Cbl-b Enhances Sensitivity to 5-Fluorouracil via EGFR- and Mitochondria-Mediated Pathways in Gastric Cancer Cells

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
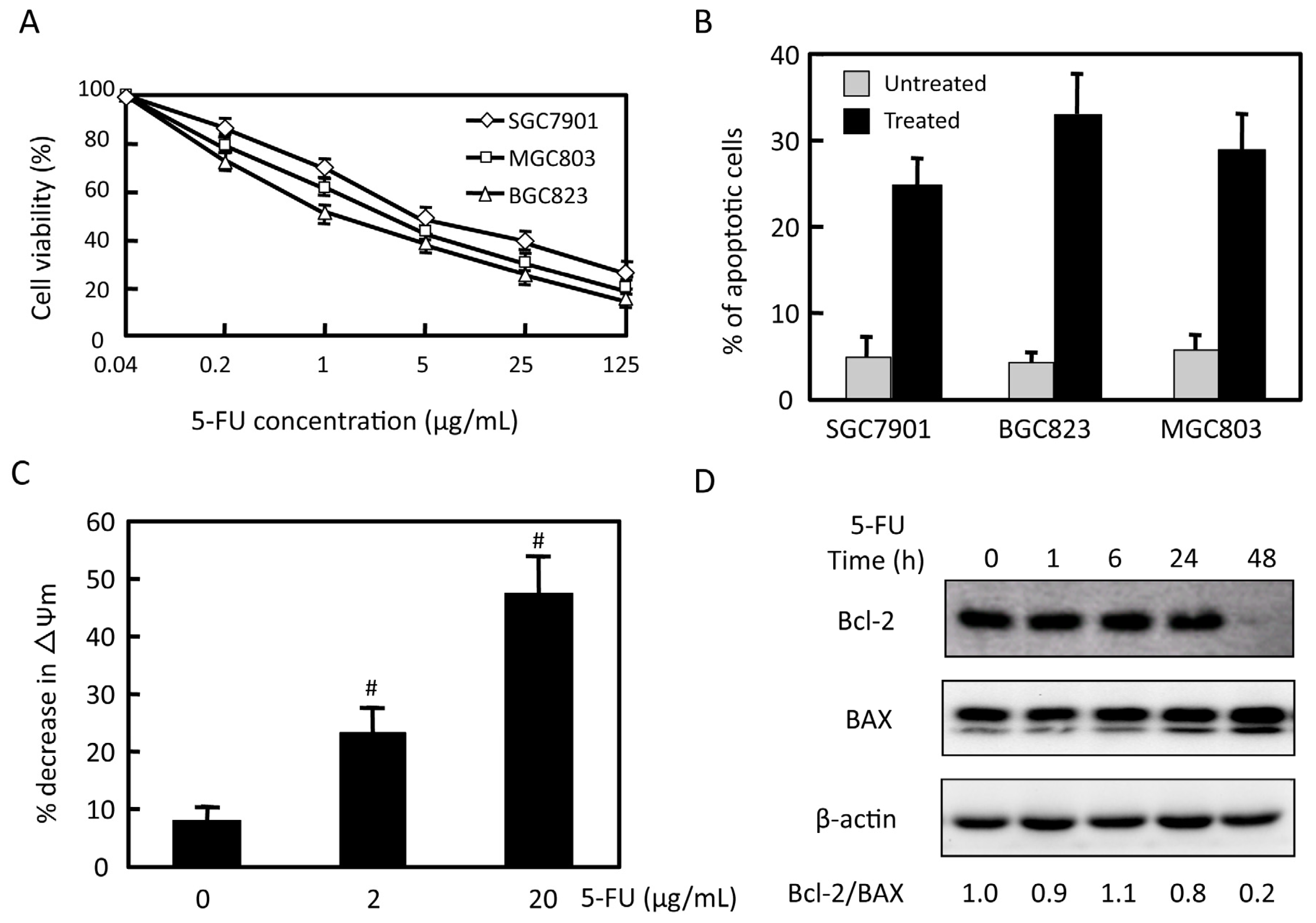
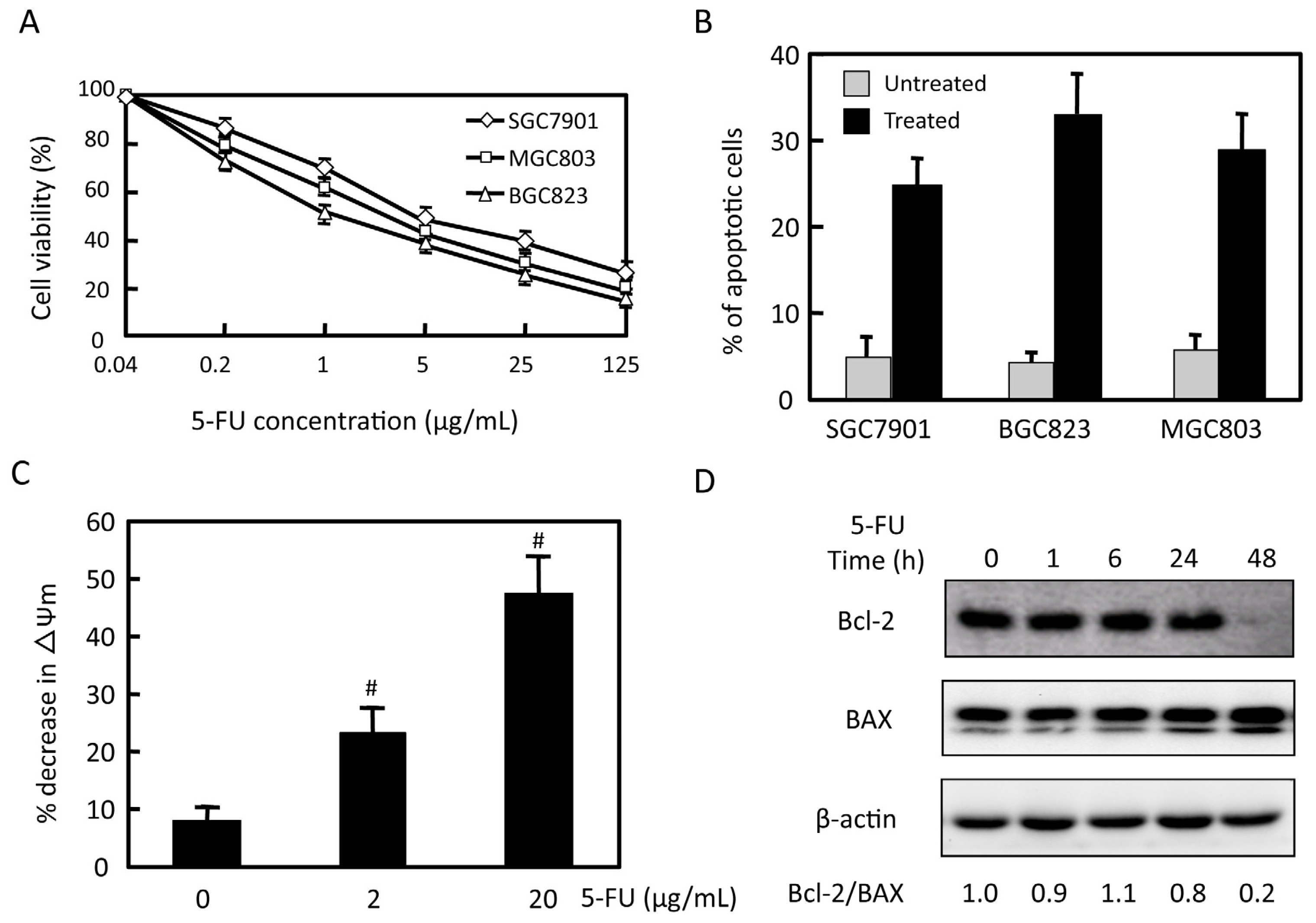
2.1. Effects of 5-FU on Cell Viability and Apoptosis in Gastric Cancer Cells
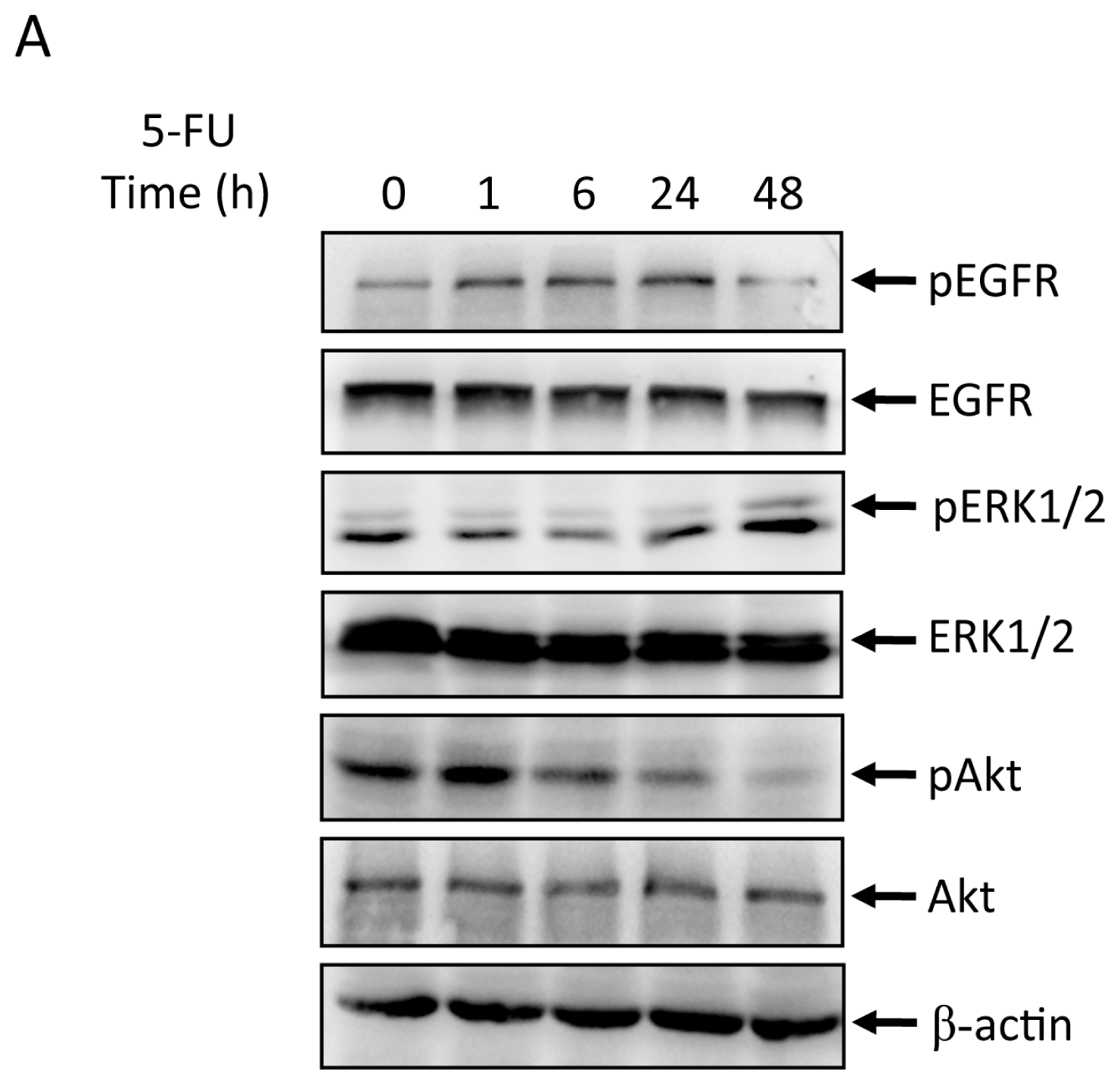
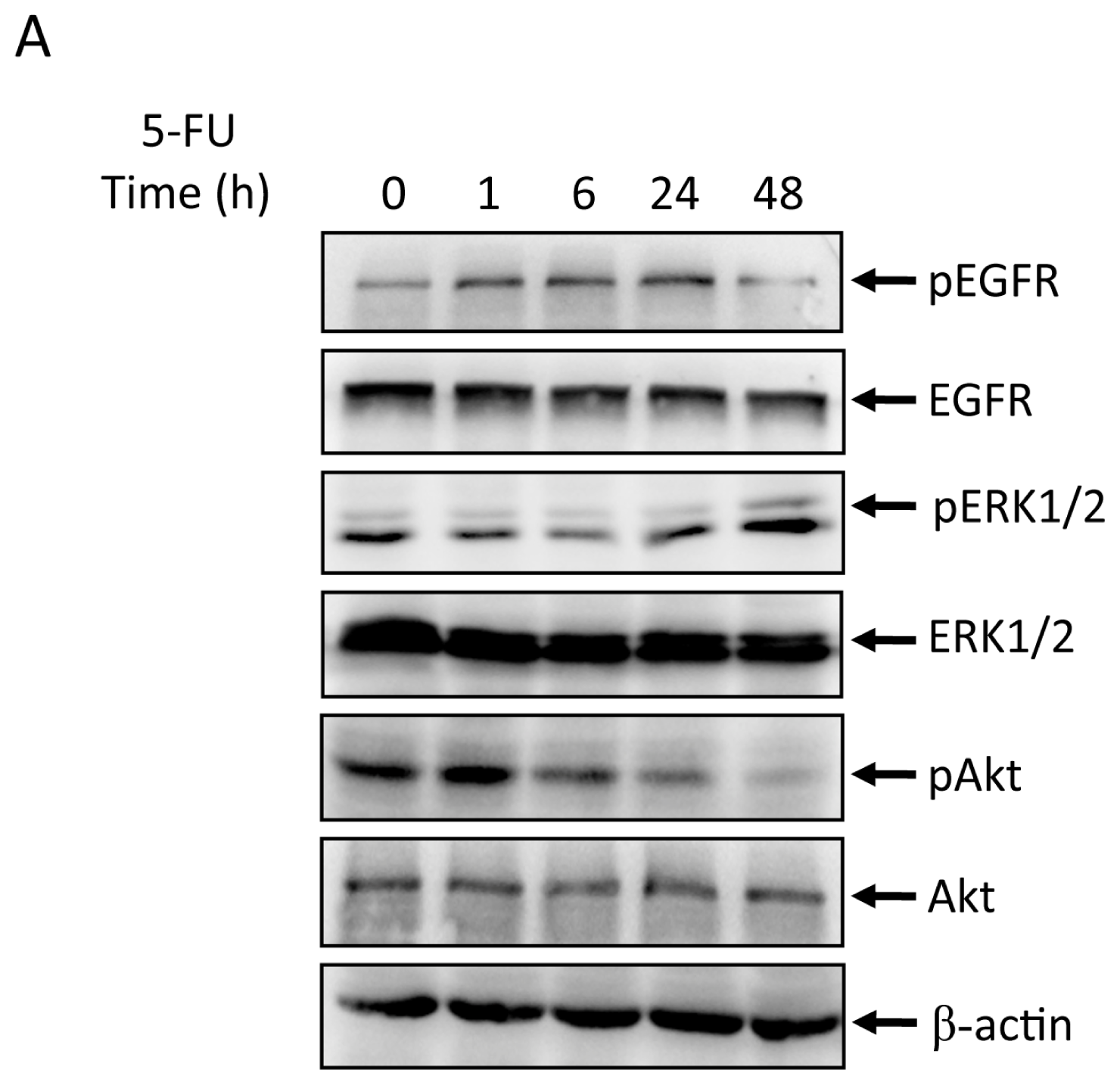
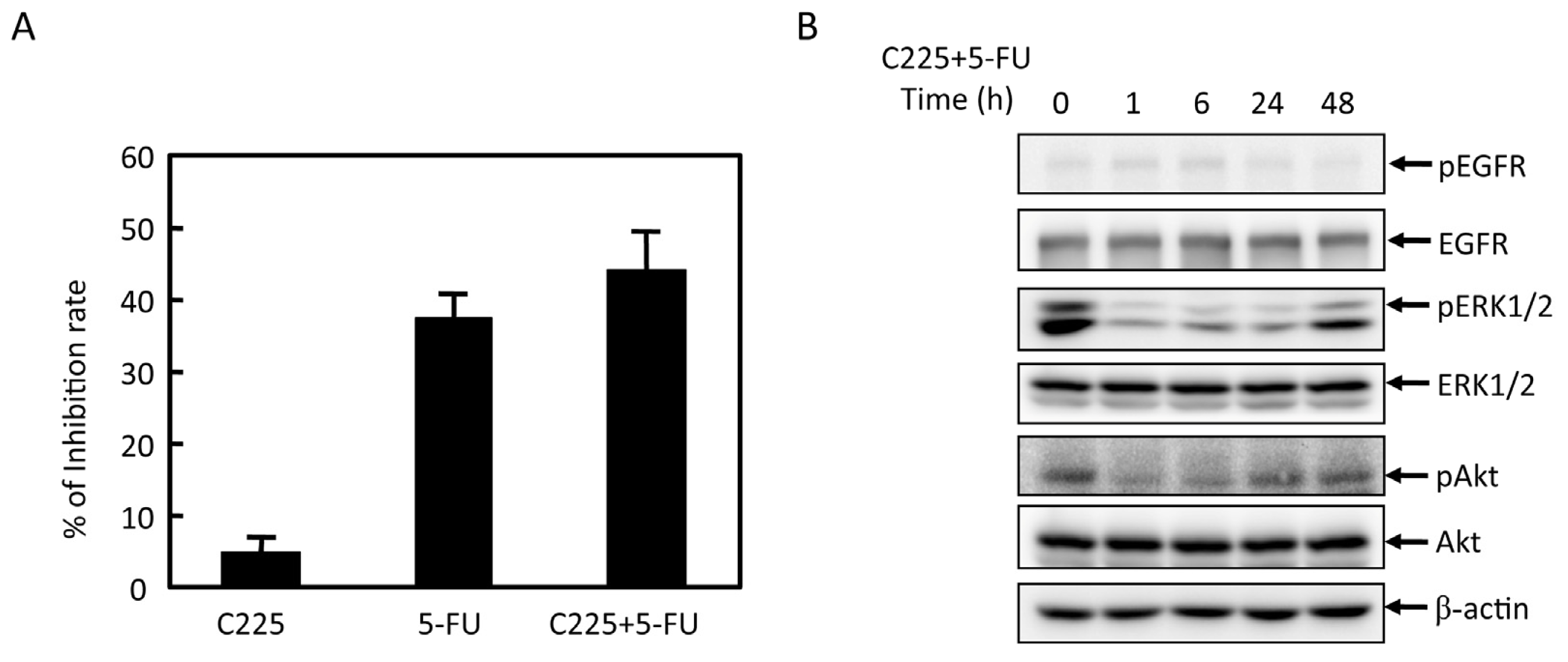
2.2. Effects of 5-FU on EGFR, ERK, and Akt Expression in MGC803 Cells
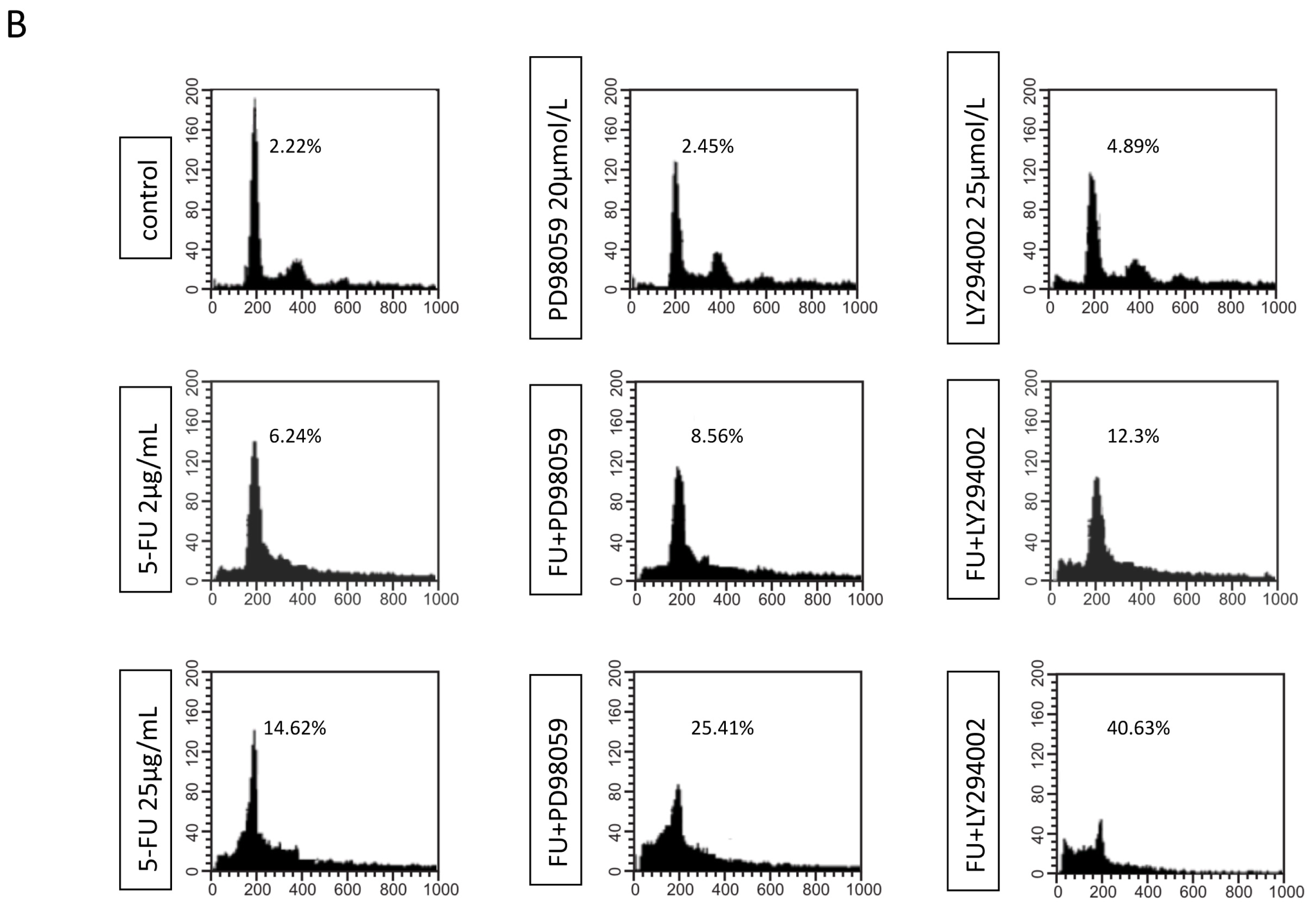
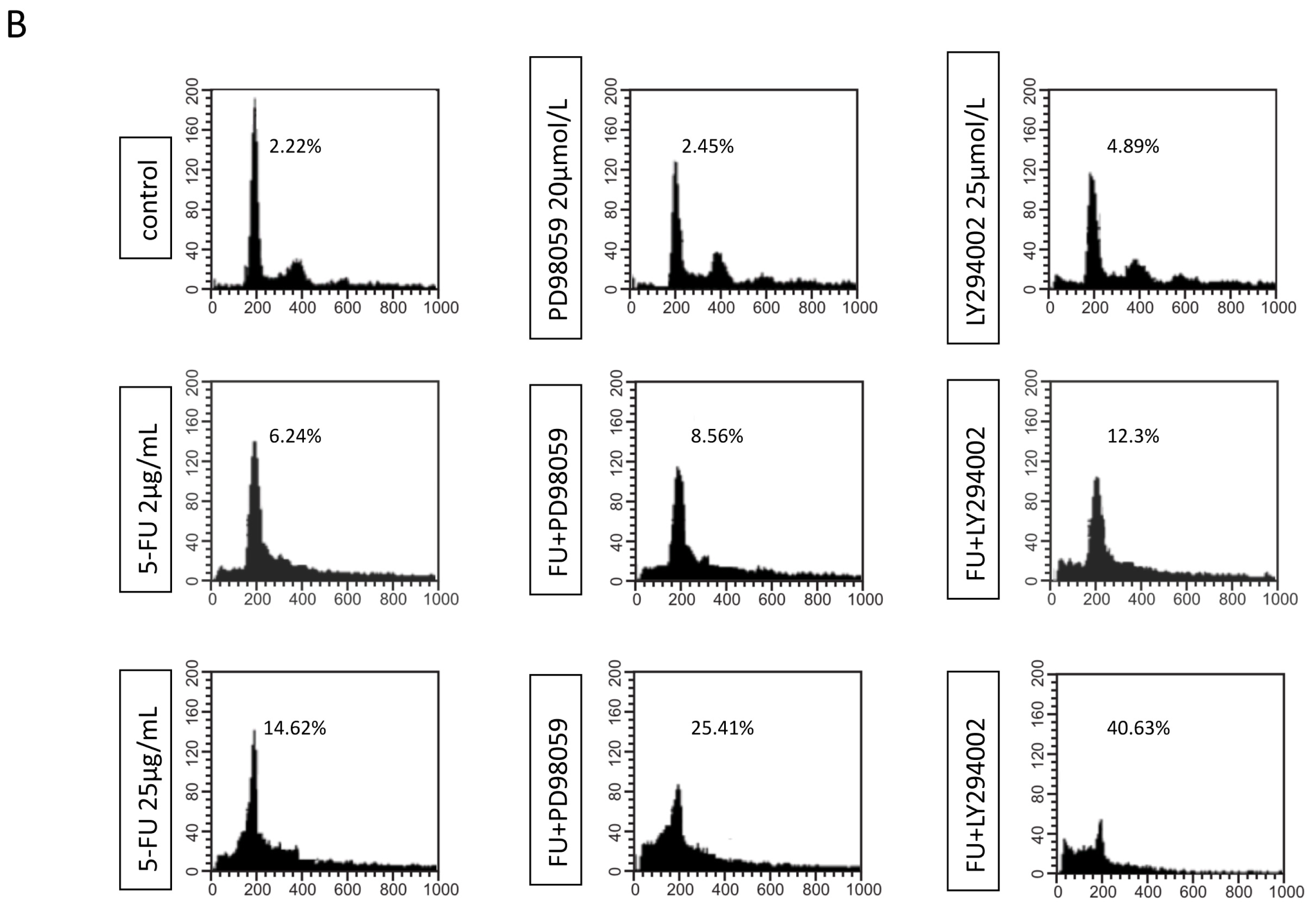
2.3. Effects of EGFR Inhibitor and 5-FU on ERK and Akt in MGC803 Cells
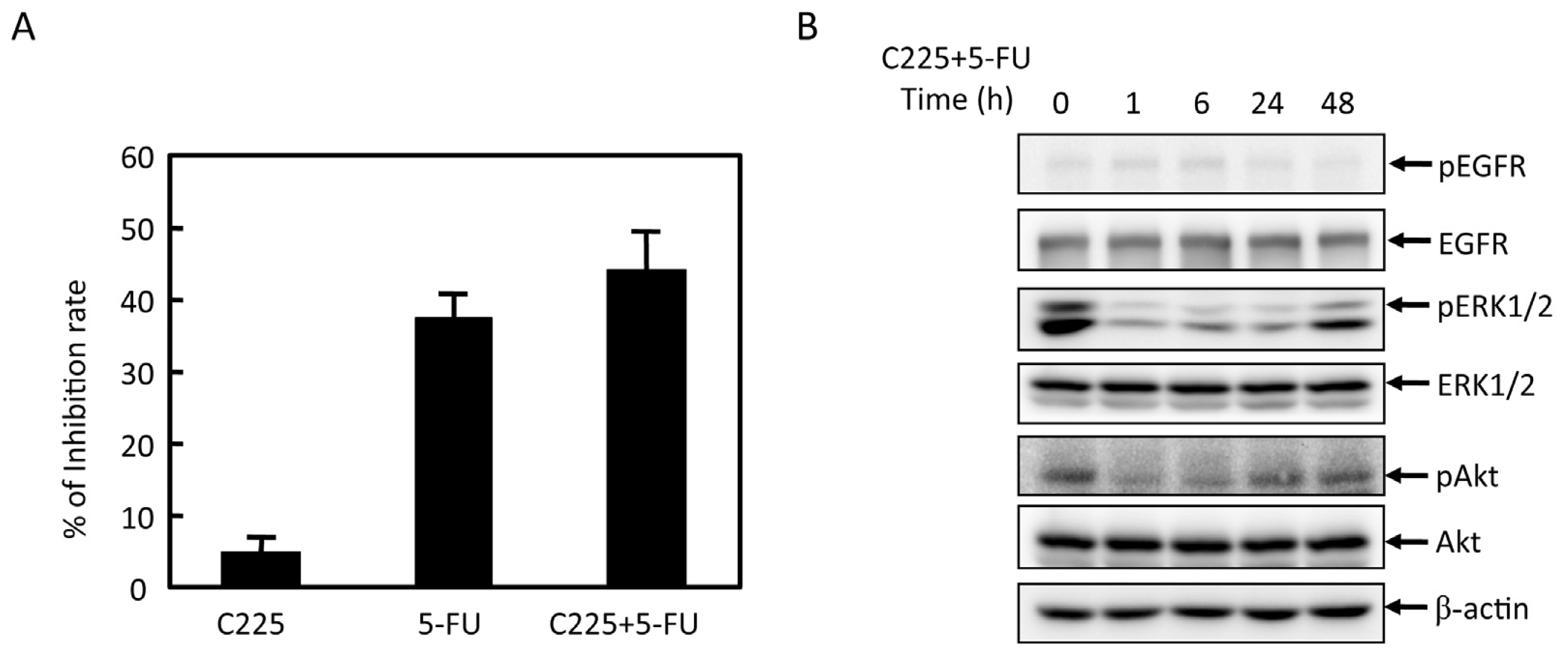
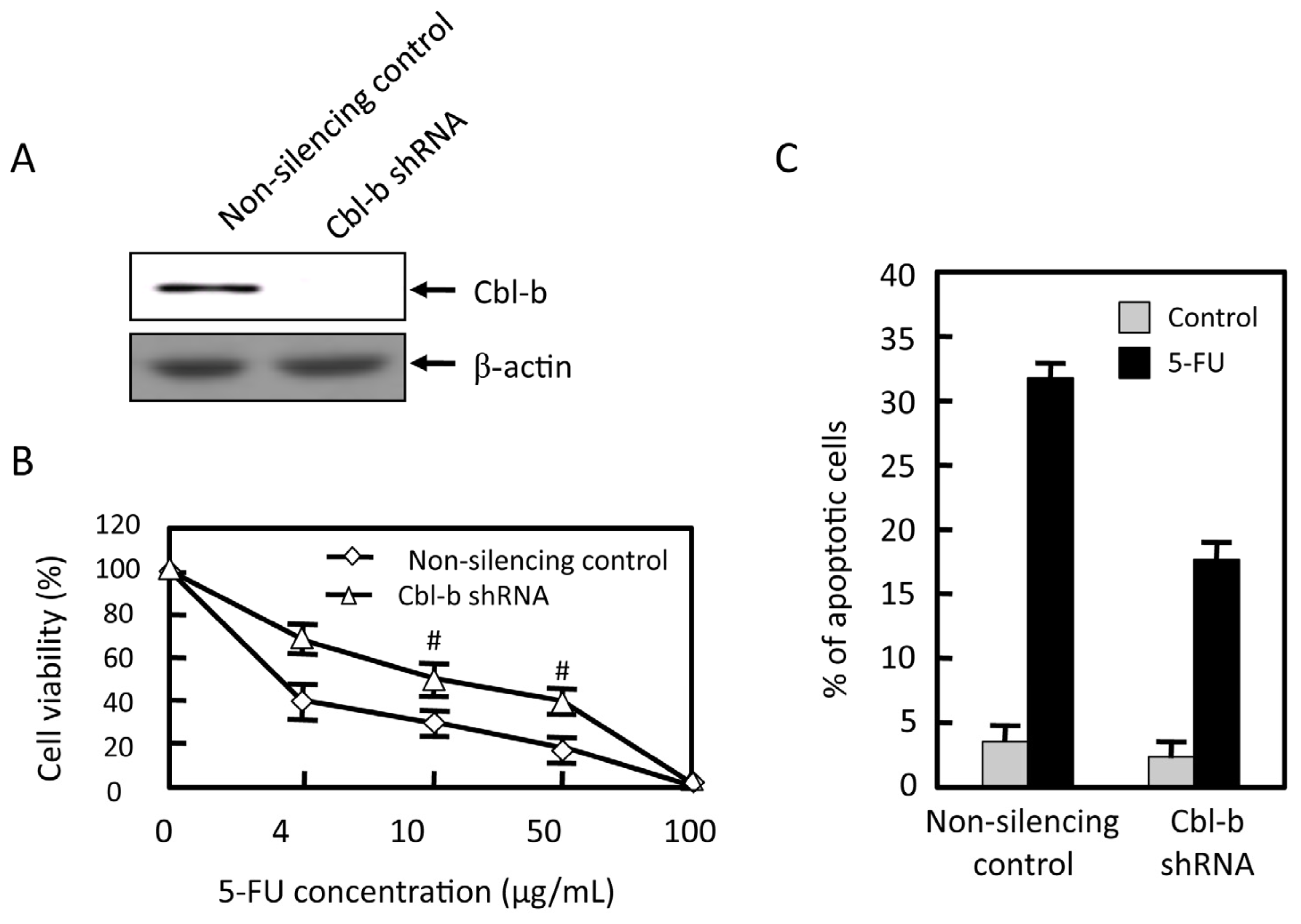
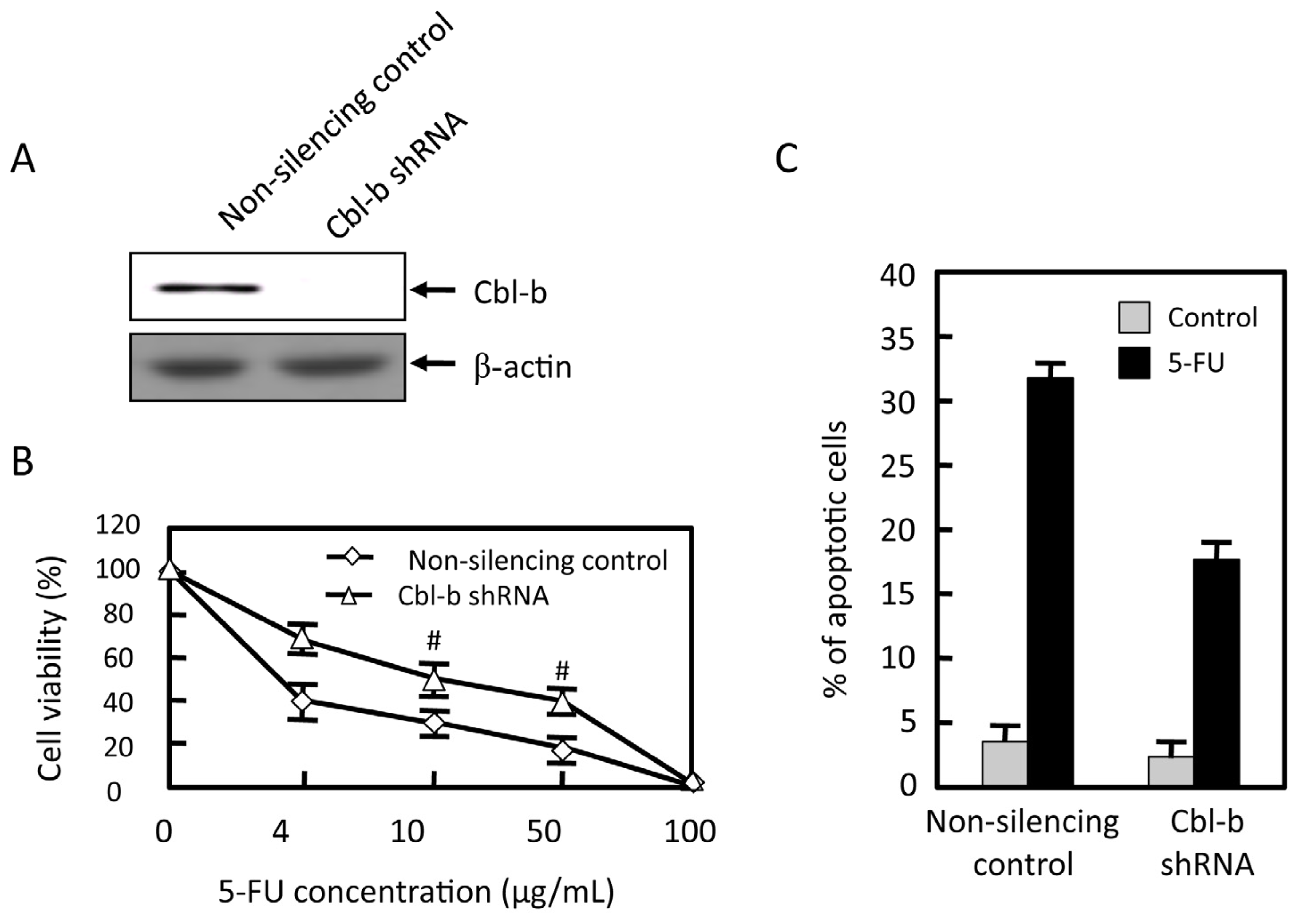
2.4. Effects of Cbl-b on 5-FU Chemosensitivity in MGC803 Cells
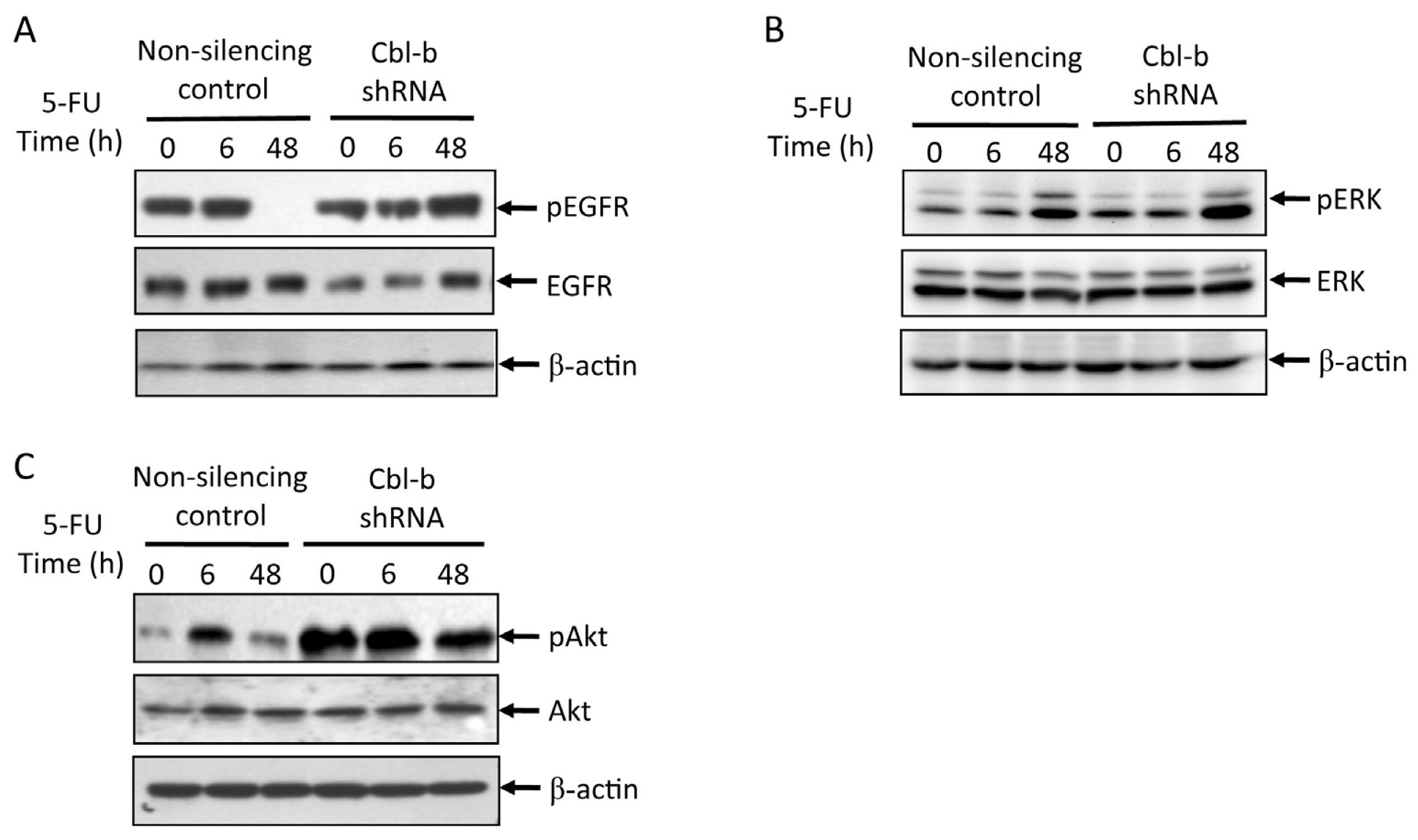
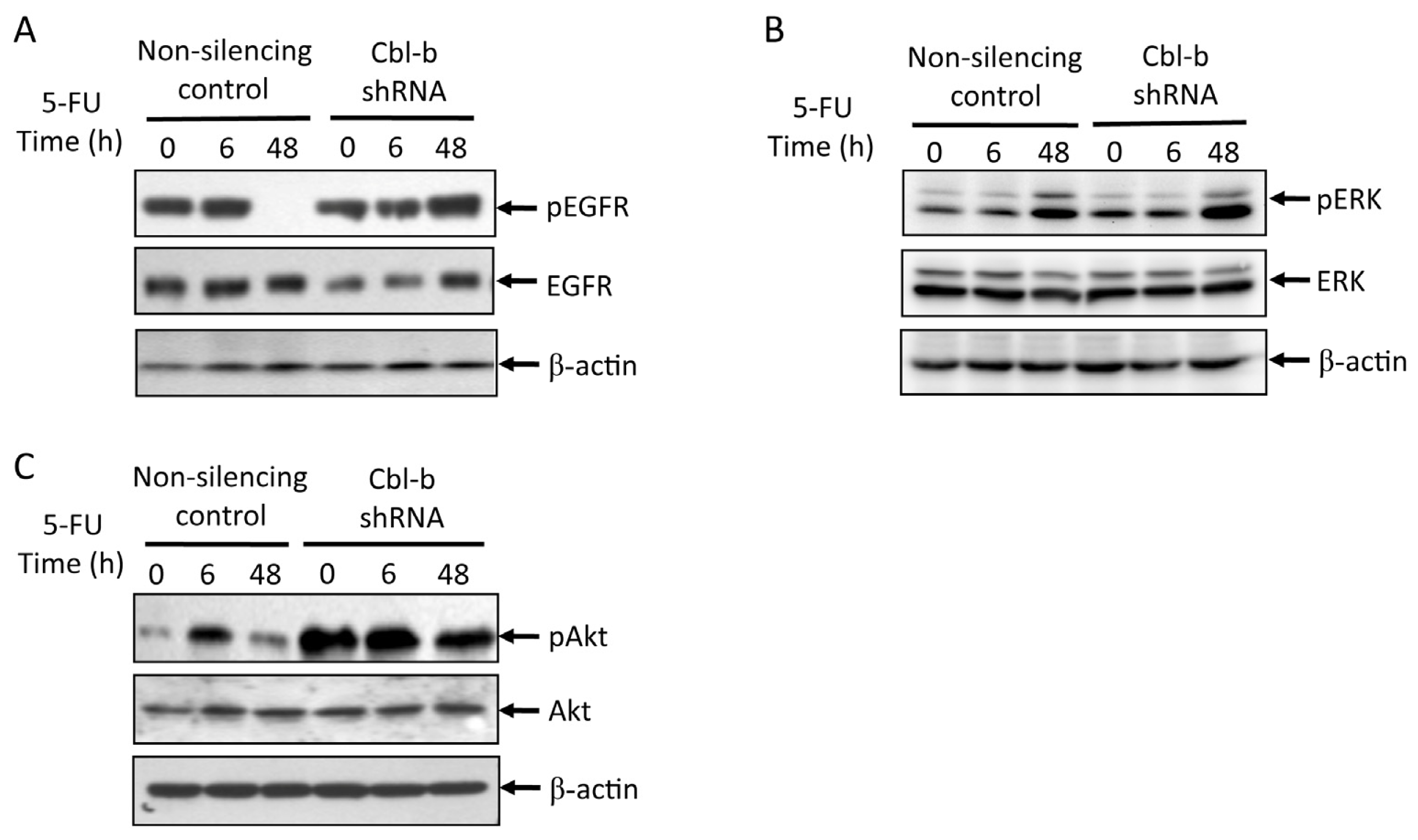
2.5. Effects of Cbl-b on 5-FU-Induced EGFR, REK, and Akt Activation
3. Experimental Section
3.1. Reagents and Antibodies
3.2. Cells and Cell Culture
3.3. RNA Interference Stable Infection
3.4. MTT Assay
3.5. Flow Cytometry Analysis
3.6. Measurement of Mitochondrial Membrane Potential
3.7. Immunoblotting
3.8. Statistical Analysis
4. Conclusions
Acknowledgments
Conflicts of Interest
References
- Cervantes, A.; Rosello, S.; Roda, D.; Rodriguez-Braun, E. The treatment of advanced gastric cancer: Current strategies and future perspectives. Ann. Oncol 2008, 19, v103–v107. [Google Scholar]
- Siegel, R.; Naishadham, D.; Jemal, A. Cancer statistics, 2012. CA Cancer J. Clin 2012, 62, 10–29. [Google Scholar]
- Longley, D.; Harkin, D.; Johnston, P. 5-Fluorouracil: Mechanisms of action and clinical strategies. Nat. Rev. Cancer 2003, 3, 330–338. [Google Scholar]
- Boeckx, C.; Baay, M.; Wouters, A.; Specenier, P.; Vermorken, J.B.; Peeters, M.; Lardon, F. Anti-epidermal growth factor receptor therapy in head and neck squamous cell carcinoma: Focus on potential molecular mechanisms of drug resistance. Oncologist 2013, 18, 850–864. [Google Scholar]
- Kim, J.W.; Kim, H.P.; Im, S.A.; Kang, S.; Hur, H.S.; Yoon, Y.K.; Oh, D.Y.; Kim, J.H.; Lee, D.S.; Kim, T.Y.; et al. The growth inhibitory effect of lapatinib, a dual inhibitor of EGFR and HER2 tyrosine kinase, in gastric cancer cell lines. Cancer Lett 2008, 272, 296–306. [Google Scholar]
- Okabe, T.; Okamoto, I.; Tsukioka, S.; Uchida, J.; Iwasa, T.; Yoshida, T.; Hatashita, E.; Yamada, Y.; Satoh, T.; Tamura, K.; et al. Synergistic antitumor effect of S-1 and the epidermal growth factor receptor inhibitor gefitinib in non-small cell lung cancer cell lines: Role of gefitinib-induced down-regulation of thymidylate synthase. Mol. Cancer Ther 2008, 7, 599–606. [Google Scholar]
- Hsu, C.; Gao, M.; Chen, C.; Yeh, P.; Cheng, A. Inhibitors of epidermoid growth factor receptor suppress cell growth and enhance chemosensitivity of nasopharyngeal cancer cells in vitro. Oncology 2005, 68, 538–547. [Google Scholar]
- Nozawa, H.; Tadakuma, T.; Ono, T.; Sato, M.; Hiroi, S.; Masumoto, K.; Sato, Y. Small interfering RNA targeting epidermal growth factor receptor enhances chemosensitivity to cisplatin, 5-fluorouracil and docetaxel in head and neck squamous cell carcinoma. Cancer Sci 2006, 97, 1115–1124. [Google Scholar]
- Wu, X.; Deng, Y.; Wang, G.; Tao, K. Combining siRNAs at two different sites in the EGFR to suppress its expression, induce apoptosis, and enhance 5-fluorouracil sensitivity of colon cancer cells. J. Surg. Res 2007, 138, 56–63. [Google Scholar]
- Van Schaeybroeck, S.; Karaiskou-McCaul, A.; Kelly, D.; Longley, D.; Galligan, L.; van Cutsem, E.; Johnston, P. Epidermal growth factor receptor activity determines response of colorectal cancer cells to gefitinib alone and in combination with chemotherapy. Clin. Cancer Res 2005, 11, 7480–7489. [Google Scholar]
- Komoto, M.; Nakata, B.; Nishii, T.; Kawajiri, H.; Shinto, O.; Amano, R.; Yamada, N.; Yashiro, M.; Hirakawa, K. In vitro and in vivo evidence that a combination of lapatinib plus S-1 is a promising treatment for pancreatic cancer. Cancer Sci 2010, 101, 468–473. [Google Scholar]
- Mohapatra, B.; Ahmad, G.; Nadeau, S.; Zutshi, N.; An, W.; Scheffe, S.; Dong, L.; Feng, D.; Goetz, B.; Arya, P.; et al. Protein tyrosine kinase regulation by ubiquitination: Critical roles of Cbl-family ubiquitin ligases. BBA Mol. Cell Res 2013, 1833, 122–139. [Google Scholar]
- Komander, D. The emerging complexity of protein ubiquitination. Biochem. Soc. Trans 2009, 37, 937–953. [Google Scholar]
- Dikic, I.; Wakatsuki, S.; Walters, K. Ubiquitin-binding domains—From structures to functions. Nat. Rev. Mol. Cell Biol 2009, 10, 659–671. [Google Scholar]
- Duan, L.; Reddi, A.L.; Ghosh, A.; Dimri, M.; Band, H. The Cbl family and other ubiquitin ligases: Destructive forces in control of antigen receptor signaling. Immunity 2004, 21, 7–17. [Google Scholar]
- Fang, D.; Wang, H.; Fang, N.; Altman, Y.; Elly, C.; Liu, Y. Cbl-b, a RING-type E3 ubiquitin ligase, targets phosphatidylinositol 3-kinase for ubiquitination in T cells. J. Biol. Chem 2001, 276, 4872–4878. [Google Scholar]
- Fang, D.; Liu, Y.C. Proteolysis-independent regulation of PI3K by Cbl-b-mediated ubiquitination in T cells. Nat. Immunol 2001, 2, 870–875. [Google Scholar]
- Zhang, R.; Zhang, N.; Mueller, D.L. Casitas B-lineage lymphoma b inhibits antigen recognition and slows cell cycle progression at late times during CD4+ T cell clonal expansion. J. Immunol 2008, 181, 5331–5339. [Google Scholar]
- Qu, X.; Zhang, Y.; Li, Y.; Hu, X.; Xu, Y.; Xu, L.; Hou, K.; Sada, K.; Liu, Y. Ubiquitin ligase Cbl-b sensitizes leukemia and gastric cancer cells to anthracyclines by activating the mitochondrial pathway and modulating Akt and ERK survival signals. FEBS Lett 2009, 583, 2255–2262. [Google Scholar]
- Qu, X.; Li, Y.; Liu, J.; Xu, L.; Zhang, Y.; Hu, X.; Hou, K.; Liu, Y. Cbl-b promotes chemotherapy-induced apoptosis in rat basophilic leukemia cells by suppressing PI3K/Akt activation and enhancing MEK/ERK activation. Mol. Cell. Biochem 2010, 340, 107–114. [Google Scholar]
- Li, Y.; Qu, X.; Qu, J.; Zhang, Y.; Liu, J.; Teng, Y.; Hu, X.; Hou, K.; Liu, Y. Arsenic trioxide induces apoptosis and G2/M phase arrest by inducing Cbl to inhibit PI3K/Akt signaling and thereby regulate p53 activation. Cancer Lett 2009, 284, 208–215. [Google Scholar]
- Ischenko, I.; Camaj, P.; Seeliger, H.; Kleespies, A.; Guba, M.; de Toni, E.N.; Schwarz, B.; Graeb, C.; Eichhorn, M.E.; Jauch, K.W.; et al. Inhibition of Src tyrosine kinase reverts chemoresistance toward 5-fluorouracil in human pancreatic carcinoma cells: An involvement of epidermal growth factor receptor signaling. Oncogene 2008, 27, 7212–7222. [Google Scholar]
- Pu, Y.; Hsieh, M.; Wang, C.; Liu, G.; Huang, C.; Lin, C.; Guan, J.; Lin, S.; Hour, T. Epidermal growth factor receptor inhibitor (PD168393) potentiates cytotoxic effects of paclitaxel against androgen-independent prostate cancer cells. Biochem. Pharmacol 2006, 71, 751–760. [Google Scholar]
- Bu, X.; Jia, F.; Wang, W.; Guo, X.; Wu, M.; Wei, L. Coupled down-regulation of mTOR and telomerase activity during fluorouracil-induced apoptosis of hepatocarcinoma cells. BMC Cancer 2007, 7, 208. [Google Scholar]
- Qin, L.; Zhang, X.; Zhang, L.; Feng, Y.; Weng, G.X.; Li, M.Z.; Kong, Q.L.; Qian, C.N.; Zeng, Y.X.; Zeng, M.S.; et al. Downregulation of BMI-1 enhances 5-fluorouracil-induced apoptosis in nasopharyngeal carcinoma cells. Biochem. Biophys. Res. Commun 2008, 371, 531–535. [Google Scholar]
- Xu, L.; Qu, X.; Zhang, Y.; Hu, X.; Yang, X.; Hou, K.; Teng, Y.; Zhang, J.; Sada, K.; Liu, Y. Oxaliplatin enhances TRAIL-induced apoptosis in gastric cancer cells by CBL-regulated death receptor redistribution in lipid rafts. FEBS Lett 2009, 583, 943–948. [Google Scholar]
- Wang, Y.; Chen, Z.; Bergmann, A. Regulation of EGFR and Notch signaling by distinct isoforms of D-cbl during Drosophila development. Dev. Biol 2010, 342, 1–10. [Google Scholar]
- Tan, Y.; Krishnaswamy, S.; Nandi, S.; Kanteti, R.; Vora, S.; Onel, K.; Hasina, R.; Lo, F.; El-Hashani, E.; Cervantes, G.; et al. CBL is frequently altered in lung cancers: Its relationship to mutations in MET and EGFR tyrosine kinases. PLoS One 2010, 5, e8972. [Google Scholar]
- Vivanco, I.; Rohle, D.; Versele, M.; Iwanami, A.; Kuga, D.; Oldrini, B.; Tanaka, K.; Dang, J.; Kubek, S.; Palaskas, N.; et al. The phosphatase and tensin homolog regulates epidermal growth factor receptor (EGFR) inhibitor response by targeting EGFR for degradation. Proc. Natl. Acad. Sci. USA 2010, 107, 6459–6464. [Google Scholar]
- Feng, Y.; Tsao, C.; Wu, C.; Chang, J.; Lu, P.; Yeh, K.; Shieh, G.; Shiau, A.; Lee, J. Sprouty2 protein enhances the response to gefitinib through epidermal growth factor receptor in colon cancer cells. Cancer Sci 2010, 101, 2033–2038. [Google Scholar]
- Ettenberg, S.; Keane, M.; Nau, M.; Frankel, M.; Wang, L.; Pierce, J.; Lipkowitz, S. cbl-b inhibits epidermal growth factor receptor signaling. Oncogene 1999, 18, 1855–1866. [Google Scholar]
- Ahsan, A.; Hiniker, S.; Ramanand, S.; Nyati, S.; Hegde, A.; Helman, A.; Menawat, R.; Bhojani, M.; Lawrence, T.; Nyati, M. Role of epidermal growth factor receptor degradation in cisplatin-induced cytotoxicity in head and neck cancer. Cancer Res 2010, 70, 2862–2869. [Google Scholar]

© 2013 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Feng, D.; Ma, Y.; Liu, J.; Xu, L.; Zhang, Y.; Qu, J.; Liu, Y.; Qu, X. Cbl-b Enhances Sensitivity to 5-Fluorouracil via EGFR- and Mitochondria-Mediated Pathways in Gastric Cancer Cells. Int. J. Mol. Sci. 2013, 14, 24399-24411. https://doi.org/10.3390/ijms141224399
Feng D, Ma Y, Liu J, Xu L, Zhang Y, Qu J, Liu Y, Qu X. Cbl-b Enhances Sensitivity to 5-Fluorouracil via EGFR- and Mitochondria-Mediated Pathways in Gastric Cancer Cells. International Journal of Molecular Sciences. 2013; 14(12):24399-24411. https://doi.org/10.3390/ijms141224399
Chicago/Turabian StyleFeng, Dan, Yanju Ma, Jing Liu, Ling Xu, Ye Zhang, Jinglei Qu, Yunpeng Liu, and Xiujuan Qu. 2013. "Cbl-b Enhances Sensitivity to 5-Fluorouracil via EGFR- and Mitochondria-Mediated Pathways in Gastric Cancer Cells" International Journal of Molecular Sciences 14, no. 12: 24399-24411. https://doi.org/10.3390/ijms141224399
APA StyleFeng, D., Ma, Y., Liu, J., Xu, L., Zhang, Y., Qu, J., Liu, Y., & Qu, X. (2013). Cbl-b Enhances Sensitivity to 5-Fluorouracil via EGFR- and Mitochondria-Mediated Pathways in Gastric Cancer Cells. International Journal of Molecular Sciences, 14(12), 24399-24411. https://doi.org/10.3390/ijms141224399