Biological Functional Relevance of Asymmetric Dimethylarginine (ADMA) in Cardiovascular Disease

 ,
, 

Abstract
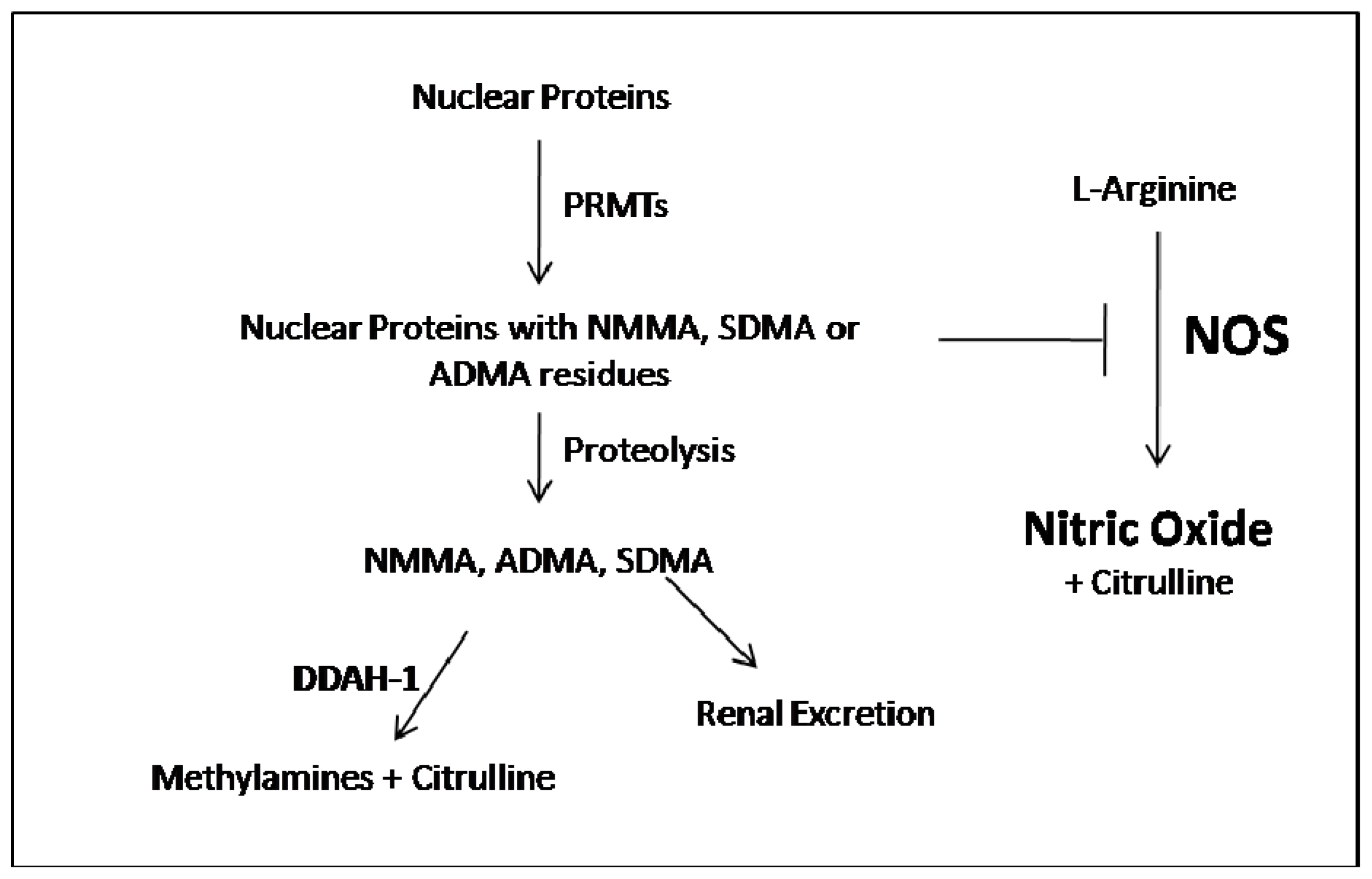
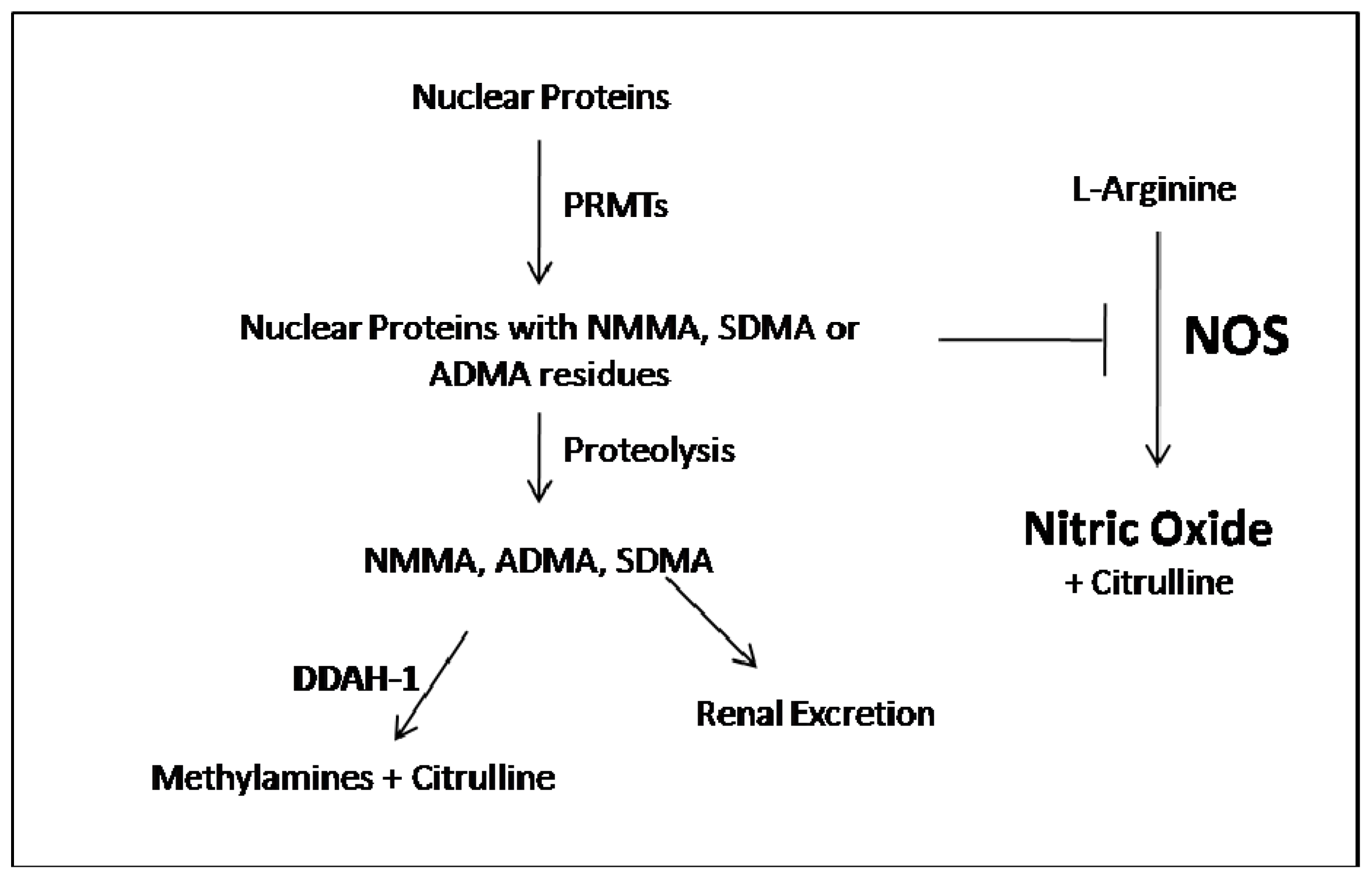
:1. ADMA Metabolism
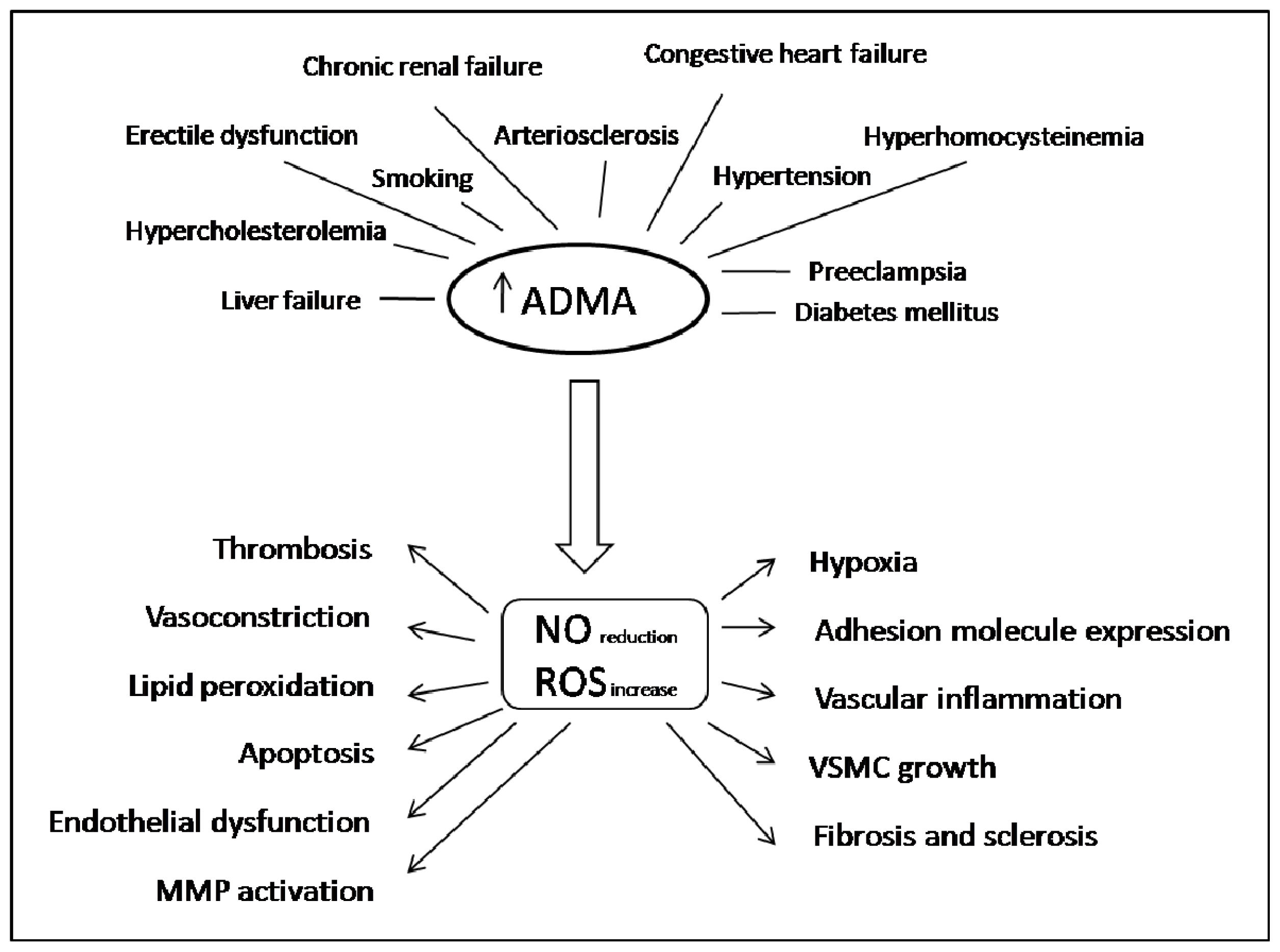
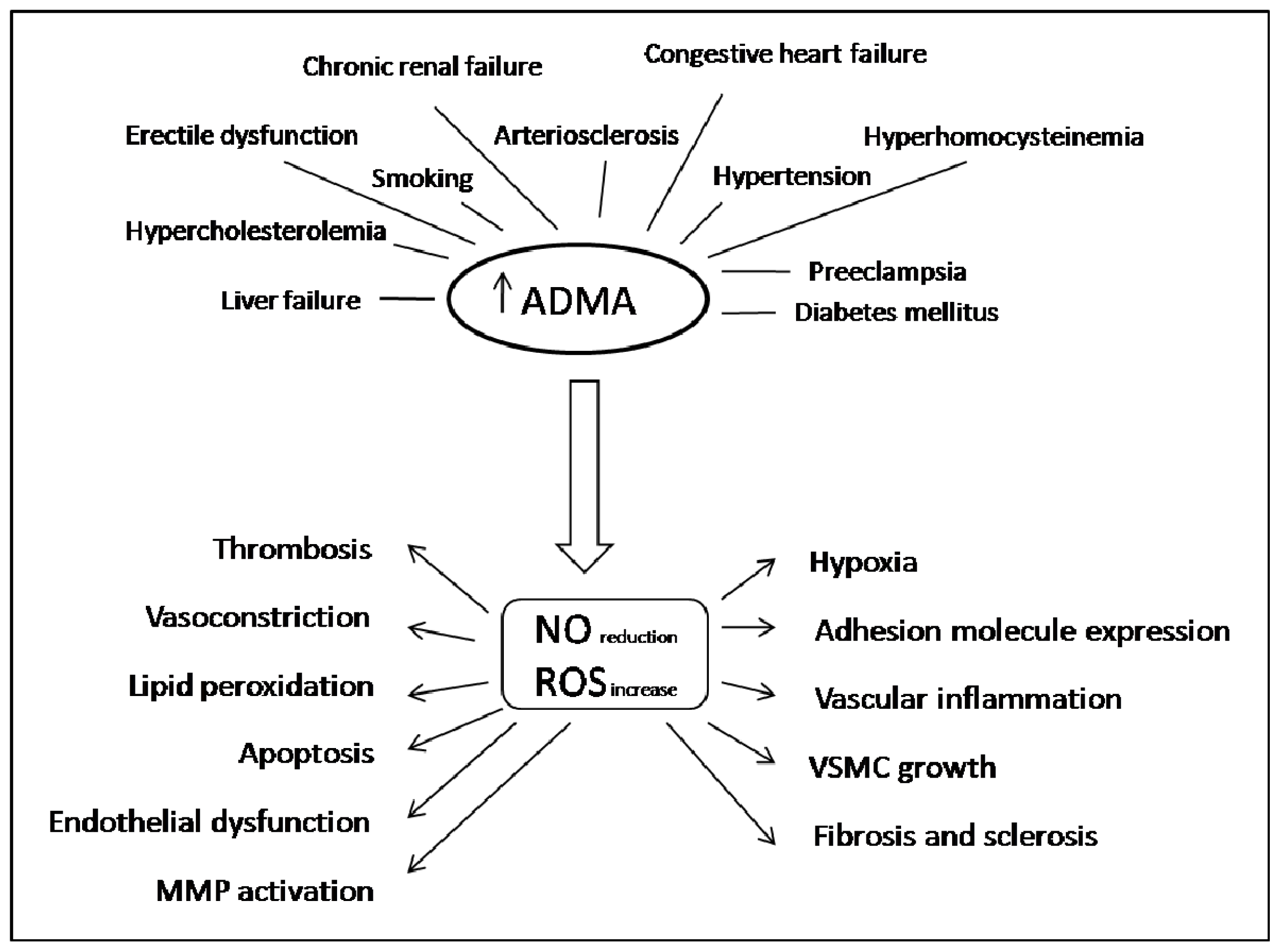
2. ADMA in Oxidative Stress, Inflammation and Cardiovascular Disease
Acknowledgments
Conflicts of Interest
References
- Naseem, K.M. The role of nitric oxide in cardiovascular diseases. Mol. Aspects Med 2005, 26, 33–65. [Google Scholar]
- Napoli, C.; Ignarro, L.J. Nitric oxide and pathogenic mechanisms involved in the development of vascular diseases. Arch. Pharm. Res 2009, 32, 1103–1108. [Google Scholar]
- Antoniades, C.; Shirodaria, C.; Leeson, P.; Antonopoulos, A.; Warrick, N.; van Assche, T.; Cunnington, C.; Tousoulis, D.; Pillai, P.; Ratnatunga, C.; et al. Association of plasma asymmetrical dimethylarginine (ADMA) with elevated vascular superoxide production and endothelial nitric oxide synthase uncoupling: Implications for endothelial function in human atherosclerosis. Eur. Heart J 2009, 30, 1142–1150. [Google Scholar]
- Olken, N.M.; Marletta, M.A. NG-methyl-l-arginine functions as an alternate substrate and mechanism-based inhibitor of nitric oxide synthase. Biochemistry 1993, 32, 9677–9685. [Google Scholar]
- Fickling, S.; Leone, A.; Nussey, S.; Vallance, P.; Whitley, G. Synthesis of NG, NG dimethylarginine by human endothelial cells. Endothelium 1993, 1, 137–140. [Google Scholar]
- Bedford, M.T.; Clarke, S.G. Protein arginine methylation in mammals: Who, what, and why. Mol. Cell 2009, 33, 1–13. [Google Scholar]
- Palm, F.; Onozato, M.L.; Luo, Z.; Wilcox, C.S. Dimethylargininedimethylaminohydrolase (DDAH): Expression, regulation, and function in the cardiovascular and renal systems. Am. J. Physiol. Heart Circ. Physiol 2007, 293, H3227–H3245. [Google Scholar]
- Rodionov, R.N.; Murry, D.J.; Vaulman, S.F.; Stevens, J.W.; Lentz, S.R. Human alanine-glyoxylate aminotransferase 2 lowers asymmetric dimethylarginine and protects from inhibition of nitric oxide production. J. Biol. Chem 2010, 285, 5385–5391. [Google Scholar]
- Closs, E.I.; Boissel, J.P.; Habermeier, A.; Rotmann, A. Structure and function of cationic amino acid transporters (CATs). J. Membr. Biol 2006, 213, 67–77. [Google Scholar]
- Davids, M.; van Hell, A.J.; Visser, M.; Nijveldt, R.J.; van Leeuwen, P.A.; Teerlink, T. Role of the human erythrocyte in generation and storage of asymmetric dimethylarginine. Am. J. Physiol. Heart Circ. Physiol 2012, 302, H1762–H1770. [Google Scholar]
- Billecke, S.S.; Kitzmiller, L.A.; Northrup, J.J.; Whitesall, S.E.; Kimoto, M.; Hinz, A.V.; D’Alecy, L.G. Contribution of whole blood to the control of plasma asymmetrical dimethylarginine. Am. J. Physiol. Heart Circ. Physiol 2006, 291, 1788–1796. [Google Scholar]
- Iarlori, C.; Gambi, D.; Lugaresi, A.; Patruno, A.; Felaco, M.; Salvatore, M.; Speranza, L.; Reale, M. Reduction of free radicals in multiple sclerosis: Effect of glatiramer acetate (Copaxone). Mult. Scler 2008, 14, 739–748. [Google Scholar]
- Felaco, M.; di Maio, F.D.; de Fazio, P.; D’Arcangelo, C.; de Lutiis, M.A.; Varvara, G.; Grilli, A.; Barbacane, R.C.; Reale, M.; Conti, P. Localization of the e-NOS enzyme in endothelial cells and odontoblasts of healthy human dental pulp. Life Sci 2000, 68, 297–306. [Google Scholar]
- Franceschelli, S.; Pesce, M.; Vinciguerra, I.; Ferrone, A.; Riccioni, G.; Patruno, A.; Grilli, A.; Felaco, M.; Speranza, L. Licocalchone-C extracted from Glycyrrhiza glabra inhibits lipopolysaccharide-interferon-γ inflammation by improving antioxidantconditions and regulating inducible nitric oxide synthase expression. Molecules 2011, 16, 5720–5734. [Google Scholar]
- Grilli, A.; de Lutiis, M.A.; Patruno, A.; Speranza, L.; Gizzi, F.; Taccardi, A.A.; di Napoli, P.; de Caterina, R.; Conti, P.; Felaco, M. Inducible nitric oxide synthase and heme oxygenase-1 in rat heart: Direct effect of chronic exposure to hypoxia. Ann. Clin. Lab. Sci 2003, 33, 208–215. [Google Scholar]
- Speranza, L.; Franceschelli, S.; Pesce, M.; Ferrone, A.; Patruno, A.; Riccioni, G.; de Lutiis, M.A.; Felaco, M.; Grilli, A. Negative feedback interaction of HO-1/INOS in PBMC of acute congestive heart failure patients. J. Biol. Regul. Homeost. Agents 2013, 27, 739–748. [Google Scholar]
- Han, J.; Shuvaev, V.V.; Muzykantov, V.R. Targeted interception of signaling reactive oxygen species in the vascular endothelium. Ther. Deliv 2012, 3, 263–276. [Google Scholar]
- Kampoli, A.M.; Tousoulis, D.; Tentolouris, C.; Stefanadis, C. Novel agents targeting nitric oxide. Curr. Vasc. Pharmacol 2012, 10, 61–76. [Google Scholar]
- Sharshar, T.; Gray, F.; Lorin de la Grandmaison, G.; Hopkinson, N.S.; Ross, E.; Dorandeu, A.; Orlikowski, D.; Raphael, J.C.; Gajdos, P.; Annane, D. Apoptosis of neurons in cardiovascular autonomic centres triggered by inducible nitric oxide synthase after death from septic shock. Lancet 2003, 362, 1799–1805. [Google Scholar]
- Speranza, L.; Franceschelli, S.; Pesce, M.; Vinciguerra, I.; de Lutiis, M.A.; Grilli, A.; Felaco, M.; Patruno, A. Phosphodiesterase type-5 inhibitor and oxidative stress. Int. J. Immunopathol. Pharmacol 2008, 21, 879–889. [Google Scholar]
- Caplin, B.; Leiper, J. Endogenous nitric oxide synthase inhibitors in the biology of disease: Markers, mediators, and regulators? Arterioscler. Thromb. Vasc. Biol 2012, 32, 1343–1353. [Google Scholar]
- Riccioni, G.; Speranza, L.; Scotti, L.; Bucciarelli, V.; di Ilio, E.; D’Orazio, N.; Pesce, M.; Aceto, A.; Sorrenti, V.; Frigiola, A.; et al. The effect of pharmacological treatment on ADMA in patients with heart failure. Front. Biosci. (Elite Ed.) 2011, 3, 1310–1314. [Google Scholar]
- Ueda, S.; Yamagishi, S.; Matsumoto, Y.; Fukami, K.; Okuda, S. Asymmetric dimethylarginine (ADMA) is a novel emerging risk factor for cardiovascular disease and the development of renal injury in chronic kidney disease. Clin. Exp. Nephrol 2007, 11, 115–121. [Google Scholar]
- Galal, O.; Podlogar, J.; Verspohl, E.J. Impact of ADMA (asymmetric dimethylarginine) on physiology with respect to diabetes mellitus and respiratory system BEAS-2B cells (human bronchial epithelial cells). J. Pharm. Pharmacol 2013, 65, 253–263. [Google Scholar]
- Ghebremariam, Y.T.; Erlanson, D.A.; Cooke, J.P. A novel and potent inhibitor of dimethylarginine dimethylaminohydrolase: A modulator of cardiovascular nitric oxide. J. Pharmacol. Exp. Ther 2013. [Google Scholar] [CrossRef]
- Labruijere, S.; van Houten, E.L.; de Vries, R.; Musterd-Bagghoe, U.M.; Garrelds, I.M.; Kramer, P.; Danser, A.H.; Villalón, C.M.; Visser, J.A.; van den Brink, A.M. Analysis of the vascular responses in a murine model of polycystic ovary syndrome. J. Endocrinol 2013, 218, 205–213. [Google Scholar]
- Beutel, G.; Perthel, R.; Suntharalingam, M.; Bode-Böger, S.M.; Martens-Lobenhoffer, J.; Kielstein, J.T.; Kielstein, H. Effect of chronic elevated asymmetric dimethylarginine (ADMA) levels on granulopoiesis. Ann. Hematol 2013, 92, 505–508. [Google Scholar]
- Ghebremariam, Y.T.; Yamada, K.; Lee, J.C.; Johnson, C.L.; Atzler, D.; Anderssohn, M.; Agrawal, R.; Higgins, J.P.; Patterson, A.J.; Böger, R.H.; et al. FXR agonist INT-747 upregulates DDAH expression and enhances insulin sensitivity in high-salt fed Dahl rats. PLoS One 2013, 8, e60653. [Google Scholar]
- Li Volti, G.; Salomone, S.; Sorrenti, V.; Mangiameli, A.; Urso, V.; Siarkos, I.; Galvano, F.; Salamone, F. Effect of silibinin on endothelial dysfunction and ADMA levels in obese diabetic mice. Cardiovasc. Diabetol 2011, 10, 62. [Google Scholar]
- Li Volti, G.; Sorrenti, V.; Acquaviva, R.; Murabito, P.; Gullo, A.; Barcellona, M.L.; Galvano, F.; Rodella, L.; Rezzani, R.; Vanella, L.; et al. Effect of ischemia-reperfusion on renal expression and activity of N(G)-N(G)-dimethylarginine dimethylaminohydrolases. Anesthesiology 2008, 109, 1054–1062. [Google Scholar]
- Speranza, L.; Franceschelli, S.; D’Orazio, N.; Gaeta, R.; Bucciarelli, T.; Felaco, M.; Grilli, A.; Riccioni, G. The biological effect of pharmacological treatment on dimethylaminohydrolases (DDAH-1) and cationic amino acid transporter-1 (CAT-1) expression in patients with acute congestive heart failure. Microvasc. Res 2011, 82, 391–396. [Google Scholar]
- Davids, M.; Teerlink, T. Plasma concentrations of arginine and asymmetric dimethylarginine do not reflect their intracellular concentrations in peripheral blood mononuclear cells. Metabolism 2013, 62, 1455–1461. [Google Scholar]
- Haghikia, A.; Podewski, E.; Libhaber, E.; Labidi, S.; Fischer, D.; Roentgen, P.; Tsikas, D.; Jordan, J.; Lichtinghagen, R.; von Kaisenberg, C.S.; et al. Phenotyping and outcome on contemporary management in a German cohort of patients with peripartum cardiomyopathy. Basic Res. Cardiol 2013, 108, 366. [Google Scholar]
- Riccioni, G.; D’Orazio, N.; Salvatore, C.; Franceschelli, S.; Pesce, M.; Speranza, L. Carotenoids and vitamins C and E in the prevention of cardiovascular disease. Int. J. Vitam. Nutr. Res 2012, 82, 15–26. [Google Scholar]
- Speranza, L.; Pesce, M.; Franceschelli, S.; Bucciarelli, T.; Gallina, S.; Riccioni, G.; Patruno, A.; Felaco, M. The biological evaluation of ADMA/SDMA and eNOS in patients with ACHF. Front. Biosci. (Elite Ed.) 2013, 5, 551–557. [Google Scholar]
- Szlachcic, A.; Krzysiek-Maczka, G.; Pajdo, R.; Targosz, A.; Magierowski, M.; Jasnos, K.; Drozdowicz, D.; Kwiecien, S.; Brzozowski, T. The impact of asymmetric dimethylarginine (ADMA), the endogenous nitric oxide (NO) synthase inhibitor, to the pathogenesis of gastric mucosal damage. Curr. Pharm. Des 2013, 19, 90–97. [Google Scholar]
- Sydow, K.; Hornig, B.; Arakawa, N.; Bode-Böger, S.M.; Tsikas, D.; Münzel, T.; Böger, R.H. Endothelial dysfunction in patients with peripheral arterial disease and chronic hyperhomocysteinemia: Potential role of ADMA. Vasc. Med 2004, 9, 93–101. [Google Scholar]
- Dimitroulas, T.; Giannakoulas, G.; Sfetsios, T.; Karvounis, H.; Dimitroula, H.; Koliakos, G.; Settas, L. Asymmetrical dimethylarginine in systemic sclerosis-related pulmonary arterial hypertension. Rheumatology 2008, 47, 1682–1685. [Google Scholar]
- Sorrenti, V.; Mazza, F.; Campisi, A.; Vanella, L.; Li Volti, G.; di Giacomo, C. High glucose-mediated imbalance of nitric oxide synthase and dimethylargininedimethylaminohydrolase expression in endothelial cells. Curr. Neurovasc. Res 2006, 3, 49–54. [Google Scholar]
- Mohan, S.; Fung, H.L. Mechanism of cellular oxidation stress induced by asymmetric dimethylarginine. Int. J. Mol. Sci 2012, 13, 7521–7531. [Google Scholar]
- Aldámiz-Echevarría, L.; Andrade, F. Asymmetric dimethylarginine, endothelial dysfunction and renal disease. Int. J. Mol. Sci 2012, 13, 11288–11311. [Google Scholar]
- Cao, Y.; Mu, J.J.; Fang, Y.; Yuan, Z.Y.; Liu, F.Q. Impact of high salt independent of blood pressure on PRMT/ADMA/DDAH. Pathway in the aorta of dahl salt-sensitive rats. Int. J. Mol. Sci 2013, 14, 8062–8072. [Google Scholar]
- Antoniades, C.; Demosthenous, M.; Tousoulis, D.; Antonopoulos, A.S.; Vlachopoulos, C.; Toutouza, M.; Marinou, K.; Bakogiannis, C.; Mavragani, K.; Lazaros, G.; et al. Role of asymmetrical dimethylarginine in inflammation-induced endothelial dysfunction in human atherosclerosis. Hypertension 2011, 58, 93–98. [Google Scholar]
- Zuccalà, A.; Fiorenza, S.; Rapanà, R.; Santoro, A. Hypertension, atherosclerosis and kidney. G. Ital. Nefrol 2005, 22(Suppl 31), S9–S14. [Google Scholar]
- Hsu, C.P.; Lin, S.J.; Chung, M.Y.; Lu, T.M. Asymmetric dimethylarginine predicts clinical outcomes in ischemic chronic heart failure. Atherosclerosis 2012, 225, 504–510. [Google Scholar]

{kind=link}
{kind=link}
| Model | ADMA results | Final result | Author | |
|---|---|---|---|---|
| In vitro | INS-1 cells BEAS-2B cells | In INS-1 cells: (1) ADMA (0.05–32 μM) increased insulin release in vitro, except at a high concentration (32 μM); (2) ADMA stimulated the production of IL-6 and MIP-2. In BEAS-2B cells ADMA did not cause any increase in IL-8 or TNF-α or RANTES secretion. | ADMA has a pathophysiological impact leading to a diabetic situation but has no impact on the respiratory system. | [24] |
| Cultured primary human vascular endothelial cells (ECs) | PD 404182 significantly increased intracellular levels of ADMA. | PD 404182 directly and dose-dependently inhibits DDAH and reduced lipopolysaccharide (LPS)-induced NO production. | [25] | |
| Animal | Mouse model of Polycystic ovary syndrome (PCOS) | DHT (dihydrotestosterone) treatment (compared with placebo) induced no change in plasma ADMA levels. | In DHT-exposed mice, hyperandrogenemia specifically decreases endothelium dependent vasorelaxation without deterioration of smooth muscle function. | [26] |
| Male Sprague–Dawley rats | Chronic endogenous infusion of ADMA leads to a significant elevation of plasma ADMA levels. | Chronic elevated plasma levels of ADMA in healthy rats did not affect the number of peripheral blood cells including leukocyte subset. | [27] | |
| Dahl salt-sensitive (SS/JrHsd) | Serum ADMA and NO concentration remained unchanged between the baseline and 6-weeks study period in the HS-fed animals despite the treatment group. Both ADMA and NO levels were not favorably affected by treatment with INT-747. | High-salt diet downregulated DDAH expression while treatment with INT-747 protected the loss in DDAH expression and enhanced insulin sensitivity compared to vehicle controls. | [28] | |
| db/db mice | Silibinin administration markedly decreased plasma ADMA; consistently, aorta ADMA was reduced in silibinin-treated animals. | Silibinin markedly improves endothelial dysfunction in db/db mice by reducing circulating and vascular ADMA levels. | [29] | |
| Rats | Both plasma and renal concentrations of ADMA increased after renal ischemia followed by reperfusion. | Ischemia-reperfusion injury leads to reduced DDAH activity and modification of different DDAH isoform expression, thus leading to increased ADMA levels, which may lead to increased cardiovascular risk. | [30] | |
| Human study | Acute congestive heart failure | ADMA and SDMA plasma levels were significantly higher after pharmacological treatment respect to baseline values. | In patients with ACHF, acute renal impairment function and the modulation of metabolism and extracellular transport by the DDAH-1/CAT-1 system determine high ADMA and SDMA levels after therapy for acute congestive heart failure. | [31] |
| Healthy humans | ADMA, SDMA, MMA and arginine levels were significantly higher in PBMC than in plasma, whereas homoarginine levels were not significantly different. | In healthy individuals, plasma levels of arginine, MMA, ADMA, and SDMA poorly reflect their intracellular levels in PBMC. | [32] | |
| Peripartum cardiomyopathy (PPCM) | ADMA was significantly higher in serum from PPCM patients compared to healthy postpartum women. | Increased levels of Cathepsin D activity, miR-146a and ADMA in serum of PPCM patients support the pathophysiological role of 16 kDa Prolactin for PPCM and may be used as a specific diagnostic marker profile. | [33] |
© 2013 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Franceschelli, S.; Ferrone, A.; Pesce, M.; Riccioni, G.; Speranza, L. Biological Functional Relevance of Asymmetric Dimethylarginine (ADMA) in Cardiovascular Disease. Int. J. Mol. Sci. 2013, 14, 24412-24421. https://doi.org/10.3390/ijms141224412
Franceschelli S, Ferrone A, Pesce M, Riccioni G, Speranza L. Biological Functional Relevance of Asymmetric Dimethylarginine (ADMA) in Cardiovascular Disease. International Journal of Molecular Sciences. 2013; 14(12):24412-24421. https://doi.org/10.3390/ijms141224412
Chicago/Turabian StyleFranceschelli, Sara, Alessio Ferrone, Mirko Pesce, Graziano Riccioni, and Lorenza Speranza. 2013. "Biological Functional Relevance of Asymmetric Dimethylarginine (ADMA) in Cardiovascular Disease" International Journal of Molecular Sciences 14, no. 12: 24412-24421. https://doi.org/10.3390/ijms141224412
APA StyleFranceschelli, S., Ferrone, A., Pesce, M., Riccioni, G., & Speranza, L. (2013). Biological Functional Relevance of Asymmetric Dimethylarginine (ADMA) in Cardiovascular Disease. International Journal of Molecular Sciences, 14(12), 24412-24421. https://doi.org/10.3390/ijms141224412