The Role of Glucose Transporters in Brain Disease: Diabetes and Alzheimer’s Disease

Abstract
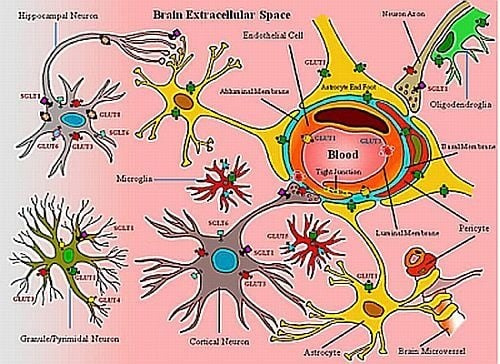
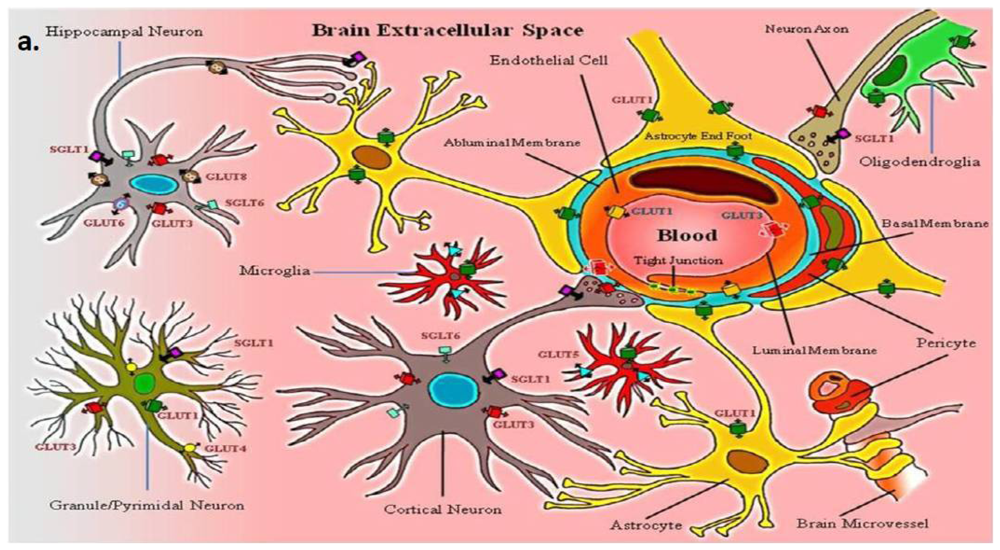
:
1. Introduction
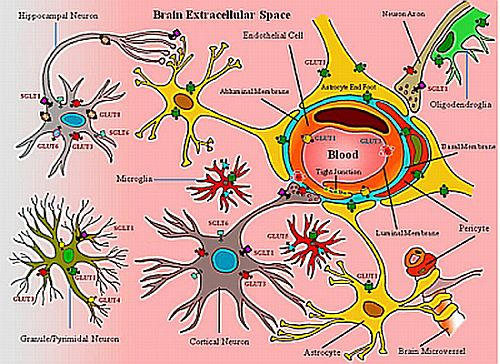
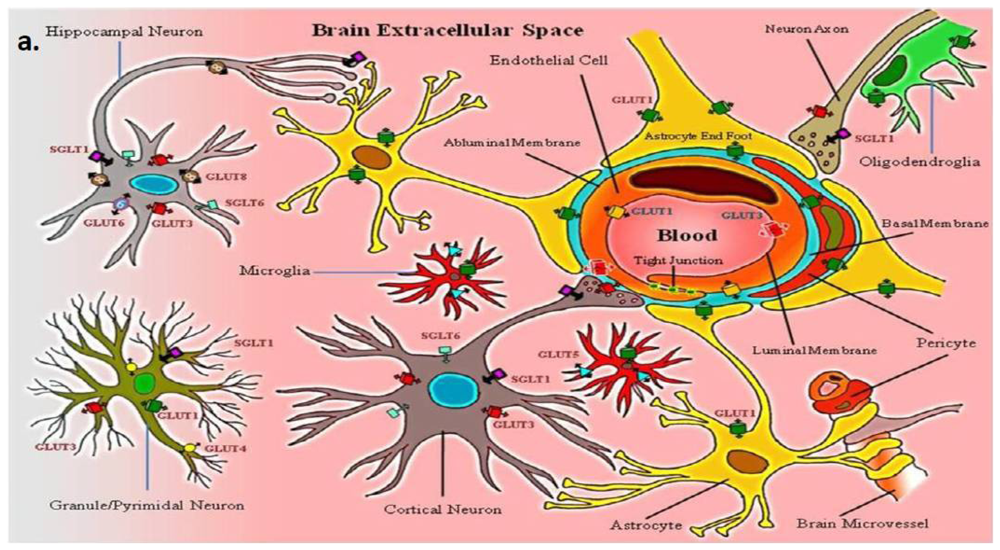
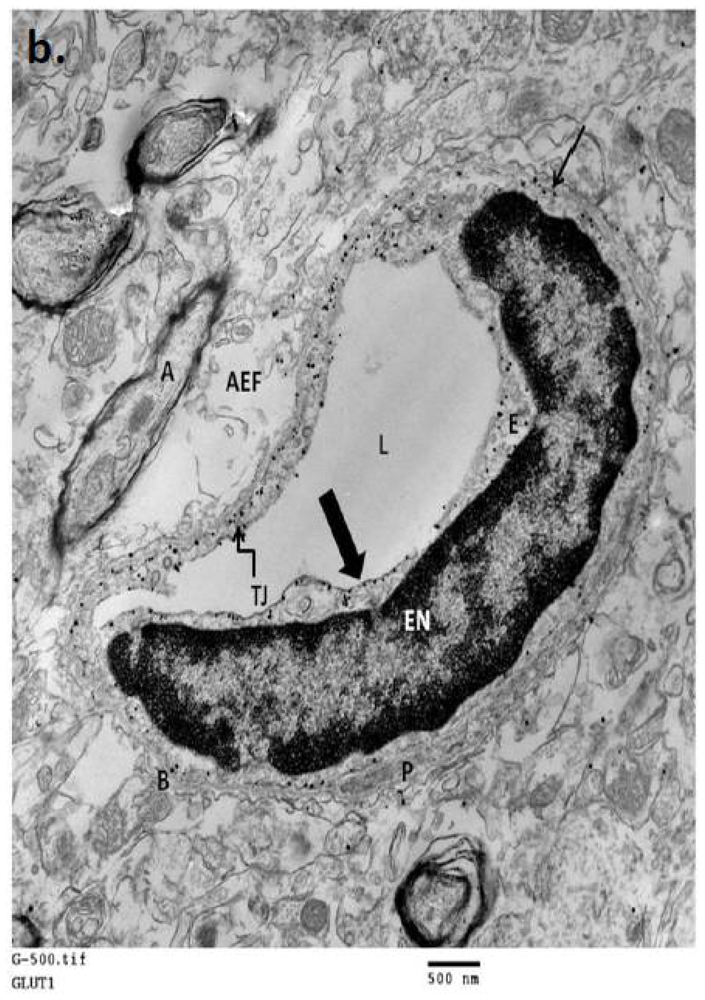
2. Neurovascular Unit: Glucose Transporters and Transport
3. Diabetes: Glucose Metabolism and Glucose Transporters in the Brain
4. Alzheimer’s Disease: Glucose Metabolism and Glucose Transporters in the Brain
5. Future Challenges
Acknowledgments
References
- Magistretti, P.J.; Pellerin, L. Cellular mechanisms of brain energy metabolism and their relevance to functional brain imaging. Philos. Trans. R. Soc. Lond. B 1999, 354, 1155–1163. [Google Scholar]
- Larrabee, M.G. Lactate metabolism and its effects on glucose metabolism in an excised neural tissue. J. Neurochem 1995, 64, 1734–1741. [Google Scholar]
- Pardridge, W.M.; Oldendorf, W.H. Transport of metabolic substrates through the blood-brain barrier. J. Neurochem 1977, 28, 5–12. [Google Scholar]
- Magistretti, P.J.; Pellerin, L. Astrocytes couple synaptic activity to glucose utilization in the brain. News Physiol. Sci 1999, 14, 177–182. [Google Scholar]
- Tekkok, S.B.; Brown, A.M.; Westenbroek, R.; Pellerin, L.; Ransom, B.R. Transfer of glycogen-derived lactate from astrocytes to axons via specific monocarboxylate transporters supports mouse optic nerve activity. J. Neurosci. Res 2005, 81, 644–652. [Google Scholar]
- Goldstein, G.W. Relation of potassium transport to oxidative metabolism in isolated brain capillaries. J. Physiol 1979, 286, 185–195. [Google Scholar]
- Guzman, M.; Blazquez, C. Is there an astrocyte-neuron ketone body shuttle? Trends Endocrinol. Metab 2001, 12, 169–173. [Google Scholar]
- Pardridge, W.M. Blood-brain barrier transport of glucose, free fatty acids, and ketone bodies. Adv. Exp. Med. Biol 1991, 291, 43–53. [Google Scholar]
- Cunnane, S.; Nugent, S.; Roy, M.; Courchesne-Loyer, A.; Croteau, E.; Tremblay, S.; Castellano, A.; Pifferi, F.; Bocti, C.; Paquet, N.; et al. Brain fuel metabolism, aging, and alzheimer’s disease. Nutrition 2011, 27, 3–20. [Google Scholar]
- Ronnemaa, E.; Zethelius, B.; Sundelof, J.; Sundstrom, J.; Degerman-Gunnarsson, M.; Lannfelt, L.; Berne, C.; Kilander, L. Glucose metabolism and the risk of alzheimer’s disease and dementia: A population-based 12 year follow-up study in 71-year-old men. Diabetologia 2009, 52, 1504–1510. [Google Scholar]
- Messier, C.; Gagnon, M. Glucose regulation and cognitive functions: Relation to alzheimer’s disease and diabetes. Behav. Brain Res 1996, 75, 1–11. [Google Scholar]
- Kroner, Z. The relationship between alzheimer’s disease and diabetes: Type 3 diabetes? Altern. Med. Rev 2009, 14, 373–379. [Google Scholar]
- Yamazaki, Y.; Miwa, T.; Sakurai, H.; Hanyu, H.; Iwamoto, T.; Odawara, M. Clinical backgrounds and morbidity of cognitive impairment in elderly diabetic patients. Endocr. J 2011, 58, 109–115. [Google Scholar]
- Akter, K.; Lanza, E.A.; Martin, S.A.; Myronyuk, N.; Rua, M.; Raffa, R.B. Diabetes mellitus and alzheimer’s disease: Shared pathology and treatment? Br. J. Clin. Pharmacol 2011, 71, 365–376. [Google Scholar]
- Meneilly, G.S.; Hill, A. Alterations in glucose metabolism in patients with alzheimer’s disease. J. Am. Geriatr. Soc 1993, 41, 710–714. [Google Scholar]
- Bell, G.I.; Kayano, T.; Buse, J.B.; Burant, C.F.; Takeda, J.; Lin, D.; Fukumoto, H.; Seino, S. Molecular biology of mammalian glucose transporters. Diabetes Care 1990, 13, 198–208. [Google Scholar]
- Mueckler, M.M. The molecular biology of mammalian glucose transporters. Curr. Opin. Nephrol. Hypertens 1992, 1, 12–20. [Google Scholar]
- Dwyer, D.S.; Vannucci, S.J.; Simpson, I.A. Expression, regulation, and functional role of glucose transporters (gluts) in brain. Int. Rev. Neurobiol 2002, 51, 159–188. [Google Scholar]
- Wright, E.M.; Turk, E.; Hager, K.; Lescale-Matys, L.; Hirayama, B.; Supplisson, S.; Loo, D.D. The na+/glucose cotransporter (sglt1). Acta Physiol. Scand. Suppl 1992, 607, 201–207. [Google Scholar]
- Maher, F.; Vannucci, S.; Takeda, J.; Simpson, I.A. Expression of mouse-glut3 and human-glut3 glucose transporter proteins in brain. Biochem. Biophys. Res. Commun 1992, 182, 703–711. [Google Scholar]
- Vannucci, S.J.; Seaman, L.B.; Brucklacher, R.M.; Vannucci, R.C. Glucose transport in developing rat brain: Glucose transporter proteins, rate constants and cerebral glucose utilization. Mol. Cell. Biochem 1994, 140, 177–184. [Google Scholar]
- Vannucci, S.J.; Maher, F.; Simpson, I.A. Glucose transporter proteins in brain: Delivery of glucose to neurons and glia. Glia 1997, 21, 2–21. [Google Scholar]
- Vorbrodt, A.W.; Dobrogowska, D.H.; Tarnawski, M. Immunogold study of interendothelial junction-associated and glucose transporter proteins during postnatal maturation of the mouse blood-brain barrier. J. Neurocytol 2001, 30, 705–716. [Google Scholar]
- Wright, E.M.; Turk, E. The sodium/glucose cotransport family slc5. Pflugers Arch 2004, 447, 510–518. [Google Scholar]
- Thorens, B.; Mueckler, M. Glucose transporters in the 21st century. Am. J. Physiol. Endocrinol. Metab 2010, 298, E141–145. [Google Scholar]
- Mueckler, M.; Caruso, C.; Baldwin, S.A.; Panico, M.; Blench, I.; Morris, H.R.; Allard, W.J.; Lienhard, G.E.; Lodish, H.F. Sequence and structure of a human glucose transporter. Science 1985, 229, 941–945. [Google Scholar]
- Morgello, S.; Uson, R.R.; Schwartz, E.J.; Haber, R.S. The human blood-brain barrier glucose transporter (glut1) is a glucose transporter of gray matter astrocytes. Glia 1995, 14, 43–54. [Google Scholar]
- Mann, G.E.; Yudilevich, D.L.; Sobrevia, L. Regulation of amino acid and glucose transporters in endothelial and smooth muscle cells. Physiol. Rev 2003, 83, 183–252. [Google Scholar]
- Devaskar, S.; Chundu, K.; Zahm, D.S.; Holtzclaw, L.; Holloran, K. The neonatal rabbit brain glucose transporter. Brain Res. Dev. Brain Res 1992, 67, 95–103. [Google Scholar]
- Vannucci, S.J. Developmental expression of glut1 and glut3 glucose transporters in rat brain. J. Neurochem 1994, 62, 240–246. [Google Scholar]
- Liu, Q.; Vera, J.C.; Peng, H.; Golde, D.W. The predicted atp-binding domains in the hexose transporter glut1 critically affect transporter activity. Biochemistry 2001, 40, 7874–7881. [Google Scholar]
- Rumsey, S.C.; Kwon, O.; Xu, G.W.; Burant, C.F.; Simpson, I.; Levine, M. Glucose transporter isoforms glut1 and glut3 transport dehydroascorbic acid. J. Biol. Chem 1997, 272, 18982–18989. [Google Scholar]
- Bondy, C.A.; Lee, W.H.; Zhou, J. Ontogeny and cellular distribution of brain glucose transporter gene expression. Mol. Cell. Neurosci 1992, 3, 305–314. [Google Scholar]
- McCall, A.L.; van Bueren, A.M.; Nipper, V.; Moholt-Siebert, M.; Downes, H.; Lessov, N. Forebrain ischemia increases glut1 protein in brain microvessels and parenchyma. J. Cereb. Blood Flow Metab 1996, 16, 69–76. [Google Scholar]
- Virgintino, D.; Robertson, D.; Monaghan, P.; Errede, M.; Bertossi, M.; Ambrosi, G.; Roncali, L. Glucose transporter glut1 in human brain microvessels revealed by ultrastructural immunocytochemistry. J. Submicrosc. Cytol. Pathol 1997, 29, 365–370. [Google Scholar]
- Gerhart, D.Z.; LeVasseur, R.J.; Broderius, M.A.; Drewes, L.R. Glucose transporter localization in brain using light and electron immunocytochemistry. J. Neurosci. Res 1989, 22, 464–472. [Google Scholar]
- Farrell, C.L.; Pardridge, W.M. Ultrastructural localization of blood-brain barrier-specific antibodies using immunogold-silver enhancement techniques. J. Neurosci. Methods 1991, 37, 103–110. [Google Scholar]
- Cornford, E.M.; Hyman, S.; Landaw, E.M. Developmental modulation of blood-brain-barrier glucose transport in the rabbit. Brain Res 1994, 663, 7–18. [Google Scholar]
- Cornford, E.M.; Hyman, S.; Swartz, B.E. The human brain glut1 glucose transporter: Ultrastructural localization to the blood-brain barrier endothelia. J. Cereb. Blood Flow Metab 1994, 14, 106–112. [Google Scholar]
- Farrell, C.L.; Pardridge, W.M. Blood-brain barrier glucose transporter is asymmetrically distributed on brain capillary endothelial lumenal and ablumenal membranes: An electron microscopic immunogold study. Proc. Natl. Acad. Sci. USA 1991, 88, 5779–5783. [Google Scholar]
- Leybaert, L.; De Bock, M.; van Moorhem, M.; Decrock, E.; de Vuyst, E. Neurobarrier coupling in the brain: Adjusting glucose entry with demand. J. Neurosci. Res 2007, 85, 3213–3220. [Google Scholar]
- Birnbaum, M.J.; Haspel, H.C.; Rosen, O.M. Cloning and characterization of a cdna encoding the rat brain glucose-transporter protein. Proc. Natl. Acad. Sci. USA 1986, 83, 5784–5788. [Google Scholar]
- Devaskar, S.; Zahm, D.S.; Holtzclaw, L.; Chundu, K.; Wadzinski, B.E. Developmental regulation of the distribution of rat brain insulin-insensitive (glut 1) glucose transporter. Endocrinology 1991, 129, 1530–1540. [Google Scholar]
- Dwyer, K.J.; Boado, R.J.; Pardridge, W.M. Cis-element/cytoplasmic protein interaction within the 3′-untranslated region of the glut1 glucose transporter mrna. J. Neurochem 1996, 66, 449–458. [Google Scholar]
- McGowan, K.M.; Long, S.D.; Pekala, P.H. Glucose transporter gene expression: Regulation of transcription and mrna stability. Pharmacol. Ther 1995, 66, 465–505. [Google Scholar]
- Devraj, K.; Klinger, M.E.; Myers, R.L.; Mokashi, A.; Hawkins, R.A.; Simpson, I.A. Glut-1 glucose transporters in the blood-brain barrier: Differential phosphorylation. J. Neurosci. Res 2011, 89, 1913–1925. [Google Scholar]
- Shawahna, R.; Uchida, Y.; Decleves, X.; Ohtsuki, S.; Yousif, S.; Dauchy, S.; Jacob, A.; Chassoux, F.; Daumas-Duport, C.; Couraud, P.O.; et al. Transcriptomic and quantitative proteomic analysis of transporters and drug metabolizing enzymes in freshly isolated human brain microvessels. Mol. Pharm 2011, 8, 1332–1341. [Google Scholar]
- Uchida, Y.; Ohtsuki, S.; Katsukura, Y.; Ikeda, C.; Suzuki, T.; Kamiie, J.; Terasaki, T. Quantitative targeted absolute proteomics of human blood-brain barrier transporters and receptors. J. Neurochem 2011, 117, 333–345. [Google Scholar]
- Ito, K.; Uchida, Y.; Ohtsuki, S.; Aizawa, S.; Kawakami, H.; Katsukura, Y.; Kamiie, J.; Terasaki, T. Quantitative membrane protein expression at the blood-brain barrier of adult and younger cynomolgus monkeys. J. Pharm. Sci 2011, 100, 3939–3950. [Google Scholar]
- Enerson, B.E.; Drewes, L.R. The rat blood-brain barrier transcriptome. J. Cereb. Blood Flow Metab 2006, 26, 959–973. [Google Scholar]
- Gerhart, D.Z.; Broderius, M.A.; Borson, N.D.; Drewes, L.R. Neurons and microvessels express the brain glucose transporter protein glut3. Proc. Natl. Acad. Sci. USA 1992, 89, 733–737. [Google Scholar]
- Ngarmukos, C.; Baur, E.L.; Kumagai, A.K. Co-localization of glut1 and glut4 in the blood-brain barrier of the rat ventromedial hypothalamus. Brain Res 2001, 900, 1–8. [Google Scholar]
- Nishizaki, T.; Matsuoka, T. Low glucose enhances na+/glucose transport in bovine brain artery endothelial cells. Stroke 1998, 29, 844–849. [Google Scholar]
- Wang, D.; Pascual, J.M.; Yang, H.; Engelstad, K.; Mao, X.; Cheng, J.; Yoo, J.; Noebels, J.L.; De Vivo, D.C. A mouse model for glut-1 haploinsufficiency. Hum. Mol. Genet 2006, 15, 1169–1179. [Google Scholar]
- Ganguly, A.; McKnight, R.A.; Raychaudhuri, S.; Shin, B.C.; Ma, Z.; Moley, K.; Devaskar, S.U. Glucose transporter isoform-3 mutations cause early pregnancy loss and fetal growth restriction. Am. J. Phys. Endocrinol. Metab 2007, 292, E1241–E1255. [Google Scholar]
- Ganguly, A.; Devaskar, S.U. Glucose transporter isoform-3-null heterozygous mutation causes sexually dimorphic adiposity with insulin resistance. Am. J. Physiol. Endocrinol. Metab 2008, 294, E1144–E1151. [Google Scholar]
- Nishizaki, T.; Kammesheidt, A.; Sumikawa, K.; Asada, T.; Okada, Y. A sodium- and energy-dependent glucose transporter with similarities to sglt1-2 is expressed in bovine cortical vessels. Neurosci. Res 1995, 22, 13–22. [Google Scholar]
- Yu, A.S.; Hirayama, B.A.; Timbol, G.; Liu, J.; Basarah, E.; Kepe, V.; Satyamurthy, N.; Huang, S.C.; Wright, E.M.; Barrio, J.R. Functional expression of sglts in rat brain. Am. J. Physiol. Cell Physiol 2010, 299, C1277–C1284. [Google Scholar]
- Vemula, S.; Roder, K.E.; Yang, T.; Bhat, G.J.; Thekkumkara, T.J.; Abbruscato, T.J. A functional role for sodium-dependent glucose transport across the blood-brain barrier during oxygen glucose deprivation. J. Pharmacol. Exp. Ther 2009, 328, 487–495. [Google Scholar]
- Simpson, I.A.; Carruthers, A.; Vannucci, S.J. Supply and demand in cerebral energy metabolism: The role of nutrient transporters. J. Cereb. Blood Flow Metab 2007, 27, 1766–1791. [Google Scholar]
- Maher, F.; Davies-Hill, T.M.; Simpson, I.A. Substrate specificity and kinetic parameters of glut3 in rat cerebellar granule neurons. Biochem. J 1996, 315, 827–831. [Google Scholar]
- Lowe, A.G.; Walmsley, A.R. The kinetics of glucose transport in human red blood cells. Biochim. Biophys. Acta 1986, 857, 146–154. [Google Scholar]
- Mantych, G.J.; James, D.E.; Chung, H.D.; Devaskar, S.U. Cellular localization and characterization of glut 3 glucose transporter isoform in human brain. Endocrinology 1992, 131, 1270–1278. [Google Scholar]
- Chen, J.; Williams, S.; Ho, S.; Loraine, H.; Hagan, D.; Whaley, J.M.; Feder, J.N. Quantitative pcr tissue expression profiling of the human sglt2 gene and related family members. Diabetes Ther 2010, 1, 57–92. [Google Scholar]
- Choeiri, C.; Staines, W.; Messier, C. Immunohistochemical localization and quantification of glucose transporters in the mouse brain. Neuroscience 2002, 111, 19–34. [Google Scholar]
- Apelt, J.; Mehlhorn, G.; Schliebs, R. Insulin-sensitive glut4 glucose transporters are colocalized with glut3-expressing cells and demonstrate a chemically distinct neuron-specific localization in rat brain. J. Neurosci. Res 1999, 57, 693–705. [Google Scholar]
- El Messari, S.; Leloup, C.; Quignon, M.; Brisorgueil, M.J.; Penicaud, L.; Arluison, M. Immunocytochemical localization of the insulin-responsive glucose transporter 4 (glut4) in the rat central nervous system. J. Comp. Neurol 1998, 399, 492–512. [Google Scholar]
- Kobayashi, M.; Nikami, H.; Morimatsu, M.; Saito, M. Expression and localization of insulin-regulatable glucose transporter (glut4) in rat brain. Neurosci. Lett 1996, 213, 103–106. [Google Scholar]
- Reagan, L.P.; Rosell, D.R.; Alves, S.E.; Hoskin, E.K.; McCall, A.L.; Charron, M.J.; McEwen, B.S. Glut8 glucose transporter is localized to excitatory and inhibitory neurons in the rat hippocampus. Brain Res 2002, 932, 129–134. [Google Scholar]
- Gomez, O.; Ballester-Lurbe, B.; Mesonero, J.E.; Terrado, J. Glucose transporters glut4 and glut8 are upregulated after facial nerve axotomy in adult mice. J. Anat 2011, 219, 525–530. [Google Scholar]
- Gomez, O.; Ballester-Lurbe, B.; Poch, E.; Mesonero, J.E.; Terrado, J. Developmental regulation of glucose transporters glut3, glut4 and glut8 in the mouse cerebellar cortex. J. Anat 2010, 217, 616–623. [Google Scholar]
- Piroli, G.G.; Grillo, C.A.; Charron, M.J.; McEwen, B.S.; Reagan, L.P. Biphasic effects of stress upon glut8 glucose transporter expression and trafficking in the diabetic rat hippocampus. Brain Res 2004, 1006, 28–35. [Google Scholar]
- Piroli, G.G.; Grillo, C.A.; Hoskin, E.K.; Znamensky, V.; Katz, E.B.; Milner, T.A.; McEwen, B.S.; Charron, M.J.; Reagan, L.P. Peripheral glucose administration stimulates the translocation of glut8 glucose transporter to the endoplasmic reticulum in the rat hippocampus. J. Com. Neurol 2002, 452, 103–114. [Google Scholar]
- Sankar, R.; Thamotharan, S.; Shin, D.; Moley, K.H.; Devaskar, S.U. Insulin-responsive glucose transporters-glut8 and glut4 are expressed in the developing mammalian brain. Brain Res. Mol. Brain Res 2002, 107, 157–165. [Google Scholar]
- Schmidt, S.; Gawlik, V.; Holter, S.M.; Augustin, R.; Scheepers, A.; Behrens, M.; Wurst, W.; Gailus-Durner, V.; Fuchs, H.; de Angelis, M.H.; et al. Deletion of glucose transporter glut8 in mice increases locomotor activity. Behav. Genet 2008, 38, 396–406. [Google Scholar]
- Schmidt, S.; Joost, H.G.; Schurmann, A. Glut8, the enigmatic intracellular hexose transporter. Am. J. Physiol. Endocrinol. Metab 2009, 296, E614–E618. [Google Scholar]
- Shin, B.C.; McKnight, R.A.; Devaskar, S.U. Glucose transporter glut8 translocation in neurons is not insulin responsive. J. Neurosci. Res 2004, 75, 835–844. [Google Scholar]
- Payne, J.; Maher, F.; Simpson, I.; Mattice, L.; Davies, P. Glucose transporter glut 5 expression in microglial cells. Glia 1997, 21, 327–331. [Google Scholar]
- Yu, S.; Ding, W.G. The 45 kda form of glucose transporter 1 (glut1) is localized in oligodendrocyte and astrocyte but not in microglia in the rat brain. Brain Res 1998, 797, 65–72. [Google Scholar]
- Brownlee, M. The pathobiology of diabetic complications: A unifying mechanism. Diabetes 2005, 54, 1615–1625. [Google Scholar]
- Blackshear, P.J.; Alberti, K.G. Experimental diabetic ketoacidosis. Sequential changes of metabolic intermediates in blood, liver, cerebrospinal fluid and brain after acute insulin deprivation in the streptozotocin-diabetic rat. Biochem. J 1974, 138, 107–117. [Google Scholar]
- Folbergrova, J.; Memezawa, H.; Smith, M.L.; Siesjo, B.K. Focal and perifocal changes in tissue energy state during middle cerebral artery occlusion in normo- and hyperglycemic rats. J. Cereb. Blood Flow Metab 1992, 12, 25–33. [Google Scholar]
- Garcia-Espinosa, M.A.; Garcia-Martin, M.L.; Cerdan, S. Role of glial metabolism in diabetic encephalopathy as detected by high resolution 13c nmr. NMR Biomed 2003, 16, 440–449. [Google Scholar]
- Hofer, R.E.; Lanier, W.L. The effects of insulin infusion on plasma and brain glucose in hyperglycemic diabetic rats. A comparison with placebo-treated diabetic and nondiabetic rats. Anesthesiology 1991, 75, 673–678. [Google Scholar]
- Hofer, R.E.; Lanier, W.L. Effects of insulin on blood, plasma, and brain glucose in hyperglycemic diabetic rats. Stroke 1991, 22, 505–509. [Google Scholar]
- Hoxworth, J.M.; Xu, K.; Zhou, Y.; Lust, W.D.; LaManna, J.C. Cerebral metabolic profile, selective neuron loss, and survival of acute and chronic hyperglycemic rats following cardiac arrest and resuscitation. Brain Res 1999, 821, 467–479. [Google Scholar]
- Jacob, R.J.; Fan, X.; Evans, M.L.; Dziura, J.; Sherwin, R.S. Brain glucose levels are elevated in chronically hyperglycemic diabetic rats: No evidence for protective adaptation by the blood brain barrier. Metabolism 2002, 51, 1522–1524. [Google Scholar]
- Mans, A.M.; DeJoseph, M.R.; Davis, D.W.; Hawkins, R.A. Brain energy metabolism in streptozotocin-diabetes. Biochem. J 1988, 249, 57–62. [Google Scholar]
- Puchowicz, M.A.; Xu, K.; Magness, D.; Miller, C.; Lust, W.D.; Kern, T.S.; LaManna, J.C. Comparison of glucose influx and blood flow in retina and brain of diabetic rats. J. Cereb. Blood Flow Metab 2004, 24, 449–457. [Google Scholar]
- Ruderman, N.B.; Ross, P.S.; Berger, M.; Goodman, M.N. Regulation of glucose and ketone-body metabolism in brain of anaesthetized rats. Biochem. J 1974, 138, 1–10. [Google Scholar]
- Shram, N.F.; Netchiporouk, L.I.; Martelet, C.; Jaffrezic-Renault, N.; Cespuglio, R. Brain glucose: Voltammetric determination in normal and hyperglycaemic rats using a glucose microsensor. Neuroreport 1997, 8, 1109–1112. [Google Scholar]
- Tang, J.; Zhu, X.W.; Lust, W.D.; Kern, T.S. Retina accumulates more glucose than does the embryologically similar cerebral cortex in diabetic rats. Diabetologia 2000, 43, 1417–1423. [Google Scholar]
- Van der Graaf, M.; Janssen, S.W.; van Asten, J.J.; Hermus, A.R.; Sweep, C.G.; Pikkemaat, J.A.; Martens, G.J.; Heerschap, A. Metabolic profile of the hippocampus of zucker diabetic fatty rats assessed by in vivo 1h magnetic resonance spectroscopy. NMR Biomed 2004, 17, 405–410. [Google Scholar]
- Wagner, S.R., IV; Lanier, W.L. Metabolism of glucose, glycogen, and high-energy phosphates during complete cerebral ischemia. A comparison of normoglycemic, chronically hyperglycemic diabetic, and acutely hyperglycemic nondiabetic rats. Anesthesiology 1994, 81, 1516–1526. [Google Scholar]
- Duarte, J.M.; Carvalho, R.A.; Cunha, R.A.; Gruetter, R. Caffeine consumption attenuates neurochemical modifications in the hippocampus of streptozotocin-induced diabetic rats. J. Neurochem 2009, 111, 368–379. [Google Scholar]
- Choi, T.B.; Boado, R.J.; Pardridge, W.M. Blood-brain barrier glucose transporter mrna is increased in experimental diabetes mellitus. Biochem. Biophys. Res. Commun 1989, 164, 375–380. [Google Scholar]
- Pardridge, W.M.; Triguero, D.; Farrell, C.R. Downregulation of blood-brain barrier glucose transporter in experimental diabetes. Diabetes 1990, 39, 1040–1044. [Google Scholar]
- Taarnhoj, J.; Alm, A. The effect of diabetes on transport through the blood-retinal and blood-brain barriers in rats. Graefes Arch. Clin. Exp. Ophthalmol 1991, 229, 291–293. [Google Scholar]
- Kainulainen, H.; Schurmann, A.; Vilja, P.; Joost, H.G. In-vivo glucose uptake and glucose transporter proteins glut1 and glut3 in brain tissue from streptozotocin-diabetic rats. Acta Physiol. Scand 1993, 149, 221–225. [Google Scholar]
- Pelligrino, D.A.; Lipa, M.D.; Albrecht, R.F. Regional blood-brain glucose transfer and glucose utilization in chronically hyperglycemic, diabetic rats following acute glycemic normalization. J. Cereb. Blood Flow Metab 1990, 10, 774–780. [Google Scholar]
- Pelligrino, D.A.; LaManna, J.C.; Duckrow, R.B.; Bryan, R.M., Jr; Harik, S.I. Hyperglycemia and blood-brain barrier glucose transport. J. Cereb. Blood Flow Metab. 1992, 12, 887–899. [Google Scholar]
- Vannucci, S.J.; Gibbs, E.M.; Simpson, I.A. Glucose utilization and glucose transporter proteins glut-1 and glut-3 in brains of diabetic (db/db) mice. Am. J. Physiol 1997, 272, E267–E274. [Google Scholar]
- Gjedde, A.; Crone, C. Blood-brain glucose transfer: Repression in chronic hyperglycemia. Science 1981, 214, 456–457. [Google Scholar]
- McCall, A.L.; Millington, W.R.; Wurtman, R.J. Metabolic fuel and amino acid transport into the brain in experimental diabetes mellitus. Proc. Natl. Acad. Sci. USA 1982, 79, 5406–5410. [Google Scholar]
- Matthaei, S.; Horuk, R.; Olefsky, J.M. Blood-brain glucose transfer in diabetes mellitus. Decreased number of glucose transporters at blood-brain barrier. Diabetes 1986, 35, 1181–1184. [Google Scholar]
- Mooradian, A.D.; Morin, A.M. Brain uptake of glucose in diabetes mellitus: The role of glucose transporters. Am. J. Med. Sci 1991, 301, 173–177. [Google Scholar]
- Cattelotte, J.; Andre, P.; Ouellet, M.; Bourasset, F.; Scherrmann, J.M.; Cisternino, S. In situ mouse carotid perfusion model: Glucose and cholesterol transport in the eye and brain. J. Cereb. Blood Flow Metab 2008, 28, 1449–1459. [Google Scholar]
- Shah, K.K.; Abbruscato, T.J. Function of sodium/glucose co-transporter at the blood-brain barrier in diabetes. In Fourth Annual Diabetes Drug Discovery: New Diabetes Drug Targets and Candidates; Cambridge Healthtech Institute: Boston, MA, USA, 2011. [Google Scholar]
- Badr, G.A.; Tang, J.; Ismail-Beigi, F.; Kern, T.S. Diabetes downregulates glut1 expression in the retina and its microvessels but not in the cerebral cortex or its microvessels. Diabetes 2000, 49, 1016–1021. [Google Scholar]
- Lutz, A.J.; Pardridge, W.M. Insulin therapy normalizes glut1 glucose transporter mrna but not immunoreactive transporter protein in streptozocin-diabetic rats. Metabolism 1993, 42, 939–944. [Google Scholar]
- Hou, W.K.; Xian, Y.X.; Zhang, L.; Lai, H.; Hou, X.G.; Xu, Y.X.; Yu, T.; Xu, F.Y.; Song, J.; Fu, C.L.; et al. Influence of blood glucose on the expression of glucose trans-porter proteins 1 and 3 in the brain of diabetic rats. Chin. Med. J 2007, 120, 1704–1709. [Google Scholar]
- Duelli, R.; Maurer, M.H.; Staudt, R.; Heiland, S.; Duembgen, L.; Kuschinsky, W. Increased cerebral glucose utilization and decreased glucose transporter glut1 during chronic hyperglycemia in rat brain. Brain Res 2000, 858, 338–347. [Google Scholar]
- Vannucci, S.J.; Koehler-Stec, E.M.; Li, K.; Reynolds, T.H.; Clark, R.; Simpson, I.A. Glut4 glucose transporter expression in rodent brain: Effect of diabetes. Brain Res 1998, 797, 1–11. [Google Scholar]
- Grillo, C.A.; Piroli, G.G.; Hendry, R.M.; Reagan, L.P. Insulin-stimulated translocation of glut4 to the plasma membrane in rat hippocampus is pi3-kinase dependent. Brain Res 2009, 1296, 35–45. [Google Scholar]
- Kreis, R.; Ross, B.D. Cerebral metabolic disturbances in patients with subacute and chronic diabetes mellitus: Detection with proton mr spectroscopy. Radiology 1992, 184, 123–130. [Google Scholar]
- Criego, A.B.; Tkac, I.; Kumar, A.; Thomas, W.; Gruetter, R.; Seaquist, E.R. Brain glucose concentrations in patients with type 1 diabetes and hypoglycemia unawareness. J. Neurosci. Res 2005, 79, 42–47. [Google Scholar]
- Seaquist, E.R.; Tkac, I.; Damberg, G.; Thomas, W.; Gruetter, R. Brain glucose concentrations in poorly controlled diabetes mellitus as measured by high-field magnetic resonance spectroscopy. Metabolism 2005, 54, 1008–1013. [Google Scholar]
- Fanelli, C.G.; Dence, C.S.; Markham, J.; Videen, T.O.; Paramore, D.S.; Cryer, P.E.; Powers, W.J. Blood-to-brain glucose transport and cerebral glucose metabolism are not reduced in poorly controlled type 1 diabetes. Diabetes 1998, 47, 1444–1450. [Google Scholar]
- Boyle, P.J.; Kempers, S.F.; O’Connor, A.M.; Nagy, R.J. Brain glucose uptake and unawareness of hypoglycemia in patients with insulin-dependent diabetes mellitus. N. Engl. J. Med 1995, 333, 1726–1731. [Google Scholar]
- Gutniak, M.; Blomqvist, G.; Widen, L.; Stone-Elander, S.; Hamberger, B.; Grill, V. D-[u-11c]glucose uptake and metabolism in the brain of insulin-dependent diabetic subjects. Am. J. Physiol 1990, 258, E805–E812. [Google Scholar]
- Van de Ven, K.C.; van der Graaf, M.; Tack, C.J.; Heerschap, A.; de Galan, B.E. Steady-state brain glucose concentrations during hypoglycemia in healthy humans and patients with type 1 diabetes mellitus. Diabetes 2012, 61, 1974–1977. [Google Scholar]
- Henry, P.G.; Criego, A.B.; Kumar, A.; Seaquist, E.R. Measurement of cerebral oxidative glucose consumption in patients with type 1 diabetes mellitus and hypoglycemia unawareness using (13)c nuclear magnetic resonance spectroscopy. Metabolism 2010, 59, 100–106. [Google Scholar]
- Guo, X.; Geng, M.; Du, G. Glucose transporter 1, distribution in the brain and in neural disorders: Its relationship with transport of neuroactive drugs through the blood-brain barrier. Biochem. Genet 2005, 43, 175–187. [Google Scholar]
- Liu, Y.; Liu, F.; Iqbal, K.; Grundke-Iqbal, I.; Gong, C.X. Decreased glucose transporters correlate to abnormal hyperphosphorylation of tau in alzheimer disease. FEBS Lett 2008, 582, 359–364. [Google Scholar]
- Arriagada, P.V.; Growdon, J.H.; Hedley-Whyte, E.T.; Hyman, B.T. Neurofibrillary tangles but not senile plaques parallel duration and severity of alzheimer’s disease. Neurology 1992, 42, 631–639. [Google Scholar]
- Riley, K.P.; Snowdon, D.A.; Markesbery, W.R. Alzheimer’s neurofibrillary pathology and the spectrum of cognitive function: Findings from the nun study. Ann. Neurol 2002, 51, 567–577. [Google Scholar]
- Alafuzoff, I.; Iqbal, K.; Friden, H.; Adolfsson, R.; Winblad, B. Histopathological criteria for progressive dementia disorders: Clinical-pathological correlation and classification by multivariate data analysis. Acta Neuropathol 1987, 74, 209–225. [Google Scholar]
- Grundke-Iqbal, I.; Iqbal, K.; Tung, Y.C.; Quinlan, M.; Wisniewski, H.M.; Binder, L.I. Abnormal phosphorylation of the microtubule-associated protein tau (tau) in alzheimer cytoskeletal pathology. Proc. Natl. Acad. Sci. USA 1986, 83, 4913–4917. [Google Scholar]
- Hardy, J.A.; Higgins, G.A. Alzheimer’s disease: The amyloid cascade hypothesis. Science 1992, 256, 184–185. [Google Scholar]
- Liu, Y.; Liu, F.; Grundke-Iqbal, I.; Iqbal, K.; Gong, C.X. Brain glucose transporters, o-glcnacylation and phosphorylation of tau in diabetes and alzheimer’s disease. J. Neurochem 2009, 111, 242–249. [Google Scholar]
- Hoyer, S. The brain insulin signal transduction system and sporadic (type ii) alzheimer disease: An update. J. Neural. Transm 2002, 109, 341–360. [Google Scholar]
- Hoyer, S. Oxidative energy metabolism in alzheimer brain. Studies in early-onset and late-onset cases. Mol. Chem. Neuropathol 1992, 16, 207–224. [Google Scholar]
- Hoyer, S.; Nitsch, R. Cerebral excess release of neurotransmitter amino acids subsequent to reduced cerebral glucose metabolism in early-onset dementia of alzheimer type. J. Neural. Transm 1989, 75, 227–232. [Google Scholar]
- Nitsch, R.M.; Blusztajn, J.K.; Pittas, A.G.; Slack, B.E.; Growdon, J.H.; Wurtman, R.J. Evidence for a membrane defect in alzheimer disease brain. Proc. Natl. Acad. Sci. USA 1992, 89, 1671–1675. [Google Scholar]
- Pettegrew, J.W.; Klunk, W.E.; Kanal, E.; Panchalingam, K.; McClure, R.J. Changes in brain membrane phospholipid and high-energy phosphate metabolism precede dementia. Neurobiol. Aging 1995, 16, 973–975. [Google Scholar]
- Hoyer, S.; Nitsch, R.; Oesterreich, K. Ammonia is endogenously generated in the brain in the presence of presumed and verified dementia of alzheimer type. Neurosci. Lett 1990, 117, 358–362. [Google Scholar]
- McGeer, E.G.; McGeer, P.L.; Akiyama, H.; Harrop, R. Cortical glutaminase, beta-glucuronidase and glucose utilization in alzheimer’s disease. Can. J. Neurol. Sci 1989, 16, 511–515. [Google Scholar]
- Heiss, W.D.; Szelies, B.; Kessler, J.; Herholz, K. Abnormalities of energy metabolism in alzheimer’s disease studied with pet. Ann. N. Y. Acad. Sci 1991, 640, 65–71. [Google Scholar]
- Smith, G.S.; de Leon, M.J.; George, A.E.; Kluger, A.; Volkow, N.D.; McRae, T.; Golomb, J.; Ferris, S.H.; Reisberg, B.; Ciaravino, J.; et al. Topography of cross-sectional and longitudinal glucose metabolic deficits in alzheimer’s disease. Pathophysiologic implications. Arch. Neurol 1992, 49, 1142–1150. [Google Scholar]
- Minoshima, S.; Frey, K.A.; Koeppe, R.A.; Foster, N.L.; Kuhl, D.E. A diagnostic approach in alzheimer’s disease using three-dimensional stereotactic surface projections of fluorine-18-fdg pet. J. Nucl. Med 1995, 36, 1238–1248. [Google Scholar]
- Owen, O.E.; Morgan, A.P.; Kemp, H.G.; Sullivan, J.M.; Herrera, M.G.; Cahill, G.F., Jr. Brain metabolism during fasting. J. Clin. Invest. 1967, 46, 1589–1595. [Google Scholar]
- Fehm, H.L.; Kern, W.; Peters, A. The selfish brain: Competition for energy resources. Prog. Brain Res 2006, 153, 129–140. [Google Scholar]
- Liu, F.; Shi, J.; Tanimukai, H.; Gu, J.; Gu, J.; Grundke-Iqbal, I.; Iqbal, K.; Gong, C.X. Reduced o-glcnacylation links lower brain glucose metabolism and tau pathology in alzheimer’s disease. Brain 2009, 132, 1820–1832. [Google Scholar]
- Kuhl, D.E.; Metter, E.J.; Riege, W.H.; Phelps, M.E. Effects of human aging on patterns of local cerebral glucose utilization determined by the [18f]fluorodeoxyglucose method. J. Cereb. Blood Flow Metab 1982, 2, 163–171. [Google Scholar]
- Moeller, J.R.; Ishikawa, T.; Dhawan, V.; Spetsieris, P.; Mandel, F.; Alexander, G.E.; Grady, C.; Pietrini, P.; Eidelberg, D. The metabolic topography of normal aging. J. Cereb. Blood Flow Metab 1996, 16, 385–398. [Google Scholar]
- Ivancevic, V.; Alavi, A.; Souder, E.; Mozley, P.D.; Gur, R.E.; Benard, F.; Munz, D.L. Regional cerebral glucose metabolism in healthy volunteers determined by fluordeoxyglucose positron emission tomography: Appearance and variance in the transaxial, coronal, and sagittal planes. Clin. Nucl. Med 2000, 25, 596–602. [Google Scholar]
- Hoyer, S. Glucose metabolism and insulin receptor signal transduction in alzheimer disease. Eur. J. Pharmacol 2004, 490, 115–125. [Google Scholar]
- Love, D.C.; Hanover, J.A. The hexosamine signaling pathway: Deciphering the “o-glcnac code”. Sci. STKE 2005, 2005, re13. [Google Scholar]
- Liu, F.; Iqbal, K.; Grundke-Iqbal, I.; Hart, G.W.; Gong, C.X. O-glcnacylation regulates phosphorylation of tau: A mechanism involved in alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2004, 101, 10804–10809. [Google Scholar]
- Li, X.; Lu, F.; Wang, J.Z.; Gong, C.X. Concurrent alterations of o-glcnacylation and phosphorylation of tau in mouse brains during fasting. Eur. J. Neurosci 2006, 23, 2078–2086. [Google Scholar]
- Perry, G.; Nunomura, A.; Raina, A.K.; Aliev, G.; Siedlak, S.L.; Harris, P.L.; Casadesus, G.; Petersen, R.B.; Bligh-Glover, W.; Balraj, E.; et al. A metabolic basis for alzheimer disease. Neurochem. Res 2003, 28, 1549–1552. [Google Scholar]
- Iqbal, K.; Grundke-Iqbal, I. Metabolic/signal transduction hypothesis of alzheimer’s disease and other tauopathies. Acta Neuropathol 2005, 109, 25–31. [Google Scholar]
- Mosconi, L.; Pupi, A.; De Leon, M.J. Brain glucose hypometabolism and oxidative stress in preclinical alzheimer’s disease. Ann. N. Y. Acad. Sci 2008, 1147, 180–195. [Google Scholar]
- Casadesus, G.; Moreira, P.I.; Nunomura, A.; Siedlak, S.L.; Bligh-Glover, W.; Balraj, E.; Petot, G.; Smith, M.A.; Perry, G. Indices of metabolic dysfunction and oxidative stress. Neurochem. Res 2007, 32, 717–722. [Google Scholar]
- Harik, S.I.; LaManna, J.C. Altered glucose metabolism in microvessels from patients with alzheimer’s disease. Ann. Neurol 1991, 29, 573. [Google Scholar]
- Jagust, W.J.; Seab, J.P.; Huesman, R.H.; Valk, P.E.; Mathis, C.A.; Reed, B.R.; Coxson, P.G.; Budinger, T.F. Diminished glucose transport in alzheimer’s disease: Dynamic pet studies. J. Cereb. Blood Flow Metab 1991, 11, 323–330. [Google Scholar]
- Piert, M.; Koeppe, R.A.; Giordani, B.; Berent, S.; Kuhl, D.E. Diminished glucose transport and phosphorylation in alzheimer’s disease determined by dynamic fdg-pet. J. Nucl. Med 1996, 37, 201–208. [Google Scholar]
- Friedland, R.P.; Jagust, W.J.; Huesman, R.H.; Koss, E.; Knittel, B.; Mathis, C.A.; Ober, B.A.; Mazoyer, B.M.; Budinger, T.F. Regional cerebral glucose transport and utilization in alzheimer’s disease. Neurology 1989, 39, 1427–1434. [Google Scholar]
- Kalaria, R.N.; Harik, S.I. Reduced glucose transporter at the blood-brain barrier and in cerebral cortex in alzheimer disease. J. Neurochem 1989, 53, 1083–1088. [Google Scholar]
- Kalaria, R.N.; Harik, S.I. Abnormalities of the glucose transporter at the blood-brain barrier and in brain in alzheimer’s disease. Prog. Clin. Biol. Res 1989, 317, 415–421. [Google Scholar]
- Reiman, E.M.; Uecker, A.; Gonzalez-Lima, F.; Minear, D.; Chen, K.; Callaway, N.L.; Berndt, J.D.; Games, D. Tracking alzheimer’s disease in transgenic mice using fluorodeoxyglucose autoradiography. Neuroreport 2000, 11, 987–991. [Google Scholar]
- Valla, J.; Gonzalez-Lima, F.; Reiman, E.M. Fdg autoradiography reveals developmental and pathological effects of mutant amyloid in pdapp transgenic mice. Int. J. Dev. Neurosci 2008, 26, 253–258. [Google Scholar]
- Nicholson, R.M.; Kusne, Y.; Nowak, L.A.; LaFerla, F.M.; Reiman, E.M.; Valla, J. Regional cerebral glucose uptake in the 3xtg model of alzheimer’s disease highlights common regional vulnerability across ad mouse models. Brain Res 2010, 1347, 179–185. [Google Scholar]
- Valla, J.; Schneider, L.; Reiman, E.M. Age- and transgene-related changes in regional cerebral metabolism in psapp mice. Brain Res 2006, 1116, 194–200. [Google Scholar]
- Mark, R.J.; Pang, Z.; Geddes, J.W.; Uchida, K.; Mattson, M.P. Amyloid beta-peptide impairs glucose transport in hippocampal and cortical neurons: Involvement of membrane lipid peroxidation. J. Neurosci 1997, 17, 1046–1054. [Google Scholar]
- Furst, A.J.; Lal, R.A. Amyloid-beta and glucose metabolism in alzheimer’s disease. J. Alzheimers Dis 2011, 26, 105–116. [Google Scholar]
- Simpson, I.A.; Chundu, K.R.; Davies-Hill, T.; Honer, W.G.; Davies, P. Decreased concentrations of glut1 and glut3 glucose transporters in the brains of patients with alzheimer’s disease. Ann. Neurol 1994, 35, 546–551. [Google Scholar]
- Harr, S.D.; Simonian, N.A.; Hyman, B.T. Functional alterations in alzheimer’s disease: Decreased glucose transporter 3 immunoreactivity in the perforant pathway terminal zone. J. Neuropathol. Exp. Neurol 1995, 54, 38–41. [Google Scholar]
- Mooradian, A.D.; Chung, H.C.; Shah, G.N. Glut-1 expression in the cerebra of patients with alzheimer’s disease. Neurobiol. Aging 1997, 18, 469–474. [Google Scholar]
- Sims, N.R.; Bowen, D.M.; Neary, D.; Davison, A.N. Metabolic processes in alzheimer’s disease: Adenine nucleotide content and production of 14co2 from [u-14c]glucose in vitro in human neocortex. J. Neurochem 1983, 41, 1329–1334. [Google Scholar]
- Sims, N.R.; Bowen, D.M.; Davison, A.N. [14c]acetylcholine synthesis and [14c]carbon dioxide production from [u-14c]glucose by tissue prisms from human neocortex. Biochem. J. 1981, 196, 867–876. [Google Scholar]
- Sorbi, S.; Bird, E.D.; Blass, J.P. Decreased pyruvate dehydrogenase complex activity in huntington and alzheimer brain. Ann. Neurol 1983, 13, 72–78. [Google Scholar]
- Bowen, D.M.; White, P.; Spillane, J.A.; Goodhardt, M.J.; Curzon, G.; Iwangoff, P.; Meier-Ruge, W.; Davison, A.N. Accelerated ageing or selective neuronal loss as an important cause of dementia? Lancet 1979, 1, 11–14. [Google Scholar]
- Watson, G.S.; Craft, S. Modulation of memory by insulin and glucose: Neuropsychological observations in alzheimer’s disease. Eur. J. Pharmacol 2004, 490, 97–113. [Google Scholar]
- Schubert, M.; Gautam, D.; Surjo, D.; Ueki, K.; Baudler, S.; Schubert, D.; Kondo, T.; Alber, J.; Galldiks, N.; Kustermann, E.; et al. Role for neuronal insulin resistance in neurodegenerative diseases. Proc. Natl. Acad. Sci. USA 2004, 101, 3100–3105. [Google Scholar]
- Hoyer, S. Is sporadic alzheimer disease the brain type of non-insulin dependent diabetes mellitus? A challenging hypothesis. J. Neural. Transm 1998, 105, 415–422. [Google Scholar]
- Frolich, L.; Blum-Degen, D.; Bernstein, H.G.; Engelsberger, S.; Humrich, J.; Laufer, S.; Muschner, D.; Thalheimer, A.; Turk, A.; Hoyer, S.; et al. Brain insulin and insulin receptors in aging and sporadic alzheimer’s disease. J. Neural. Transm 1998, 105, 423–438. [Google Scholar]
- Havrankova, J.; Roth, J.; Brownstein, M. Insulin receptors are widely distributed in the central nervous system of the rat. Nature 1978, 272, 827–829. [Google Scholar]
- Van Houten, M.; Posner, B.I.; Kopriwa, B.M.; Brawer, J.R. Insulin-binding sites in the rat brain: In vivo localization to the circumventricular organs by quantitative radioautography. Endocrinology 1979, 105, 666–673. [Google Scholar]
- Liu, Y.; Liu, F.; Grundke-Iqbal, I.; Iqbal, K.; Gong, C.X. Deficient brain insulin signalling pathway in alzheimer’s disease and diabetes. J. Pathol 2011, 225, 54–62. [Google Scholar]
- Gerozissis, K. Brain insulin, energy and glucose homeostasis; genes, environment and metabolic pathologies. Eur. J. Pharmacol 2008, 585, 38–49. [Google Scholar]
- Cardoso, S.; Correia, S.; Santos, R.X.; Carvalho, C.; Santos, M.S.; Oliveira, C.R.; Perry, G.; Smith, M.A.; Zhu, X.; Moreira, P.I. Insulin is a two-edged knife on the brain. J. Alzheimers Dis 2009, 18, 483–507. [Google Scholar]
- Schechter, R.; Abboud, M. Neuronal synthesized insulin roles on neural differentiation within fetal rat neuron cell cultures. Brain Res. Dev. Brain Res 2001, 127, 41–49. [Google Scholar]
- Schechter, R.; Beju, D.; Gaffney, T.; Schaefer, F.; Whetsell, L. Preproinsulin i and ii mrnas and insulin electron microscopic immunoreaction are present within the rat fetal nervous system. Brain Res 1996, 736, 16–27. [Google Scholar]
- Banks, W.A. The source of cerebral insulin. Eur. J. Pharmacol 2004, 490, 5–12. [Google Scholar]
- Watson, G.S.; Cholerton, B.A.; Reger, M.A.; Baker, L.D.; Plymate, S.R.; Asthana, S.; Fishel, M.A.; Kulstad, J.J.; Green, P.S.; Cook, D.G.; et al. Preserved cognition in patients with early alzheimer disease and amnestic mild cognitive impairment during treatment with rosiglitazone: A preliminary study. Am. J. Geriatr. Psychiatry 2005, 13, 950–958. [Google Scholar]
- Reger, M.A.; Watson, G.S.; Green, P.S.; Baker, L.D.; Cholerton, B.; Fishel, M.A.; Plymate, S.R.; Cherrier, M.M.; Schellenberg, G.D.; Frey, W.H., II; et al. Intranasal insulin administration dose-dependently modulates verbal memory and plasma amyloid-beta in memory-impaired older adults. J. Alzheimers Dis 2008, 13, 323–331. [Google Scholar]
- Reger, M.A.; Watson, G.S.; Frey, W.H., II; Baker, L.D.; Cholerton, B.; Keeling, M.L.; Belongia, D.A.; Fishel, M.A.; Plymate, S.R.; Schellenberg, G.D.; et al. Effects of intranasal insulin on cognition in memory-impaired older adults: Modulation by apoe genotype. Neurobiol. Aging 2006, 27, 451–458. [Google Scholar]
- Pedersen, W.A.; McMillan, P.J.; Kulstad, J.J.; Leverenz, J.B.; Craft, S.; Haynatzki, G.R. Rosiglitazone attenuates learning and memory deficits in tg2576 alzheimer mice. Exp. Neurol 2006, 199, 265–273. [Google Scholar]
- Francis, G.J.; Martinez, J.A.; Liu, W.Q.; Xu, K.; Ayer, A.; Fine, J.; Tuor, U.I.; Glazner, G.; Hanson, L.R.; Frey, W.H., II; et al. Intranasal insulin prevents cognitive decline, cerebral atrophy and white matter changes in murine type i diabetic encephalopathy. Brain 2008, 131, 3311–3334. [Google Scholar]

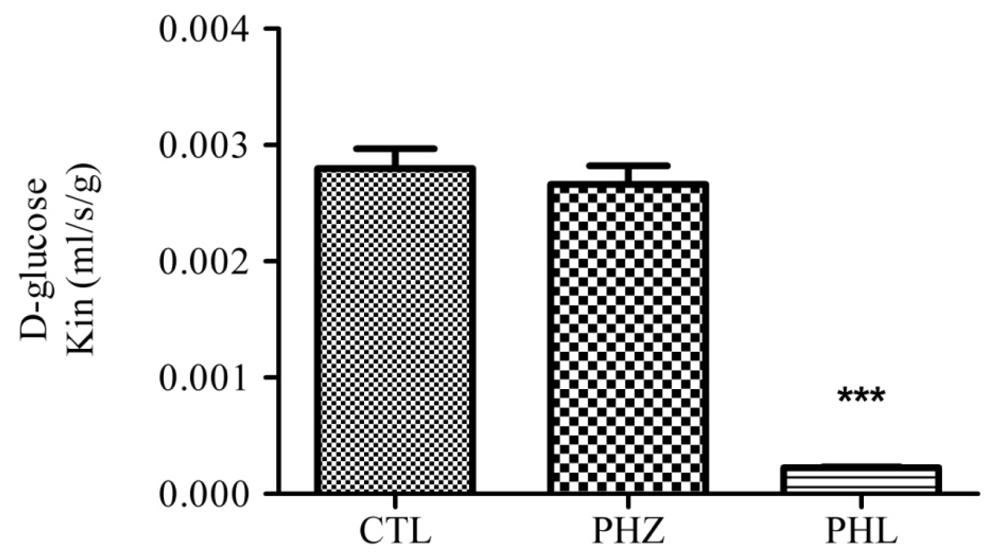
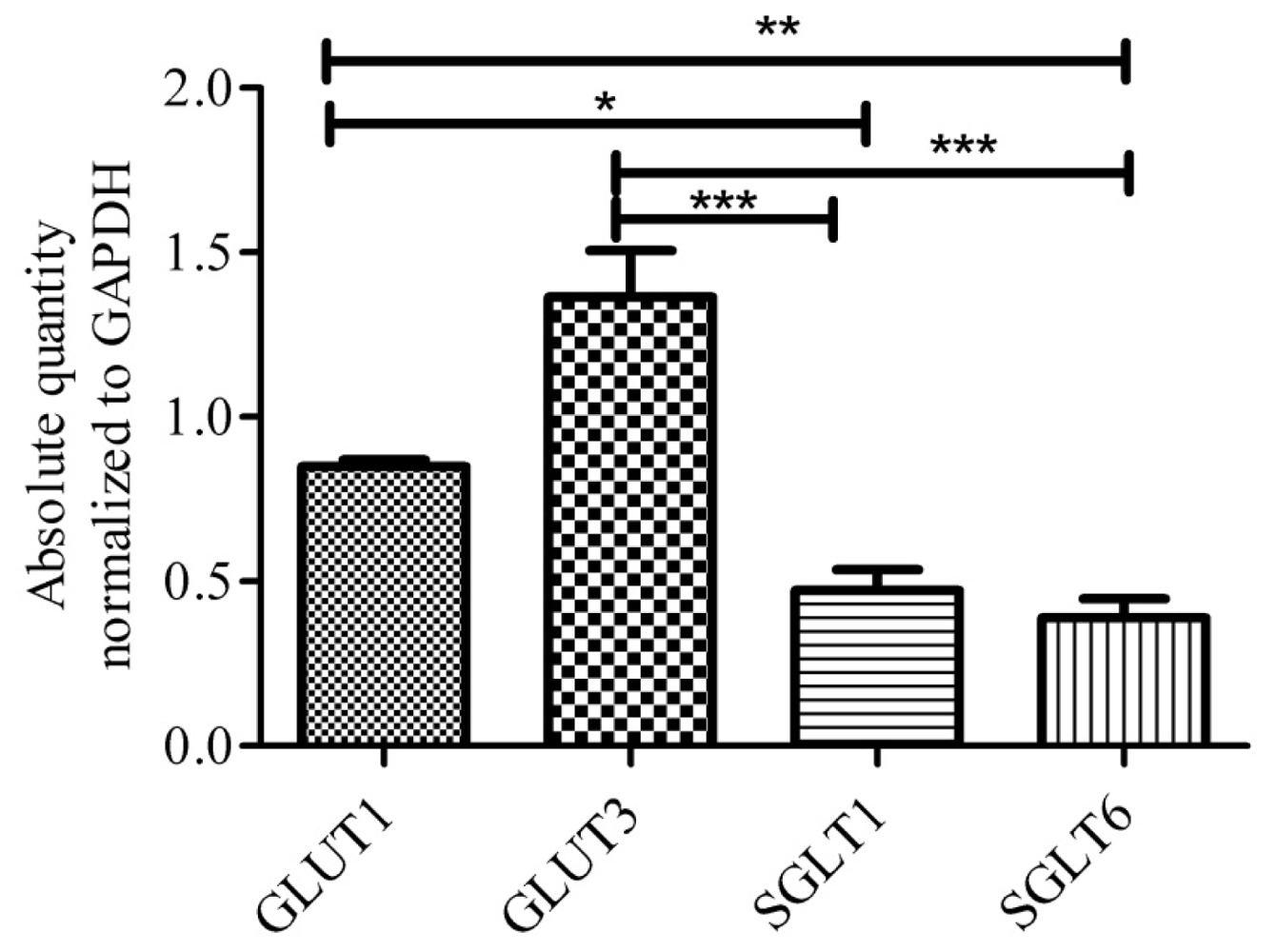

{kind=link}
{kind=link}
{kind=link}
{kind=link}
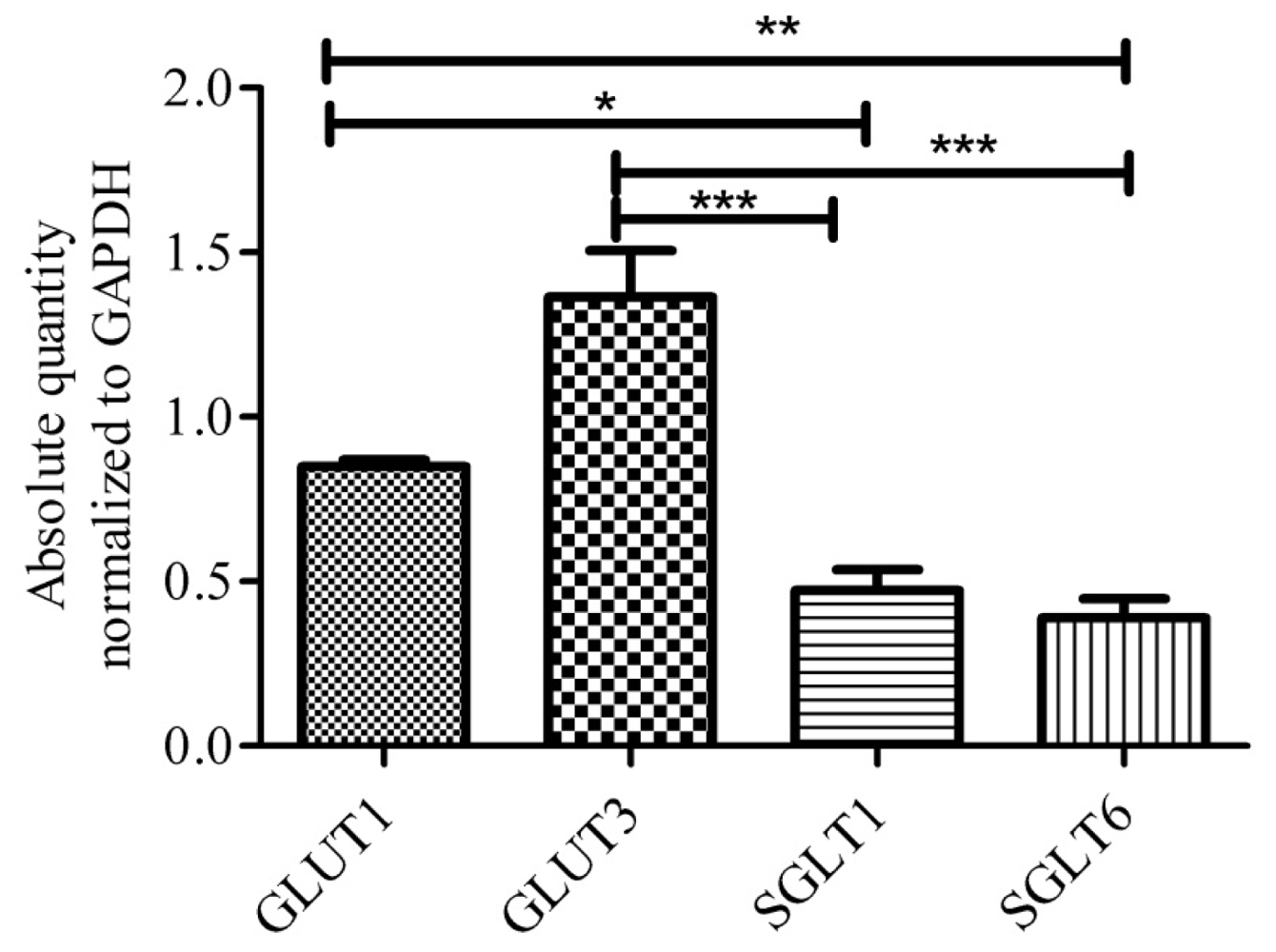{kind=link}
| Type | Protein (gene) | Sites expressed | Substrate/transports |
|---|---|---|---|
| Facilitative/Sodium-independent | GLUT1 (SLC2A1) | Brain endothelial and epithelial-like brain barriers, glial cells, blood-tissue barriers, eye, pheripheral nerves, placenta, lactating mammary gland (Ubiquitous distribution in most mammalian cells) | >>Glucose, galactose, mannose, glucosamine, ascorbic acid |
| GLUT2 (SLC2A2) | Kidney, small intestine (epithelial cells), liver, pancreas (islets), brain (astrocytes) | Mannose, galactose, fructose, glucose, glucosamine | |
| GLUT3 (SLC2A3) | Neurons, testis, placenta, brain endothelial cells? | Glucose, galactose, mannose, xylose, dehydroascorbic acid | |
| GLUT4 (SLC2A4) (Insulin-sensitive) | Brown and white adipose tissue, muscle (skeletal), fat, heart (myocardium), hippocampal neurons, cerebellar neurons | Glucose, dehydroascorbic acid, glucosamine | |
| GLUT5 (SLC2A5) | Intestine (jejunum), kidney, testis, brain microglia | Fructose | |
| GLUT6 (SLC2A6) | Brain, peripheral and spleen (leukocytes) | Glucose | |
| GLUT7 (SLC2A7) | Small intestine (mainly in brush border membrane-enterocytes), colon, testis, prostate, liver (associated with endoplasmic reticulum) | >Fructose, glucose | |
| GLUT8 (SLC2A8) (Insulin-responsive?) | Blastocytes, testis, brain (neurons), muscle, adipocytes, mammary gland? | Glucose | |
| GLUT9 (SLC2A9) | Liver, kidney (proximal tubule of epithelial cells), placenta? | Glucose, urate | |
| GLUT10 (SLC2A10) | Liver, pancreas, heart, lung, brain, skeletal muscle, placenta | Glucose, galactose | |
| GLUT11 (SLC2A11) | Iso-form A: Heart, skeletal muscle, kidney | Fructose, glucose | |
| Iso-form B: Placenta, adipose tissue, kidney | |||
| Iso-form C: Adipose tissue, heart, skeletal muscle, pancreas | |||
| GLUT12 (SLC2A12) (Insulin-sensitive?) | Heart, skeletal muscle, fat, prostrate, lactating mammary gland ?, spleen ?, breast cancer (Ductal cell carcinoma) tissue | Glucose | |
| HMIT (SLC2A13) (co-transporter) | Brain (neurons intracellular vesicles) | H+/myo-inositol | |
| Sodium-Glucose Co-transporter/Sodium-dependent | SGLT1 (SLC5A1) | Small Intestine (brush-border membrane), trachea, kidney, heart, brain (cortical, pyramidal and purkinje neuronal cells), testis, prostrate, mammary gland | >Glucose, ≥ galactose, water |
| SGLT2 (SLC5A2) | Kidney (cortex/proximal tubules), brain, liver, thyroid, muscle, heart | Glucose, galactose | |
| SGLT3 (SLC5A4/SAAT1) | (Gluco-sensor) Small intestine, testis, uterus, lung, brain, thyroid, kidney | Glucose, Na+ (H+) | |
| SGLT4 (SLC5A9) | Intestine, kidney, liver, brain, lung, trachea, uterus, pancreas | Glucose, mannose, fructose | |
| SGLT5 (SLCA10) | Kidney (cortex and medulla) | Glucose, galactose | |
| SGLT6 (SLCA11/KST1/SMIT2) | Brain (neurons), spinal cord, small intestine (ileum and jejunum), Kidney (cortex and medulla), skeletal muscle | Myo-inositol, glucose | |
| SMIT (SLC5A3) | Kidney (medulla), choroid plexus blood vessel, thyroid gland, pineal gland, dorsal root ganglion, testes | Myo-inositol, glucose | |
| NIS (SLC5A5) (sympoter) | Thyroid, breast, colon, ovary | Iodine (Na+/I−) | |
| SMVT (SLC5A6) | Brain, heart, kidney, lung, placenta | Multivitamins (Biotin, lipoate, pantothenate) | |
| CHT (SLC5A7/CHT1) | Spinal cord and medulla (intracellular vesicles) | Choline | |
© 2012 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Shah, K.; DeSilva, S.; Abbruscato, T. The Role of Glucose Transporters in Brain Disease: Diabetes and Alzheimer’s Disease. Int. J. Mol. Sci. 2012, 13, 12629-12655. https://doi.org/10.3390/ijms131012629
Shah K, DeSilva S, Abbruscato T. The Role of Glucose Transporters in Brain Disease: Diabetes and Alzheimer’s Disease. International Journal of Molecular Sciences. 2012; 13(10):12629-12655. https://doi.org/10.3390/ijms131012629
Chicago/Turabian StyleShah, Kaushik, Shanal DeSilva, and Thomas Abbruscato. 2012. "The Role of Glucose Transporters in Brain Disease: Diabetes and Alzheimer’s Disease" International Journal of Molecular Sciences 13, no. 10: 12629-12655. https://doi.org/10.3390/ijms131012629
APA StyleShah, K., DeSilva, S., & Abbruscato, T. (2012). The Role of Glucose Transporters in Brain Disease: Diabetes and Alzheimer’s Disease. International Journal of Molecular Sciences, 13(10), 12629-12655. https://doi.org/10.3390/ijms131012629