Crosstalk between Oxidative Stress and SIRT1: Impact on the Aging Process



{kind=link}
Abstract
:1. Introduction
2. Falling of the Aging Dogmas
2.1. Free Radical Theory of Aging
2.2. Sirtuins as an Elixir of Longevity
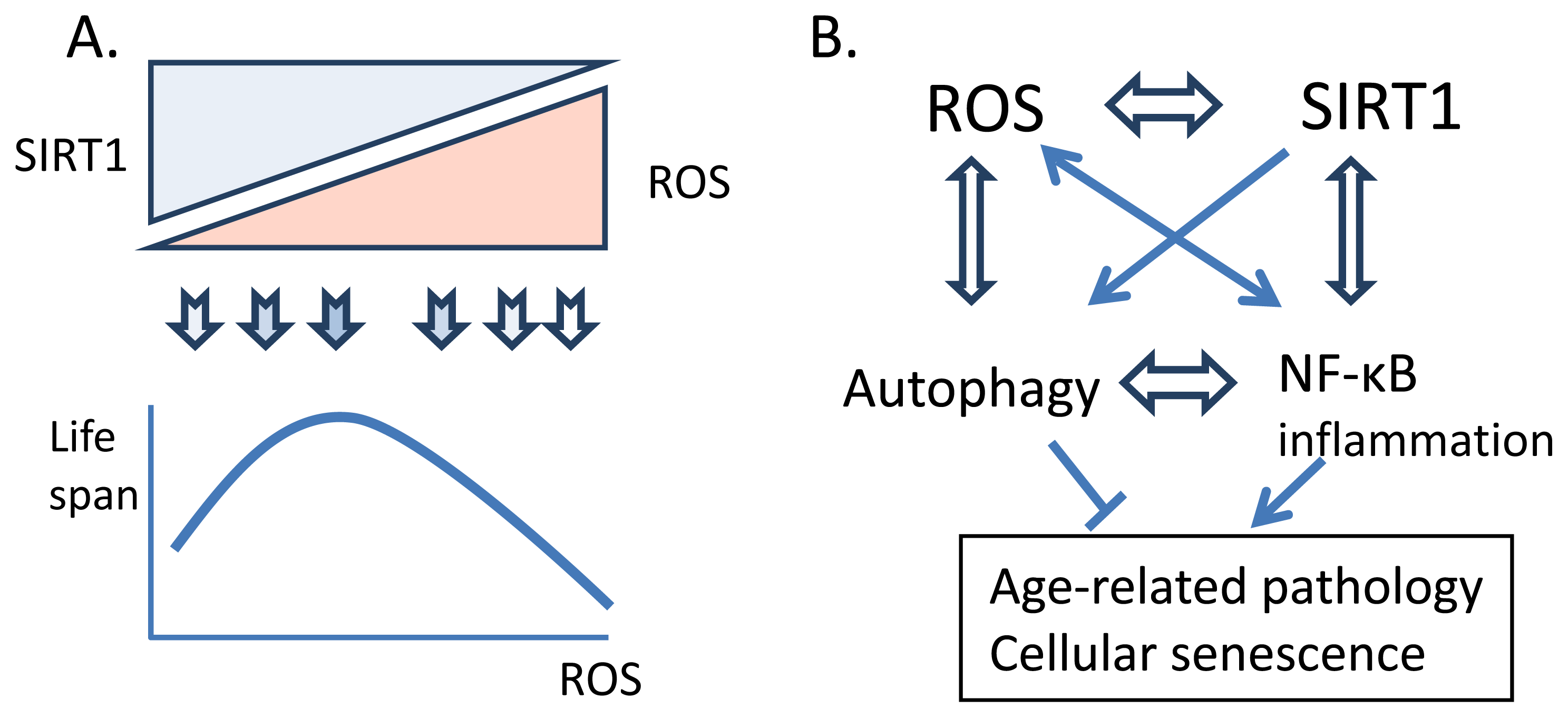
3. Signaling Crosstalk between ROS and SIRT1
3.1. SIRT1 Controls ROS Level
3.2. ROS Control SIRT1 Activity
4. Age-Related Effects of the Crosstalk between ROS and SIRT1
4.1. Autophagy
4.2. NF-κB and Inflammation
4.3. SIRT1, ROS and Insulin/IGF-1 Paradox of Aging
5. Conclusions
Acknowledgments
References
- Green, D.R.; Galluzzi, L.; Kroemer, G. Mitochondria and the autophagy-inflammation-cell death axis in organismal aging. Science 2011, 333, 1109–1112. [Google Scholar]
- Koga, H.; Kaushik, S.; Cuervo, A.M. Protein homeostasis and aging: the importance of exquisite quality control. Ageing Res. Rev 2011, 10, 205–215. [Google Scholar]
- Baraibar, M.A.; Liu, L.; Ahmed, E.K.; Friguet, B. Protein oxidative damage at the crossroads of cellular senescence, aging, and age-related diseases. Oxid. Med. Cell. Longev 2012, 2012, 919832. [Google Scholar]
- Cui, H.; Kong, Y.; Zhang, H. Oxidative stress, mitochondrial dysfunction, and aging. J. Signal. Trans 2012, 2012, 646354. [Google Scholar]
- Salminen, A.; Kaarniranta, K. Regulation of the aging process by autophagy. Trends Mol. Med 2009, 15, 217–224. [Google Scholar]
- Tschopp, J.; Schroder, K. NLRP3 inflammasome activation: The convergence of multiple signalling pathways on ROS production? Nat. Rev. Immunol 2010, 10, 210–215. [Google Scholar]
- Gross, O.; Thomas, C.J.; Guarda, G.; Tschopp, J. The inflammasome: An integrated view. Immunol. Rev 2011, 243, 136–151. [Google Scholar]
- Salminen, A.; Kaarniranta, K.; Kauppinen, A. Inflammaging: Disturbed interplay between autophagy and inflammasomes. Aging (Albany NY) 2012, 4, 166–175. [Google Scholar]
- Salminen, A.; Ojala, J.; Kaarniranta, K.; Kauppinen, A. Mitochondrial dysfunction and oxidative stress activate inflammasomes: Impact on the aging process and age-related diseases. Cell. Mol. Life Sci 2012, 69, 2999–3013. [Google Scholar]
- Canto, C.; Auwerx, J. PGC-1α, SIRT1 and AMPK, an energy sensing network that controls energy expenditure. Curr. Opin. Lipidol 2009, 20, 98–105. [Google Scholar]
- Salminen, A.; Kaarniranta, K. SIRT1: Regulation of longevity via autophagy. Cell. Signal 2009, 21, 1356–1360. [Google Scholar]
- Haigis, M.C.; Sinclair, D.A. Mammalian Sirtuins: Biological insights and disease relevance. Annu. Rev. Pathol. Mech. Dis 2010, 5, 253–295. [Google Scholar]
- Yu, J.; Auwerx, J. Protein deacetylation by SIRT1: An emerging key post-translational modification in metabolic regulation. Pharmacol. Res 2010, 62, 35–41. [Google Scholar]
- Harman, D. Aging: A theory based on free radical and radiation chemistry. J. Gerontol 1956, 11, 298–300. [Google Scholar]
- Harman, D. The biologic clock: The mitochondria? J. Am. Geriatr. Soc 1972, 20, 145–147. [Google Scholar]
- Hulbert, A.J.; Pamplona, R.; Buffenstein, R.; Buttemer, W.A. Life and death: Metabolic rate, membrane composition, and life span of animals. Physiol. Rev 2007, 87, 1175–1213. [Google Scholar]
- Berlett, B.S.; Stadtman, E.R. Protein oxidation in aging, disease, and oxidative stress. J. Biol. Chem 1997, 272, 20313–20316. [Google Scholar]
- Beckman, K.B.; Ames, B.N. The free radical theory of aging matures. Physiol. Rev 1998, 78, 547–581. [Google Scholar]
- Cadenas, E.; Davies, K.J. Mitochondrial free radical generation, oxidative stress, and aging. Free Radic. Biol. Med 2000, 29, 222–230. [Google Scholar]
- Dröge, W. Free radicals in the physiological control of cell function. Physiol. Rev. 2002, 82, 47–95. [Google Scholar]
- Hekimi, S.; Lapointe, J.; Wen, Y. Taking a “good” look at free radicals in the aging process. Trends Cell. Biol 2011, 21, 569–576. [Google Scholar]
- Ristow, M.; Schmeisser, S. Extending life span by increasing oxidative stress. Free Radic. Biol. Med 2011, 51, 327–336. [Google Scholar]
- Andziak, B.; O’Connor, T.P.; Qi, W.; DeWaal, E.M.; Pierce, A.; Chaudhuri, A.R.; van Remmen, H.; Buffenstein, R. High oxidative damage levels in the longest-living rodent, the naked mole-rat. Aging Cell 2006, 5, 463–471. [Google Scholar]
- Buffenstein, R.; Edrey, Y.H.; Yang, T.; Mele, J. The oxidative stress theory of aging: Embattled or invincible? Insights from non-traditional model organisms. AGE 2008, 30, 99–109. [Google Scholar]
- Van Raamsdonk, J.M.; Hekimi, S. Reactive oxygen species and aging in Caenorhabditis elegans: Causal or casual relationship? Antioxid. Redox Signal 2010, 13, 1911–1953. [Google Scholar]
- Lewis, K.N.; Mele, J.; Hornsby, P.J.; Buffenstein, R. Stress resistance in the naked mole-rat: The bare essentials—A mini-review. Gerontology 2012, 58, 453–462. [Google Scholar]
- Buffenstein, R. Negligible senescence in the longest living rodent, the naked mole-rat: Insights from a successfully aging species. J. Comp. Physiol. B 2008, 178, 439–445. [Google Scholar]
- Van Raamsdonk, J.M.; Hekimi, S. Deletion of the mitochondrial superoxide dismutase sod-2 extends lifespan in Caenorhabditis elegans. PLoS Genet 2009, 5, e1000361. [Google Scholar]
- Van Raamsdonk, J.M.; Hekimi, S. Superoxide dismutase is dispensable for normal animal lifespan. Proc. Nat. Acad. Sci. USA 2012, 109, 5785–5790. [Google Scholar]
- Trifunovic, A.; Hansson, A.; Wredenberg, A.; Rovio, A.T.; Dufour, E.; Khvorostov, I.; Spelbrink, J.N.; Wibom, R.; Jacobs, H.T.; Larsson, N.G. Somatic mtDNA mutations cause aging phenotypes without affecting reactive oxygen species production. Proc. Natl. Acad. Sci. USA 2005, 102, 17993–17998. [Google Scholar]
- Sanz, A.; Fernandez-Ayala, D.J.M.; Stefanatos, R.K.A.; Jacobs, H.T. Mitochondrial ROS production correlates with, but does not directly regulate lifespan in drosophila. Aging (Albany NY) 2010, 2, 200–217. [Google Scholar]
- Berger, R.G.; Lunkenbein, S.; Ströhle, A.; Hahn, A. Antioxidants in food: Mere myth or magic medicine? Crit. Rev. Food Sci. Nutr 2012, 52, 162–171. [Google Scholar]
- Poljsak, B.; Milisav, I. The neglected significance of “antioxidative stress”. Oxid. Med. Cell. Longev 2012, 2012, 480895. [Google Scholar]
- Hamanaka, R.B.; Chandel, N.S. Mitochondrial reactive oxygen species regulate cellular signaling and dictate biological outcomes. Trends Biochem. Sci 2010, 35, 505–513. [Google Scholar]
- Gough, D.R.; Cotter, T.G. Hydrogen peroxide: A Jekyll and Hyde signalling molecule. Cell Death Dis 2011, 2, e213. [Google Scholar]
- Ray, P.D.; Huang, B.W.; Tsuji, Y. Reactive oxygen species (ROS) homeostasis and redox regulation in cellular signaling. Cell Signal 2012, 24, 981–990. [Google Scholar]
- Salmeen, A.; Barford, D. Functions and mechanisms of redox regulation of cysteine-based phosphatases. Antioxid. Redox Signal 2005, 7, 560–577. [Google Scholar]
- Blander, G.; Guarente, L. The Sir2 family of protein deacetylases. Annu. Rev. Biochem 2004, 73, 417–435. [Google Scholar]
- Kaeberlein, M.; McVey, M.; Guarente, L. The SIR2/3/4 complex and SIR2 alone promote longevity in Saccharomycetes cerevisiae by two different mechanisms. Genes Dev 1999, 13, 2570–2580. [Google Scholar]
- Sinclair, D.A.; Guarente, L. Extrachromosomal rDNA circles—A cause of aging in yeast. Cell 1997, 91, 1033–1042. [Google Scholar]
- Tissenbaum, H.A.; Guarente, L. Increased dosage of a sir-2 gene extends lifespan in Caenorhabditis elegans. Nature 2001, 410, 227–230. [Google Scholar]
- Rogina, B.; Helfand, S.L. Sir2 mediates longevity in the fly through a pathway related to caloric restriction. Proc. Natl. Acad. Sci. USA 2004, 101, 15998–16003. [Google Scholar]
- Howitz, K.T.; Bitterman, K.J.; Cohen, H.Y.; Lamming, D.W.; Lavu, S.; Wood, J.G.; Zipkin, R.E.; Chung, P.; Kisielewski, A.; Zhang, L.L.; et al. Small molecule activators of Sirtuins extend Saccharomycetes cerevisiae lifespan. Nature 2003, 425, 191–196. [Google Scholar]
- Bauer, J.H.; Goupil, S.; Garber, G.B.; Helfand, S.L. An accelerated assay for the identification of lifespan-extending interventions in Drosophila melanogaster. Proc. Natl. Acad. Sci. USA 2004, 101, 12980–12985. [Google Scholar]
- Viswanathan, M.; Kim, S.K.; Berdichevsky, A.; Guarente, L. A role for SIR-2.1 regulation of ER stress response genes in determining C. elegans life span. Dev. Cell 2005, 9, 605–615. [Google Scholar]
- Pearson, K.J.; Baur, J.A.; Lewis, K.N.; Peshkin, L.; Price, N.L.; Labinskyy, N.; Swindell, W.R.; Kamara, D.; Minor, R.K.; Perez, E.; et al. Resveratrol delays age-related deterioration and mimics transcriptional aspects of dietary restriction without extending life span. Cell Metab 2008, 8, 157–168. [Google Scholar]
- Burnett, C.; Valentini, S.; Cabreiro, F.; Goss, M.; Somogyvári, M.; Piper, M.D.; Hoddinott, M.; Sutphin, G.L.; Leko, V.; McElwee, J.J.; et al. Absence of effects of Sir2 overexpression on lifespan in C. elegans and Drosophila. Nature 2011, 477, 482–485. [Google Scholar]
- Fabrizio, P.; Gattazzo, C.; Battistella, L.; Wei, M.; Cheng, C.; McGrew, K.; Longo, V.D. Sir2 blocks extreme life-span extension. Cell 2005, 123, 655–667. [Google Scholar]
- Park, S.J.; Ahmad, F.; Philp, A.; Baar, K.; Williams, T.; Luo, H.; Ke, H.; Rehmann, H.; Taussig, R.; Brown, A.L.; et al. Resveratrol ameliorates aging-related metabolic phenotypes by inhibiting cAMP phosphodiesterases. Cell 2012, 148, 421–433. [Google Scholar]
- Gerhart-Hines, Z.; Dominy, J.E., Jr.; Blättler, S.M.; Jedrychowski, M.P.; Banks, A.S.; Lim, J.H.; Chim, H.; Gygi, S.P.; Puigserver, P. The cAMP/PKA pathway rapidly activates SIRT1 to promote fatty acid oxidation independently of changes in NAD+. Mol. Cell 2011, 44, 851–863. [Google Scholar]
- Kanfi, Y.; Naiman, S.; Amir, G.; Peshti, V.; Zinman, G.; Nahum, L.; Bar-Joseph, Z.; Cohen, H.Y. The sirtuin SIRT6 regulates lifespan in male mice. Nature 2012, 483, 218–221. [Google Scholar]
- Tennen, R.I.; Chua, K.F. Chromatin regulation and genome maintenance by mammalian SIRT6. Trends Biochem. Sci 2011, 36, 39–46. [Google Scholar]
- Mostoslavsky, R.; Chua, K.F.; Lombard, D.B.; Pang, W.W.; Fischer, M.R.; Gellon, L.; Liu, P.; Mostoslavsky, G.; Franco, S.; Murphy, M.M.; et al. Genomic instability and aging-like phenotype in the absence of mammalian SIRT6. Cell 2006, 124, 315–329. [Google Scholar]
- Guarente, L. Sirtuins, aging, and medicine. N. Engl. J. Med 2011, 364, 2235–2244. [Google Scholar]
- Houtkooper, R.H.; Pirinen, E.; Auwerx, J. Sirtuins as regulators of metabolism and healthspan. Nat. Rev. Mol. Cell Biol 2012, 13, 225–238. [Google Scholar]
- Longo, V.D.; Kennedy, B.K. Sirtuins in aging and age-related disease. Cell 2006, 126, 257–268. [Google Scholar]
- Baur, J.A.; Ungvari, Z.; Minor, R.K.; Le Couteur, D.G.; de Cabo, R. Are sirtuins viable targets for improving healthspan and lifespan. Nat. Rev. Drug Disc 2012, 11, 443–461. [Google Scholar]
- Alcendor, R.R.; Gao, S.; Zhai, P.; Zablocki, D.; Holle, E.; Yu, X.; Tian, B.; Wagner, T.; Vatner, S.F.; Sadoshima, J. Sirt1 regulates aging and resistance to oxidative stress in the heart. Circ. Res 2007, 100, 1512–1521. [Google Scholar]
- Brunet, A.; Sweeney, L.B.; Sturgill, J.F.; Chua, K.F.; Greer, P.L.; Lin, Y.; Tran, H.; Ross, S.E.; Mostoslavsky, R.; Cohen, H.Y.; et al. Stress-dependent regulation of FOXO transcription factors by the SIRT1 deacetylase. Science 2004, 303, 2011–2015. [Google Scholar]
- Van der Horst, A.; Tertoolen, L.G.; de Vries-Smits, L.M.; Frye, R.A.; Medema, R.H.; Burgering, B.M. FOXO4 is acetylated upon peroxide stress and deacetylated by the longevity protein hSir2SIRT1. J. Biol. Chem 2004, 279, 28873–28879. [Google Scholar]
- Sengupta, A.; Molkentin, J.D.; Paik, J.H.; DePinho, R.A.; Yutzey, K.E. FoxO transcription factors promote cardiomyce survival upon induction of oxidative stress. J. Biol. Chem 2011, 286, 7468–7478. [Google Scholar]
- Xiong, S.; Salazar, G.; Patrushev, N.; Alexander, R.W. FoxO1 mediates an autofeedback loop regulating SIRT1 expression. J. Biol. Chem 2011, 286, 5289–5299. [Google Scholar]
- Yamamoto, T.; Sadoshima, J. Protection of the heart against ischemia/reperfusion by silent information regulator 1. Trends Cardiovasc. Med 2011, 21, 27–32. [Google Scholar]
- Yeung, F.; Hoberg, J.E.; Ramsey, C.S.; Keller, M.D.; Jones, D.R.; Frye, R.A.; Mayo, M.W. Modulation of NF-κB-dependent transcription and cell survival by the SIRT1 deacetylase. EMBO J 2004, 23, 2369–2380. [Google Scholar]
- Salminen, A.; Kauppinen, A.; Suuronen, T.; Kaarniranta, K. SIRT1 longevity factor suppresses NF-κB-driven immune responses: Regulation of aging via NF-κB acetylation? BioEssays 2008, 30, 939–942. [Google Scholar]
- Rajendran, R.; Garva, R.; Krstic-Demonacos, M.; Demonacos, C. Sirtuins: Molecular traffic lights in the crossroad of oxidative stress, chromatin remodeling, and transcription. J. Biomed. Biotechnol 2011, 2011, 368276. [Google Scholar]
- Salminen, A.; Hyttinen, J.M.T.; Kaarniranta, K. AMP-activated protein kinase inhibits NF-κB signaling and inflammation: Impact on healthspan and lifespan. J. Mol. Med 2011, 89, 667–676. [Google Scholar]
- Sumimoto, H. Structure, regulation and evolution of Nox-family NADPH oxidases that produce reactive oxygen species. FEBS J 2008, 275, 3249–3277. [Google Scholar]
- Anrather, J.; Racchumi, G.; Iadecola, C. NF-κB regulates phagocytic NADPH oxidase by inducing expression of gp91phox. J. Biol. Chem 2006, 281, 5657–5667. [Google Scholar]
- Manea, A.; Manea, S.A.; Gafencu, A.V.; Raicu, M. Regulation of NADPH oxidase subunit p22(phox) by NF-κB in human aortic smooth muscle cells. Arch. Physiol. Biochem 2007, 113, 163–172. [Google Scholar]
- Manea, A.; Tanase, L.I.; Raicu, M.; Simionescu, M. Transcriptional regulation of NADPH oxidase isoforms, Nox1 and Nox4, by nuclear factor-κB in human aortic smooth muscle cells. Biochem. Biophys. Res. Commun 2010, 396, 901–907. [Google Scholar]
- Xie, Q.; Kashiwabara, Y.; Nathan, C. Role of transcription factor NF-κB/Rel in induction of nitric oxide synthase. J. Biol. Chem 1994, 269, 4705–4708. [Google Scholar]
- Sakitani, K.; Nishizawa, M.; Inoue, K.; Masu, Y.; Okumura, T.; Ito, S. Synergistic regulation of inducible nitric oxide synthase gene by CCAAT/enhancer-binding protein β and nuclear factor-κB in hepatocytes. Genes Cells 1998, 3, 321–330. [Google Scholar]
- Morgan, M.J.; Liu, Z. Crosstalk of reactive oxygen species and NF-κB signaling. Cell Res 2011, 21, 103–115. [Google Scholar]
- Kawai, Y.; Garduno, L.; Theodore, M.; Yang, J.; Arinze, I.J. Acetylation-deacetylation of the transcription factor Nrf2 (nuclear factor erythroid 2-related factor 2) regulates its transcriptional activity and nucleocytoplasmic localization. J. Biol. Chem 2011, 286, 7629–7640. [Google Scholar]
- Nguyen, T.; Sherratt, P.J.; Pickett, C.B. Regulatory mechanisms controlling gene expression mediated by the antioxidant response element. Annu. Rev. Pharmacol. Toxicol 2003, 43, 233–260. [Google Scholar]
- Ishii, T.; Yanagawa, T. Stress-induced peroxiredoxins. Subcell. Biochem 2007, 44, 375–384. [Google Scholar]
- Tanito, M.; Agbaga, M.P.; Anderson, R.E. Upregulation of thioredoxin system via Nrf2-antioxidant responsive element pathway in adaptive-retinal neuroprotection in vivo and in vitro. Free Radic. Biol. Med 2007, 42, 1838–1850. [Google Scholar]
- Zhang, D.D.; Hannink, M. Distinct cysteine residues in Keap1 are required for Keap1-dependent ubiquitination of Nrf2 and for stabilization of Nrf2 by chemopreventive agents and oxidative stress. Mol. Cell. Biol 2003, 23, 8137–8151. [Google Scholar]
- Taguchi, K.; Fujikawa, N.; Komatsu, M.; Ishii, T.; Unno, M.; Akaike, T.; Motohashi, H.; Yamamoto, M. Keap1 degradation by autophagy for the maintenance of redox homeostasis. Proc. Natl. Acad. Sci. USA 2012, 109, 13561–13566. [Google Scholar]
- Komatsu, M.; Kurokawa, H.; Waguri, S.; Taguchi, K.; Kobayashi, A.; Ichimura, Y.; Sou, Y.S..; Ueno, I.; Sakamoto, A.; Tong, K.I.; et al. The selective autophagy substrate p62 activates the stress responsive transcription factor Nrf2 through inactivation of Keap1. Nat. Cell Biol. 2010, 12, 213–223. [Google Scholar]
- Jain, A.; Lamark, T.; Sjottem, E.; Larsen, K.B.; Awuh, J.A.; Overvatn, A.; McMahon, M.; Hayes, J.D.; Johansen, T. p62/SQSTM1 is a target gene for transcription factor NRF2 and creates a positive feedback loop by inducing antioxidant response element-driven gene transcription. J. Biol. Chem. 2010, 285, 22576–22591. [Google Scholar]
- Murphy, M.P. How mitochondria produce reactive oxygen species. Biochem. J 2009, 417, 1–13. [Google Scholar]
- Fernandez-Marcos, P.J.; Auwerx, J. Regulation of PGC-1α, a nodal regulator of mitochondrial biogenesis. Am. J. Clin. Nutr 2011, 93, 884S–890S. [Google Scholar]
- Scarpulla, R.C. Metabolic control of mitochondrial biogenesis through the PGC-1 family regulatory network. Biochim. Biophys. Acta 2011, 1813, 1269–1278. [Google Scholar]
- Nemoto, S.; Fergusson, M.M.; Finkel, T. SIRT1 functionally interacts with the metabolic regulator and transcriptional coactivator PGC-1α. J. Biol. Chem 2005, 280, 16456–16460. [Google Scholar]
- Rodgers, J.T.; Lerin, C.; Haas, W.; Gygi, S.P.; Spiegelman, B.M.; Puigserver, P. Nutrient control of glucose homeostasis through a complex of PGC-1α and SIRT1. Nature 2005, 434, 113–118. [Google Scholar]
- Gurd, B.J. Deacetylation of PGC-1α by SIRT1: Importance for skeletal muscle function and exercise-induced mitochondrial biogenesis. Appl. Physiol. Nutr. Metab 2011, 36, 589–597. [Google Scholar]
- Philp, A.; Chen, A.; Lan, D.; Meyer, G.A.; Murphy, A.N.; Knapp, A.E.; Olfert, I.M.; McCurdy, C.E.; Marcotte, G.R.; Hogan, M.C.; et al. Sirtuin 1 (SIRT1) deacetylase activity is not required for mitochondrial biogenesis or peroxisome proliferator-activated receptor-γ coactivator-1α (PGC-1α) deacetylation following endurance exercise. J. Biol. Chem 2011, 286, 30561–30570. [Google Scholar]
- Amat, R.; Planavila, A.; Chen, S.L.; Iglesias, R.; Giralt, M.; Villarroya, F. SIRT1 controls the transcription of the peroxisome proliferator-activated receptor-γ co-activator-1α (PGC-1α) gene in skeletal muscle through the PGC-1α autoregulatory loop and interaction with MyoD. J. Biol. Chem 2009, 284, 21872–21880. [Google Scholar]
- Brand, M.D.; Esteves, T.C. Physiological functions of the mitochondrial uncoupling proteins UCP2 and UCP3. Cell Metab 2005, 2, 85–93. [Google Scholar]
- Mailloux, R.J.; Harper, M.E. Uncoupling proteins and the control of mitochondrial reactive oxygen species production. Free Radic. Biol. Med 2011, 51, 1106–1115. [Google Scholar]
- Andrews, Z.B.; Horvath, T.L. Uncoupling protein-2 regulates lifespan in mice. Am. J. Physiol. Endocrinol. Metab 2009, 296, E621–E627. [Google Scholar]
- Vidal-Puig, A.J.; Grujic, D.; Zhang, C.Y.; Hagen, T.; Boss, O.; Ido, Y.; Szczepanik, A.; Wade, J.; Mootha, V.; Cortright, R.; et al. Energy metabolism in uncoupling protein 3 gene knockout mice. J. Biol. Chem 2000, 275, 16258–16266. [Google Scholar]
- Bordone, L.; Motta, M.C.; Picard, F.; Robinson, A.; Jhala, U.S.; Apfeld, J.; McDonagh, T.; Lemieux, M.; McBurney, M.; Szilvasi, A.; et al. Sirt1 regulates insulin secretion by repressing UCP2 in pancreatic beta cells. PLoS Biol 2006, 4, e31. [Google Scholar]
- Amat, R.; Solanes, G.; Giralt, M.; Villarroya, F. SIRT1 is involved in glucocorticoids-mediated control of uncoupling protein-3 gene transcription. J. Biol. Chem 2007, 282, 34066–34076. [Google Scholar]
- Diano, S.; Horvath, T.L. Mitochondrial uncoupling protein 2 (UCP2) in glucose and lipid metabolism. Trends Mol. Med 2012, 18, 52–58. [Google Scholar]
- Kamata, H.; Honda, S.; Maeda, S.; Chang, L.; Hirata, H.; Karin, M. Reactive oxygen species promote TNF-α-induced death and sustained JNK activation by inhibiting MAP kinase phosphatases. Cell 2005, 120, 649–661. [Google Scholar]
- Matsukawa, J.; Matsuzawa, A.; Takeda, K.; Ichijo, H. The ASK1-MAP kinase cascades in mammalian stress response. J. Biochem 2004, 136, 261–265. [Google Scholar]
- Nasrin, N.; Kaushik, V.K.; Fortier, E.; Wall, D.; Pearson, K.J.; de Cabo, R.; Bordone, L. JNK1 phosphorylates SIRT1 and promotes its enzymatic activity. PLoS One 2009, 4, e8414. [Google Scholar]
- Gao, Z.; Zhang, J.; Kheterpal, I.; Kennedy, N.; Davis, R.J.; Ye, J. Sirtuin 1 (SIRT1) protein degradation in response to persistent c-Jun N-terminal kinase 1 (JNK1) activation contributes to hepatic steatosis in obesity. J. Biol. Chem 2011, 286, 22227–22234. [Google Scholar]
- Wang, M.C.; Bohmann, D.; Jasper, H. JNK signaling confers tolerance to oxidative stress and extends lifespan in Drosophila. Dev Cell 2003, 5, 811–816. [Google Scholar]
- Biteau, B.; Karpac, J.; Hwangbo, D.; Jasper, H. Regulation of Drosophila lifespan by JNK signaling. Exp. Gerontol 2011, 46, 349–354. [Google Scholar]
- Cardaci, S.; Filomeni, G.; Ciriolo, M.R. Redox implications of AMPK-mediated signal transduction beyond energetic clues. J. Cell Sci 2012, 125, 2115–2125. [Google Scholar]
- Canto, C.; Gerhart-Hines, Z.; Feige, J.N.; Lagouge, M.; Noriega, L.; Molne, J.C.; Elliott, P.J.; Puigserver, P.; Auwerx, J. AMPK regulates energy expenditure by modulating NAD+ metabolism and SIRT1 activity. Nature 2009, 458, 1056–1060. [Google Scholar]
- Zmijewski, J.W.; Banerjee, S.; Bae, H.; Friggeri, A.; Lazarowski, E.R.; Abraham, E. Exposure to hydrogen peroxide induces oxidation and activation of AMP-activated protein kinase. J. Biol. Chem 2010, 285, 33154–33164. [Google Scholar]
- Mungai, P.T.; Waypa, G.B.; Jairaman, A.; Prakriya, M.; Dokic, D.; Ball, M.K.; Schumacker, P.T. Hypoxia triggers AMPK activation through reactive oxygen species-mediated activation of calcium release-activated calcium channels. Mol. Cell. Biol 2011, 31, 3531–3545. [Google Scholar]
- Emerling, B.M.; Weinberg, F.; Snyder, C.; Burgess, Z.; Mutlu, G.M.; Viollet, B.; Budinger, G.R.S.; Chandel, N.S. Hypoxic activation of AMPK is dependent on mitochondrial ROS but independent of an increase in AMP/ATP ratio. Free Radic. Biol. Med 2009, 46, 1386–1391. [Google Scholar]
- Zhang, J.; Xie, Z.; Dong, Y.; Wang, S.; Liu, C.; Zou, M.H. Identification of nitric oxide as an endogenous activator of the AMP-activated protein kinase in vascular endothelial cells. J. Biol. Chem 2008, 283, 27452–27461. [Google Scholar]
- Salminen, A.; Kaarniranta, K. AMP-activated protein kinase (AMPK) controls the aging process via an integrated signaling network. Ageing Res. Rev. 2012, 11, 230–241. [Google Scholar]
- Yamakuchi, M.; Ferlito, M.; Lowenstein, C.J. miR-34a repression of SIRT1 regulates apoptosis. Proc. Natl. Acad. Sci. USA 2008, 105, 13421–13426. [Google Scholar]
- Chen, F.; Hu, S.J. Effect of microRNAs-34a in cell cycle, differentiation, and apoptosis: A review. J. Biochem. Mol. Toxicol 2012, 26, 79–86. [Google Scholar]
- Bai, X.Y.; Ma, Y.; Ding, R.; Fu, B.; Shi, S.; Chen, X.M. miR-335 and miR-34a promote renal senescence by suppressing mitochondrial antioxidative enzymes. J. Am. Soc. Nephrol 2011, 22, 1252–1261. [Google Scholar]
- Li, N.; Muthusamy, S.; Liang, R.; Sarojini, H.; Wang, E. Increased expression of miR-34a and miR-93 in rat liver during aging, and their impact on the expression of Mgst1 and Sirt1. Mech. Ageing Dev 2011, 132, 75–85. [Google Scholar]
- Ito, T.; Yagi, S.; Yamakuchi, M. MicroRNA-34a regulation of endothelial senescence. Biochem. Biophys. Res. Commun 2010, 398, 735–740. [Google Scholar]
- Liu, B.; Chen, Y.; St. Clair, D.K. ROS and p53: A versatile partnership. Free Radic. Biol. Med 2008, 44, 1529–1535. [Google Scholar]
- Ladelfa, M.F.; Toledo, M.F.; Laiseca, J.E.; Monte, M. Interaction of p53 with tumor suppressive and oncogenic signaling pathways to control cellular reactive oxygen species production. Antioxid. Redox Signal 2011, 15, 1749–1761. [Google Scholar]
- Guo, Z.; Kozlov, S.; Lavin, M.F.; Person, M.D.; Paull, T.T. ATM activation by oxidative stress. Science 2010, 330, 517–521. [Google Scholar]
- Maillet, A.; Pervaiz, S. Redox regulation of p53, redox effectors regulated by p53: A subtle balance. Antioxid. Redox Signal 2012, 16, 1285–1294. [Google Scholar]
- Furukawa, A.; Tada-Oikawa, S.; Kawanishi, S.; Oikawa, S. H2O2 accelerates cellular senescence by accumulation of acetylated p53 via decrease in the function of SIRT1 by NAD+ depletion. Cell. Physiol. Biochem 2007, 20, 45–54. [Google Scholar]
- Caito, S.; Rajendrasozhan, S.; Cook, S.; Chung, S.; Yao, H.; Friedman, A.E.; Brookes, P.S.; Rahman, I. SIRT1 is a redox-sensitive deacetylase that is post-translationally modified by oxidants and carbonyl stress. FASEB J 2010, 24, 3145–3159. [Google Scholar]
- Cai, W.; Ramdas, M.; Zhu, L.; Chen, X.; Striker, G.E.; Vlassara, H. Oral advanced glycation endproducts (AGEs) promote insulin resistance and diabetes by depleting the antioxidant defenses AGE receptor-1 and sirtuin 1. Proc. Natl. Acad. Sci. USA 2012, 109, 15888–15893. [Google Scholar]
- Rajendrasozhan, S.; Yang, S.R.; Kinnula, V.L.; Rahman, I. SIRT1, an antiinflammatory and antiaging protein, is decreased in lungs of patients with chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med 2008, 177, 861–870. [Google Scholar]
- Zee, R.S.; Yoo, C.B.; Pimentel, D.R.; Perlman, D.H.; Burgoyne, J.R.; Hou, X.; McComb, M.E.; Costello, C.E.; Cohen, R.A.; Bachschmid, M.M. Redox regulation of Sirtuin-1 by S-glutathiolation. Antioxid. Redox Signal 2010, 13, 1023–1031. [Google Scholar]
- Kornberg, M.D.; Sen, N.; Hara, M.R.; Juluri, K.R.; Nguyen, J.V.; Snowman, A.M.; Law, L.; Hester, L.D.; Snyder, S.H. GAPDH mediates nitrosylation of nuclear proteins. Nat. Cell Biol 2010, 12, 1094–1100. [Google Scholar]
- Tong, C.; Morrison, A.; Mattison, S.; Qian, S.; Bryniarski, M.; Rankin, B.; Wang, J.; Thomas, D.P.; Li, J. Impaired SIRT1 nucleocytoplasmic shuttling in the senescent heart during ischemic stress. FASEB J. 2013, 27. fj.12-216474. [Google Scholar]
- Yang, Y.; Fu, W.; Chen, J.; Olashaw, N.; Zhang, X.; Nicosia, S.V.; Bhalla, K.; Bai, W. SIRT1 sumoylation regulates its deacetylase activity and cellular response to genotoxic stress. Nat. Cell Biol 2007, 9, 1253–1262. [Google Scholar]
- Cuervo, A.M. Autophagy and aging: Keeping that old broom working. Trends Genet 2008, 24, 604–612. [Google Scholar]
- De Magalhaes, J.P.; Curado, J.; Church, G.M. Meta-analysis of age-related gene expression profiles identifies common signatures of aging. Bioinformatics 2009, 25, 875–881. [Google Scholar]
- Salminen, A.; Kaarniranta, K. NF-κB signaling in the aging process. J. Clin. Immunol 2009, 29, 397–405. [Google Scholar]
- Cannizzo, E.S.; Clement, C.C.; Sahu, R.; Follo, C.; Santambrogio, L. Oxidative stress, inflammaging and immunosenescence. J. Proteomics 2011, 74, 2313–2323. [Google Scholar]
- Mizushima, N.; Klionsky, D.J. Protein turnover via autophagy: Implications for metabolism. Annu. Rev. Nutr 2007, 27, 19–40. [Google Scholar]
- Mizushima, N.; Komatsu, M. Autophagy: Renovation of cells and tissues. Cell 2011, 147, 728–741. [Google Scholar]
- Lee, I.H.; Cao, L.; Mostoslavsky, R.; Lombard, D.B.; Liu, J.; Bruns, N.E.; Tsokos, M.; Alt, F.W.; Finkel, T. A role for the NAD-dependent deacetylase Sirt1 in the regulation of autophagy. Proc. Natl. Acad. Sci. USA 2008, 105, 3374–3379. [Google Scholar]
- Wang, P.; Guan, Y.F.; Du, H.; Zhai, Q.W.; Su, D.F.; Miao, C.Y. Induction of autophagy contributes to the neuroprotection of nicotinamide phosphoribosyltransferase in cerebral ischemia. Autophagy 2012, 8, 77–87. [Google Scholar]
- Hariharan, N.; Maejima, Y.; Nakae, J.; Paik, J.; DePinho, R.A.; Sadoshima, J. Deacetylation of FoxO by Sirt1 plays an essential role in mediating starvation-induced autophagy in cardiac myocytes. Circ. Res 2010, 107, 1470–1482. [Google Scholar]
- Hyttinen, J.M.T.; Niittykoski, M.; Salminen, A.; Kaarniranta, K. Maturation of autophagosomes and endosomes: A key role for Rab7. Biochim. Biophys. Acta 2013, 1833, 503–510. [Google Scholar]
- Mammucari, C.; Milan, G.; Romanello, V.; Masiero, E.; Rudolf, R.; Del Piccolo, P.; Burden, S.J.; Di Lisi, R.; Sandri, C.; Zhao, J.; et al. FoxO3 controls autophagy in skeletal muscle in vivo. Cell Metab 2007, 6, 458–471. [Google Scholar]
- Nadtochiy, S.M.; Yao, H.; McBurney, M.W.; Gu, W.; Guarente, L.; Rahman, I.; Brookes, P.S. SIRT1-mediated acute cardioprotection. Am. J. Physiol. Heart Circ. Physiol 2011, 301, H1506–H1512. [Google Scholar]
- Jeong, J.K.; Moon, M.H.; Lee, Y.J.; Seol, J.W.; Park, S.Y. Autophagy induced by the class III histone deacetylase Sirt1 prevents prion peptide neurotoxicity. Neurobiol. Aging 2013, 34, 146–156. [Google Scholar]
- Gurd, B.J.; Yoshida, Y.; Lally, J.; Holloway, G.P.; Bonen, A. The deacetylase enzyme SIRT1 is not associated with oxidative capacity in rat heart and skeletal muscle and its overexpression reduces mitochondrial biogenesis. J. Physiol 2009, 587, 1817–1828. [Google Scholar]
- Kawashima, T.; Inuzuka, Y.; Okuda, J.; Kato, T.; Niizuma, S.; Tamaki, Y.; Iwanaga, Y.; Kawamoto, A.; Narazaki, M.; Matsuda, T.; et al. Constitutive SIRT1 overexpression impairs mitochondria and reduces cardiac function in mice. J. Mol. Cell. Cardiol 2011, 51, 1026–1036. [Google Scholar]
- Huang, J.; Lam, G.Y.; Brumell, J.H. Autophagy signaling through reactive oxygen species. Antioxid. Redox Signal 2011, 14, 2215–2231. [Google Scholar]
- Scherz-Shouval, R.; Elazar, Z. Regulation of autophagy by ROS: Physiology and pathology. Trends Biochem. Sci 2011, 36, 30–38. [Google Scholar]
- Lee, J.; Giordano, S.; Zhang, J. Autophagy, mitochondria and oxidative stress: Cross-talk and redox signalling. Biochem. J 2012, 441, 523–540. [Google Scholar]
- Chen, Y.; Azad, M.B.; Gibson, S.B. Superoxide is the major reactive oxygen species regulating autophagy. Cell Death Differ 2009, 16, 1040–1052. [Google Scholar]
- Scherz-Shouval, R.; Shvets, E.; Fass, E.; Shorer, H.; Gil, L.; Elazar, Z. Reactive oxygen species are essential for autophagy and specifically regulate the activity of Atg4. EMBO J 2007, 26, 1749–1760. [Google Scholar]
- Wei, Y.; Pattingre, S.; Sinha, S.; Bassik, M.; Levine, B. JNK1-mediated phosphorylation of Bcl-2 regulates starvation-induced autophagy. Mol. Cell 2008, 30, 678–688. [Google Scholar]
- Park, K.J.; Lee, S.H.; Lee, C.H.; Jang, J.Y.; Chung, J.; Kwon, M.H.; Kim, Y.S. Upregulation of Beclin-1 expression and phosphorylation of Bcl-2 and p53 are involved in the JNK-mediated autophagic cell death. Biochem. Biophys. Res. Commun. 2009, 382, 726–729. [Google Scholar]
- Zhang, X.Y.; Wu, X.Q.; Deng, R.; Sun, T.; Feng, G.K.; Zhu, X.F. Upregulation of Sestrin 2 expression via JNK pathway activation contributes to autophagy induction in cancer cells. Cell Signal 2013, 25, 150–158. [Google Scholar]
- Xu, P.; Das, M.; Reilly, J.; Davis, R.J. JNK regulates FoxO-dependent autophagy in neurons. Genes Dev 2011, 25, 310–322. [Google Scholar]
- Sarkar, S.; Korolchuk, V.I.; Renna, M.; Imarisio, S.; Fleming, A.; Williams, A.; Garcia-Arencibia, M.; Rose, C.; Luo, S.; Underwood, B.R.; et al. Complex inhibitory effects of nitric oxide on autophagy. Mol. Cell 2011, 43, 19–32. [Google Scholar]
- Alexander, A.; Cai, S.L.; Kim, J.; Nanez, A.; Sahin, M.; MacLean, K.H.; Inoki, K.; Guan, K.L.; Shen, J.; Person, M.D.; et al. ATM signals to TSC2 in the cytoplasm to regulate mTORC1 in response to ROS. Proc. Natl. Acad. Sci. USA 2010, 107, 4153–4158. [Google Scholar]
- Li, L.; Chen, Y.; Gibson, S.B. Starvation-induced autophagy is regulated by mitochondrial reactive oxygen species leading to AMPK activation. Cell Signal 2013, 25, 50–65. [Google Scholar]
- McClung, J.M.; Judge, A.R.; Powers, S.K.; Yan, Z. p38 MAPK links oxidative stress to autophagy-related gene expression in cachectic muscle wasting. Am. J. Physiol. Cell. Physiol 2010, 298, C542–C549. [Google Scholar]
- Luo, Y.; Zou, P.; Zou, J.; Wang, J.; Zhou, D.; Liu, L. Autophagy regulates ROS-induced cellular senescence via p21 in a p38 MAPKα dependent manner. Exp. Gerontol 2011, 46, 860–867. [Google Scholar]
- Lee, J.W.; Park, S.; Takahashi, Y.; Wang, H.G. The association of AMPK with ULK1 regulates autophagy. PLoS One 2010, 5, e15394. [Google Scholar]
- Sanchez, A.M.; Csibi, A.; Raibon, A.; Cornille, K.; Gay, S.; Bernardi, H.; Candau, R. AMPK promotes skeletal muscle autophagy through activation of forkhead FoxO3a and interaction with Ulk1. J. Cell. Biochem 2012, 113, 695–710. [Google Scholar]
- Bensimon, A.; Aebersold, R.; Shiloh, Y. Beyond ATM: The protein kinase landscape of the DNA damage response. FEBS Lett 2011, 585, 1625–1639. [Google Scholar]
- Wang, Y.; Nartiss, Y.; Steipe, B.; McQuibban, G.A.; Kim, P.K. ROS-induced mitochondrial depolarization initiates PARK2/PARKIN-dependent mitochondrial degradation by autophagy. Autophagy 2012, 8, 1462–1476. [Google Scholar]
- Vallabhapurapu, S.; Karin, M. Regulation and function of NF-κB transcription factors in the immune system. Annu. Rev. Immunol 2009, 27, 693–733. [Google Scholar]
- Karin, M.; Lin, A. NF-κB at the crossroads of life and death. Nat. Immunol 2002, 3, 221–227. [Google Scholar]
- Perkins, N.D. Integrating cell-signalling pathways with NF-κB and IKK function. Nat. Rev. Mol. Cell Biol 2007, 8, 49–62. [Google Scholar]
- Johnson, R.F.; Perkins, N.D. Nuclear factor-κB, p53, and mitochondria: Regulation of cellular metabolism and the Warburg effect. Trends Biochem. Sci 2012, 37, 317–324. [Google Scholar]
- Helenius, M.; Hänninen, M.; Lehtinen, S.K.; Salminen, A. Changes associated with aging and replicative senescence in the regulation of transcription factor nuclear factor-κB. Biochem. J 1996, 318, 603–608. [Google Scholar]
- Helenius, M.; Kyrylenko, S.; Vehviläinen, P.; Salminen, A. Characterization of aging-associated up-regulation of constitutive nuclear factor-κB binding activity. Antioxid. Redox Signal 2001, 3, 147–156. [Google Scholar]
- Adler, A.S.; Sinha, S.; Kawahara, T.L.; Zhang, J.Y.; Segal, E.; Chang, H.Y. Motif module map reveals enforcement of aging by continual NF-κB activity. Genes Dev 2007, 21, 3244–3257. [Google Scholar]
- Csiszar, A.; Wang, M.; Lakatta, E.G.; Ungvari, Z. Inflammation and endothelial dysfunction during aging: Role of NF-κB. J. Appl. Physiol 2008, 105, 1333–1341. [Google Scholar]
- Cribbs, D.H.; Berchtold, N.C.; Perreau, V.; Coleman, P.D.; Rogers, J.; Tenner, A.J.; Cotman, C.W. Extensive innate immune gene activation accompanies brain aging, increasing vulnerability to cognitive decline and neurodegeneration: A microarray study. J. Neuroinflamm 2012, 9, 179. [Google Scholar]
- Lin, L.; Hron, J.D.; Peng, S.L. Regulation of NF-κB, Th activation, and autoinflammation by the forkhead transcription factor Foxo3a. Immunity 2004, 21, 203–213. [Google Scholar]
- Gillum, M.P.; Kotas, M.E.; Erion, D.M.; Kursawe, R.; Chatterjee, P.; Nead, K.T.; Muise, E.S.; Hsiao, J.J.; Frederick, D.W.; Yonemitsu, S.; et al. SirT1 regulates adipose tissue inflammation. Diabetes 2011, 60, 3235–3245. [Google Scholar]
- Liu, T.F.; Yoza, B.K.; Gazzar, M.E.; Vachharajani, V.T.; McCall, C.E. NAD+-dependent SIRT1 deacetylase participates in epigenetic reprogramming during endotoxin tolerance. J. Biol. Chem 2011, 286, 9856–9864. [Google Scholar]
- Peters, T.; Weiss, J.M.; Sindrilaru, A.; Wang, H.; Oreshkova, T.; Wlaschek, M.; Maity, P.; Reimann, J.; Scharffetter-Kochanek, K. Reactive oxygen intermediate-induced pathomechanisms contribute to immunosenescence, chronic inflammation and autoimmunity. Mech. Ageing Dev 2009, 130, 564–587. [Google Scholar]
- Otani, H. Oxidative stress as pathogenesis of cardiovascular risk associated with metabolic syndrome. Antioxid. Redox Signal 2011, 15, 1911–1925. [Google Scholar]
- Gloire, G.; Legrand-Poels, S.; Piette, J. NF-κB activation by reactive oxygen species: Fifteen years later. Biochem. Pharmacol 2006, 72, 1493–1505. [Google Scholar]
- Zhou, R.; Tardivel, A.; Thorens, B.; Choi, I.; Tschopp, J. Thioredoxin-interacting protein links oxidative stress to inflammasome activation. Nat. Immunol 2010, 11, 136–140. [Google Scholar]
- Rahman, I.; Biswas, S.K.; Kirkham, P.A. Regulation of inflammation and redox signalling by dietary polyphenols. Biochem. Pharmacol 2006, 72, 1439–1452. [Google Scholar]
- Salminen, A.; Lehtonen, M.; Suuronen, T.; Kaarniranta, K.; Huuskonen, J. Terpenoids: Natural inhibitors of NF-κB signaling with anti-inflammatory and anticancer potential. Cell. Mol. Life Sci 2008, 65, 2979–2999. [Google Scholar]
- Salminen, A.; Hyttinen, J.M.; Kauppinen, A.; Kaarniranta, K. Context-dependent regulation of autophagy by IKK-NF-κB signaling: Impact on the aging process. Int. J. Cell Biol 2012, 2012, 849541. [Google Scholar]
- Kenyon, C. The first long-lived mutants: Discovery of the insulin/IGF-1 pathway for ageing. Phil. Trans. R. Soc. B 2011, 366, 9–16. [Google Scholar]
- Braeckman, B.P.; Vanfleteren, J.R. Genetic control of longevity in C. elegans. Exp. Gerontol 2007, 42, 90–98. [Google Scholar]
- Bartke, A. Pleiotropic effects of growth hormone signaling in aging. Trends Endocrinol. Metab 2011, 22, 437–42. [Google Scholar]
- Brown-Borg, H.M. Hormonal control of aging in rodents: The somatotropic axis. Mol. Cell. Endocrinol 2009, 299, 64–71. [Google Scholar]
- Bartke, A.; Brown-Borg, H. Life extension in the dwarf mouse. Curr. Top. Dev. Biol 2004, 63, 189–225. [Google Scholar]
- Lithgow, G.J.; Walker, G.A. Stress resistance as a determinate of C. elegans lifespan. Mech. Ageing Dev 2002, 123, 765–771. [Google Scholar]
- Salminen, A.; Kaarniranta, K. Insulin/IGF-1 paradox of aging: Regulation via AKT/IKK/NF-κB signaling. Cell. Signal 2010, 22, 573–577. [Google Scholar]
- Iwasaki, Y.; Nishiyama, M.; Taguchi, T.; Asai, M.; Yoshida, M.; Kambayashi, M.; Terada, Y.; Hashimoto, K. Insulin exhibits short-term anti-inflammatory but long-term proinflammatory effects in vitro. Mol. Cell. Endocrinol 2009, 298, 25–32. [Google Scholar]
- Arkan, M.C.; Hevener, A.L.; Greten, F.R.; Maeda, S.; Li, Z.W.; Long, J.M.; Wynshaw-Boris, A.; Poli, G.; Olefsky, J.; Karin, M. IKK-β links inflammation to obesity-induced insulin resistance. Nat. Med 2005, 11, 191–198. [Google Scholar]
- Musarò, A.; Dobrowolny, G.; Rosenthal, N. The neuroprotective effects of a locally acting IGF-1 isoform. Exp. Gerontol 2007, 42, 76–80. [Google Scholar]
- Pelosi, L.; Giacinti, C.; Nardis, C.; Borsellino, G.; Rizzuto, E.; Nicoletti, C.; Wannenes, F.; Battistini, L.; Rosenthal, N.; Molinaro, M.; et al. Local expression of IGF-1 accelerates muscle regeneration by rapidly modulating inflammatory cytokines and chemokines. FASEB J 2007, 21, 1393–1402. [Google Scholar]
- Vinciguerra, M.; Santini, M.P.; Claycomb, W.C.; Ladurner, A.G.; Rosenthal, N. LocalIGF-1isoformprotects cardiomyocytes from hypertrophic and oxidative stresses via SirT1 activity. Aging (Albany NY) 2010, 2, 1075–1093. [Google Scholar]
- Bolasco, G.; Calogero, R.; Carrara, M.; Banchaabouchi, M.A.; Bilbao, D.; Mazzoccoli, G.; Vinciguerra, M. Cardioprotective mIGF-1/SIRT1 signaling induces hypertension, leukocytosis and fear response in mice. Aging (Albany NY) 2012, 4, 402–416. [Google Scholar]
- Vinciguerra, M.; Santini, M.P.; Martinez, C.; Pazienza, V.; Claycomb, W.C.; Giuliani, A.; Rosenthal, N. mIGF-1/JNK1/SirT1 signaling confers protection against oxidative stress in the heart. Aging Cell 2012, 11, 139–149. [Google Scholar]
- Tang, B.L. SIRT1, neuronal cell survival and the insulin/IGF-1 aging paradox. Neurobiol. Aging 2006, 27, 501–505. [Google Scholar]
- Li, Y.; Xu, W.; McBurney, M.W.; Longo, V.D. SirT1 inhibition reduces IGF-I/IRS-2/Ras/ERK1/2 signaling and protects neurons. Cell Metab 2008, 8, 38–48. [Google Scholar]
- Wu, D.; Qiu, Y.; Gao, X.; Yuan, X.B.; Zhai, Q. Overexpression of SIRT1 in mouse forebrain impairs lipid/glucose metabolism and motor function. PLoS One 2011, 6, e21759. [Google Scholar]
- Zhang, J. The direct involvement of SirT1 in insulin-induced insulin receptor substrate-2 tyrosine phosphorylation. J. Biol. Chem 2007, 282, 34356–34364. [Google Scholar]
© 2013 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Salminen, A.; Kaarniranta, K.; Kauppinen, A. Crosstalk between Oxidative Stress and SIRT1: Impact on the Aging Process. Int. J. Mol. Sci. 2013, 14, 3834-3859. https://doi.org/10.3390/ijms14023834
Salminen A, Kaarniranta K, Kauppinen A. Crosstalk between Oxidative Stress and SIRT1: Impact on the Aging Process. International Journal of Molecular Sciences. 2013; 14(2):3834-3859. https://doi.org/10.3390/ijms14023834
Chicago/Turabian StyleSalminen, Antero, Kai Kaarniranta, and Anu Kauppinen. 2013. "Crosstalk between Oxidative Stress and SIRT1: Impact on the Aging Process" International Journal of Molecular Sciences 14, no. 2: 3834-3859. https://doi.org/10.3390/ijms14023834
APA StyleSalminen, A., Kaarniranta, K., & Kauppinen, A. (2013). Crosstalk between Oxidative Stress and SIRT1: Impact on the Aging Process. International Journal of Molecular Sciences, 14(2), 3834-3859. https://doi.org/10.3390/ijms14023834