Hypoxia-Induced Collagen Synthesis of Human Lung Fibroblasts by Activating the Angiotensin System

Abstract
:1. Introduction
2. Results
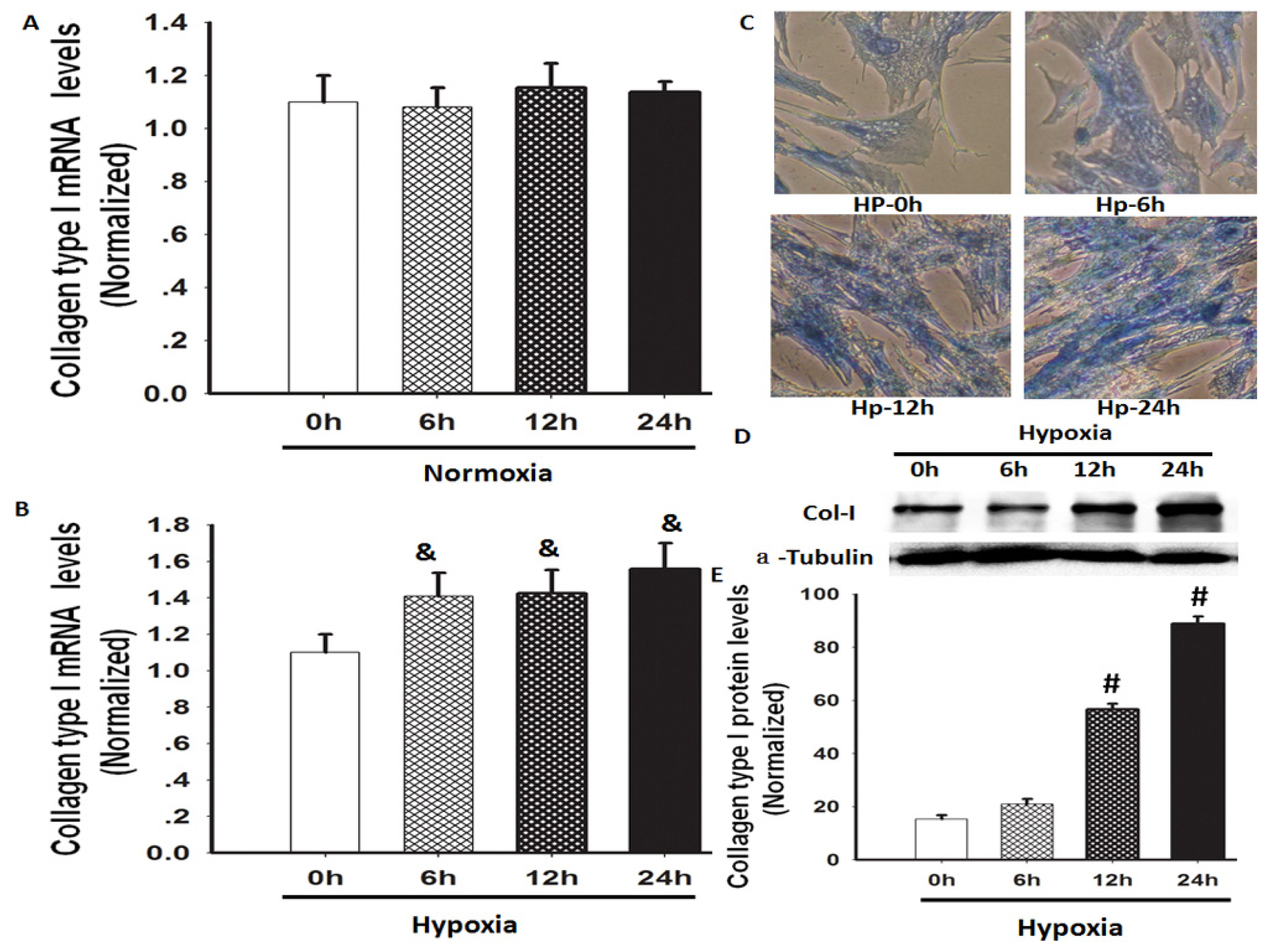
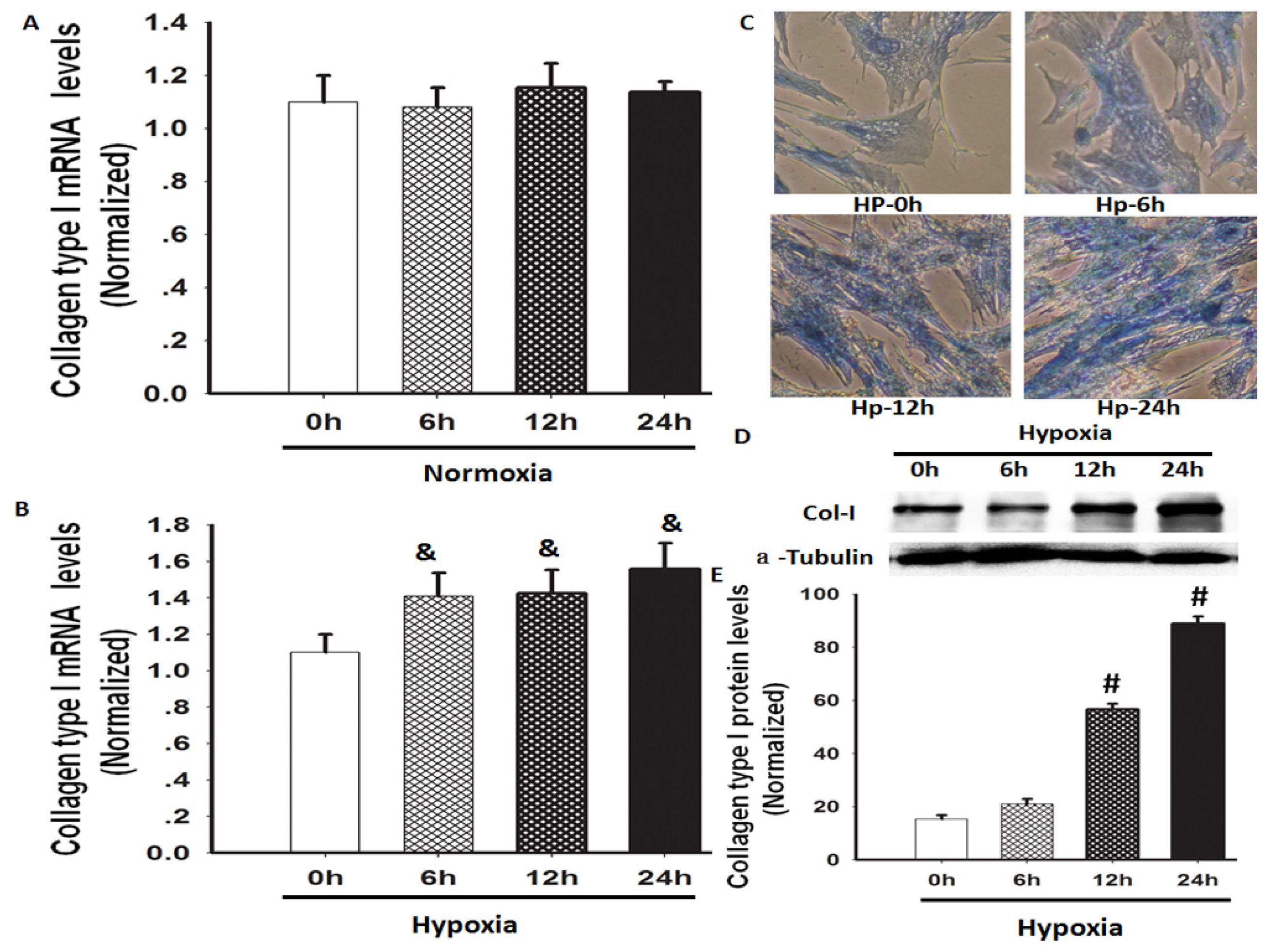
2.1. Hypoxia Increased Total Collagen and Col-I mRNA and Protein Expression
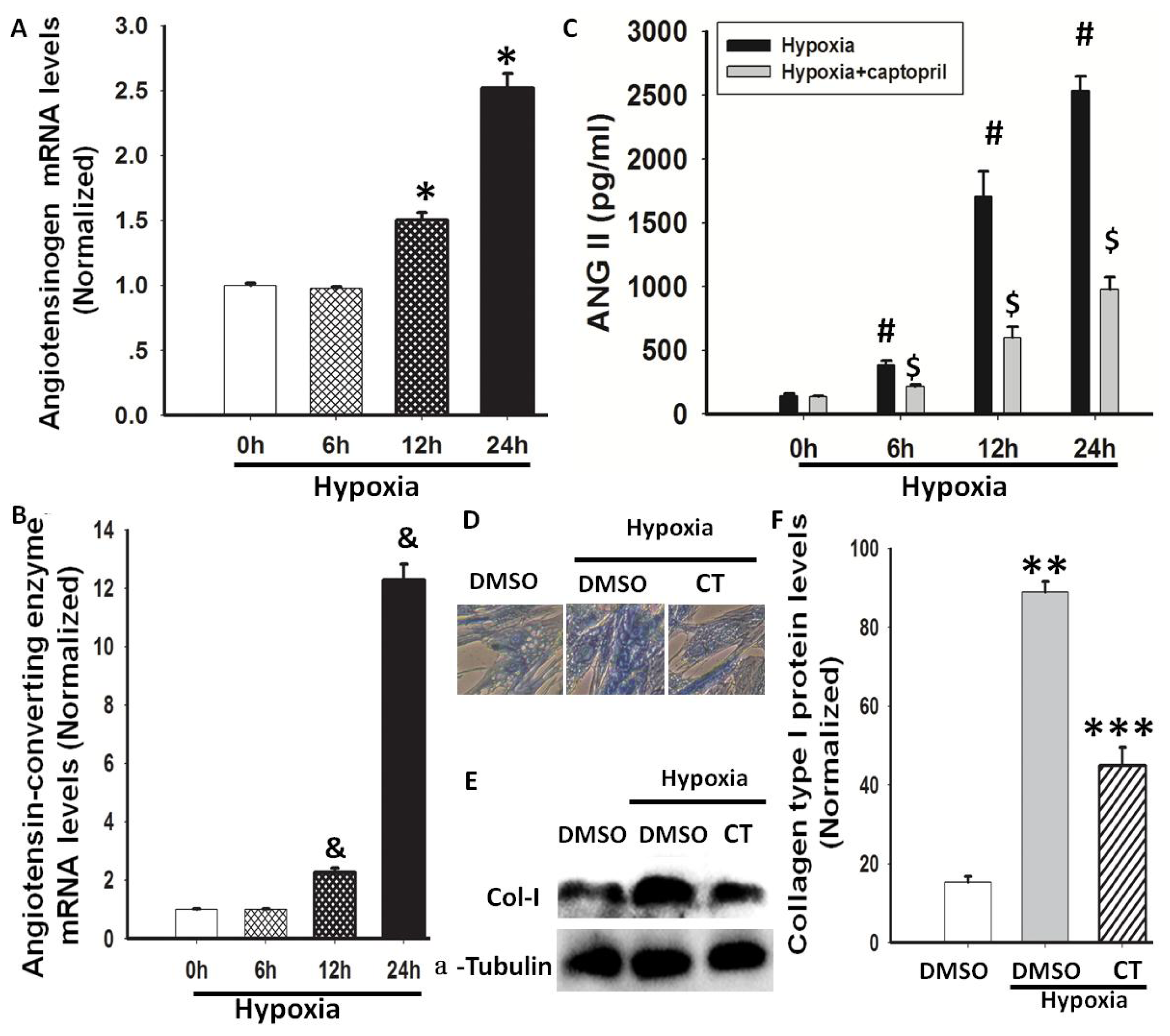
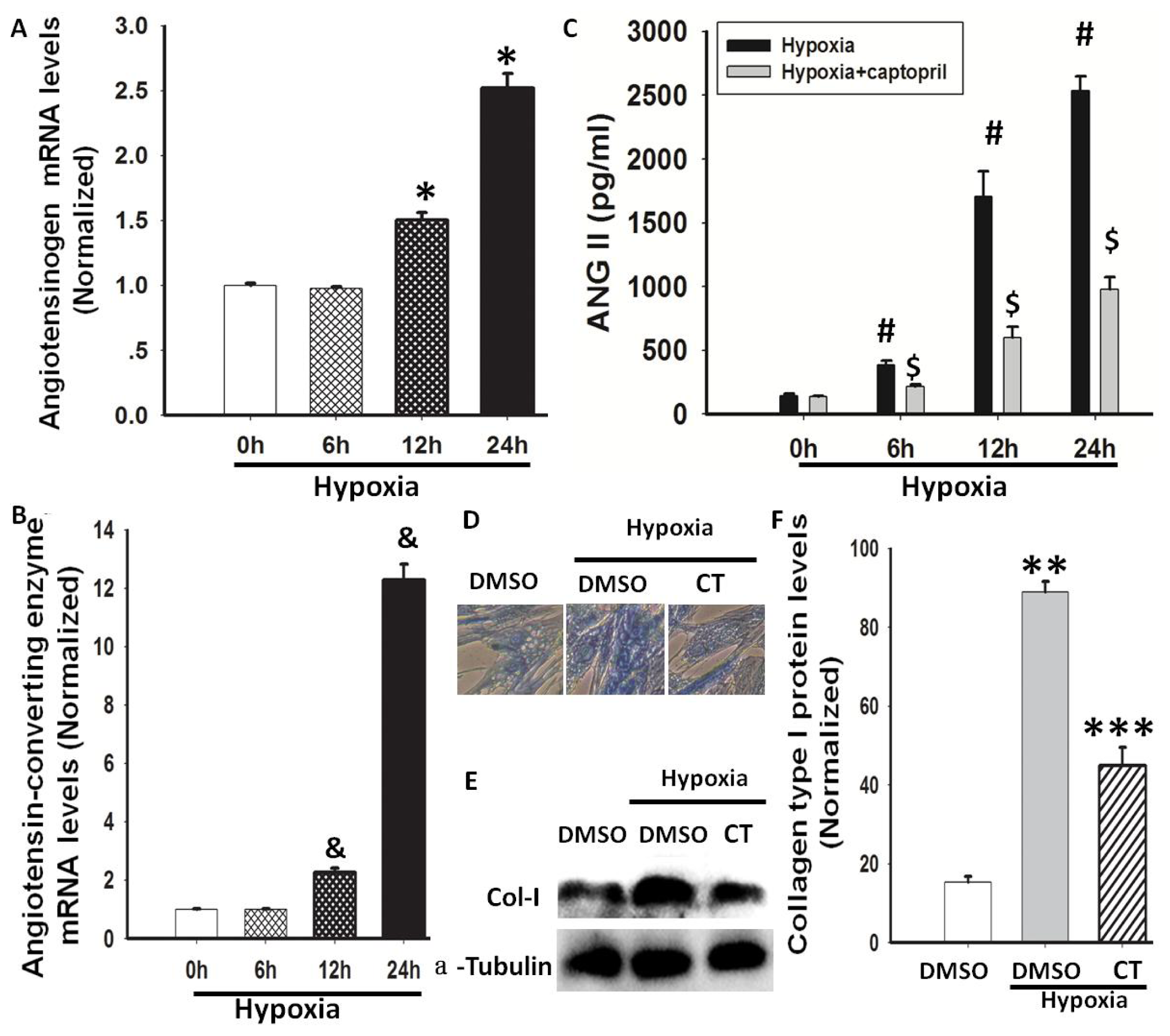
2.2. Hypoxia-Induced AGT and ACE Expression and Ang II Production
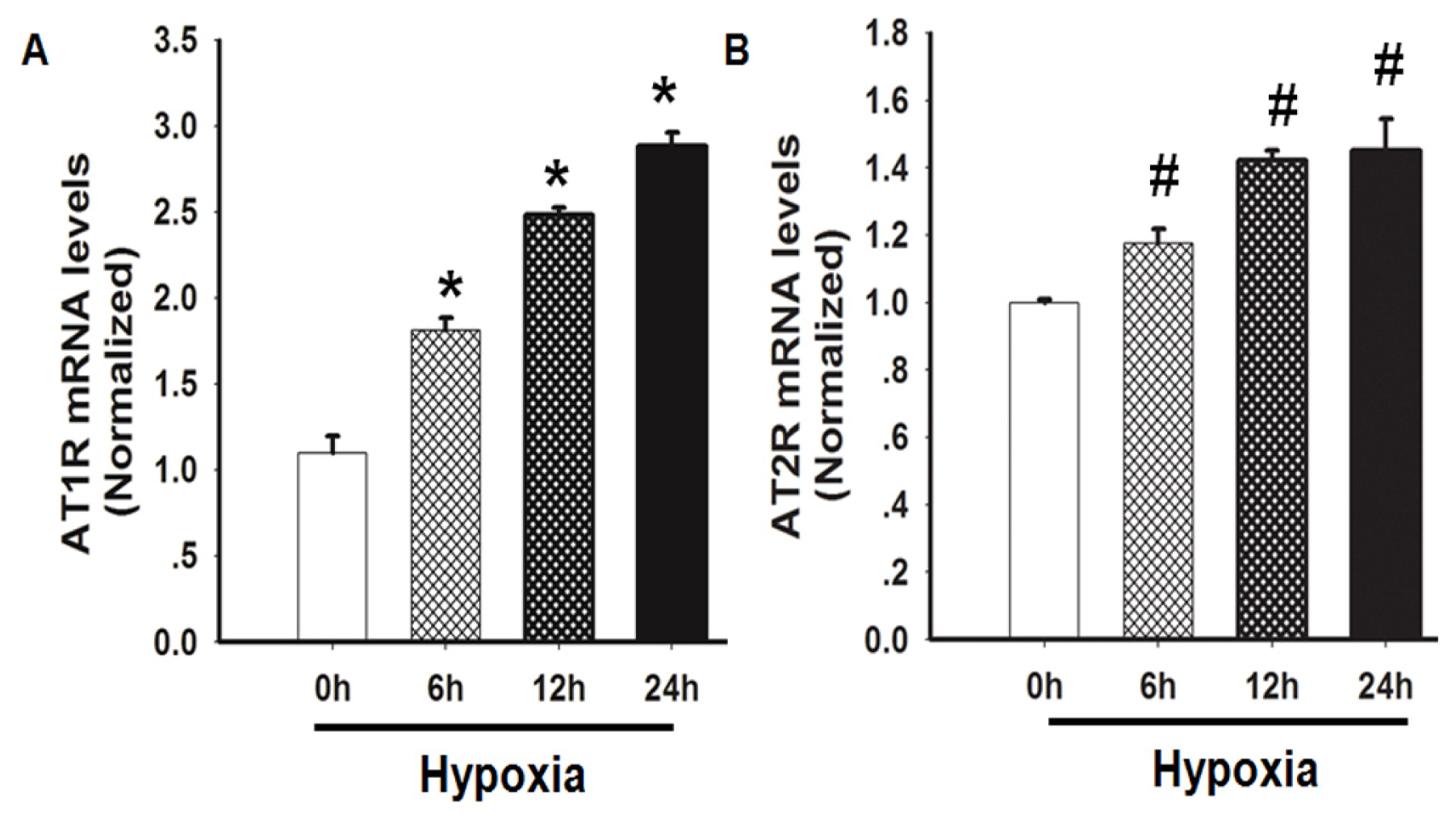
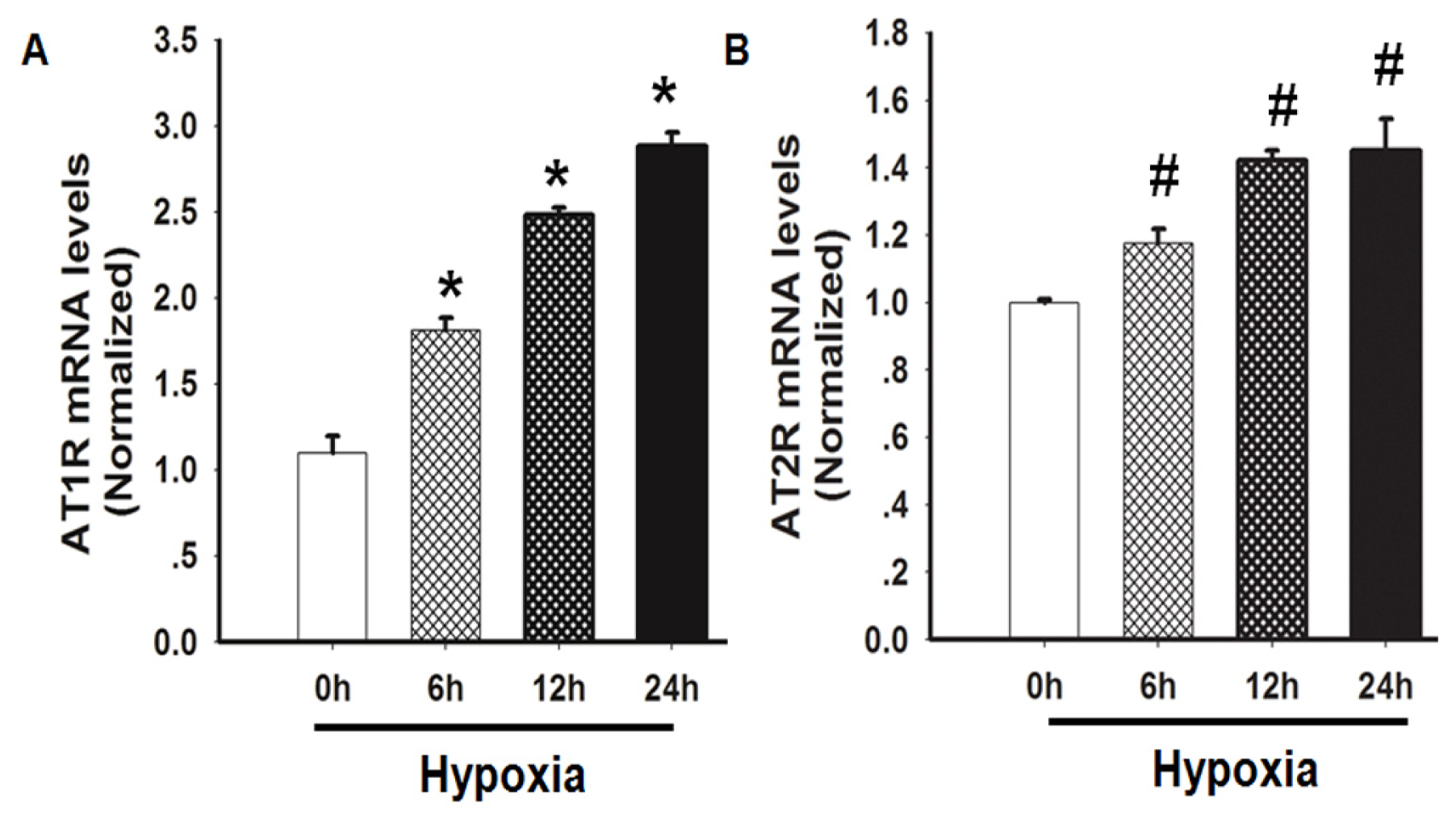
2.3. Hypoxia Induced both AT1R and AT2R mRNA Expression
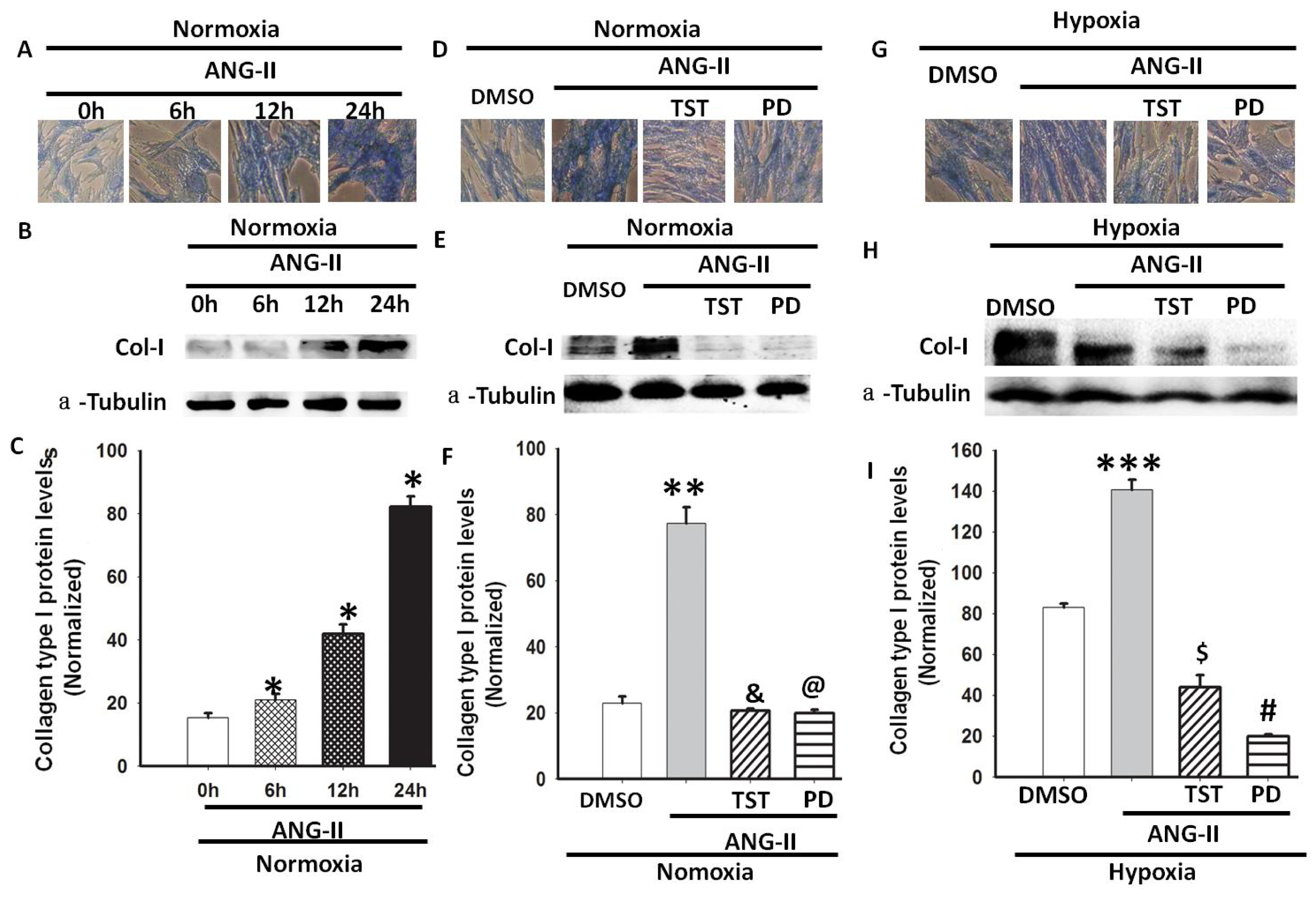
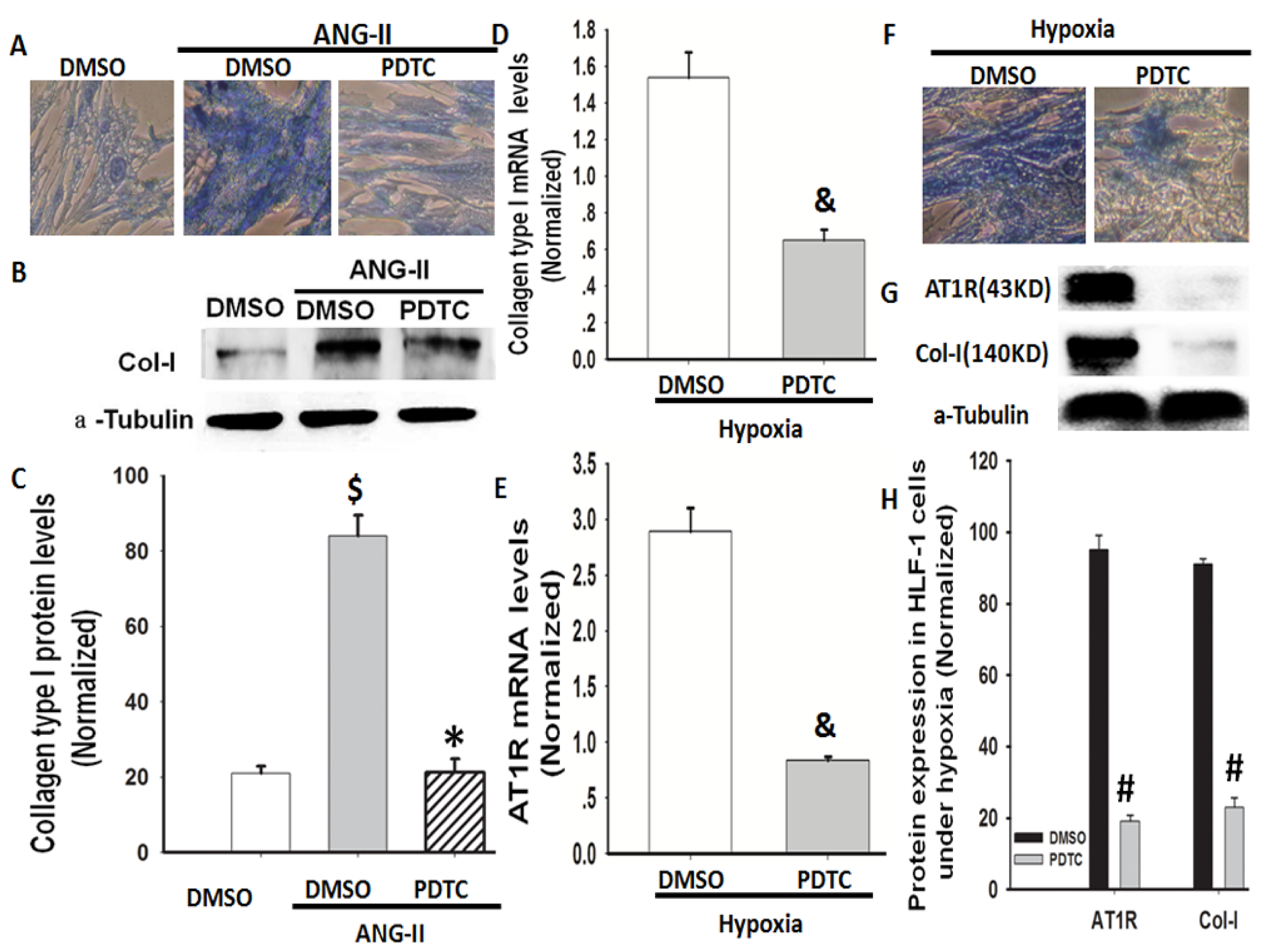
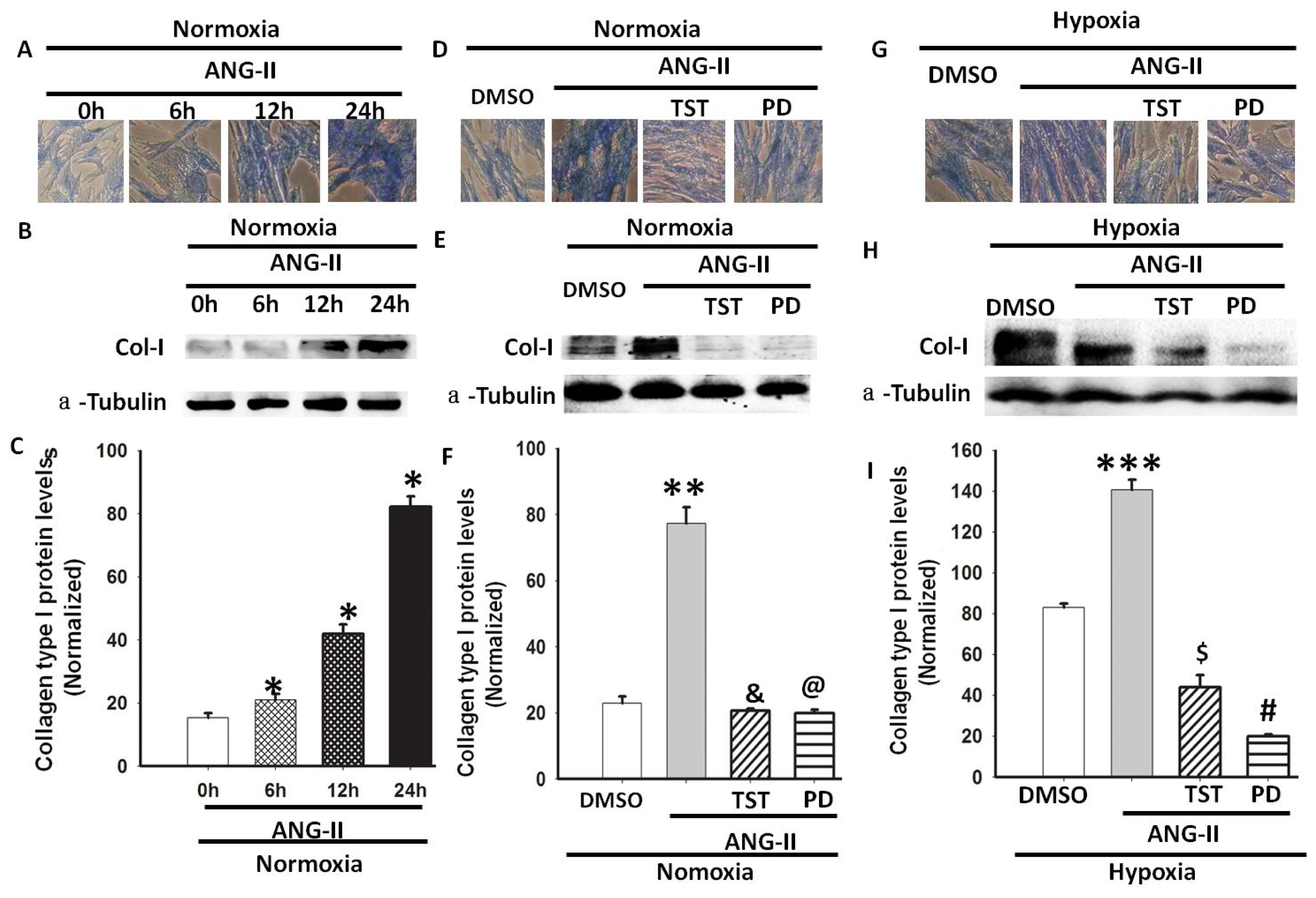
2.4. Hypoxia-Induced Col-I mRNA and Protein Expression via Ang II/ATR Signalling
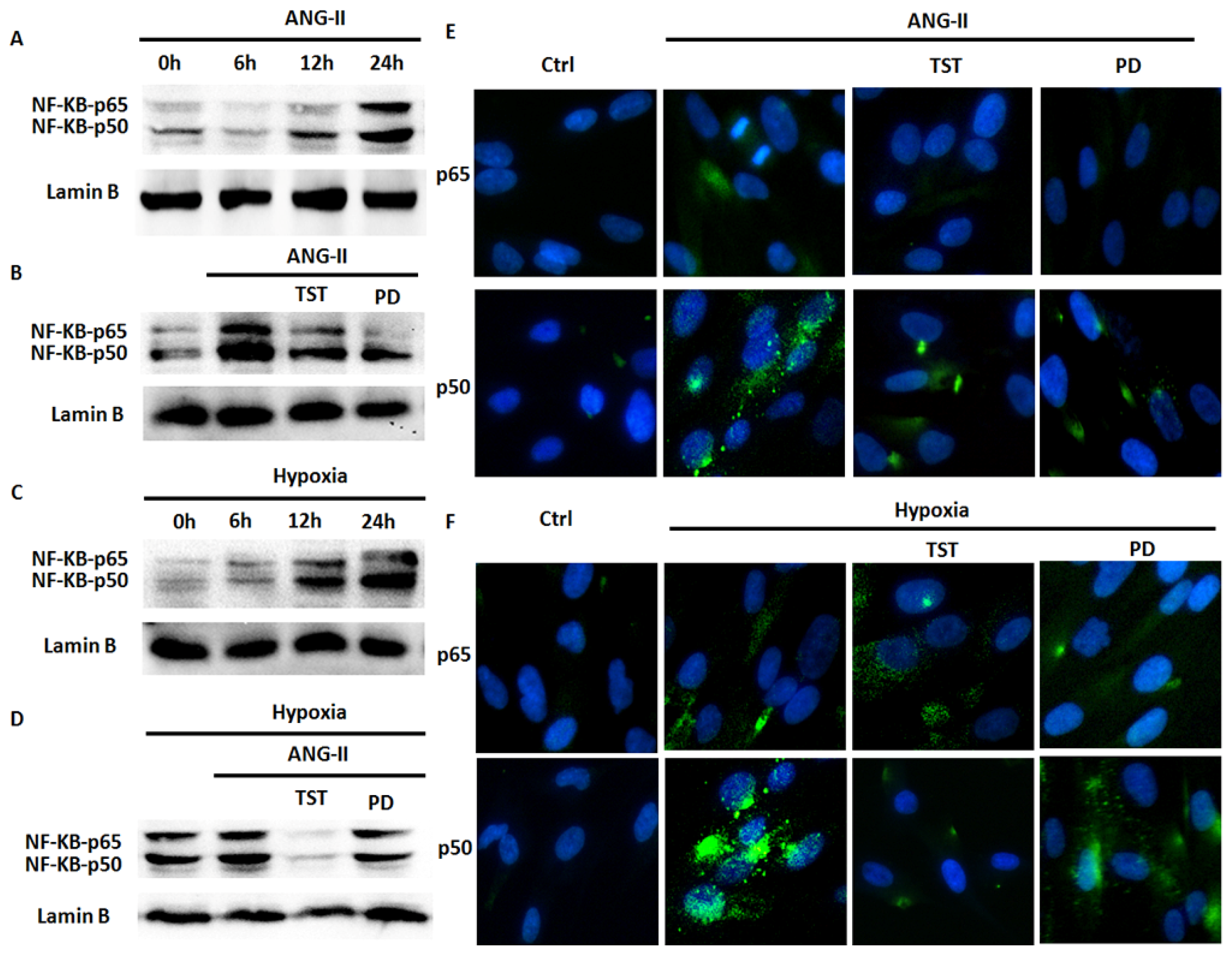
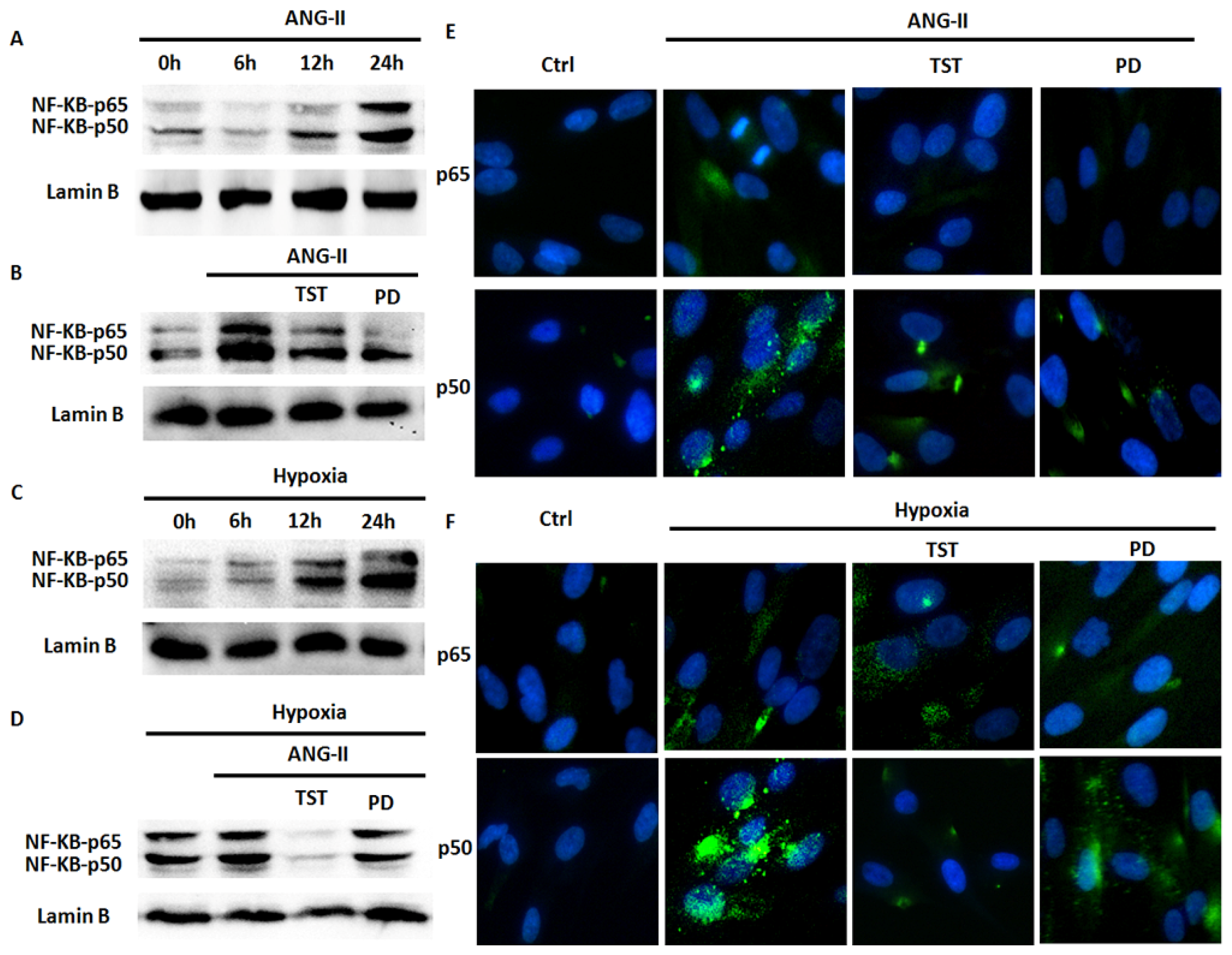
2.5. Hypoxia-Induced NF-κB Expression Involved in the Angiotensin System
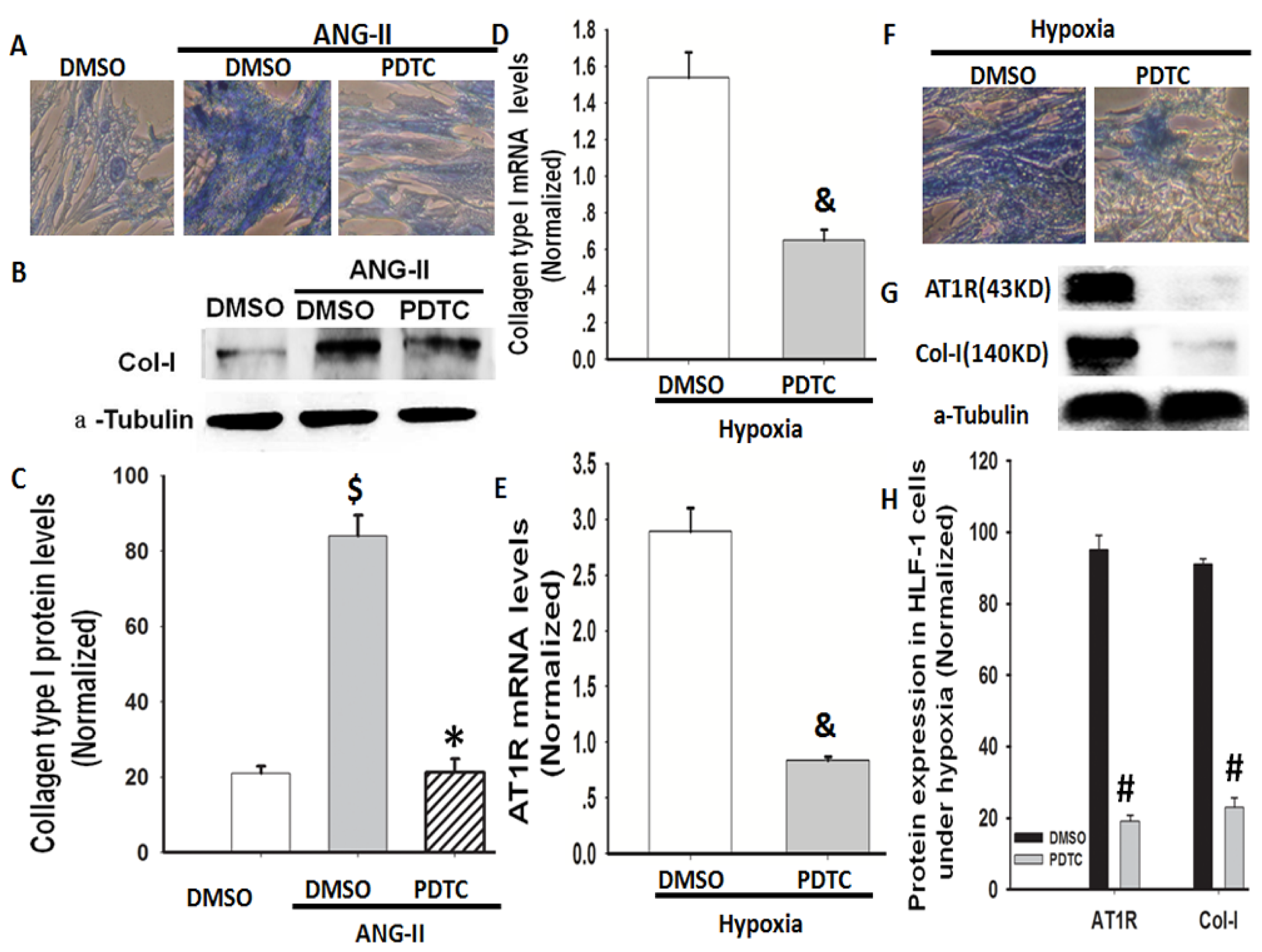
2.6. NF-κB Is Involved in Hypoxia-Induced Col-I Protein Expression Mediated by Ang II/AT1R Signalling
3. Discussion
4. Materials and Methods
4.1. Materials and Reagent
4.2. Cell Culture
4.3. RT-PCR Analysis
4.4. Western Blot Analysis
4.5. Ang II Enzyme Immunoassay
4.6. Collagen Assay
4.7. Immunofluorescence Analysis
4.8. Statistical Analysis
5. Conclusions
Acknowledgments
Conflicts of Interest
References
- Robinson, C.M.; Neary, R.; Levendale, A.; Watson, CJ.; Baugh, J.A. Hypoxia-induced DNA hypermethylation in human pulmonary fibroblasts is associated with Thy-1 promoter methylation and the development of a pro-fibrotic phenotype. Respir. Res 2012, 13, 74. [Google Scholar]
- Tzouvelekis, A.; Harokopos, V.; Paparountas, T.; Oikonomou, N.; Chatziioannou, A.; Vilaras, G.; Tsiambas, E.; Karameris, A.; Bouros, D.; Aidinis, V. Comparative expression profiling in pulmonary fibrosis suggests a role of hypoxia-inducible factor-1alpha in disease pathogenesis. Am. J. Respir. Crit. Care Med 2007, 176, 1108–1119. [Google Scholar]
- Tuder, R.M.; Yun, J.H.; Bhunia, A.; Fijalkowska, I. Hypoxia and chronic lung disease. J. Mol. Med 2007, 85, 1317–1324. [Google Scholar]
- Urquhart, D.S.; Montgomery, H.; Jaffé, A. Assessment of hypoxia in children with cystic fibrosis. Arch. Dis. Child 2005, 90, 1138–1143. [Google Scholar]
- Ahmedat, A.; Warnken, M.; Seemann, W.; Mohr, K.; Kostenis, E.; Juergens, U.; Racké, K. Pro-fibrotic processes in human lung fibroblasts are driven by an autocrine/paracrine endothelinergic system. Br. J. Pharmacol 2013, 168, 471–487. [Google Scholar]
- Wynn, T.A. Cellular and molecular mechanisms of fibrosis. J. Pathol 2008, 214, 199–210. [Google Scholar]
- Bullock, G.; Steyaert, I.; Bilbe, G.; Carey, R.M.; Kips, J.; de Paepe, B.; Pauwels, R.; Praet, M.; Siragy, H.M.; de Gasparo, M. Distribution of type-1 and type-2 angiotensin receptors in the normal human lung and in lungs from patients with chronic obstructive pulmonary disease. Histochem. Cell. Biol 2001, 115, 117–124. [Google Scholar]
- Cassis, L.; Shenoy, U.; Lipke, D.; Baughn, J.; Fettinger, M.; Gillespie, M. Lung angiotensin receptor binding characteristics during the development of monocrotaline-induced pulmonary hypertension. Biochem. Pharmacol 1997, 54, 27–31. [Google Scholar]
- Specks, U.; Martin, W.J., 2nd; Rohrbach, M.S. Bronchoalveolar lavage fluid angiotensin-converting enzyme in interstitial lung diseases. Am. Rev. Respir. Dis 1990, 141, 117–123. [Google Scholar]
- Filippatos, G.; Tilak, M.; Pinillos, H.; Uhal, B.D. Regulation of apoptosis by angiotensin II in the heart and lungs. Int. J. Mol. Med 2001, 7, 273–280. [Google Scholar]
- Marshall, R.P.; McAnulty, R.J.; Laurent, G.J. Angiotensin II is mitogenic for human lung fibroblasts via activation of the type 1 receptor. Am. J. Respir. Crit. Care. Med 2000, 161, 1999–2004. [Google Scholar]
- Rassler, B.; Marx, G.; Reissig, C.; Rohling, M.A.; Tannapfel, A.; Wenger, R.H.; Zimmer, H.G. Time course of hypoxia-induced lung injury in rats. Respir. Physiol. Neurobiol 2007, 159, 45–54. [Google Scholar]
- Kalayarasan, S.; Sriram, N.; Sudhandiran, G. Diallyl sulfide attenuates bleomycin-induced pulmonary fibrosis: Critical role of iNOS, NF-κB, TNF-α and IL-1β. Life Sci 2008, 82, 1142–1153. [Google Scholar]
- Madonna, G.; Ullman, C.D.; Gentilcore, G.; Palmieri, G.; Ascierto, P.A. NF-κB as potential target in the treatment of melanoma. J. Transl. Med 2012, 10, 53. [Google Scholar]
- Ruiz-Ortega, M.; Rupérez, M.; Esteban, V.; Rodríguez-Vita, J.; Sánchez-López, E.; Carvajal, G.; Egido, J.; Angiotensin, I.I. A key factor in the inflammatory and fibrotic response in kidney diseases. Nephrol. Dial. Transplant 2006, 21, 16–20. [Google Scholar]
- Pastor, M.D.; Nogal, A.; Molina-Pinelo, S.; Meléndez, R.; Romero-Romero, B.; Mediano, M.D.; López-Campos, J.L.; García-Carbonero, R.; Sanchez-Gastaldo, A.; Carnero, A.; et al. Identification of oxidative stress related proteins as biomarkers for lung cancer and chronic obstructive pulmonary disease in bronchoalveolar lavage. Int. J. Mol. Sci 2013, 14, 3440–3455. [Google Scholar]
- Mancini, G.B.; Etminan, M.; Zhang, B.; Levesque, L.E.; FitzGerald, J.M.; Brophy, J.M. Reduction of morbidity and mortality by statins, angiotensin-converting enzyme inhibitors, and angiotensin receptor blockers in patients with chronic obstructive pulmonary disease. J. Am. Coll. Cardiol 2006, 47, 2554–2560. [Google Scholar]
- Waseda, Y.; Yasui, M.; Nishizawa, Y.; Inuzuka, K.; Takato, H.; Ichikawa, Y.; Tagami, A.; Fujimura, M.; Nakao, S. Angiotensin II type 2 receptor antagonist reduces bleomycin-induced pulmonary fibrosis in mice. Respir. Res 2008, 9, 43. [Google Scholar]
- Ruiz-Ortega, M.; Ruperez, M.; Esteban, V.; Egido, J. Molecular mechanisms of angiotensin II-induced vascular injury. Curr. Hypertens. Rep 2003, 5, 73–79. [Google Scholar]
- Edwards, M.R.; Bartlett, N.W.; Clarke, D.; Birrell, M.; Belvisi, M.; Johnston, S.L. Targeting the NF-κB pathway in asthma and chronic obstructive pulmonary disease. Pharmacol. Ther 2009, 121, 1–13. [Google Scholar]
- Tamada, S.; Asai, T.; Kuwabara, N.; Iwai, T.; Uchida, J.; Teramoto, K.; Kaneda, N.; Yukimura, T.; Komiya, T.; Nakatani, T.; Miura, K. Molecular mechanisms and therapeutic strategies of chronic renal injury: the role of nuclear factor kappaB activation in the development of renal fibrosis. J. Pharmacol. Sci 2006, 100, 17–21. [Google Scholar]
- Muller, D.N.; Dechend, R.; Mervaala, E.M.; Park, J.K.; Schmidt, F.; Fiebeler, A.; Theuer, J.; Breu, V.; Ganten, D.; Haller, H.; et al. NF-kappaB inhibition ameliorates angiotensin II-induced inflammatory damage in rats. Hypertension 2000, 35, 193–201. [Google Scholar]
- Kirk, J.M.; Heard, B.E.; Kerr, I.; Turner-Warwick, M.; Laurent, G.J. Quantitation of types I and III collagen in biopsy lung samples from patients with cryptogenic fibrosing alveolitis. Coll. Relat. Res 1984, 4, 169–182. [Google Scholar]
- Park, S.J.; Lee, K.S.; Lee, S.J.; Kim, S.R.; Park, S.Y.; Jeon, M.S.; Lee, H.B.; Lee, Y.C. l-2-Oxothiazolidine-4-Carboxylic acid or α-Lipoic acid attenuates airway remodeling: Involvement of nuclear factor-κB (NF-κB), nuclear factor erythroid 2p45-related factor-2 (Nrf2), and hypoxia-inducible factor (HIF). Int. J. Mol. Sci 2012, 13, 7915–7937. [Google Scholar]
- Re, R.N. Implications of intracrine hormone action for physiology and medicine. Am. J. Physiol. Heart. Circ. Physiol 2003, 284, H751–H757. [Google Scholar]
- Krick, S.; Hänze, J.; Eul, B.; Savai, R.; Seay, U.; Grimminger, F.; Lohmeyer, J.; Klepetko, W.; Seeger, W.; Rose, F. Hypoxia-driven proliferation of human pulmonary artery fibroblasts: Cross-talk between HIF-1α and an autocrine angiotensin system. FASEB J 2005, 19, 857–859. [Google Scholar]
- Pendergrass, K.D.; Gwathmey, T.M.; Michalek, R.D.; Grayson, J.M.; Chappell, M.C. The angiotensin II-AT1 receptor stimulates reactive oxygen species within the cell nucleus. Biochem. Biophys. Res. Commun 2009, 384, 149–154. [Google Scholar]
- Brasier, A.R.; Jamaluddin, M.; Han, Y.; Patterson, C.; Runge, M.S. Angiotensin II induces gene transcription through cell-type-dependent effects on the nuclear factor-kappaB (NF-κB) transcription factor. Mol. Cell. Biochem 2000, 212, 155–169. [Google Scholar]
- Ihara, M.; Urata, H.; Kinoshita, A.; Suzumiya, J.; Sasaguri, M.; Kikuchi, M.; Ideishi, M.; Arakawa, K. Increased chymase-dependent angiotensin II formation in human atherosclerotic aorta. Hypertension 1999, 33, 1399–1405. [Google Scholar]
- Orito, K.; Suzuki, Y.; Matsuda, H.; Shirai, M.; Akahori, F. Chymase is activated in the pulmonary inflammation and fibrosis induced by paraquat in hamsters. Tohoku J. Exp. Med 2004, 203, 287–294. [Google Scholar]
- Lang, Y.D.; Chang, S.F.; Wang, L.F.; Chen, C.M. Chymase mediates paraquat-induced collagen production in human lung fibroblasts. Toxicol. Lett 2010, 193, 19–25. [Google Scholar]
- Li, X.; Meng, Y.; Wu, P.; Zhang, Z.; Yang, X. Angiotensin II and Aldosterone stimulating NF-κB and AP-1 activation in hepatic fibrosis of rat. Regul. Pept 2007, 138, 15–25. [Google Scholar]
- Sármán, B.; Skoumal, R.; Leskinen, H.; Rysä, J.; Ilves, M.; Soini, Y.; Tuukkanen, J.; Pikkarainen, S.; Lakó-Futó, Z.; Sármán, B.; et al. Nuclear factor-kappaB signaling contributes to severe, but not moderate, angiotensin II-induced left ventricular remodeling. J. Hypertens 2007, 25, 1927–1939. [Google Scholar]
- Gómez-Garre, D.; Largo, R.; Tejera, N.; Fortes, J.; Manzarbeitia, F.; Egido, J. Activation of NF-κB in tubular epithelial cells of rats with intense proteinuria: role of angiotensin II and endothelin-1. Hypertension 2001, 37, 1171–1178. [Google Scholar]
- Wolf, G.; Wenzel, U.; Burns, K.D.; Harris, R.C.; Stahl, R.A.; Thaiss, F. Angiotensin II activates nuclear transcription factor-kappaB through AT1 and AT2 receptors. Kidney Int 2002, 61, 1986–1995. [Google Scholar]
- Ruiz-Ortega, M.; Lorenzo, O.; Rupérez, M.; König, S.; Wittig, B.; Egido, J. Angiotensin II activates nuclear transcription factor kappaB through AT(1) and AT(2) in vascular smooth muscle cells: molecular mechanisms. Circ. Res 2000, 86, 1266–1272. [Google Scholar]
- Cairns, J.A.; Walls, A.F. Mast cell tryptase stimulates the synthesis of type I collagen in human lung fibroblasts. J. Clin. Invest 1997, 99, 1313–1321. [Google Scholar]
- Zhou, T.B.; Qin, Y.H.; Li, Z.Y.; Xu, H.L.; Zhao, Y.J.; Lei, F.Y. All-trans retinoic acid treatment is associated with prohibitin expression in renal interstitial fibrosis rats. Int. J. Mol. Sci 2012, 13, 2769–2782. [Google Scholar]
- Mifune, M.; Sasamura, H.; Shimizu-Hirota, R.; Miyazaki, H.; Saruta, T. Angiotensin II type 2 receptors stimulate collagen synthesis in cultured vascular smooth muscle cells. Hypertension 2000, 36, 845–850. [Google Scholar]
- Uhal, B.D.; Li, X.; Piasecki, C.C.; Molina-Molina, M. Angiotensin signalling in pulmonary fibrosis. Int. J. Biochem. Cell Biol 2012, 44, 465–468. [Google Scholar]
- Tang, J.M.; Wang, J.N.; Zhang, L.; Zheng, F.; Yang, J.Y.; Kong, X.; Guo, L.Y.; Chen, L.; Huang, Y.Z.; Wan, Y.; et al. VEGF/SDF-1 promotes cardiac stem cell mobilization and myocardial repair in the infarcted heart. Cardiovasc. Res 2011, 91, 402–411. [Google Scholar]
- Tang, J.; Wang, J.; Kong, X.; Yang, J.; Guo, L.; Zheng, F.; Zhang, L.; Huang, Y.; Wan, Y. Vascular endothelial growth factor promotes cardiac stem cell migration via the PI3K/Akt pathway. Exp. Cell. Res 2009, 315, 3521–3531. [Google Scholar]
- Lang, Y.D.; Hung, C.L.; Wu, T.Y.; Wang, L.F.; Chen, C.M. The renin-angiotensin system mediates hyperoxia-induced collagen production in human lung fibroblasts. Free Radic. Biol. Med 2010, 49, 88–95. [Google Scholar]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Target name | Primer name | Sequence (5′-3′) | Amplicon length |
|---|---|---|---|
| Col-I | Collagen type I | CTTCACCTACAGCGTCACTG GGATGGAGGGAGTTTACAGG | 194 |
| AGT | Angiotensinogen | CACCTCGTCATCCACAATGAGA GATGTCTTGGCCTGAATTGG | 107 |
| ACE | Angiotensin-converting enzyme | CGACGAGCATGACATCAACT TCTCCTTGGTGATGCTTCCAT | 122 |
| AT1R | Angiotensin II receptor type 1 | ATCCACCAAGAAGCCTGCAC TGAAGTGCTGCAGAGGAATG | 112 |
| AT2R | Angiotensin II receptor type 2 | CCTCGCTGTGGCTGATTTACTCCTT TTGCACATCACAGGTCCAA | 109 |
| β-actin | GTCCACCGCAAATGCTTCTA TGCTGTCACCTTCACCGTTC | 190 |
© 2013 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Liu, S.-S.; Wang, H.-Y.; Tang, J.-M.; Zhou, X.-M. Hypoxia-Induced Collagen Synthesis of Human Lung Fibroblasts by Activating the Angiotensin System. Int. J. Mol. Sci. 2013, 14, 24029-24045. https://doi.org/10.3390/ijms141224029
Liu S-S, Wang H-Y, Tang J-M, Zhou X-M. Hypoxia-Induced Collagen Synthesis of Human Lung Fibroblasts by Activating the Angiotensin System. International Journal of Molecular Sciences. 2013; 14(12):24029-24045. https://doi.org/10.3390/ijms141224029
Chicago/Turabian StyleLiu, Shan-Shan, Hao-Yan Wang, Jun-Ming Tang, and Xiu-Mei Zhou. 2013. "Hypoxia-Induced Collagen Synthesis of Human Lung Fibroblasts by Activating the Angiotensin System" International Journal of Molecular Sciences 14, no. 12: 24029-24045. https://doi.org/10.3390/ijms141224029
APA StyleLiu, S.-S., Wang, H.-Y., Tang, J.-M., & Zhou, X.-M. (2013). Hypoxia-Induced Collagen Synthesis of Human Lung Fibroblasts by Activating the Angiotensin System. International Journal of Molecular Sciences, 14(12), 24029-24045. https://doi.org/10.3390/ijms141224029