Oncomir | Promotes cell survival | [164] |
| miR-497 | Human colon cancer | Downregulation of miR-497 activates mTOR signalling through upregulation of IGF1-R | Downregulated | Tumour suppressor | Suppresses cell proliferation and invasion | [144] |
| miR-128 | Glioblastoma | Decreases the Bmi-1 oncogene by binding its 3′-UTR, leading to decreased Akt phosphorylation and reduced mTORC1 activity | Downregulated | Tumour suppressor | Reduces proliferation and self-renewal | [165] |
| miR-218 | Oral squamous cell carcinoma | Epigenetic silencing of miR-218 suppresses Akt S473 phosphorylation and reduces Rictor levels | Downregulated | Tumour suppressor | Induces growth inhibition | [166] |
| miR-155 | Waldenström macroglobulinemia | Regulates mTOR by AKT phosphorylation | Overexpressed | Oncomir | Regulates cell proliferation cell-cycle, migration and adhesion | [168] |
© 2013 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
AlQurashi, N.; Hashimi, S.M.; Wei, M.Q. Chemical Inhibitors and microRNAs (miRNA) Targeting the Mammalian Target of Rapamycin (mTOR) Pathway: Potential for Novel Anticancer Therapeutics. Int. J. Mol. Sci. 2013, 14, 3874-3900. https://doi.org/10.3390/ijms14023874
AlQurashi N, Hashimi SM, Wei MQ. Chemical Inhibitors and microRNAs (miRNA) Targeting the Mammalian Target of Rapamycin (mTOR) Pathway: Potential for Novel Anticancer Therapeutics. International Journal of Molecular Sciences. 2013; 14(2):3874-3900. https://doi.org/10.3390/ijms14023874
Chicago/Turabian StyleAlQurashi, Naif, Saeed M. Hashimi, and Ming Q. Wei. 2013. "Chemical Inhibitors and microRNAs (miRNA) Targeting the Mammalian Target of Rapamycin (mTOR) Pathway: Potential for Novel Anticancer Therapeutics" International Journal of Molecular Sciences 14, no. 2: 3874-3900. https://doi.org/10.3390/ijms14023874
APA StyleAlQurashi, N., Hashimi, S. M., & Wei, M. Q. (2013). Chemical Inhibitors and microRNAs (miRNA) Targeting the Mammalian Target of Rapamycin (mTOR) Pathway: Potential for Novel Anticancer Therapeutics. International Journal of Molecular Sciences, 14(2), 3874-3900. https://doi.org/10.3390/ijms14023874