Mechanisms of Oxidative Damage in Multiple Sclerosis and Neurodegenerative Diseases: Therapeutic Modulation via Fumaric Acid Esters

{kind=link}
{kind=link}
Abstract
:1. Introduction: Oxidative Stress in Neurologic Diseases
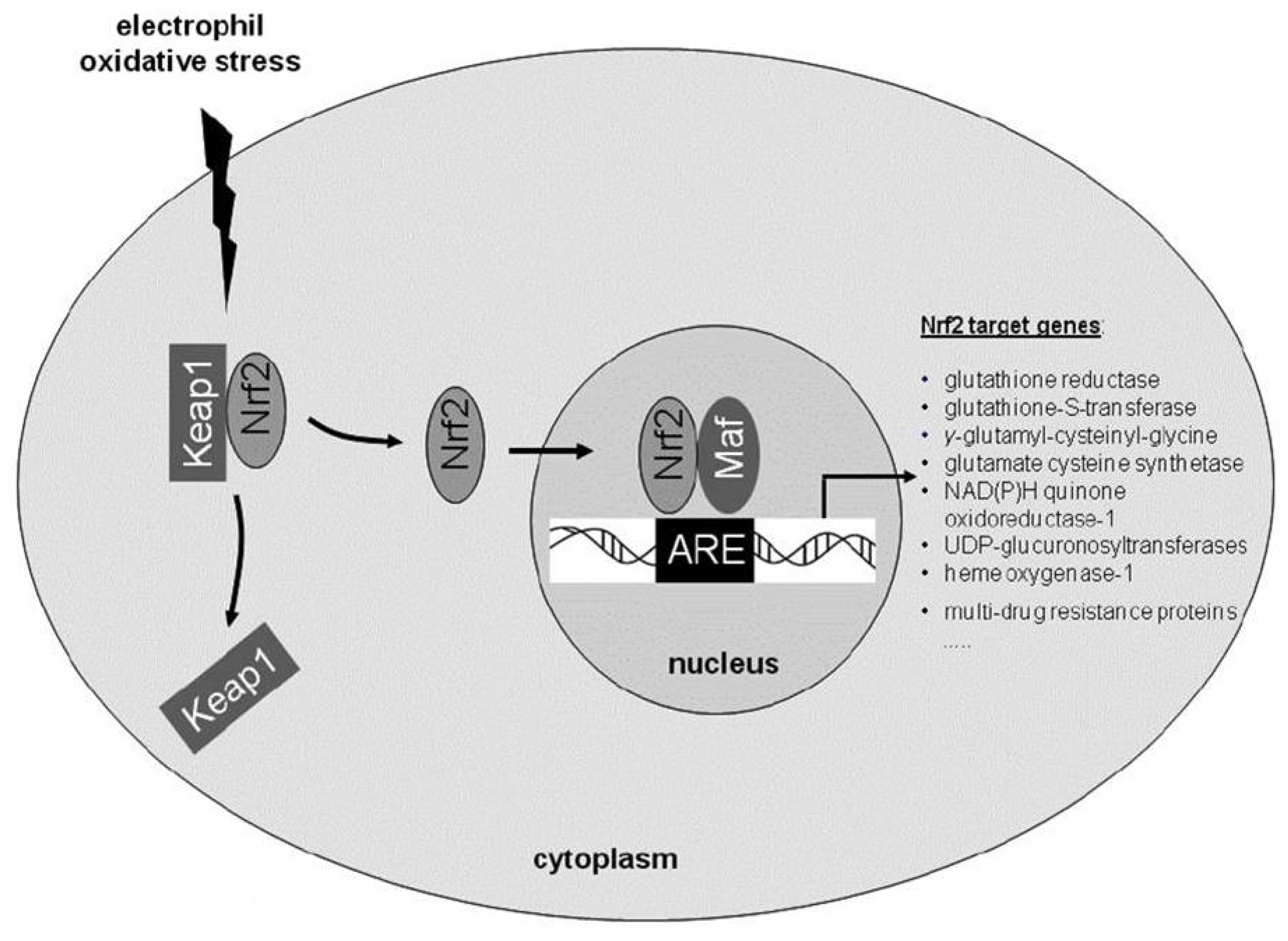
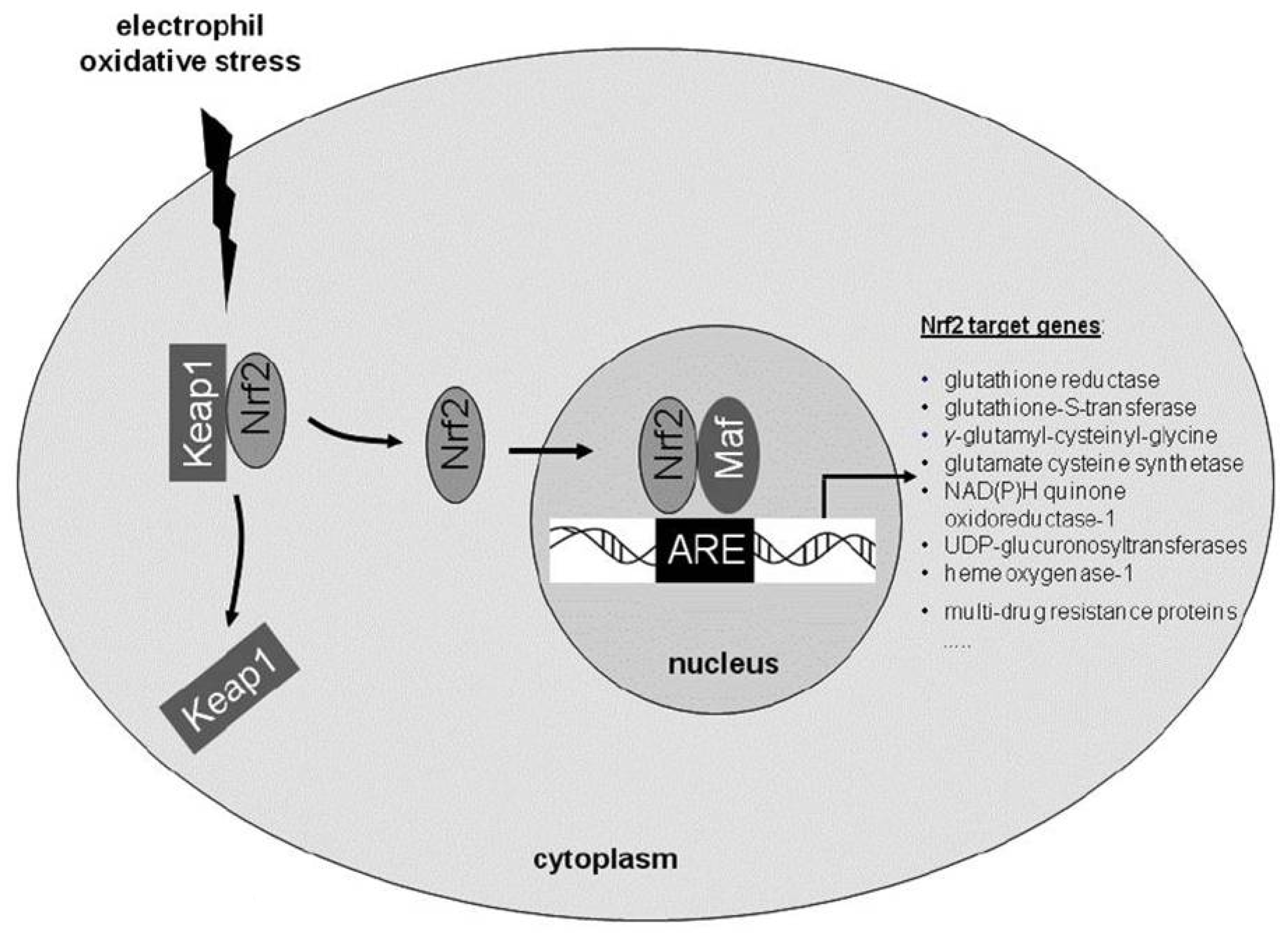
2. Role of Endogenous Anti-Oxidative Pathways: The Importance of the Transcription Factor Nuclear Factor (Erythroid-Derived 2)-Related Factor 2 (Nrf2)
3. Oxidative Stress and the Nrf2 Pathways in Neurodegenerative Diseases
3.1. Alzheimer’s Disease
3.2. Amyotrophic Lateral Sclerosis
3.3. Parkinson’s Disease
3.4. Huntington’s Disease
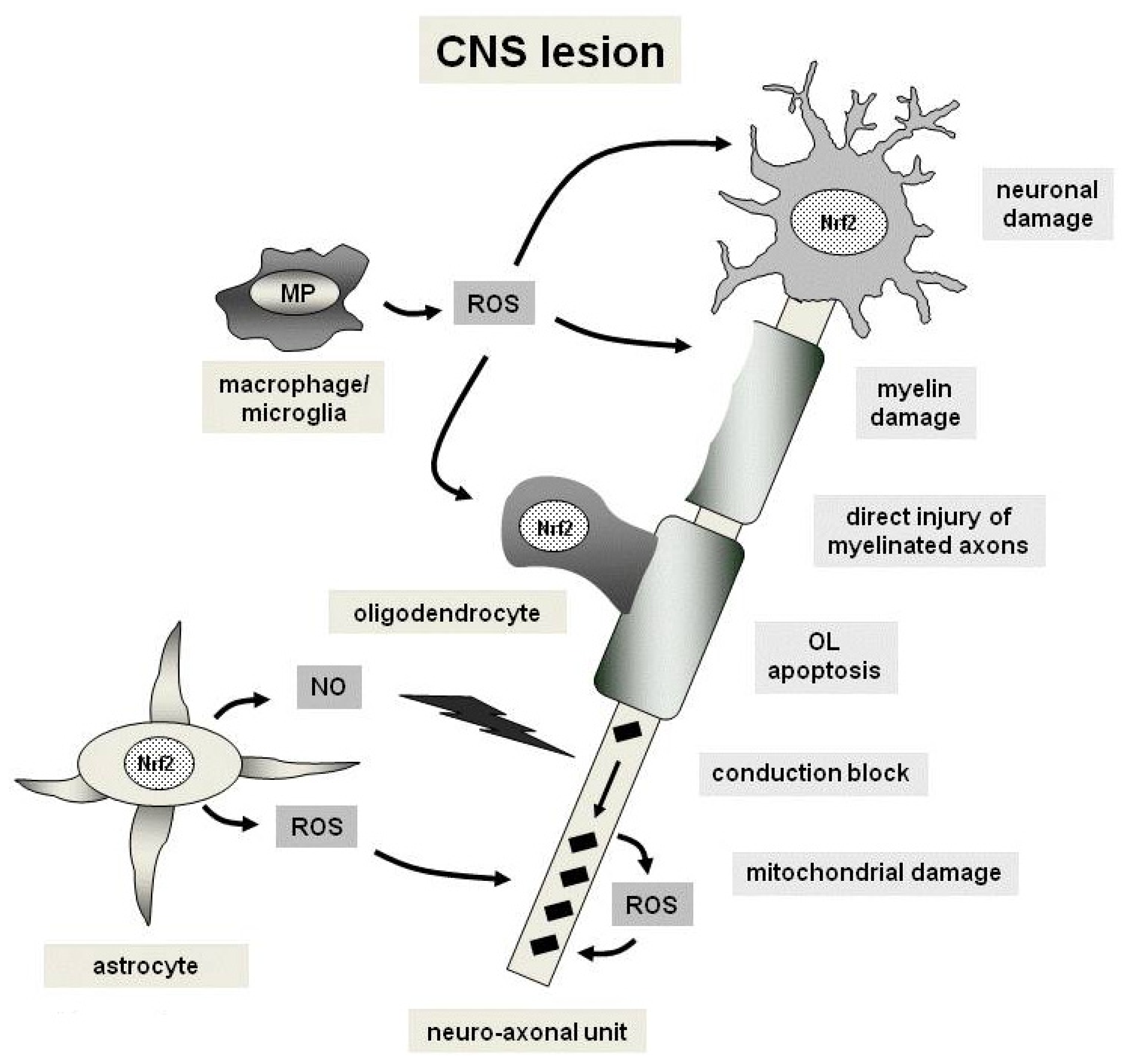
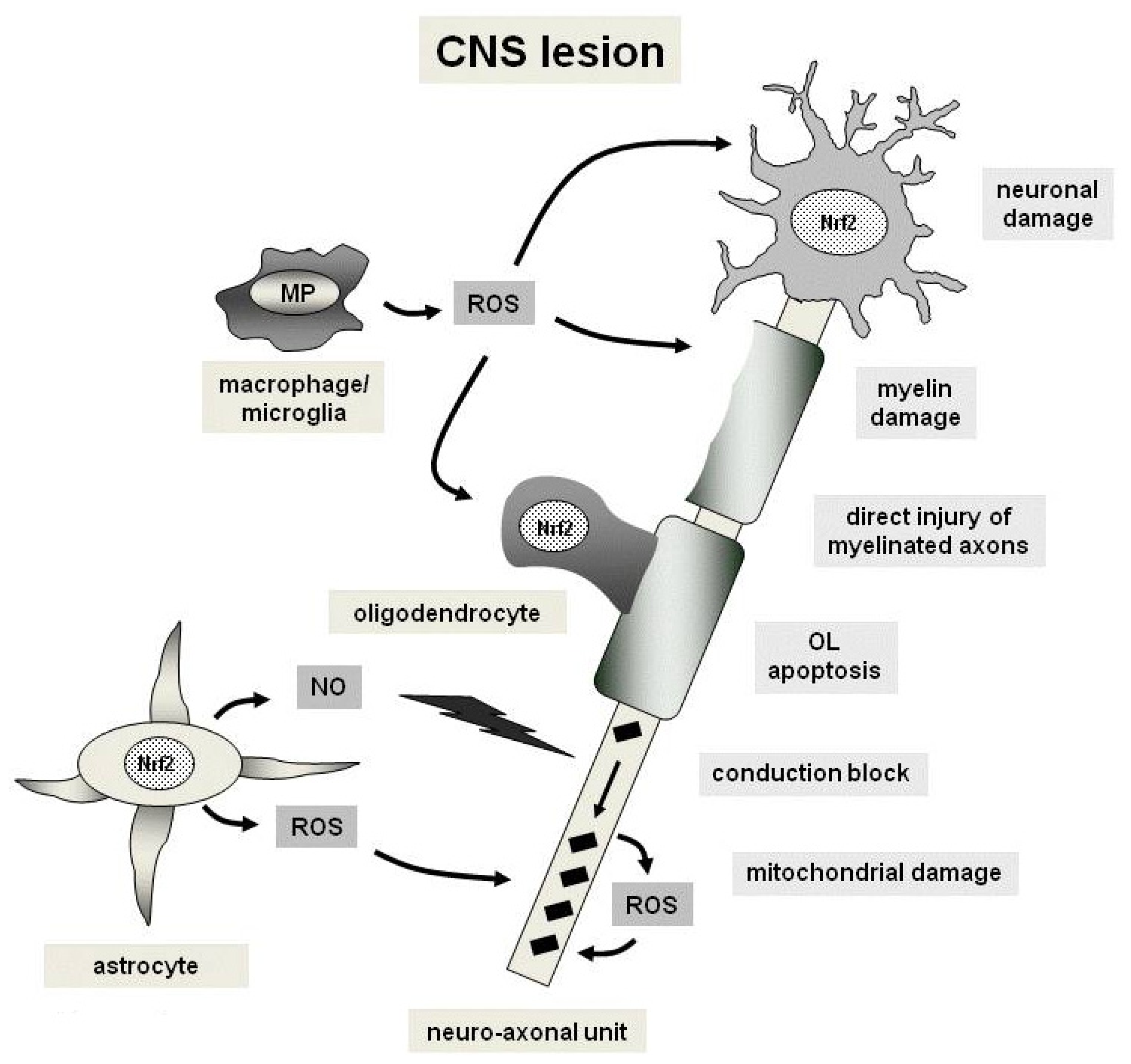
4. Oxidative Stress and the Nrf2 Pathways in Multiple Sclerosis
4.1. Mitochondrial Dysfunction
- Energy failure
- Induction of apoptosis: mitochondrial injury can elicit pro-apoptotic events via liberation of apoptosis induced factor or cytochrome C [93], and
- Enhanced production of reactive oxygen species.
4.2. Free Radicals
5. Anti-Oxidative Effects of Fumaric Acid Esters in Autoimmune Demyelination
6. Discussion
7. Conclusions
References
- Langlais, P.J.; Anderson, G.; Guo, S.X.; Bondy, S.C. Increased cerebral free radical production during thiamine deficiency. Metab. Brain Dis 1997, 12, 137–143. [Google Scholar]
- Gibson, G.E.; Zhang, H. Interactions of oxidative stress with thiamine homeostasis promote neurodegeneration. Neurochem. Int 2002, 40, 493–504. [Google Scholar]
- Todd, K.G.; Butterworth, R.F. Early microglial response in experimental thiamine deficiency: An immunohistochemical analysis. Glia 1999, 25, 190–198. [Google Scholar]
- Itoh, K.; Wakabayashi, N.; Katoh, Y.; Ishii, T.; Igarashi, K.; Engel, J.D.; Yamamoto, M. Keap1 represses nuclear activation of antioxidant responsive elements by Nrf2 through binding to the amino-terminal Neh2 domain. Gene. Dev 1999, 13, 76–86. [Google Scholar]
- Wakabayashi, N.; Itoh, K.; Wakabayashi, J.; Motohashi, H.; Noda, S.; Takahashi, S.; Imakado, S.; Kotsuji, T.; Otsuka, F.; Roop, D.R.; et al. Keap1-null mutation leads to postnatal lethality due to constitutive Nrf2 activation. Nat. Genet 2003, 35, 238–245. [Google Scholar]
- Alam, J.; Killeen, E.; Gong, P.; Naquin, R.; Hu, B.; Stewart, D.; Ingelfinger, J.R.; Nath, K.A. Heme activates the heme oxygenase-1 gene in renal epithelial cells by stabilizing Nrf2. Am. J. Physiol. Renal Physiol 2003, 284, F743–F752. [Google Scholar]
- Stewart, D.; Killeen, E.; Naquin, R.; Alam, S.; Alam, J. Degradation of transcription factor Nrf2 via the ubiquitin-proteasome pathway and stabilization by cadmium. J. Biol. Chem 2003, 278, 2396–2402. [Google Scholar]
- Lee, J.M.; Li, J.; Johnson, D.A.; Stein, T.D.; Kraft, A.D.; Calkins, M.J.; Jakel, R.J.; Johnson, J.A. Nrf2, a multi-organ protector? FASEB J 2005, 19, 1061–1066. [Google Scholar]
- Maines, M.D.; Trakshel, G.M.; Kutty, R.K. Characterization of two constitutive forms of rat liver microsomal heme oxygenase. Only one molecular species of the enzyme is inducible. J. Biol. Chem 1986, 261, 411–419. [Google Scholar]
- Shibahara, S.; Muller, R.; Taguchi, H.; Yoshida, T. Cloning and expression of cDNA for rat heme oxygenase. Proc. Nat. Acad. Sci. USA 1985, 82, 7865–7869. [Google Scholar]
- Baranano, D.E.; Snyder, S.H. Neural roles for heme oxygenase: Contrasts to nitric oxide synthase. Proc. Nat. Acad. Sci. USA 2001, 98, 10996–11002. [Google Scholar]
- Chen, K.; Gunter, K.; Maines, M.D. Neurons overexpressing heme oxygenase-1 resist oxidative stress-mediated cell death. J. Neurochem 2000, 75, 304–313. [Google Scholar]
- Poss, K.D.; Tonegawa, S. Reduced stress defense in heme oxygenase 1-deficient cells. Proc. Nat. Acad. Sci. USA 1997, 94, 10925–10930. [Google Scholar]
- Yachie, A.; Niida, Y.; Wada, T.; Igarashi, N.; Kaneda, H.; Toma, T.; Ohta, K.; Kasahara, Y.; Koizumi, S. Oxidative stress causes enhanced endothelial cell injury in human heme oxygenase-1 deficiency. J. Clin. Invest 1999, 103, 129–135. [Google Scholar]
- Mehindate, K.; Sahlas, D.J.; Frankel, D.; Mawal, Y.; Liberman, A.; Corcos, J.; Dion, S.; Schipper, H.M. Proinflammatory cytokines promote glial heme oxygenase-1 expression and mitochondrial iron deposition: Implications for multiple sclerosis. J. Neurochem 2001, 77, 1386–1395. [Google Scholar]
- Regan, R.F.; Guo, Y.; Kumar, N. Heme oxygenase-1 induction protects murine cortical astrocytes from hemoglobin toxicity. Neurosci. Lett 2000, 282, 1–4. [Google Scholar]
- Schipper, H.M.; Bernier, L.; Mehindate, K.; Frankel, D. Mitochondrial iron sequestration in dopamine-challenged astroglia: Role of heme oxygenase-1 and the permeability transition pore. J. Neurochem 1999, 72, 1802–1811. [Google Scholar]
- Ho, Y.S.; Magnenat, J.L.; Bronson, R.T.; Cao, J.; Gargano, M.; Sugawara, M.; Funk, C.D. Mice deficient in cellular glutathione peroxidase develop normally and show no increased sensitivity to hyperoxia. J. Biol. Chem 1997, 272, 16644–16651. [Google Scholar]
- Ho, Y.S.; Xiong, Y.; Ma, W.; Spector, A.; Ho, D.S. Mice lacking catalase develop normally but show differential sensitivity to oxidant tissue injury. J. Biol. Chem 2004, 279, 32804–32812. [Google Scholar]
- Yoh, K.; Itoh, K.; Enomoto, A.; Hirayama, A.; Yamaguchi, N.; Kobayashi, M.; Morito, N.; Koyama, A.; Yamamoto, M.; Takahashi, S. Nrf2-deficient female mice develop lupus-like autoimmune nephritis 1. Kidney Int 2001, 60, 1343–1353. [Google Scholar]
- Thimmulappa, R.K.; Lee, H.; Rangasamy, T.; Reddy, S.P.; Yamamoto, M.; Kensler, T.W.; Biswal, S. Nrf2 is a critical regulator of the innate immune response and survival during experimental sepsis. J. Clin. Invest 2006, 116, 984–995. [Google Scholar]
- Kensler, T.W.; Wakabayashi, N.; Biswal, S. Cell survival responses to environmental stresses via the Keap1-Nrf2-ARE pathway. Annu. Rev. Pharmacol. Toxicol 2007, 47, 89–116. [Google Scholar]
- Kotlo, K.U.; Yehiely, F.; Efimova, E.; Harasty, H.; Hesabi, B.; Shchors, K.; Einat, P.; Rozen, A.; Berent, E.; Deiss, L.P. Nrf2 is an inhibitor of the Fas pathway as identified by Achilles’ Heel Method, a new function-based approach to gene identification in human cells. Oncogene 2003, 22, 797–806. [Google Scholar]
- Morito, N.; Yoh, K.; Itoh, K.; Hirayama, A.; Koyama, A.; Yamamoto, M.; Takahashi, S. Nrf2 regulates the sensitivity of death receptor signals by affecting intracellular glutathione levels. Oncogene 2003, 22, 9275–9281. [Google Scholar]
- Pehar, M.; Vargas, M.R.; Robinson, K.M.; Cassina, P.; Díaz-Amarilla, P.J.; Hagen, T.M.; Radi, R.; Barbeito, L.; Beckman, J.S. Mitochondrial superoxide production and nuclear factor erythroid 2-related factor 2 activation in p75 neurotrophin receptor-induced motor neuron apoptosis. J. Neurosci 2007, 27, 7777–7785. [Google Scholar]
- Bertram, L.; Tanzi, R.E. Thirty years of Alzheimer’s disease genetics: The implications of systematic meta-analyses. Nat. Rev. Neurosci 2008, 9, 768–778. [Google Scholar]
- Marshak, D.R.; Pesce, S.A.; Stanley, L.C.; Griffin, W.S. Increased S100 beta neurotrophic activity in Alzheimer’s disease temporal lobe. Neurobiol. Aging 1992, 13, 1–7. [Google Scholar]
- Schipper, H.M.; Bennett, D.A.; Liberman, A.; Bienias, J.L.; Schneider, J.A.; Kelly, J.; Arvanitakis, Z. Glial heme oxygenase-1 expression in Alzheimer disease and mild cognitive impairment. Neurobiol. Aging 2006, 27, 252–261. [Google Scholar]
- Shaftel, S.S.; Griffin, W.S.; O’Banion, M.K. The role of interleukin-1 in neuroinflammation and Alzheimer disease: An evolving perspective. J. Neuroinflamm 2008, 5, 7–15. [Google Scholar]
- Smith, M.A.; Harris, P.L.; Sayre, L.M.; Perry, G. Iron accumulation in Alzheimer disease is a source of redox-generated free radicals. Proc. Nat. Acad. Sci. USA 1997, 94, 9866–9868. [Google Scholar]
- Wang, X.; Su, B.; Perry, G.; Smith, M.A.; Zhu, X. Insights into amyloid-beta-induced mitochondrial dysfunction in Alzheimer disease. Free Radic. Biol. Med 2007, 43, 1569–1573. [Google Scholar]
- Stys, P.K. General mechanisms of axonal damage and its prevention. J. Neurol. Sci 2005, 233, 3–13. [Google Scholar]
- Raina, A.K.; Templeton, D.J.; Deak, J.C.; Perry, G.; Smith, M.A. Quinone reductase (NQO1), a sensitive redox indicator, is increased in Alzheimer’s disease. Redox. Rep 1999, 4, 23–27. [Google Scholar]
- Wang, Y.; Santa-Cruz, K.; DeCarli, C.; Johnson, J.A. NAD(P)H: Quinone oxidoreductase activity is increased in hippocampal pyramidal neurons of patients with Aalzheimer’s disease. Neurobiol. Aging 2000, 21, 525–531. [Google Scholar]
- Ramsey, C.P.; Glass, C.A.; Montgomery, M.B.; Lindl, K.A.; Ritson, G.P.; Chia, L.A.; Hamilton, R.L.; Chu, C.T.; Jordan-Sciutto, K.L. Expression of Nrf2 in neurodegenerative diseases. J. Neuropathol. Exp. Neurol 2007, 66, 75–85. [Google Scholar]
- Kanninen, K.; Malm, T.M.; Jyrkkanen, H.K.; Goldsteins, G.; Keksa-Goldsteine, V.; Tanila, H.; Yamamoto, M.; Yla-Herttuala, S.; Levonen, A.L.; Koistinaho, J. Nuclear factor erythroid 2-related factor 2 protects against beta amyloid. Mol. Cell Neurosci 2008, 39, 302–313. [Google Scholar]
- Rowland, L.P.; Shneider, N.A. Amyotrophic lateral sclerosis. N. Engl. J. Med 2001, 344, 1688–1700. [Google Scholar]
- Rosen, D.R. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 1993, 364, 362–372. [Google Scholar]
- Gurney, M.E. Transgenic-mouse model of amyotrophic lateral sclerosis. N. Engl. J. Med 1994, 331, 1721–1722. [Google Scholar]
- Howland, D.S.; Liu, J.; She, Y.; Goad, B.; Maragakis, N.J.; Kim, B.; Erickson, J.; Kulik, J.; DeVito, L.; Psaltis, G.; et al. Focal loss of the glutamate transporter EAAT2 in a transgenic rat model of SOD1 mutant-mediated amyotrophic lateral sclerosis (ALS). Proc. Nat. Acad. Sci. USA 2002, 99, 1604–1609. [Google Scholar]
- Beckman, J.S.; Estevez, A.G.; Crow, J.P.; Barbeito, L. Superoxide dismutase and the death of motoneurons in ALS. Tr. Neurosci 2001, 24, S15–S20. [Google Scholar]
- Bruijn, L.I.; Miller, T.M.; Cleveland, D.W. Unraveling the mechanisms involved in motor neuron degeneration in ALS. Annu. Rev. Neurosci 2004, 27, 723–749. [Google Scholar]
- Cleveland, D.W.; Rothstein, J.D. From Charcot to Lou Gehrig: Deciphering selective motor neuron death in ALS. Nat. Rev. Neurosci 2001, 2, 806–819. [Google Scholar]
- Manfredi, G.; Xu, Z. Mitochondrial dysfunction and its role in motor neuron degeneration in ALS. Mitochondrion 2005, 5, 77–87. [Google Scholar]
- Goodall, E.F.; Morrison, K.E. Amyotrophic lateral sclerosis (motor neuron disease): Proposed mechanisms and pathways to treatment. Expert Rev. Mol. Med 2006, 8, 1–22. [Google Scholar]
- Ischiropoulos, H.; Beckman, J.S. Oxidative stress and nitration in neurodegeneration: Cause, effect, or association? J. Clin. Invest 2003, 111, 163–169. [Google Scholar]
- Pacher, P.; Beckman, J.S.; Liaudet, L. Nitric oxide and peroxynitrite in health and disease. Physiol. Rev 2007, 87, 315–424. [Google Scholar]
- Estevez, A.G.; Spear, N.; Manuel, S.M.; Radi, R.; Henderson, C.E.; Barbeito, L.; Beckman, J.S. Nitric oxide and superoxide contribute to motor neuron apoptosis induced by trophic factor deprivation. J. Neurosci 1998, 18, 923–931. [Google Scholar]
- Pehar, M.; Cassina, P.; Vargas, M.R.; Castellanos, R.; Viera, L.; Beckman, J.S.; Estevez, A.G.; Barbeito, L. Astrocytic production of nerve growth factor in motor neuron apoptosis: Implications for amyotrophic lateral sclerosis. J. Neurochem 2004, 89, 464–473. [Google Scholar]
- Raoul, C.; Estevez, A.G.; Nishimune, H.; Cleveland, D.W.; deLapeyriere, O.; Henderson, C.E.; Haase, G.; Pettmann, B. Motoneuron death triggered by a specific pathway downstream of Fas. potentiation by ALS-linked SOD1 mutations. Neuron 2002, 35, 1067–1083. [Google Scholar]
- Barbeito, L.H.; Pehar, M.; Cassina, P.; Vargas, M.R.; Peluffo, H.; Viera, L.; Estevez, A.G.; Beckman, J.S. A role for astrocytes in motor neuron loss in amyotrophic lateral sclerosis. Brain Res. Rev 2004, 47, 263–274. [Google Scholar]
- Ferraiuolo, L.; Heath, P.R.; Holden, H.; Kasher, P.; Kirby, J.; Shaw, P.J. Microarray analysis of the cellular pathways involved in the adaptation to and progression of motor neuron injury in the SOD1 G93A mouse model of familial ALS. J. Neurosci 2007, 27, 9201–9219. [Google Scholar]
- Vargas, M.R.; Pehar, M.; Cassina, P.; Martinez-Palma, L.; Thompson, J.A.; Beckman, J.S.; Barbeito, L. Fibroblast growth factor-1 induces heme oxygenase-1 via nuclear factor erythroid 2-related factor 2 (Nrf2) in spinal cord astrocytes: Consequences for motor neuron survival. J. Biol. Chem 2005, 280, 25571–25579. [Google Scholar]
- Gong, Y.H.; Parsadanian, A.S.; Andreeva, A.; Snider, W.D.; Elliott, J.L. Restricted expression of G86R Cu/Zn superoxide dismutase in astrocytes results in astrocytosis but does not cause motoneuron degeneration. J. Neurosci 2000, 20, 660–665. [Google Scholar]
- Vargas, M.R.; Pehar, M.; Díaz-Amarilla, P.J.; Beckman, J.S.; Barbeito, L. Transcriptional profile of primary astrocytes expressing ALS-linked mutant SOD1. J. Neurosci. Res 2008, 86, 3515–3525. [Google Scholar]
- Alam, Z.I.; Daniel, S.E.; Lees, A.J.; Marsden, D.C.; Jenner, P.; Halliwell, B. A generalised increase in protein carbonyls in the brain in Parkinson’s but not incidental Lewy body disease. J. Neurochem 1997, 69, 1326–1329. [Google Scholar]
- Alam, Z.I.; Jenner, A.; Daniel, S.E.; Lees, A.J.; Cairns, N.; Marsden, C.D.; Jenner, P.; Halliwell, B. Oxidative DNA damage in the parkinsonian brain: An apparent selective increase in 8-hydroxyguanine levels in substantia nigra. J. Neurochem 1997, 69, 1196–1203. [Google Scholar]
- Dexter, D.T.; Holley, A.E.; Flitter, W.D.; Slater, T.F.; Wells, F.R.; Daniel, S.E.; Lees, A.J.; Jenner, P.; Marsden, C.D. Increased levels of lipid hydroperoxides in the parkinsonian substantia nigra: An HPLC and ESR study. Mov. Disord 1994, 9, 92–97. [Google Scholar]
- Burton, N.C.; Kensler, T.W.; Guilarte, T.R. In vivo modulation of the Parkinsonian phenotype by Nrf2. Neurotoxicology 2006, 27, 1094–1100. [Google Scholar]
- Jakel, R.J.; Townsend, J.A.; Kraft, A.D.; Johnson, J.A. Nrf2-mediated protection against 6-hydroxydopamine. Brain Res 2007, 1144, 192–201. [Google Scholar]
- Chen, P.C.; Vargas, M.R.; Pani, A.K.; Smeyne, R.J.; Johnson, D.A.; Kan, Y.W.; Johnson, J.A. Nrf2-mediated neuroprotection in the MPTP mouse model of Parkinson’s disease: Critical role for the astrocyte. Proc. Nat. Acad. Sci. USA 2009, 106, 2933–2938. [Google Scholar]
- Wruck, C.J.; Claussen, M.; Fuhrmann, G.; Romer, L.; Schulz, A.; Pufe, T.; Waetzig, V.; Peipp, M.; Herdegen, T.; Gotz, M.E. Luteolin protects rat PC12 and C6 cells against MPP+ induced toxicity via an ERK dependent Keap1-Nrf2-ARE pathway. J. Neural Transm. Suppl 2007, 72, 57–67. [Google Scholar]
- Yamamoto, N.; Sawada, H.; Izumi, Y.; Kume, T.; Katsuki, H.; Shimohama, S.; Akaike, A. Proteasome inhibition induces glutathione synthesis and protects cells from oxidative stress: Relevance to Parkinson disease. J. Biol. Chem 2007, 282, 4364–4372. [Google Scholar]
- Schipper, H.M. Heme oxygenase expression in human central nervous system disorders. Free Radic. Biol. Med 2004, 37, 1995–2011. [Google Scholar]
- Van Muiswinkel, F.L.; de Vos, R.A.; Bol, J.G.; Andringa, G.; Jansen Steur, E.N.; Ross, D.; Siegel, D.; Drukarch, B. Expression of NAD(P)H: Quinone oxidoreductase in the normal and Parkinsonian substantia nigra. Neurobiol. Aging 2004, 25, 1253–1262. [Google Scholar]
- Brennan, W.A., Jr; Bird, E.D.; Aprille, J.R. Regional mitochondrial respiratory activity in Huntington’s disease brain. J. Neurochem. 1985, 44, 1948–1950. [Google Scholar]
- Gu, M.; Gash, M.T.; Mann, V.M.; Javoy-Agid, F.; Cooper, J.M.; Schapira, A.H. Mitochondrial defect in Huntington’s disease caudate nucleus. Ann. Neurol 1996, 39, 385–389. [Google Scholar]
- Calkins, M.J.; Jakel, R.J.; Johnson, D.A.; Chan, K.; Kan, Y.W.; Johnson, J.A. Protection from mitochondrial complex II inhibition in vitro and in vivo by Nrf2-mediated transcription. Proc. Nat. Acad. Sci. USA 2005, 102, 244–249. [Google Scholar]
- Shih, A.Y.; Imbeault, S.; Barakauskas, V.; Erb, H.; Jiang, L.; Li, P.; Murphy, T.H. Induction of the Nrf2-driven antioxidant response confers neuroprotection during mitochondrial stress in vivo. J. Biol. Chem 2005, 280, 22925–22936. [Google Scholar]
- Shih, A.Y.; Li, P.; Murphy, T.H. A small-molecule-inducible Nrf2-mediated antioxidant response provides effective prophylaxis against cerebral ischemia in vivo. J. Neurosci 2005, 25, 10321–10335. [Google Scholar]
- Bogdanov, M.B.; Andreassen, O.A.; Dedeoglu, A.; Ferrante, R.J.; Beal, M.F. Increased oxidative damage to DNA in a transgenic mouse model of Huntington’s disease. J. Neurochem 2001, 79, 1246–1249. [Google Scholar]
- Tabrizi, S.J.; Workman, J.; Hart, P.E.; Mangiarini, L.; Mahal, A.; Bates, G.; Cooper, J.M.; Schapira, A.H. Mitochondrial dysfunction and free radical damage in the Huntington R6/2 transgenic mouse. Ann. Neurol 2000, 47, 80–86. [Google Scholar]
- Ellrichmann, G.; Petrasch-Parwez, E.; Lee, D.H.; Reick, C.; Arning, L.; Saft, C.; Gold, R.; Linker, R.A. Efficacy of fumaric acid esters in the R6/2 and YAC128 models of Huntington’s disease. PLoS One 2011, 6, e16172–e16180. [Google Scholar]
- Frohman, E.M.; Racke, M.K.; Raine, C.S. Multiple sclerosis—The plaque and its pathogenesis. N. Engl. J. Med 2006, 354, 942–955. [Google Scholar]
- Lucchinetti, C.; Bruck, W.; Parisi, J.; Scheithauer, B.; Rodriguez, M.; Lassmann, H. Heterogeneity of multiple sclerosis lesions: Implications for the pathogenesis of demyelination. Ann. Neurol 2000, 47, 707–717. [Google Scholar]
- Witte, M.E.; Bo, L.; Rodenburg, R.J.; Belien, J.A.; Musters, R.; Hazes, T.; Wintjes, L.T.; Smeitink, J.A.; Geurts, J.J.; de Vries, H.E.; et al. Enhanced number and activity of mitochondria in multiple sclerosis lesions. J. Pathol 2009, 219, 193–204. [Google Scholar]
- Hendriks, J.J.A.; Alblas, J.; van der Pol, S.M.A.; van Tol, E.A.F.; Dijkstra, C.D.; de Vries, H.E. Flavonoids influence monocytic GTPase activity and are protective in experimental allergic encephalitis. J. Exp. Med 2004, 200, 1667–1672. [Google Scholar]
- Smith, K.J.; Kapoor, R.; Felts, P.A. Demyelination: The role of reactive oxygen and nitrogen species. Brain Pathol 1999, 9, 69–92. [Google Scholar]
- Van Meeteren, M.E.; Hendriks, J.J.; Dijkstra, C.D.; van Tol, E.A. Dietary compounds prevent oxidative damage and nitric oxide production by cells involved in demyelinating disease. Biochem. Pharmacol 2004, 67, 967–975. [Google Scholar]
- Vladimirova, O.; O’Connor, J.; Cahill, A.; Alder, H.; Butunoi, C.; Kalman, B. Oxidative damage to DNA in plaques of MS brains. Mult. Scler. J 1998, 4, 413–418. [Google Scholar]
- Boul-Enein, F.; Rauschka, H.; Kornek, B.; Stadelmann, C.; Stefferl, A.; Bruck, W.; Lucchinetti, C.; Schmidbauer, M.; Jellinger, K.; Lassmann, H. Preferential loss of myelin-associated glycoprotein reflects hypoxia-like white matter damage in stroke and inflammatory brain diseases. J. Neuropathol. Exp. Neurol 2003, 62, 25–33. [Google Scholar]
- Mahad, D.; Ziabreva, I.; Lassmann, H.; Turnbull, D. Mitochondrial defects in acute multiple sclerosis lesions. Brain 2008, 131, 1722–1735. [Google Scholar]
- Trapp, B.D.; Stys, P.K. Virtual hypoxia and chronic necrosis of demyelinated axons in multiple sclerosis. Lancet Neurol 2009, 8, 280–291. [Google Scholar]
- Witte, M.E.; Geurts, J.J.; de Vries, H.E.; van der Valk, P.; van Horssen, J. Mitochondrial dysfunction: A potential link between neuroinflammation and neurodegeneration? Mitochondrion 2010, 10, 411–418. [Google Scholar]
- Dutta, R.; McDonough, J.; Yin, X.; Peterson, J.; Chang, A.; Torres, T.; Gudz, T.; Macklin, W.B.; Lewis, D.A.; Fox, R.J.; et al. Mitochondrial dysfunction as a cause of axonal degeneration in multiple sclerosis patients. Ann. Neurol 2006, 59, 478–489. [Google Scholar]
- Dutta, R.; Trapp, B.D. Mechanisms of neuronal dysfunction and degeneration in multiple sclerosis. Prog. Neurobiol 2011, 93, 1–12. [Google Scholar]
- Mahad, D.; Lassmann, H.; Turnbull, D. Review: Mitochondria and disease progression in multiple sclerosis. Neuropathol. Appl. Neurobiol 2008, 34, 577–589. [Google Scholar]
- Paling, D.; Golay, X.; Wheeler-Kingshott, C.; Kapoor, R.; Miller, D. Energy failure in multiple sclerosis and its investigation using MR techniques. J. Neurol 2011, 258, 2113–2127. [Google Scholar]
- Mahad, D.J.; Ziabreva, I.; Campbell, G.; Lax, N.; White, K.; Hanson, P.S.; Lassmann, H.; Turnbull, D.M. Mitochondrial changes within axons in multiple sclerosis. Brain 2009, 132, 1161–1174. [Google Scholar]
- Tavazzi, B.; Batocchi, A.P.; Amorini, A.M.; Nociti, V.; D’Urso, S.; Longo, S.; Gullotta, S.; Picardi, M.; Lazzarino, G. Serum metabolic profile in multiple sclerosis patients. Mult. Scler. Int 2011, 2011, 167156–167163. [Google Scholar]
- Regenold, W.T.; Phatak, P.; Makley, M.J.; Stone, R.D.; Kling, M.A. Cerebrospinal fluid evidence of increased extra-mitochondrial glucose metabolism implicates mitochondrial dysfunction in multiple sclerosis disease progression. J. Neurol. Sci 2008, 275, 106–112. [Google Scholar]
- Lazzarino, G.; Amorini, A.M.; Eikelenboom, M.J.; Killestein, J.; Belli, A.; Di Pietro, V.; Tavazzi, B.; Barkhof, F.; Polman, C.H.; Uitdehaag, B.M.; et al. Cerebrospinal fluid ATP metabolites in multiple sclerosis. Mult. Scler. J. 2010, 16, 549–554. [Google Scholar]
- Veto, S.; Acs, P.; Bauer, J.; Lassmann, H.; Berente, Z.; Setalo, G., Jr; Borgulya, G.; Sumegi, B.; Komoly, S.; Gallyas, F., Jr; et al. Brain 2010, 133, 822–834.
- Smith, K.J.; Lassmann, H. The role of nitric oxide in multiple sclerosis. Lancet Neurol 2002, 1, 232–241. [Google Scholar]
- Van Horssen, J.; Witte, M.E.; Schreibelt, G.; De Vries, H.E. Radical changes in multiple sclerosis pathogenesis. Biochim. Biophys. Acta 2011, 1812, 141–150. [Google Scholar]
- Love, S. Oxidative stress in brain ischemia. Brain Pathol 1999, 9, 119–131. [Google Scholar]
- Bedard, K.; Krause, K.H. The NOX family of ROS-generating NADPH oxidases: Physiology and pathophysiology. Physiol. Rev 2007, 87, 245–313. [Google Scholar]
- Murphy, M.P. How mitochondria produce reactive oxygen species. Biochem. J 2009, 417, 1–13. [Google Scholar]
- Cross, A.H.; Manning, P.T.; Keeling, R.M.; Schmidt, R.E.; Misko, T.P. Peroxynitrite formation within the central nervous system in active multiple sclerosis. J. Neuroimmunol 1998, 88, 45–56. [Google Scholar]
- Liu, J.S.; Zhao, M.L.; Brosnan, C.F.; Lee, S.C. Expression of inducible nitric oxide synthase and nitrotyrosine in multiple sclerosis lesions. Am. J. Pathol 2001, 158, 2057–2066. [Google Scholar]
- Gray, E.; Thomas, T.L.; Betmouni, S.; Scolding, N.; Love, S. Elevated myeloperoxidase activity in white matter in multiple sclerosis. Neurosci. Lett 2008, 444, 195–198. [Google Scholar]
- Gray, E.; Thomas, T.L.; Betmouni, S.; Scolding, N.; Love, S. Elevated activity and microglial expression of myeloperoxidase in demyelinated cerebral cortex in multiple sclerosis. Brain Pathol 2008, 18, 86–95. [Google Scholar]
- Marik, C.; Felts, P.A.; Bauer, J.; Lassmann, H.; Smith, K.J. Lesion genesis in a subset of patients with multiple sclerosis: A role for innate immunity? Brain 2007, 130, 2800–2815. [Google Scholar]
- Cheret, C.; Gervais, A.; Lelli, A.; Colin, C.; Amar, L.; Ravassard, P.; Mallet, J.; Cumano, A.; Krause, K.H.; Mallat, M. Neurotoxic activation of microglia is promoted by a nox1-dependent NADPH oxidase. J. Neurosci 2008, 28, 12039–12051. [Google Scholar]
- Nikic, I.; Merkler, D.; Sorbara, C.; Brinkoetter, M.; Kreutzfeldt, M.; Bareyre, F.M.; Bruck, W.; Bishop, D.; Misgeld, T.; Kerschensteiner, M. A reversible form of axon damage in experimental autoimmune encephalomyelitis and multiple sclerosis. Nat. Med 2011, 17, 495–499. [Google Scholar]
- Zambonin, J.L.; Zhao, C.; Ohno, N.; Campbell, G.R.; Engeham, S.; Ziabreva, I.; Schwarz, N.; Lee, S.E.; Frischer, J.M.; Turnbull, D.M.; et al. Increased mitochondrial content in remyelinated axons: Implications for multiple sclerosis. Brain 2011, 134, 1901–1913. [Google Scholar]
- Aktas, O.; Prozorovski, T.; Smorodchenko, A.; Savaskan, N.E.; Lauster, R.; Kloetzel, P.M.; Infante-Duarte, C.; Brocke, S.; Zipp, F. Green tea epigallocatechin-3-gallate mediates T cellular NF-kappa B inhibition and exerts neuroprotection in autoimmune encephalomyelitis. J. Immunol 2004, 173, 5794–5800. [Google Scholar]
- Bechtold, D.A.; Kapoor, R.; Smith, K.J. Axonal protection using flecainide in experimental autoimmune encephalomyelitis. Ann. Neurol 2004, 55, 607–616. [Google Scholar]
- Kappos, L.; Gold, R.; Miller, D.H.; MacManus, D.G.; Havrdova, E.; Limmroth, V.; Polman, C.H.; Schmierer, K.; Yousry, T.A.; Eraksoy, M.; et al. Effect of BG-12 on contrast-enhanced lesions in patients with relapsing—Remitting multiple sclerosis: Subgroup analyses from the phase 2b study. Mult. Scler 2012, 18, 314–321. [Google Scholar]
- Bittner, S.; Meuth, S.G.; Gobel, K.; Melzer, N.; Herrmann, A.M.; Simon, O.J.; Weishaupt, A.; Budde, T.; Bayliss, D.A.; Bendszus, M.; et al. TASK1 modulates inflammation and neurodegeneration in autoimmune inflammation of the central nervous system. Brain 2009, 132, 2501–2516. [Google Scholar]
- Duffy, S.; So, A.; Murphy, T.H. Activation of endogenous antioxidant defenses in neuronal cells prevents free radical-mediated damage. J. Neurochem 1998, 71, 69–77. [Google Scholar]
- Hubbs, A.F.; Benkovic, S.A.; Miller, D.B.; O’Callaghan, J.P.; Battelli, L.; Schwegler-Berry, D.; Ma, Q. Vacuolar leukoencephalopathy with widespread astrogliosis in mice lacking transcription factor Nrf2. Am. J. Pathol 2007, 170, 2068–2076. [Google Scholar]
- Kappos, L.; Gold, R.; Miller, D.H.; Macmanus, D.G.; Havrdova, E.; Limmroth, V.; Polman, C.H.; Schmierer, K.; Yousry, T.A.; Yang, M.; et al. Efficacy and safety of oral fumarate in patients with relapsing-remitting multiple sclerosis: A multicentre, randomised, double-blind, placebo-controlled phase IIb study. Lancet 2008, 372, 1463–1472. [Google Scholar]
- Khodagholi, F.; Eftekharzadeh, B.; Maghsoudi, N.; Rezaei, P.F. Chitosan prevents oxidative stress-induced amyloid beta formation and cytotoxicity in NT2 neurons: Involvement of transcription factors Nrf2 and NF-kappaB. Mol. Cell Biochem 2010, 337, 39–51. [Google Scholar]
- Kraft, A.D.; Johnson, D.A.; Johnson, J.A. Nuclear factor E2-related factor 2-dependent antioxidant response element activation by tert-butylhydroquinone and sulforaphane occurring preferentially in astrocytes conditions neurons against oxidative insult. J. Neurosci 2004, 24, 1101–1112. [Google Scholar]
- Lee, J.M.; Calkins, M.J.; Chan, K.; Kan, Y.W.; Johnson, J.A. Identification of the NF-E2-related factor-2-dependent genes conferring protection against oxidative stress in primary cortical astrocytes using oligonucleotide microarray analysis. J. Biol. Chem 2003, 278, 12029–12038. [Google Scholar]
- Lewerenz, J.; Albrecht, P.; Tien, M.L.; Henke, N.; Karumbayaram, S.; Kornblum, H.I.; Wiedau-Pazos, M.; Schubert, D.; Maher, P.; Methner, A. Induction of Nrf2 and xCT are involved in the action of the neuroprotective antibiotic ceftriaxone in vitro. J. Neurochem 2009, 111, 332–343. [Google Scholar]
- Liu, Y.; Kern, J.T.; Walker, J.R.; Johnson, J.A.; Schultz, P.G.; Luesch, H. A genomic screen for activators of the antioxidant response element. Proc. Nat. Acad. Sci. USA 2007, 104, 5205–5210. [Google Scholar]
- Rachakonda, G.; Xiong, Y.; Sekhar, K.R.; Stamer, S.L.; Liebler, D.C.; Freeman, M.L. Covalent modification at Cys151 dissociates the electrophile sensor Keap1 from the ubiquitin ligase CUL3. Chem. Res. Toxicol 2008, 21, 705–710. [Google Scholar]
- Satoh, T.; Okamoto, S.I.; Cui, J.; Watanabe, Y.; Furuta, K.; Suzuki, M.; Tohyama, K.; Lipton, S.A. Activation of the Keap1/Nrf2 pathway for neuroprotection by electrophilic [correction of electrophillic] phase II inducers. Proc. Nat. Acad. Sci. USA 2006, 103, 768–773. [Google Scholar]
- Satoh, T.; Harada, N.; Hosoya, T.; Tohyama, K.; Yamamoto, M.; Itoh, K. Keap1/Nrf2 system regulates neuronal survival as revealed through study of keap1 gene-knockout mice. Biochem. Biophys. Res. Commun 2009, 380, 298–302. [Google Scholar]
- Schilling, S.; Goelz, S.; Linker, R.; Luehder, F.; Gold, R. Fumaric acid esters are effective in chronic experimental autoimmune encephalomyelitis and suppress macrophage infiltration. Clin. Exp. Immunol 2006, 145, 101–107. [Google Scholar]
- Su, J.Y.; Duffy, S.; Murphy, T.H. Reduction of H2O2-evoked, intracellular calcium increases in the rat N18-RE-105 neuronal cell line by pretreatment with an electrophilic antioxidant inducer. Neurosci. Lett 1999, 273, 109–112. [Google Scholar]
- Thiessen, A.; Schmidt, M.M.; Dringen, R. Fumaric acid dialkyl esters deprive cultured rat oligodendroglial cells of glutathione and upregulate the expression of heme oxygenase 1. Neurosci. Lett 2010, 475, 56–60. [Google Scholar]
- Yang, L.; Calingasan, N.Y.; Thomas, B.; Chaturvedi, R.K.; Kiaei, M.; Wille, E.J.; Liby, K.T.; Williams, C.; Royce, D.; Risingsong, R.; et al. Neuroprotective effects of the triterpenoid, CDDO methyl amide, a potent inducer of Nrf2-mediated transcription. PLoS One 2009, 4, e5757. [Google Scholar]
- Linker, R.A.; Lee, D.H.; Ryan, S.; van Dam, A.M.; Conrad, R.; Bista, P.; Zeng, W.; Hronowsky, X.; Buko, A.; Chollate, S.; et al. Fumaric acid esters exert neuroprotective effects in neuroinflammation via activation of the Nrf2 antioxidant pathway. Brain 2011, 134, 678–692. [Google Scholar]
- Schmidt, M.M.; Dringen, R. Fumaric acid diesters deprive cultured primary astrocytes rapidly of glutathione. Neurochem. Int 2010, 57, 460–467. [Google Scholar]
- Scannevin, R.H.; Chollate, S.; Jung, M.Y.; Shackett, M.; Patel, H.; Bista, P.; Zeng, W.; Ryan, S.; Yamamoto, M.; Lukashev, M.; et al. Fumarates promote cytoprotection of central nervous system cells against oxidative stress via the nuclear factor (erythroid-derived 2)-like 2 pathway. J. Pharmacol. Exp. Ther 2012, 341, 274–284. [Google Scholar]
- Albrecht, P.; Bouchachia, I.; Zimmermann, C.; Hofstetter, H.H.; Kovacs, Z.; Henke, N.; Lisak, D.; Issberner, A.; Lewerenz, J.; Maher, P.; et al. Effects of dimethyl fumarate on neuroprotection and immunomodulation. J. Neuroinflamm 2012, 9, 163. [Google Scholar]
- Ashrafian, H.; Czibik, G.; Bellahcene, M.; Aksentijevic, D.; Smith, A.C.; Mitchell, S.J.; Dodd, M.S.; Kirwan, J.; Byrne, J.J.; Ludwig, C.; et al. Fumarate is cardioprotective via activation of the Nrf2 antioxidant pathway. Cell Metab 2012, 15, 361–371. [Google Scholar]
© 2012 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Lee, D.-H.; Gold, R.; Linker, R.A. Mechanisms of Oxidative Damage in Multiple Sclerosis and Neurodegenerative Diseases: Therapeutic Modulation via Fumaric Acid Esters. Int. J. Mol. Sci. 2012, 13, 11783-11803. https://doi.org/10.3390/ijms130911783
Lee D-H, Gold R, Linker RA. Mechanisms of Oxidative Damage in Multiple Sclerosis and Neurodegenerative Diseases: Therapeutic Modulation via Fumaric Acid Esters. International Journal of Molecular Sciences. 2012; 13(9):11783-11803. https://doi.org/10.3390/ijms130911783
Chicago/Turabian StyleLee, De-Hyung, Ralf Gold, and Ralf A. Linker. 2012. "Mechanisms of Oxidative Damage in Multiple Sclerosis and Neurodegenerative Diseases: Therapeutic Modulation via Fumaric Acid Esters" International Journal of Molecular Sciences 13, no. 9: 11783-11803. https://doi.org/10.3390/ijms130911783
APA StyleLee, D.-H., Gold, R., & Linker, R. A. (2012). Mechanisms of Oxidative Damage in Multiple Sclerosis and Neurodegenerative Diseases: Therapeutic Modulation via Fumaric Acid Esters. International Journal of Molecular Sciences, 13(9), 11783-11803. https://doi.org/10.3390/ijms130911783