Identification of Combined Genetic Determinants of Liver Stiffness within the SREBP1c-PNPLA3 Pathway

Abstract
:1. Introduction
2. Results
2.1. Phenotypic Characterization by Transient Elastography (TE)
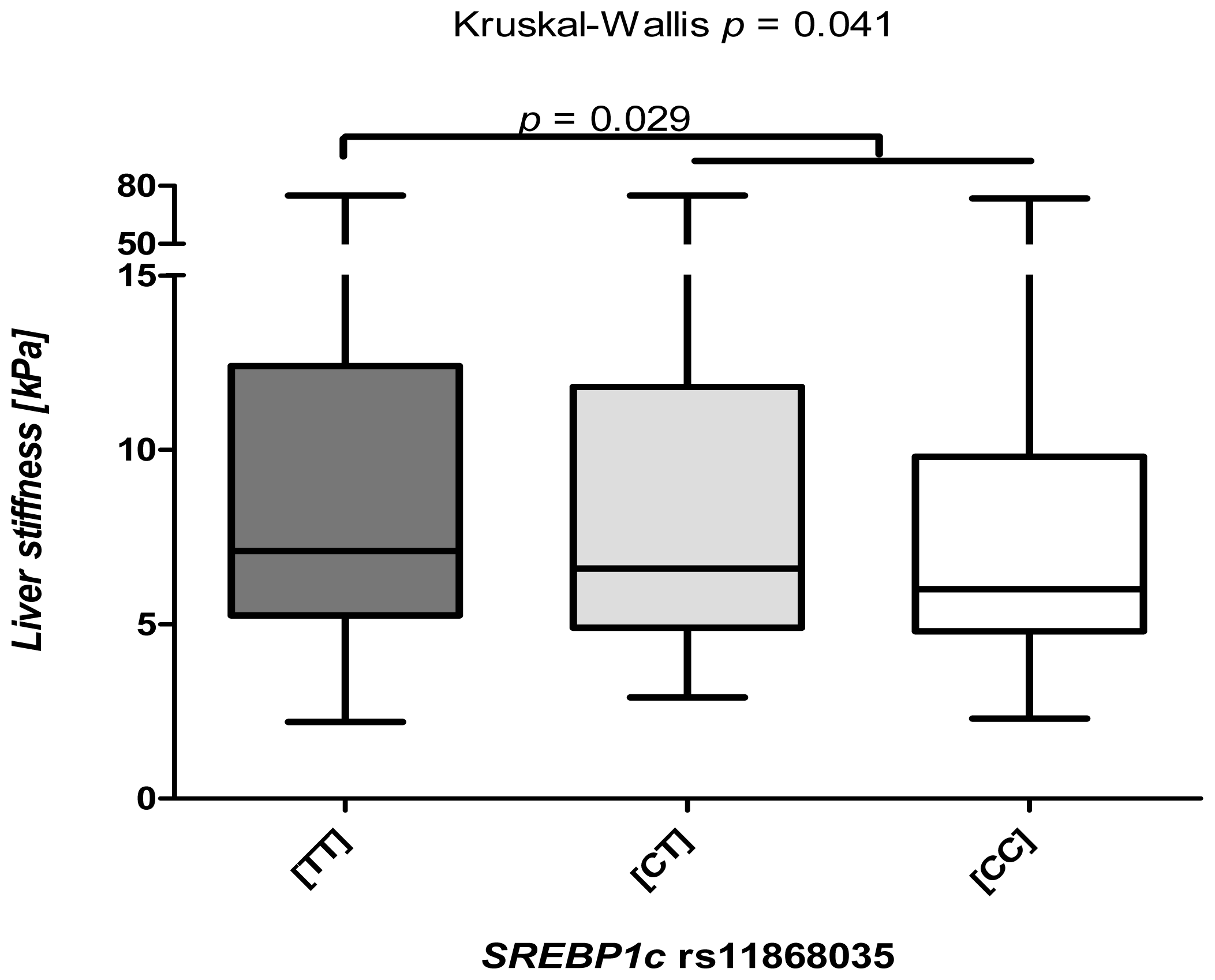
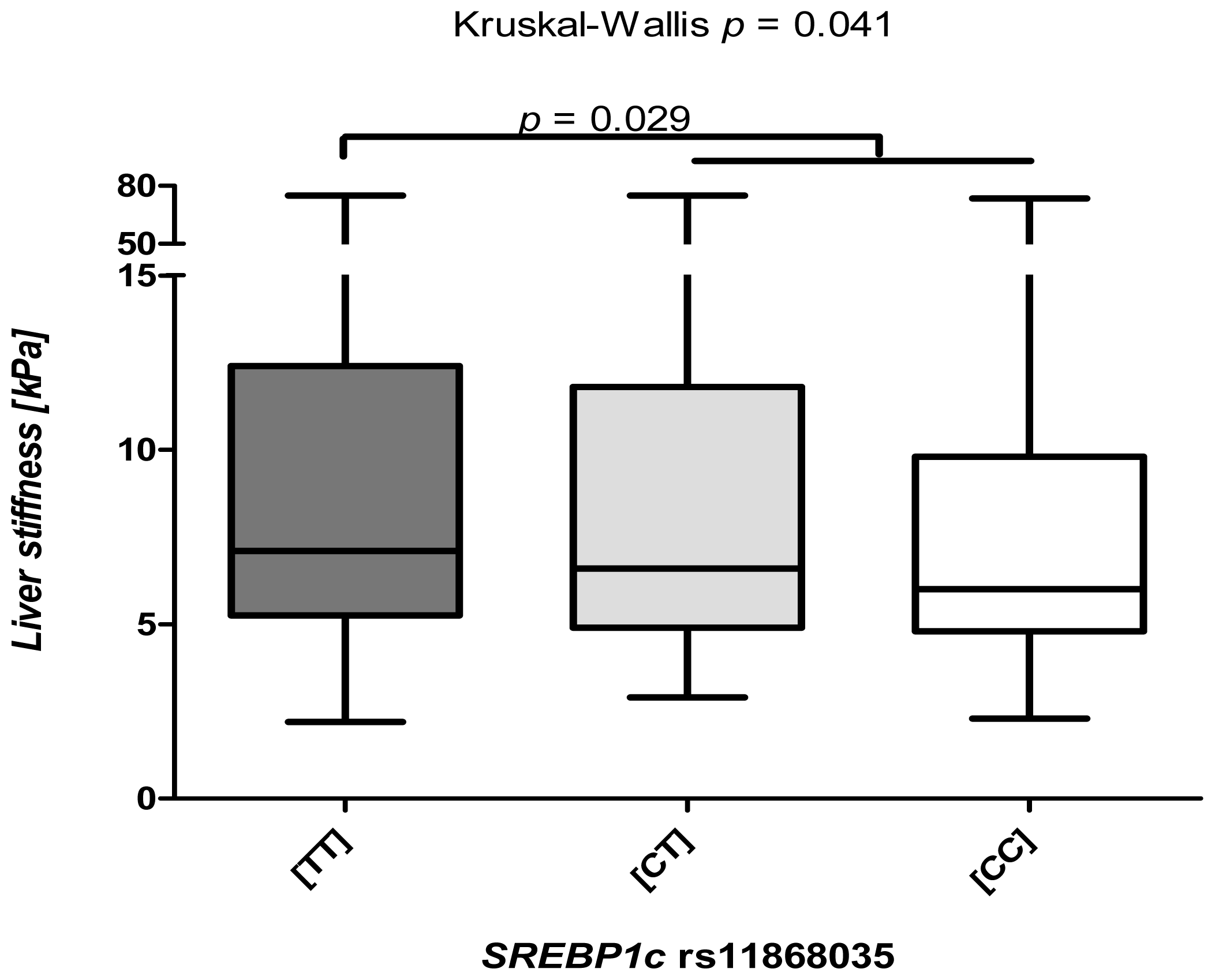
2.2. The SREBP1c rs11868038 Variant Increases Liver Fibrosis
2.3. The SREBP1c Polymorphism Is Associated Predominantly with Low TE Levels
2.4. The Common SREBP1c Variant Increases Liver Stiffness Independently of Other Profibrogenic Factors
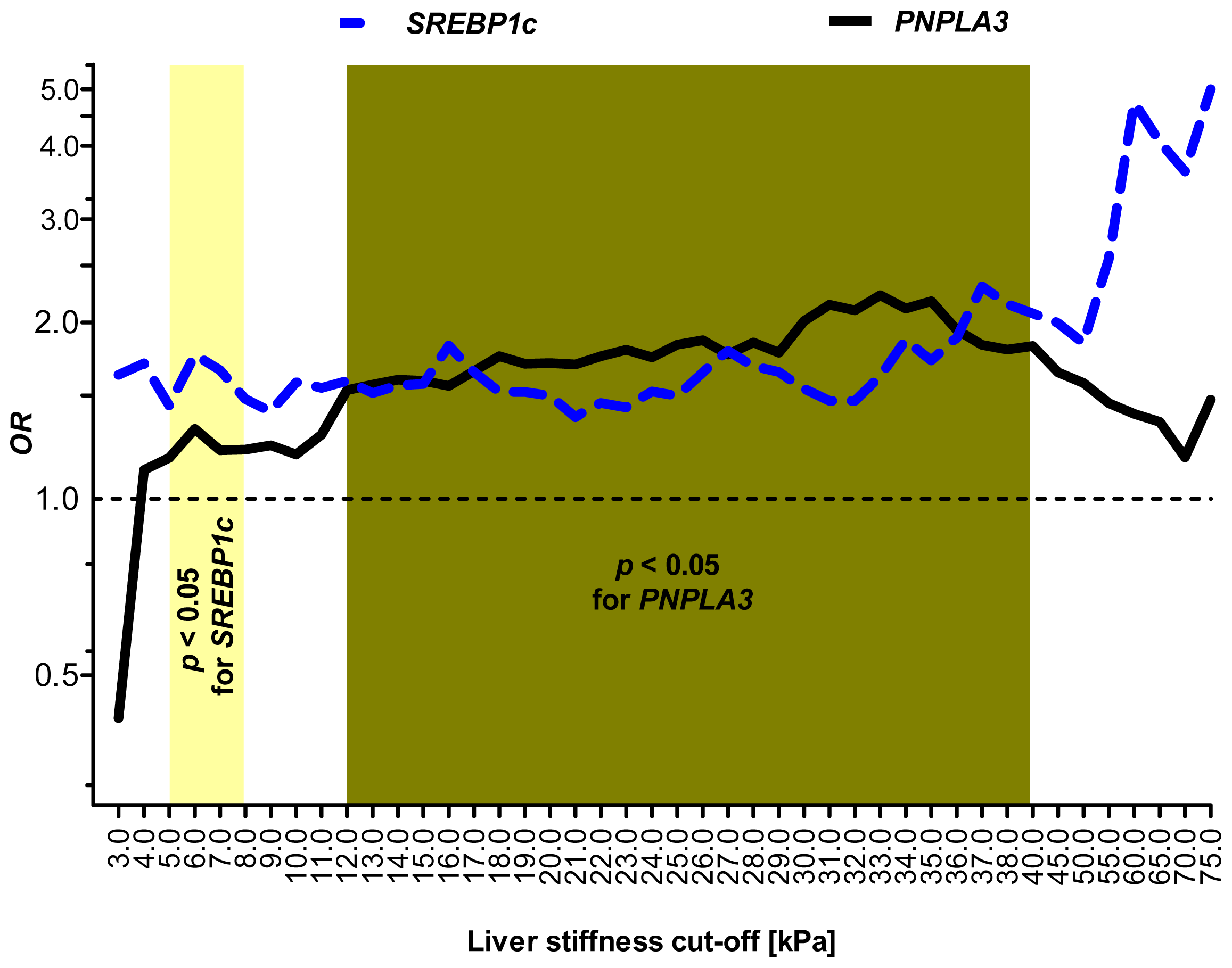
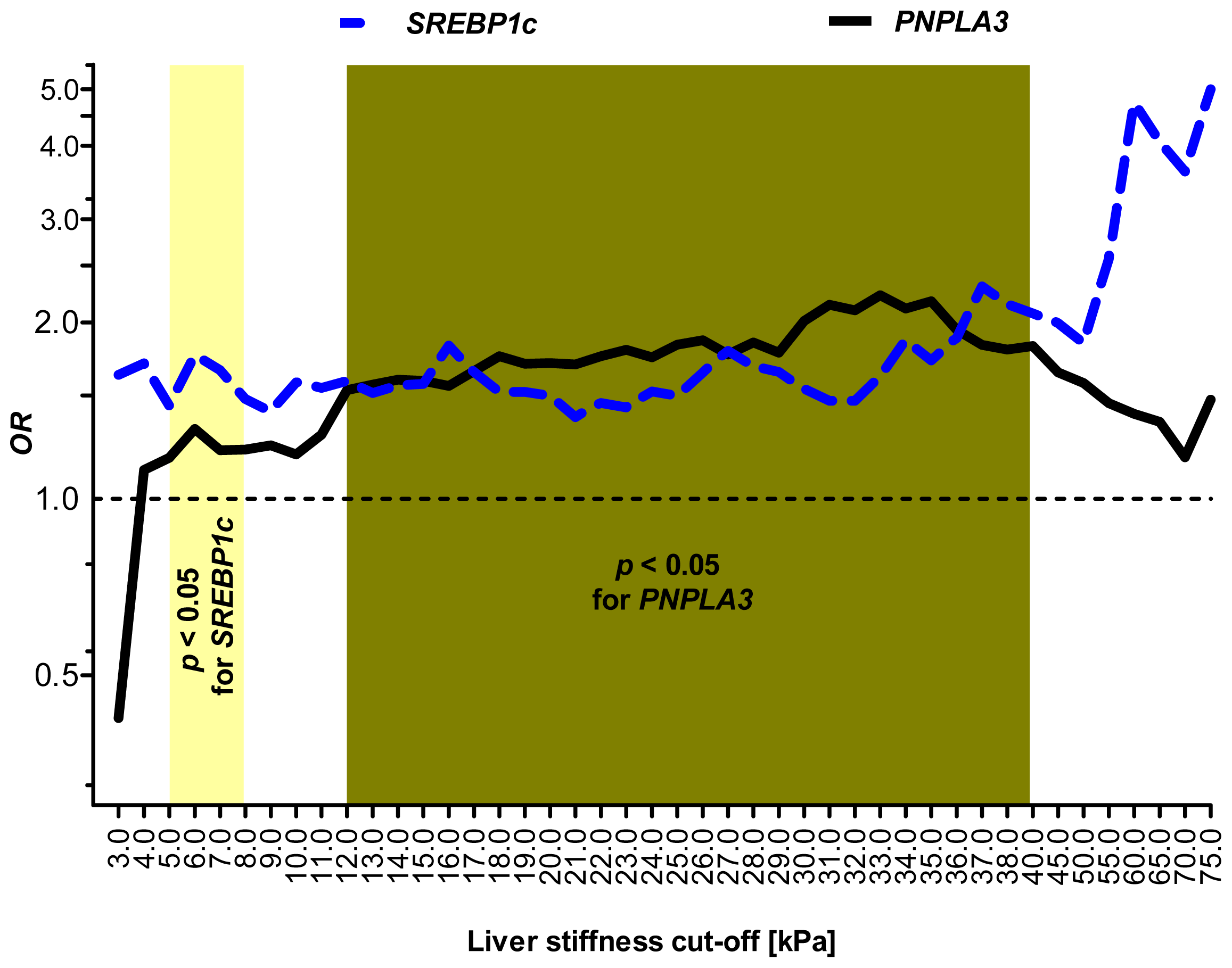
2.5. Sensitivity Analysis: SREBP1c and PNPLA3 Variants Are Associated with Distinct Stages of Liver Injury
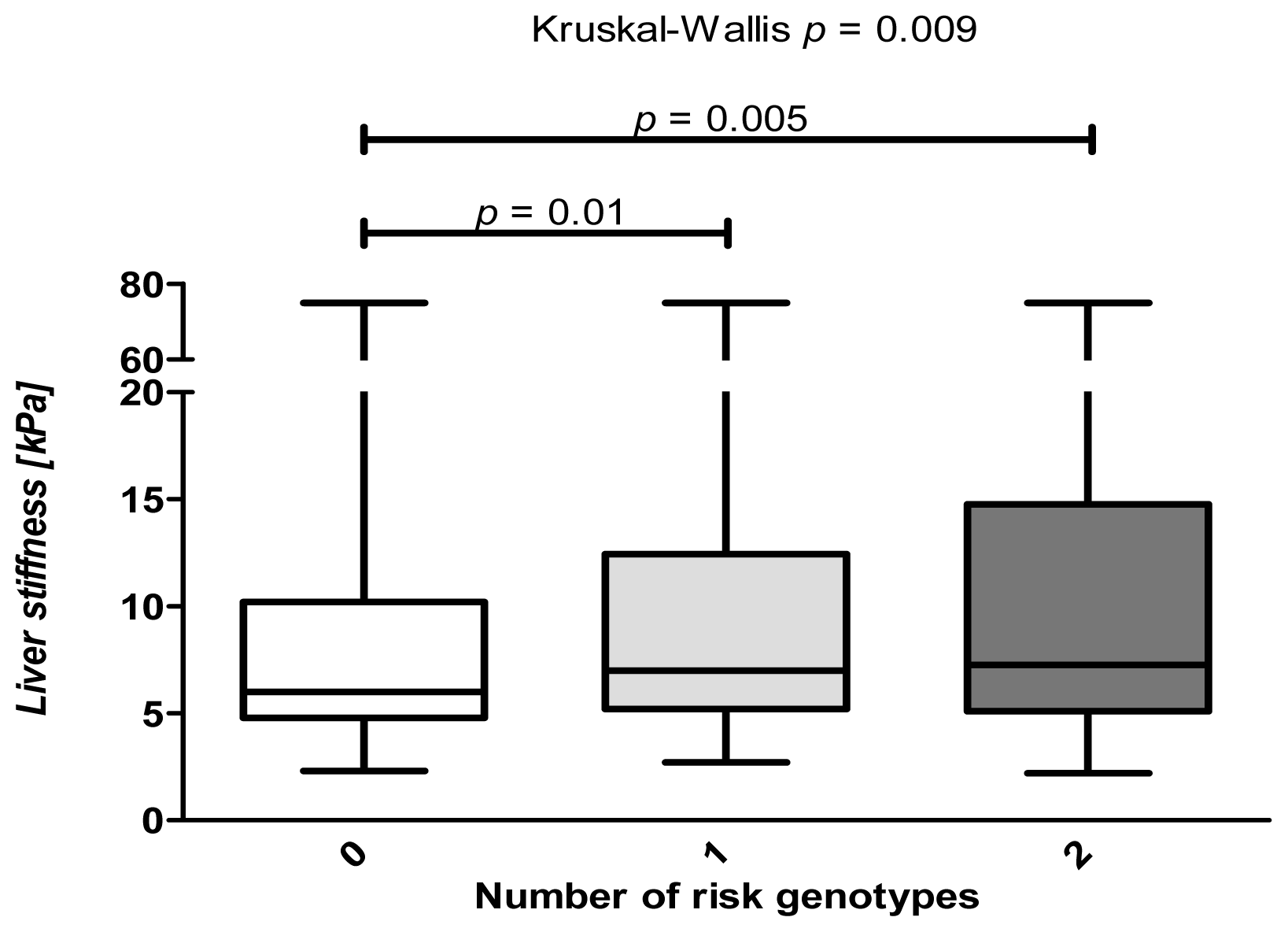
2.6. Combined Analysis of the Common PNPLA3 and SREBP1c Variants
3. Discussion
4. Experimental Section
4.1. Study Cohort
4.2. DNA Isolation and Genotyping of the SREBP1c Variants
4.3. Statistical Analysis
5. Conclusions
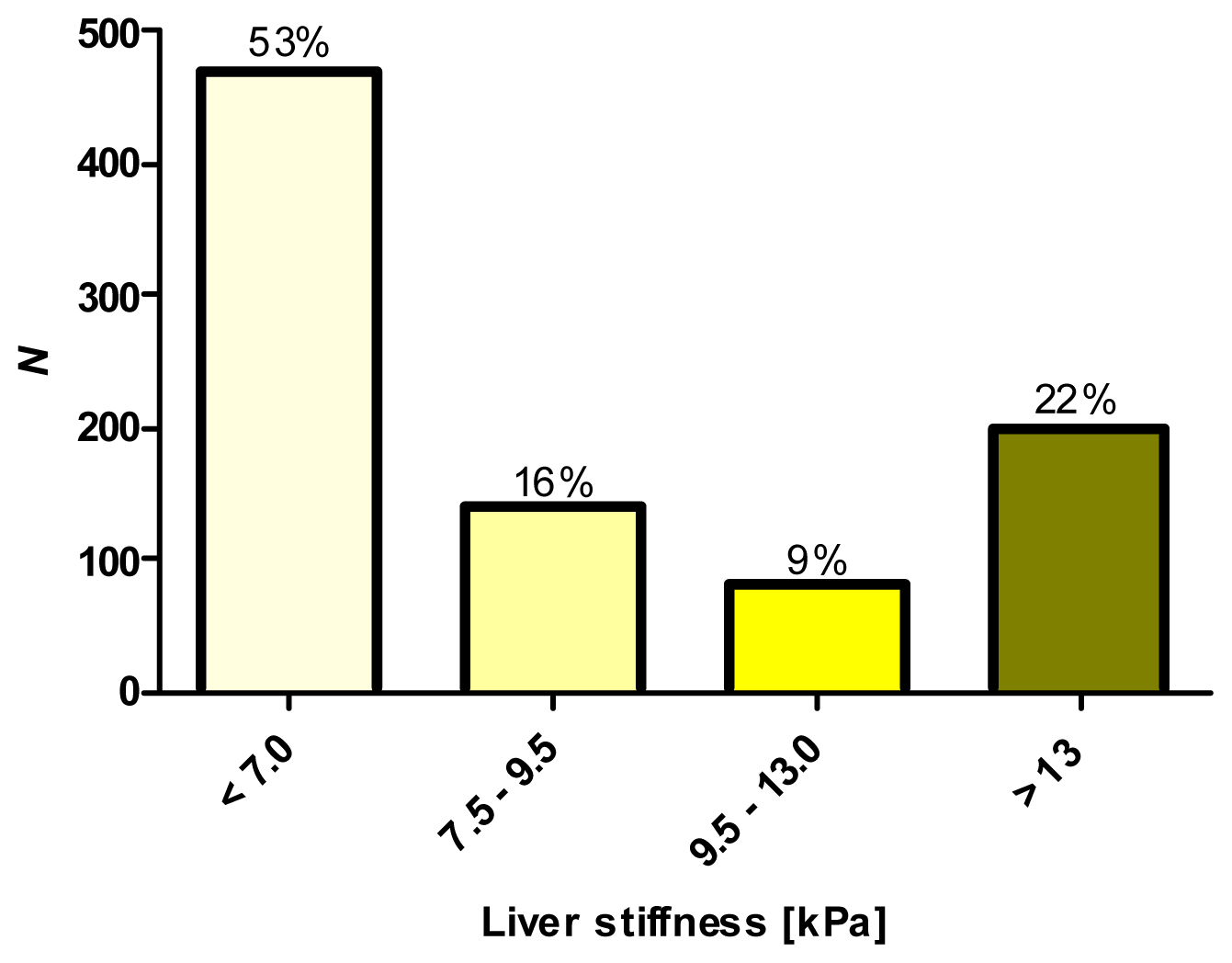

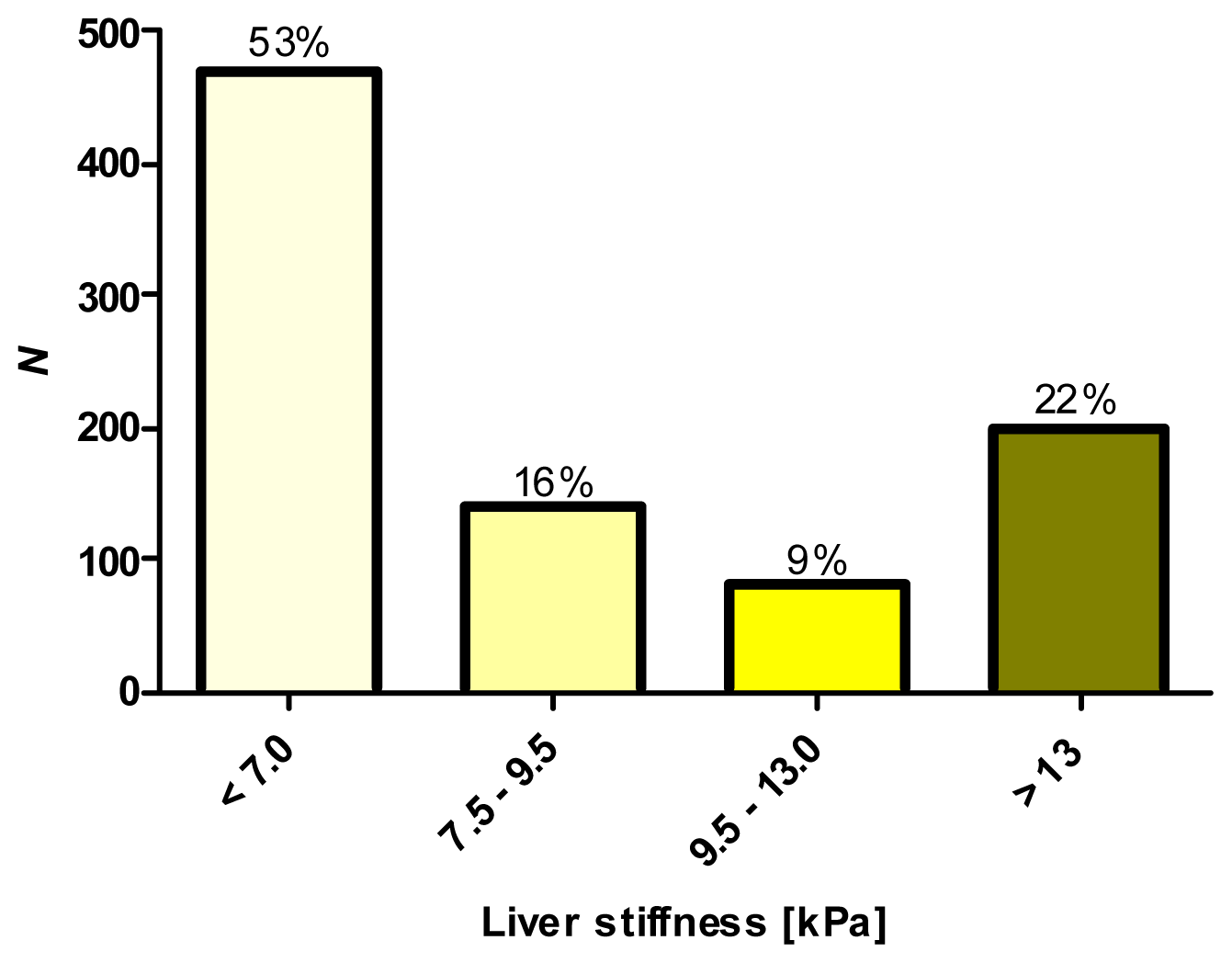{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Allele counts | |||
|---|---|---|---|
| Transient elastography | (T) | (C) | p |
| <7 kPa | 622 (66.1%) | 320 (33.9%) | |
| ≥7 kPa | 604 (71.7%) | 238 (28.3%) | 0.009 |
| Genotype counts | ||||
|---|---|---|---|---|
| Transient elastography | (TT) | (CT) | (CC) | p |
| <7 kPa | 204 (43.3%) | 214 (45.4%) | 53 (11.3%) | |
| ≥7 kPa | 217 (51.5%) | 170 (40.4%) | 34 (8.1%) | 0.009 |
| Univariate analysis | |||
|---|---|---|---|
| Factor | OR | 95% CI | p |
| SREBP1c (TT) | 1.658 | 1.035–2.656 | 0.035 |
| Age | 1.031 | 1.020–1.042 | <0.001 |
| Alcohol consumption | 1.001 | 0.998–1.004 | >0.05 |
| PNPLA3 (IM) + (MM) | 1.179 | 0.955–1.456 | >0.05 |
| BMI | 1.008 | 0.995–1.020 | >0.05 |
| Gender | 0.971 | 0.933–1.010 | >0.05 |
| Multivariate analysis | |||
|---|---|---|---|
| Factor | OR | 95% CI | p |
| SREBP1c (TT) | 1.670 | 1.033–2.700 | 0.036 |
| Age | 1.031 | 1.020–1.042 | <0.001 |
| Variables | Subject characteristics |
|---|---|
| N (male/female) | 899 (547/352) |
| BMI (kg/m2) | 24.6 (14.9–45.2) |
| Age (years) | 50 (17–83) |
| Liver disease (N) | |
| HCV | 541 (60.2%) |
| ALD | 112 (12.5%) |
| HBV | 67 (7.5%) |
| PBC/PSC/AIH | 67 (7.5%) |
| NAFLD/NASH | 64 (7.1%) |
| Hemochromatosis | 25 (2.8%) |
| Other liver diseases # | 23 (2.6%) |
| Transient elastography | |
| Liver stiffness (kPa) | 6.8 (2.2–75.0) |
Acknowledgments
Conflicts of Interest
References
- Bataller, R.; Brenner, D.A. Liver fibrosis. J. Clin. Invest 2005, 115, 209–218. [Google Scholar]
- Krawczyk, M.; Müllenbach, R.; Weber, S.N.; Zimmer, V.; Lammert, F. Genome-wide association studies and genetic risk assessment of liver diseases. Nat. Rev. Gastroenterol. Hepatol 2010, 7, 669–681. [Google Scholar]
- Terjung, B.; Lemnitzer, I.; Dumoulin, F.L.; Effenberger, W.; Brackmann, H.H.; Sauerbruch, T.; Spengler, U. Bleeding complications after percutaneous liver biopsy. An analysis of risk factors. Digestion 2003, 67, 138–145. [Google Scholar]
- Abecasis, G.R.; Auton, A.; Brooks, L.D.; DePristo, M.A.; Durbin, R.M.; Handsaker, R.E.; Kang, H.M.; Marth, G.T.; McVean, G.A. 1000 Genomes Project Consortium. An integrated map of genetic variation from 1092 human genomes. Nature 2012, 491, 56–65. [Google Scholar]
- Krawczyk, M.; Grünhage, F.; Zimmer, V.; Lammert, F. Variant adiponutrin (PNPLA3) represents a common fibrosis risk gene: Non-invasive elastography-based study in chronic liver disease. J. Hepatol 2011, 55, 299–306. [Google Scholar]
- Romeo, S.; Kozlitina, J.; Xing, C.; Pertsemlidis, A.; Cox, D.; Pennacchio, L.A.; Boerwinkle, E.; Cohen, J.C.; Hobbs, H.H. Genetic variation in PNPLA3 confers susceptibility to nonalcoholic fatty liver disease. Nat. Genet 2008, 40, 1461–1465. [Google Scholar]
- Kantartzis, K.; Peter, A.; Machicao, F.; Machann, J.; Wagner, S.; Königsrainer, I.; Königsrainer, A.; Schick, F.; Fritsche, A.; Häring, H.U.; Stefan, N. Dissociation between fatty liver and insulin resistance in humans carrying a variant of the patatin-like phospholipase 3 gene. Diabetes 2009, 58, 2616–2623. [Google Scholar]
- Kotronen, A.; Johansson, L.E.; Johansson, L.M.; Roos, C.; Westerbacka, J.; Hamsten, A.; Bergholm, R.; Arkkila, P.; Arola, J.; Kiviluoto, T.; et al. A common variant in PNPLA3, which encodes adiponutrin, is associated with liver fat content in humans. Diabetologia 2009, 52, 1056–1060. [Google Scholar]
- Sookoian, S.; Castano, G.O.; Burgueno, A.L.; Gianotti, T.F.; Rosselli, M.S.; Pirola, C.J. A nonsynonymous gene variant in the adiponutrin gene is associated with nonalcoholic fatty liver disease severity. J. Lipid. Res 2009, 50, 2111–2116. [Google Scholar]
- Romeo, S.; Sentinelli, F.; Dash, S.; Yeo, G.S.; Savage, D.B.; Leonetti, F.; Capoccia, D.; Incani, M.; Maglio, C.; Iacovino, M.; et al. Morbid obesity exposes the association between PNPLA3 I148M (rs738409) and indices of hepatic injury in individuals of European descent. Int. J. Obes. (Lond.) 2010, 34, 190–194. [Google Scholar]
- Valenti, L.; Alisi, A.; Galmozzi, E.; Bartuli, A.; del Menico, B.; Alterio, A.; Dongiovanni, P.; Fargion, S.; Nobili, V. I148M patatin-like phospholipase domain-containing 3 gene variant and severity of pediatric nonalcoholic fatty liver disease. Hepatology 2010, 52, 1274–1280. [Google Scholar]
- Wagenknecht, L.E.; Palmer, N.D.; Bowden, D.W.; Rotter, J.I.; Norris, J.M.; Ziegler, J.; Chen, Y.D.; Haffner, S.; Scherzinger, A.; Langefeld, C.D. Association of PNPLA3 with non-alcoholic fatty liver disease in a minority cohort: The insulin resistance atherosclerosis family study. Liver. Int 2011, 31, 412–416. [Google Scholar]
- Tian, C.; Stokowski, R.P.; Kershenobich, D.; Ballinger, D.G.; Hinds, D.A. Variant in PNPLA3 is associated with alcoholic liver disease. Nat. Genet 2010, 42, 21–23. [Google Scholar]
- Stickel, F.; Buch, S.; Lau, K.; Meyerzu Schwabedissen, H.; Berg, T.; Ridinger, M.; Rietschel, M.; Schafmayer, C.; Braun, F.; Hinrichsen, H.; et al. Genetic variation in the PNPLA3 gene is associated with alcoholic liver injury in caucasians. Hepatology 2011, 53, 86–95. [Google Scholar]
- Trépo, E.; Gustot, T.; Degré, D.; Lemmers, A.; Verset, L.; Demetter, P.; Ouziel, R.; Quertinmont, E.; Vercruysse, V.; Amininejad, L.; et al. Common polymorphism in the PNPLA3/adiponutrin gene confers higher risk of cirrhosis and liver damage in alcoholic liver disease. J. Hepatol 2011, 55, 906–912. [Google Scholar]
- Trépo, E.; Pradat, P.; Potthoff, A.; Momozawa, Y.; Quertinmont, E.; Gustot, T.; Lemmers, A.; Berthillon, P.; Amininejad, L.; Chevallier, M.; et al. Impact of patatin-like phospholipase-3 (rs738409 C > G) polymorphism on fibrosis progression and steatosis in chronic hepatitis C. Hepatology 2011, 54, 60–69. [Google Scholar]
- Valenti, L.; Alisi, A.; Nobili, V. I148M PNPLA3 variant and progressive liver disease: A new paradigm in hepatology. Hepatology 2012, 56, 1883–1889. [Google Scholar]
- Nischalke, H.D.; Berger, C.; Luda, C.; Berg, T.; Müller, T.; Grünhage, F.; Lammert, F.; Coenen, M.; Krämer, B.; Körner, C.; et al. The PNPLA3 rs738409 148M/M genotype is a risk factor for liver cancer in alcoholic cirrhosis but shows no or weak association in hepatitis C cirrhosis. PLoS One 2011, 6, e27087. [Google Scholar]
- Valenti, L.; Rumi, M.; Galmozzi, E.; Aghemo, A.; del Menico, B.; de Nicola, S.; Dongiovanni, P.; Maggioni, M.; Fracanzani, A.L.; Rametta, R.; et al. Patatin-like phospholipase domain-containing 3 I148M polymorphism, steatosis, and liver damage in chronic hepatitis C. Hepatology 2011, 53, 791–799. [Google Scholar]
- Falleti, E.; Fabris, C.; Cmet, S.; Cussigh, A.; Bitetto, D.; Fontanini, E.; Fornasiere, E.; Bignulin, S.; Fumolo, E.; Bignulin, E.; et al. PNPLA3 rs738409C/G polymorphism in cirrhosis: Relationship with the aetiology of liver disease and hepatocellular carcinoma occurrence. Liver Int 2011, 31, 1137–1143. [Google Scholar]
- Burza, M.A.; Pirazzi, C.; Maglio, C.; Sjöholm, K.; Mancina, R.M.; Svensson, P.A.; Jacobson, P.; Adiels, M.; Baroni, M.G.; Borén, J.; et al. PNPLA3 I148M (rs738409) genetic variant is associated with hepatocellular carcinoma in obese individuals. Dig. Liver Dis 2012, 44, 1037–1041. [Google Scholar]
- Hassan, M.M.; Kaseb, A.; Etzel, C.J.; El-Serag, H.; Spitz, M.R.; Chang, P.; Hale, K.S.; Liu, M.; Rashid, A.; Shama, M.; et al. Genetic variation in the PNPLA3 gene and hepatocellular carcinoma in USA: Risk and prognosis prediction. Mol. Carcinog 2013. [Google Scholar] [CrossRef]
- Sookoian, S.; Pirola, C.J. Meta-analysis of the influence of I148M variant of patatin-like phospholipase domain containing 3 gene (PNPLA3) on the susceptibility and histological severity of nonalcoholic fatty liver disease. Hepatology 2011, 53, 1883–1894. [Google Scholar]
- Zimmer, V.; Lammert, F. Genetics and epigenetics in the fibrogenic evolution of chronic liver diseases. Best. Pract. Res. Clin. Gastroenterol 2011, 25, 269–280. [Google Scholar]
- Kumari, M.; Schoiswohl, G.; Chitraju, C.; Paar, M.; Cornaciu, I.; Rangrez, A.Y.; Wongsiriroj, N.; Nagy, H.M.; Ivanova, P.T.; Scott, S.A.; et al. Adiponutrin functions as a nutritionally regulated lysophosphatidic acid acyltransferase. Cell Metab 2012, 15, 691–702. [Google Scholar]
- Kollerits, B.; Coassin, S.; Beckmann, N.D.; Teumer, A.; Kiechl, S.; Döring, A.; Kavousi, M.; Hunt, S.C.; Lamina, C.; Paulweber, B.; et al. Genetic evidence for a role of adiponutrin in the metabolism of apolipoprotein B-containing lipoproteins. Hum. Mol. Genet 2009, 18, 4669–4676. [Google Scholar]
- Krawczyk, M.; Grünhage, F.; Mahler, M.; Tirziu, S.; Acalovschi, M.; Lammert, F. The common adiponutrin variant p.I148M does not confer gallstone risk but affects fasting glucose and triglyceride levels. J. Physiol. Pharmacol 2011, 62, 369–375. [Google Scholar]
- Palmer, C.N.; Maglio, C.; Pirazzi, C.; Burza, M.A.; Adiels, M.; Burch, L.; Donnelly, L.A.; Colhoun, H.; Doney, A.S.; Dillon, J.F.; et al. Paradoxical lower serum triglyceride levels and higher type 2 diabetes mellitus susceptibility in obese individuals with the PNPLA3 148M variant. PLoS One 2012, 7, e39362. [Google Scholar]
- Pirazzi, C.; Adiels, M.; Burza, M.A.; Mancina, R.M.; Levin, M.; Ståhlman, M.; Taskinen, M.R.; Orho-Melander, M.; Perman, J.; Pujia, A.; et al. Patatin-like phospholipase domain-containing 3 (PNPLA3) I148M (rs738409) affects hepatic VLDL secretion in humans and in vitro. J. Hepatol. 2012, 57, 1276–1282. [Google Scholar]
- Mehal, W.Z.; Iredale, J.; Friedman, S.L. Scraping fibrosis: Expressway to the core of fibrosis. Nat. Med 2011, 17, 552–553. [Google Scholar]
- Huang, Y.; He, S.; Li, J.Z.; Seo, Y.K.; Osborne, T.F.; Cohen, J.C.; Hobbs, H.H. A feed-forward loop amplifies nutritional regulation of PNPLA3. Proc. Natl. Acad. Sci. USA 2010, 107, 7892–7897. [Google Scholar]
- Dubuquoy, C.; Robichon, C.; Lasnier, F.; Langlois, C.; Dugail, I.; Foufelle, F.; Girard, J.; Burnol, A.F.; Postic, C.; Moldes, M. Distinct regulation of adiponutrin/PNPLA3 gene expression by the transcription factors ChREBP and SREBP1c in mouse and human hepatocytes. J. Hepatol 2011, 55, 145–153. [Google Scholar]
- Brown, M.S.; Goldstein, J.L. The SREBP pathway: Regulation of cholesterol metabolism by proteolysis of a membrane-bound transcription factor. Cell 1997, 89, 331–340. [Google Scholar]
- Horton, J.D.; Shimomura, I.; Brown, M.S.; Hammer, R.E.; Goldstein, J.L.; Shimano, H. Activation of cholesterol synthesis in preference to fatty acid synthesis in liver and adipose tissue of transgenic mice overproducing sterol regulatory element-binding protein-2. J. Clin. Invest 1998, 101, 2331–2339. [Google Scholar]
- Shimano, H.; Horton, J.D.; Hammer, R.E.; Shimomura, I.; Brown, M.S.; Goldstein, J.L. Overproduction of cholesterol and fatty acids causes massive liver enlargement in transgenic mice expressing truncated SREBP-1a. J. Clin. Invest 1996, 98, 1575–1584. [Google Scholar]
- Bommer, G.T.; MacDougald, O.A. Regulation of lipid homeostasis by the bifunctional SREBF2-miR33a locus. Cell Metable 2011, 13, 241–247. [Google Scholar]
- Hua, X.; Wu, J.; Goldstein, J.L.; Brown, M.S.; Hobbs, H.H. Structure of the human gene encoding sterol regulatory element binding protein-1 (SREBF1) and localization of SREBF1 and SREBF2 to chromosomes 17p11.2 and 22q13. Genomics 1995, 25, 667–673. [Google Scholar]
- Liu, J.X.; Liu, J.; Li, P.Q.; Xie, X.D.; Guo, Q.; Tian, L.M.; Ma, X.Q.; Zhang, J.P.; Liu, J.; Gao, J.Y. Association of sterol regulatory element-binding protein-1c gene polymorphism with type 2 diabetes mellitus, insulin resistance and blood lipid levels in Chinese population. Diabetes Res. Clin. Pract 2008, 82, 42–47. [Google Scholar]
- Harding, A.H.; Loos, R.J.; Luan, J.; O’Rahilly, S.; Wareham, N.J.; Barroso, I. Polymorphisms in the gene encoding sterol regulatory element-binding factor-1c are associated with type 2 diabetes. Diabetologia 2006, 49, 2642–2648. [Google Scholar]
- Laudes, M.; Barroso, I.; Luan, J.; Soos, M.A.; Yeo, G.; Meirhaeghe, A.; Logie, L.; Vidal-Puig, A.; Schafer, A.J.; Wareham, N.J.; et al. Genetic variants in human sterol regulatory element binding protein-1c in syndromes of severe insulin resistance and type 2 diabetes. Diabetes 2004, 53, 842–846. [Google Scholar]
- Felder, T.K.; Oberkofler, H.; Weitgasser, R.; Mackevics, V.; Krempler, F.; Paulweber, B.; Patsch, W. The SREBF-1 locus is associated with type 2 diabetes and plasma adiponectin levels in a middle-aged Austrian population. Int. J. Obes 2007, 31, 1099–1103. [Google Scholar]
- Friedrich-Rust, M.; Ong, M.F.; Martens, S.; Sarrazin, C.; Bojunga, J.; Zeuzem, S.; Herrmann, E. Performance of transient elastography for the staging of liver fibrosis: A meta-analysis. Gastroenterology 2008, 134, 960–974. [Google Scholar]
- Grünhage, F.; Acalovschi, M.; Tirziu, S.; Walier, M.; Wienker, T.F.; Ciocan, A.; Mosteanu, O.; Sauerbruch, T.; Lammert, F. Increased gallstone risk in humans conferred by common variant of hepatic ATP-binding cassette transporter for cholesterol. Hepatology 2007, 46, 793–801. [Google Scholar]
- Barabási, A.L.; Gulbahce, N.; Loscalzo, J. Network medicine: A network-based approach to human disease. Nat. Rev. Genet 2011, 12, 56–68. [Google Scholar]
- Hartwell, L.H.; Hopfield, J.J.; Leibler, S.; Murray, A.W. From molecular to modular cell biology. Nature 1999, 402, C47–C52. [Google Scholar]
- Shimomura, I.; Bashmakov, Y.; Horton, J.D. Increased levels of nuclear SREBP-1c associated with fatty livers in two mouse models of diabetes mellitus. J. Biol. Chem 1999, 274, 30028–30032. [Google Scholar]
- Knebel, B.; Haas, J.; Hartwig, S.; Jacob, S.; Köllmer, C.; Nitzgen, U.; Muller-Wieland, D.; Kotzka, J. Liver-specific expression of transcriptionally active sREBP-1c is associated with fatty liver and increased visceral fat mass. PLoS One 2012, 7, e31812. [Google Scholar]
- Higuchi, N.; Kato, M.; Shundo, Y.; Tajiri, H.; Tanaka, M.; Yamashita, N.; Kohjima, M.; Kotoh, K.; Nakamuta, M.; Takayanagi, R.; et al. Liver X receptor in cooperation with SREBP-1c is a major lipid synthesis regulator in nonalcoholic fatty liver disease. Hepatol. Res 2008, 38, 1122–1129. [Google Scholar]
- Moon, Y.A.; Liang, G.; Xie, X.; Frank-Kamenetsky, M.; Fitzgerald, K.; Koteliansky, V.; Brown, M.S.; Goldstein, J.L.; Horton, J.D. The scap/SREBP pathway is essential for developing diabetic fatty liver and carbohydrate-induced hypertriglyceridemia in animals. Cell Metab 2012, 15, 240–246. [Google Scholar]
- Desmet, V.J.; Gerber, M.; Hoofnagle, J.H.; Manns, M.; Scheuer, P.J. Classification of chronic hepatitis: Diagnosis, grading and staging. Hepatology 1994, 19, 1513–1520. [Google Scholar]
© 2013 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Krawczyk, M.; Grünhage, F.; Lammert, F. Identification of Combined Genetic Determinants of Liver Stiffness within the SREBP1c-PNPLA3 Pathway. Int. J. Mol. Sci. 2013, 14, 21153-21166. https://doi.org/10.3390/ijms141021153
Krawczyk M, Grünhage F, Lammert F. Identification of Combined Genetic Determinants of Liver Stiffness within the SREBP1c-PNPLA3 Pathway. International Journal of Molecular Sciences. 2013; 14(10):21153-21166. https://doi.org/10.3390/ijms141021153
Chicago/Turabian StyleKrawczyk, Marcin, Frank Grünhage, and Frank Lammert. 2013. "Identification of Combined Genetic Determinants of Liver Stiffness within the SREBP1c-PNPLA3 Pathway" International Journal of Molecular Sciences 14, no. 10: 21153-21166. https://doi.org/10.3390/ijms141021153
APA StyleKrawczyk, M., Grünhage, F., & Lammert, F. (2013). Identification of Combined Genetic Determinants of Liver Stiffness within the SREBP1c-PNPLA3 Pathway. International Journal of Molecular Sciences, 14(10), 21153-21166. https://doi.org/10.3390/ijms141021153