Helicobacter pylori Disrupts Host Cell Membranes, Initiating a Repair Response and Cell Proliferation


Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
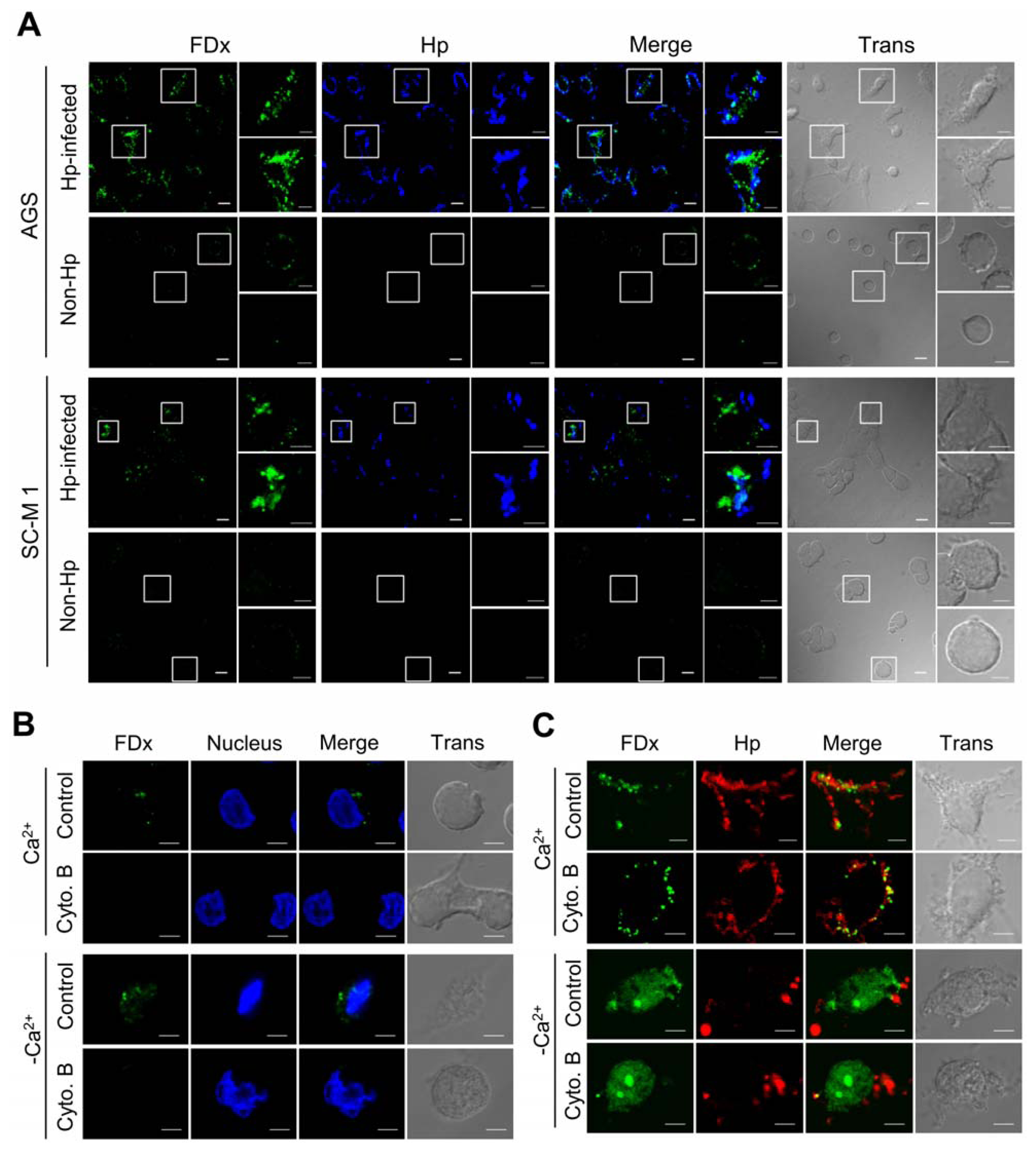
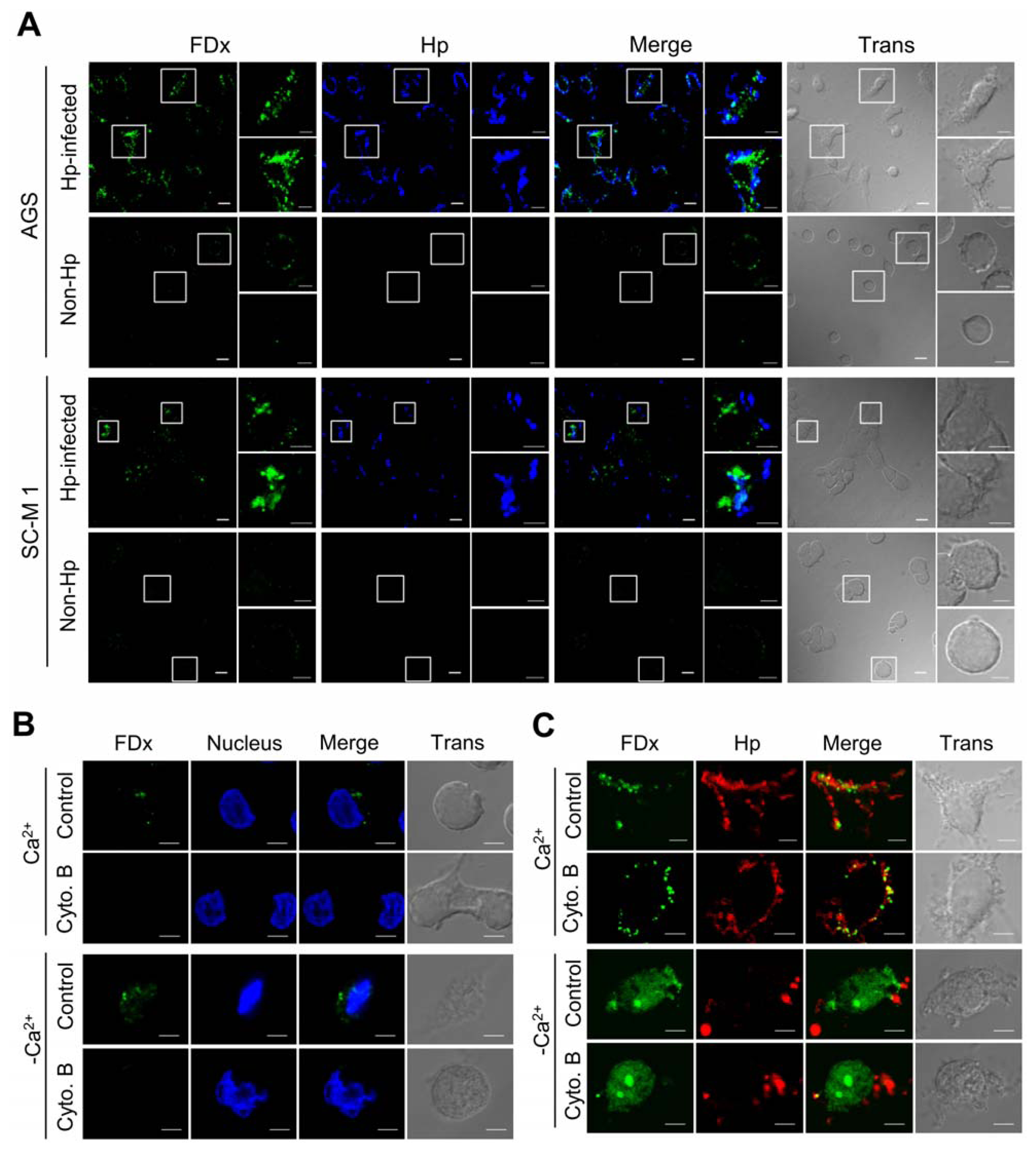
2.1. H. pylori Infection Causes Plasma Membrane Microinjury
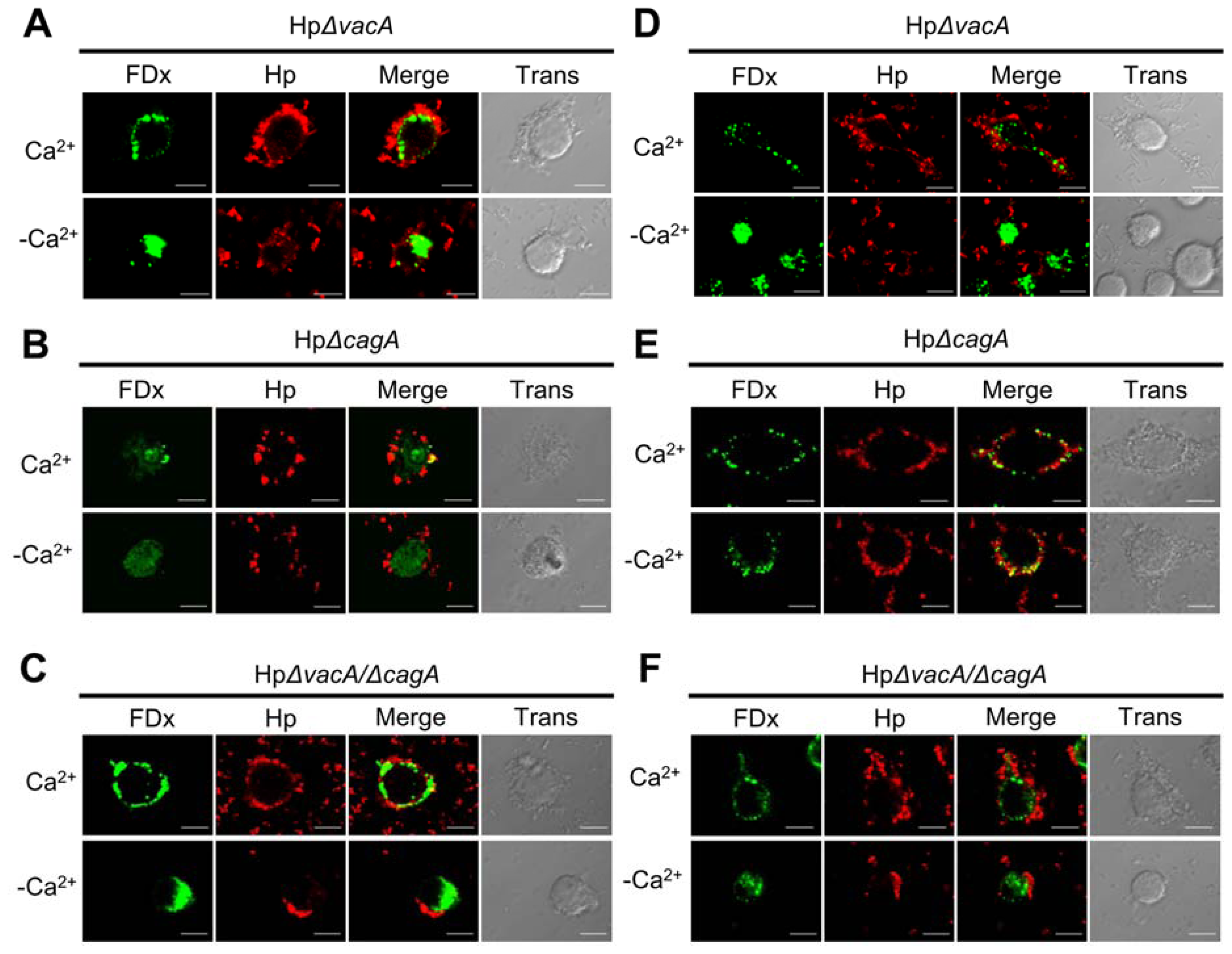
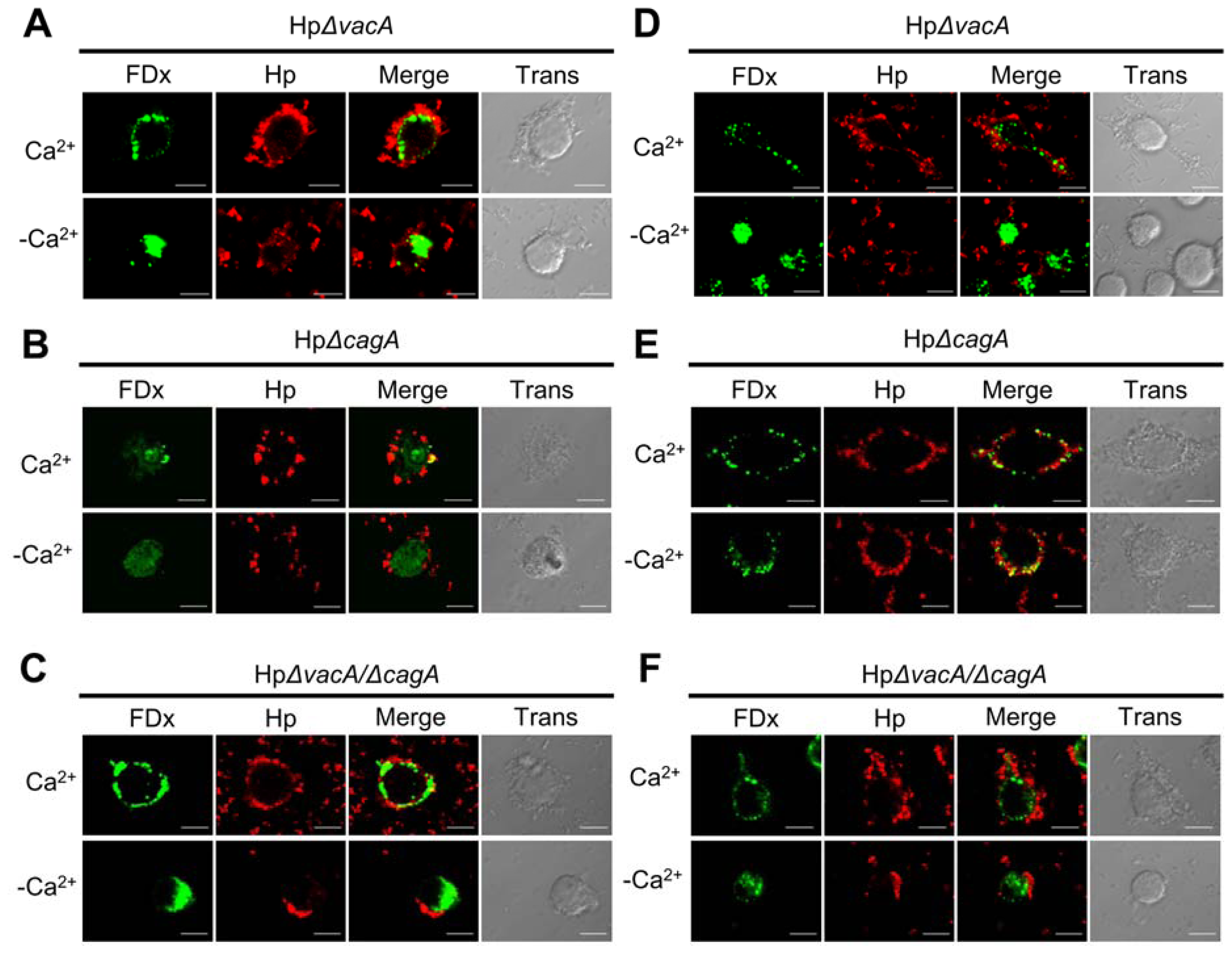
2.2. Membrane Injury by H. pylori Infection Is Independent of VacA and CagA
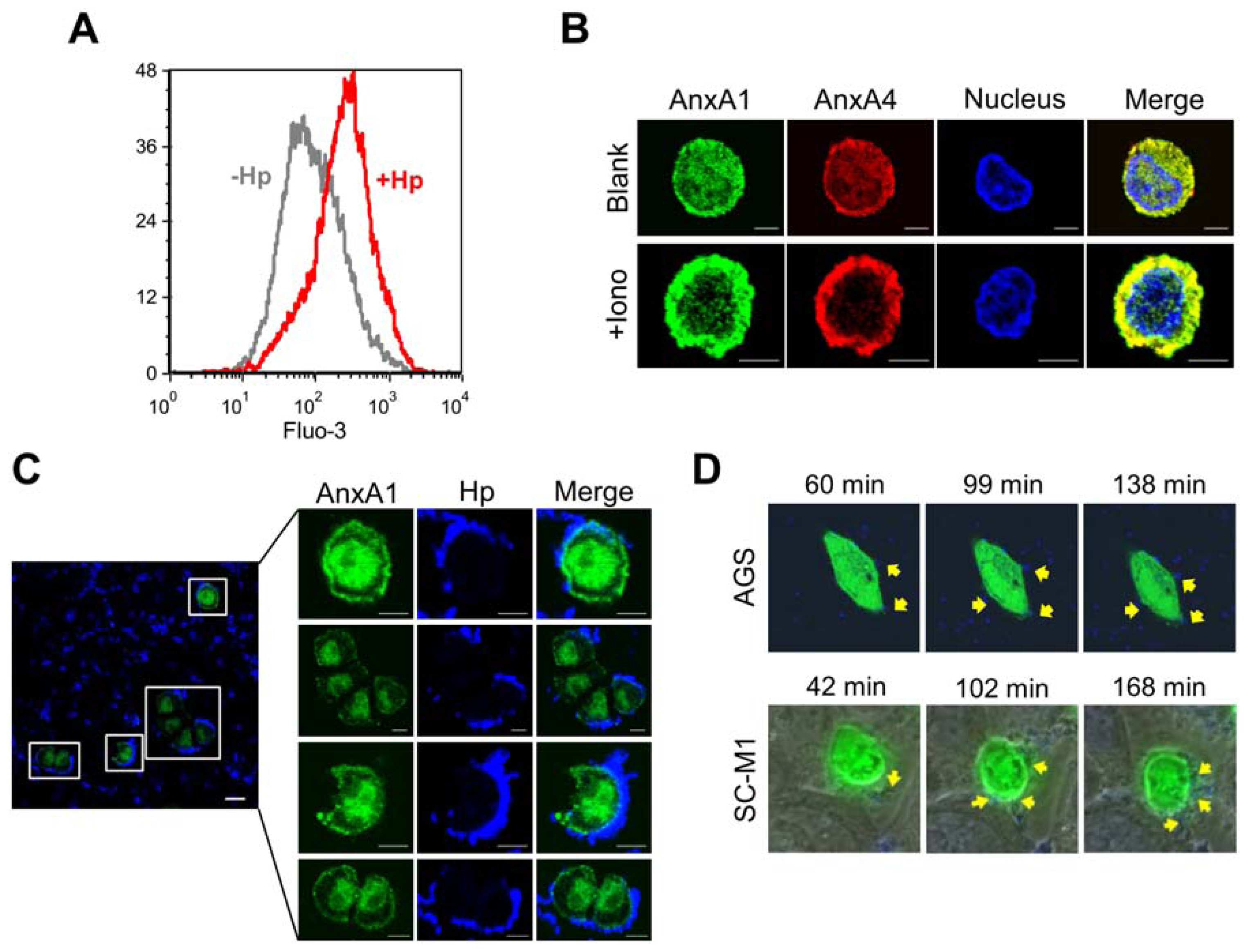
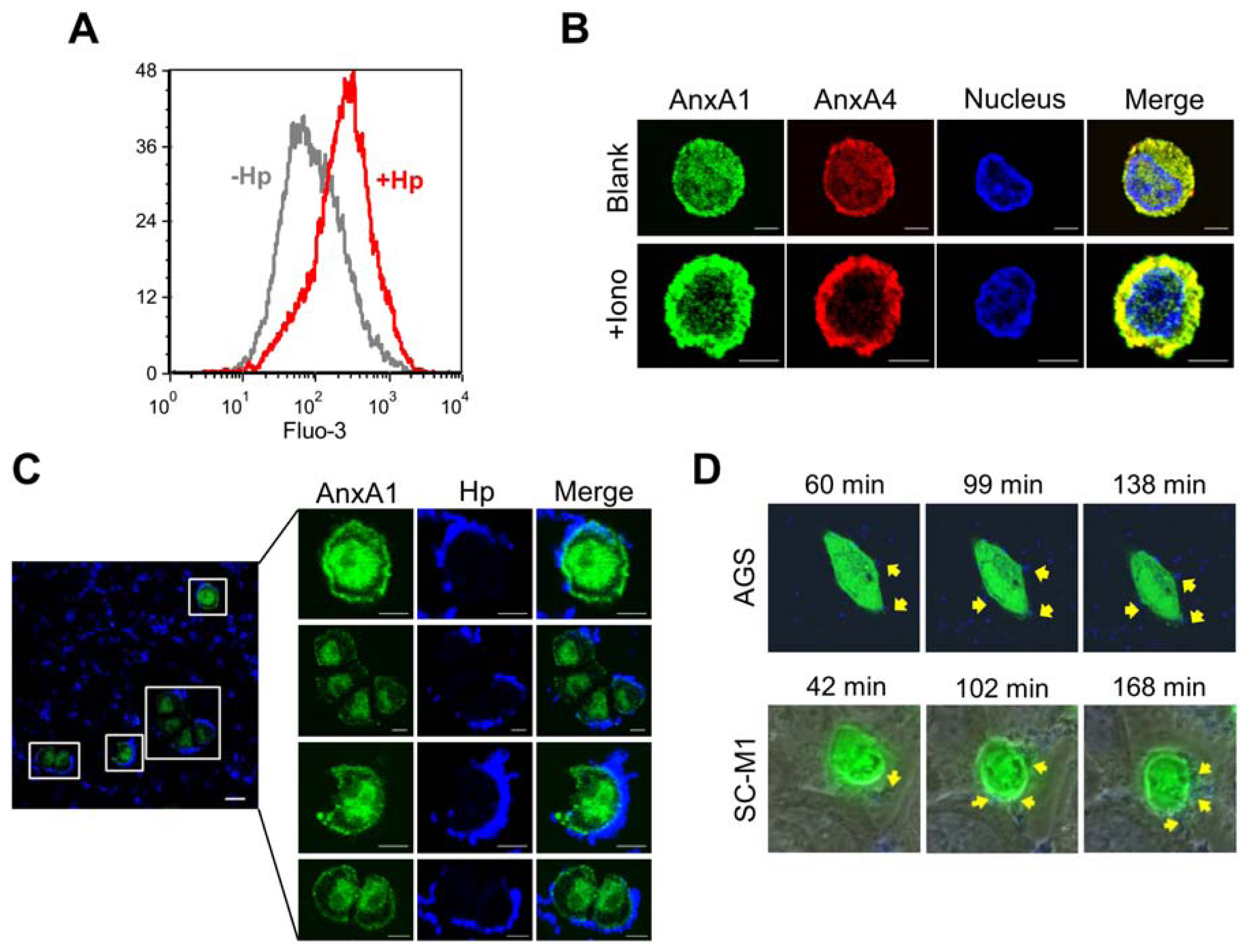
2.3. H. pylori Infection Causes Intracellular Ca2+ Increase and AnxA1 Translocation
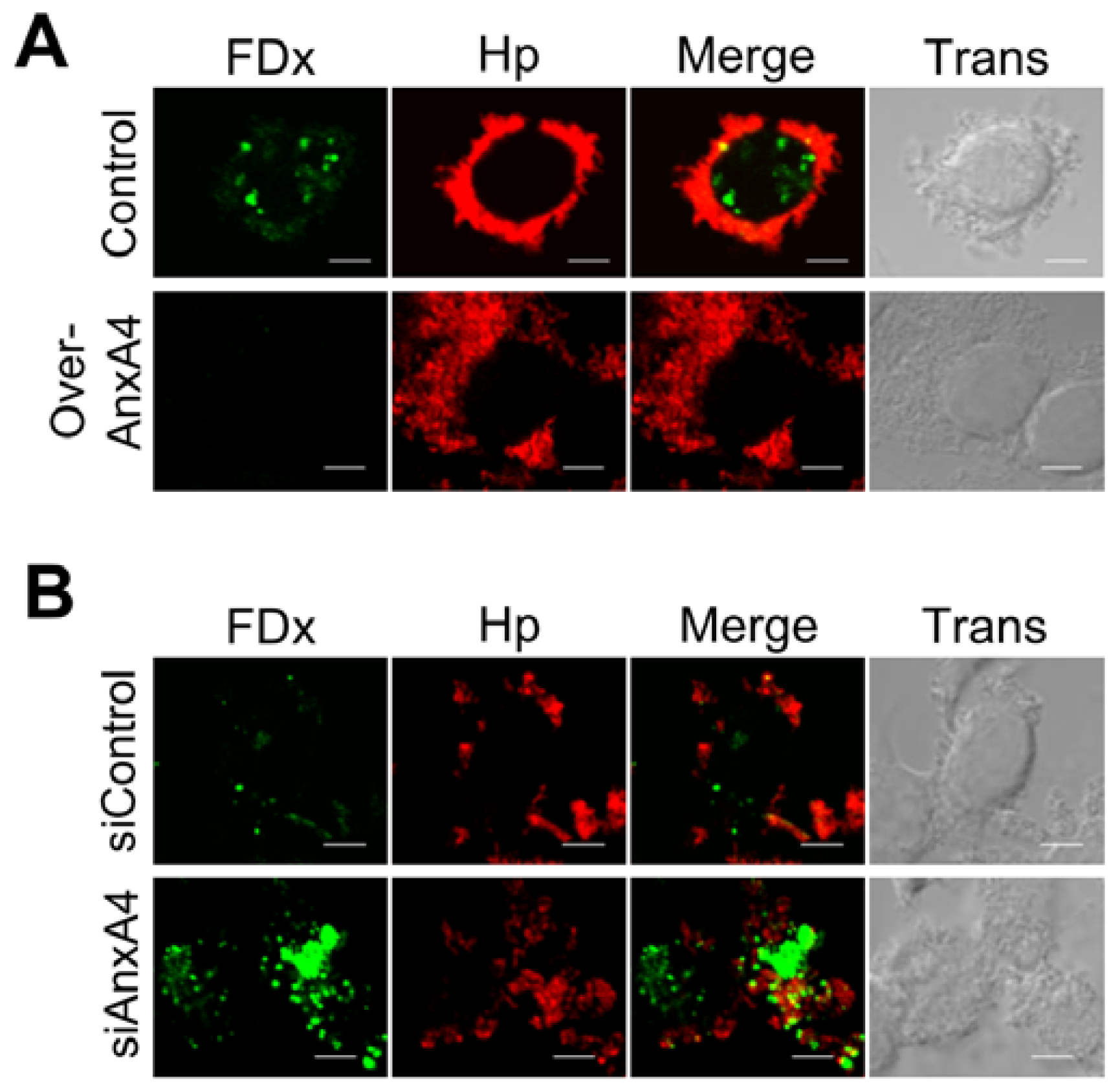
2.4. AnxA4 also Localizes to the Plasma Membrane after H. pylori Infection
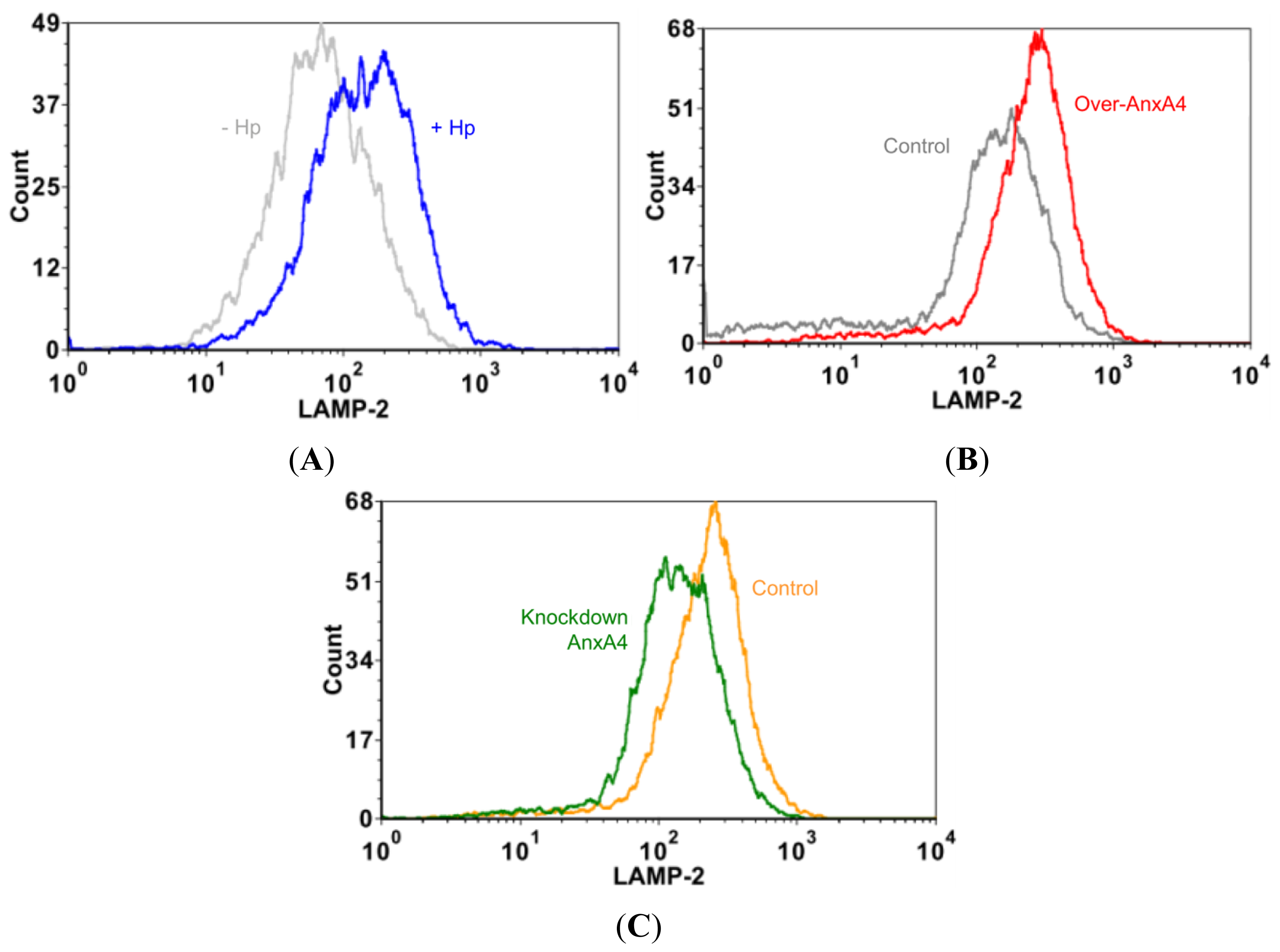
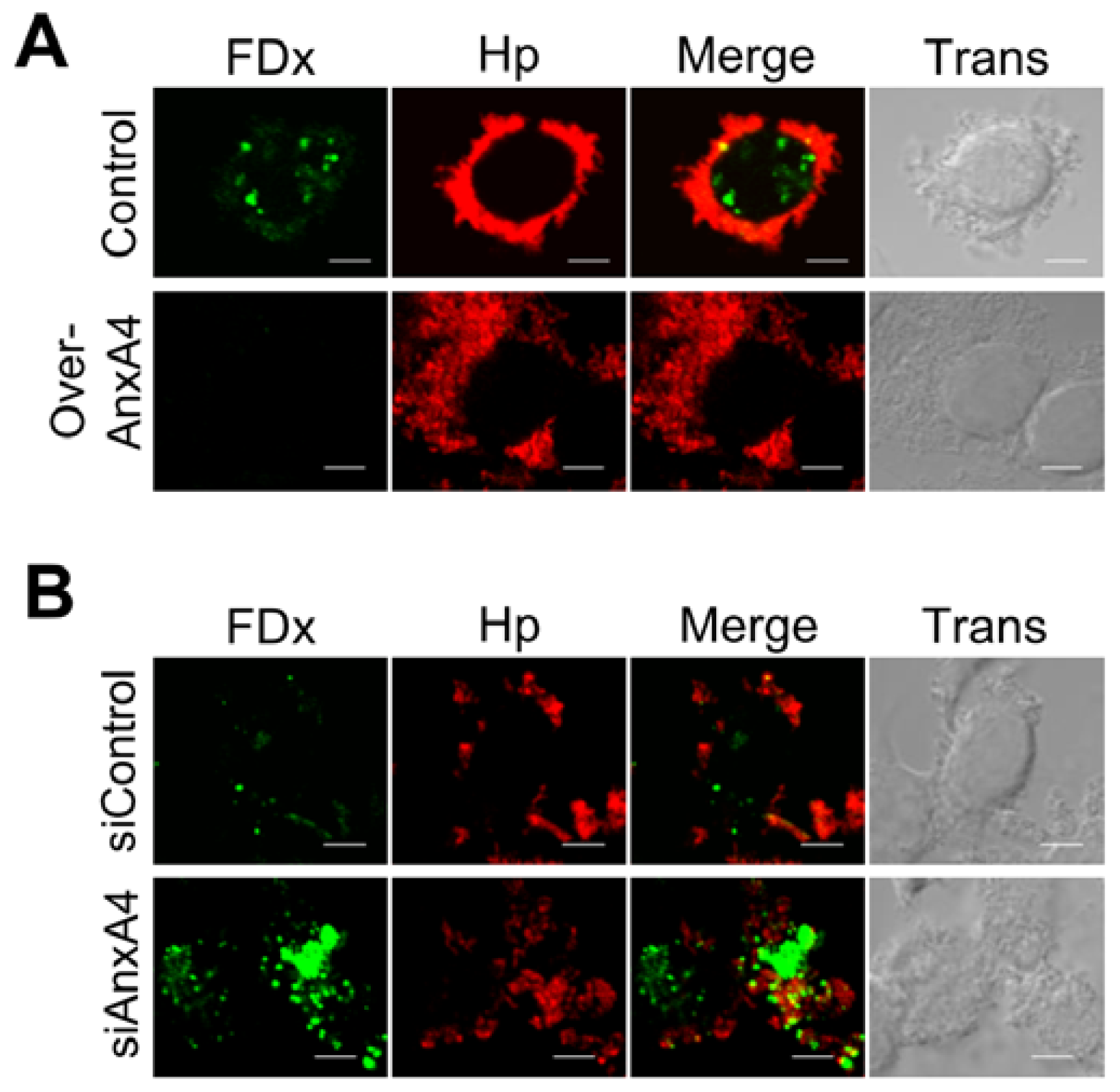
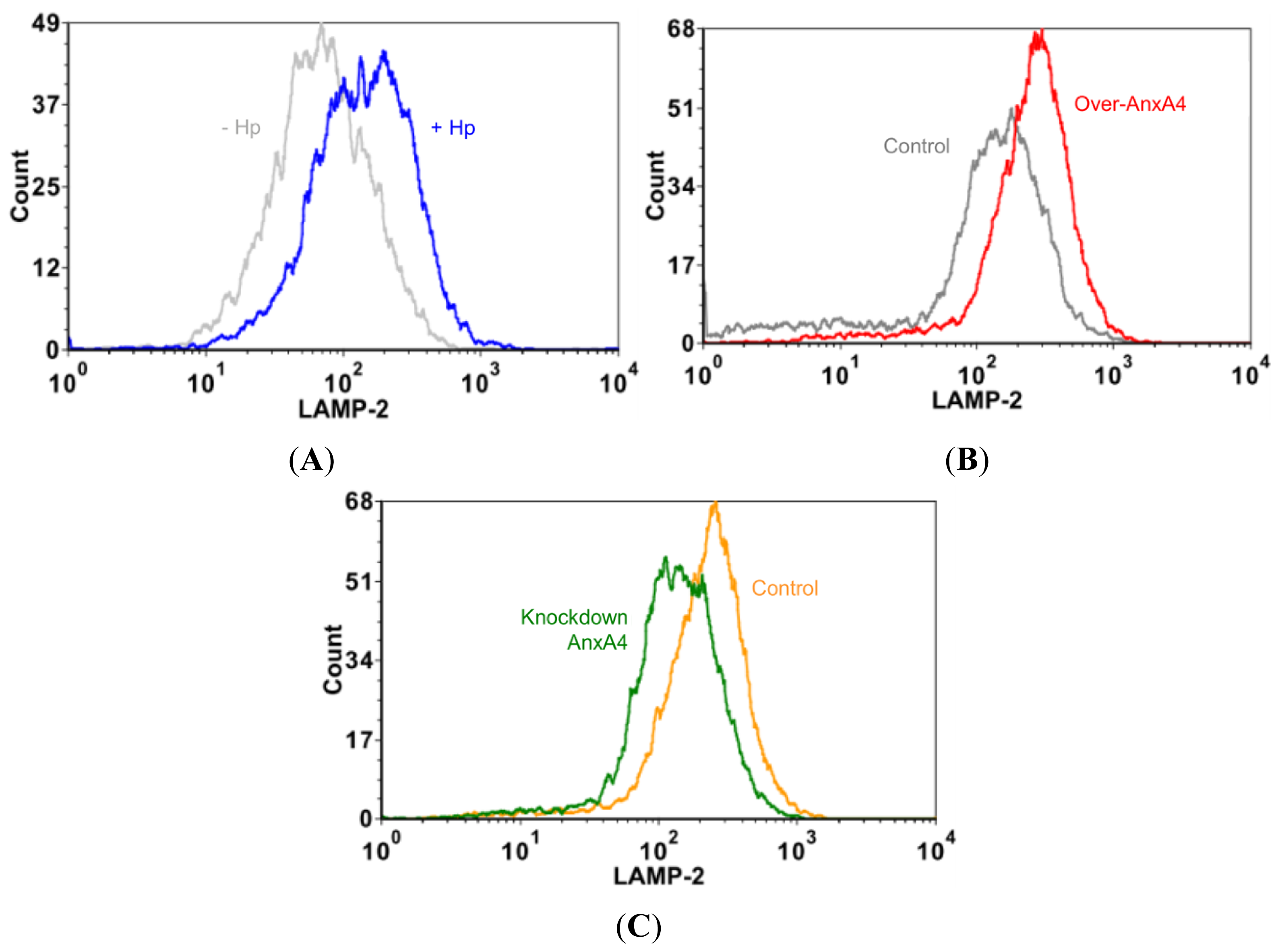
2.5. AnxA4 Inhibits Plasma Membrane Lesions Caused by H. pylori
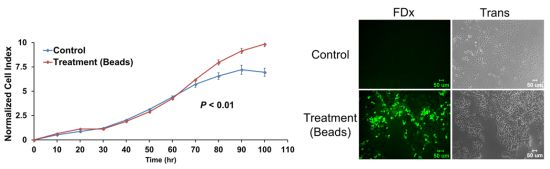
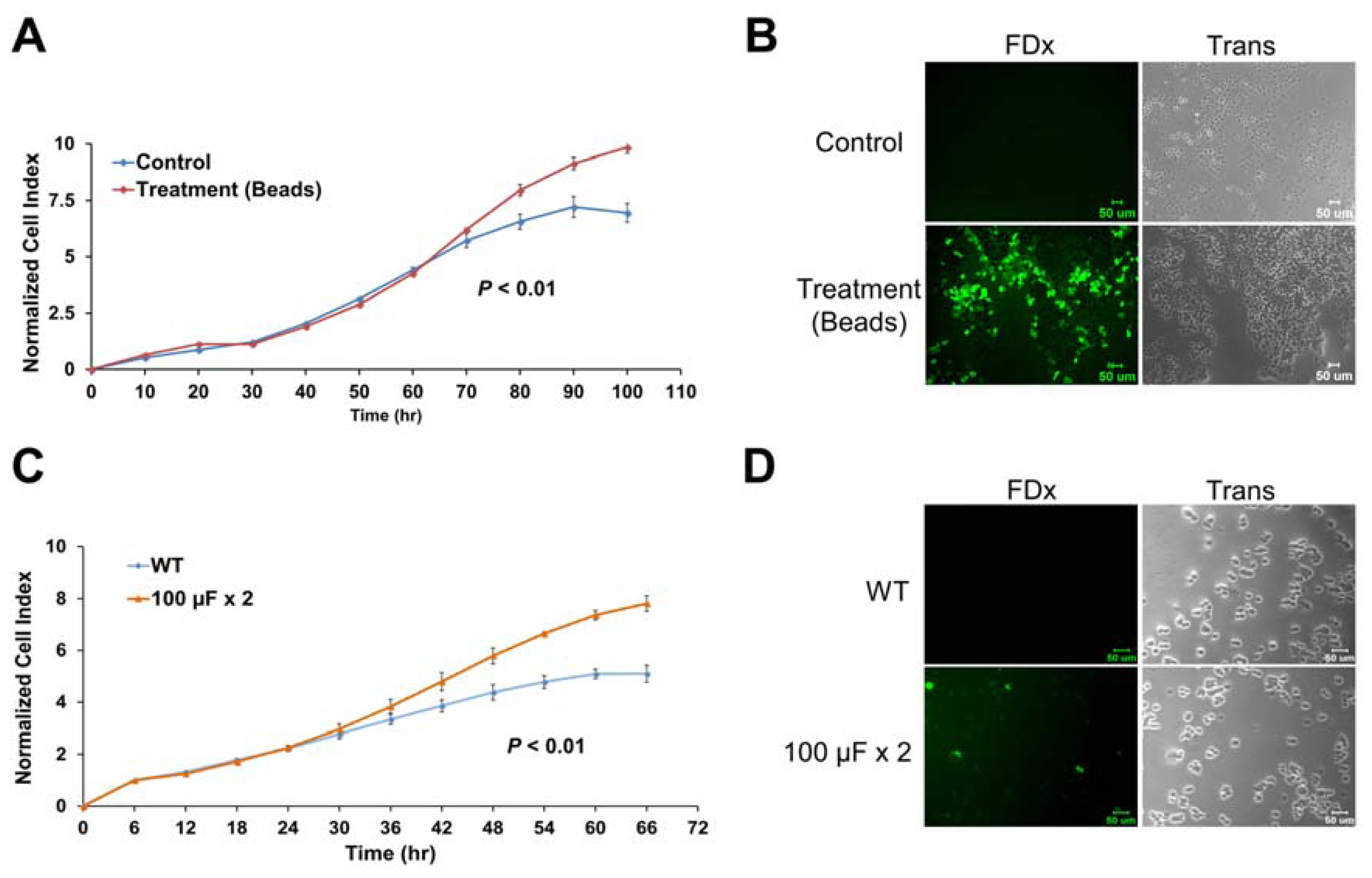
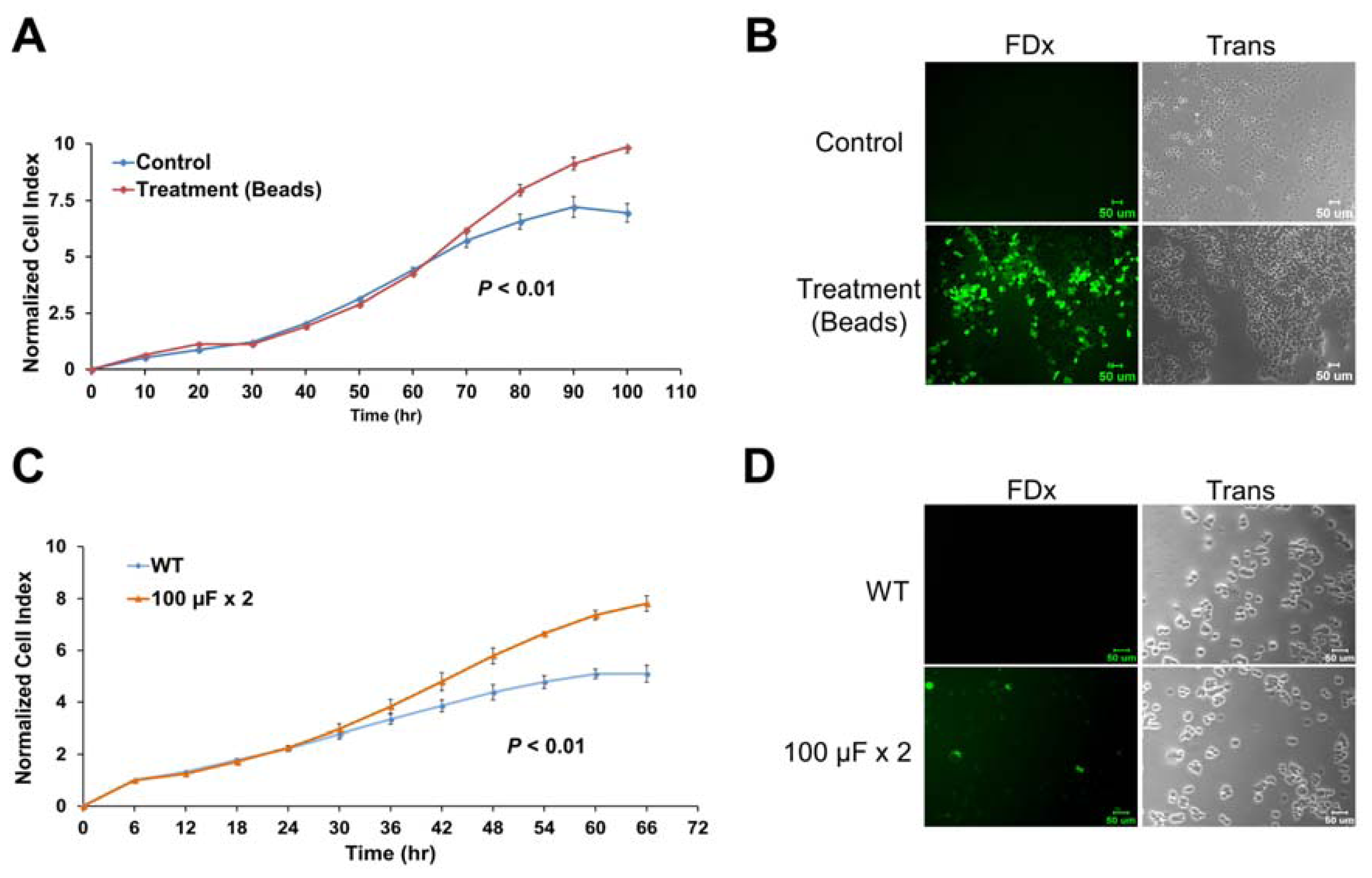
2.6. Membrane Disruption Promotes Cell Proliferation
2.7. Discussion
3. Experimental Section
3.1. Bacterial Strains and Culture Conditions
3.2. Cell Lines and Culture Conditions
3.3. Construction of Insertion Mutants of H. pylori
3.4. Plasmids and Transfections
3.5. Antibodies
3.6. Live Cell Imaging
3.7. Confocal Microscopy
3.8. Flow Cytometry
3.9. Cell Proliferation Assay
3.10. Statistical Analysis
4. Conclusions
Supplementary Materials
ijms-13-10176-s001.pdfAcknowledgments
References
- Divangahi, M.; Chen, M.; Gan, H.; Desjardins, D.; Hickman, T.T.; Lee, D.M.; Fortune, S.; Behar, S.M.; Remold, H.G. Mycobacterium tuberculosis evades macrophage defenses by inhibiting plasma membrane repair. Nat. Immunol 2009, 10, 899–906. [Google Scholar]
- McNeil, P.L.; Kirchhausen, T. An emergency response team for membrane repair. Nat. Rev. Mol. Cell Biol 2005, 6, 499–505. [Google Scholar]
- Roy, D.; Liston, D.R.; Idone, V.J.; Di, A.; Nelson, D.J.; Pujol, C.; Bliska, J.B.; Chakrabarti, S.; Andrews, N.W. A process for controlling intracellular bacterial infections induced by membrane injury. Science 2004, 304, 1515–1518. [Google Scholar]
- Bi, G.Q.; Alderton, J.M.; Steinhardt, R.A. Calcium-regulated exocytosis is required for cell membrane resealing. J. Cell Biol 1995, 131, 1747–1758. [Google Scholar]
- McNeil, A.K.; Rescher, U.; Gerke, V.; McNeil, P.L. Requirement for annexin A1 in plasma membrane repair. J. Biol. Chem 2006, 281, 35202–35207. [Google Scholar]
- Babbin, B.A.; Laukoetter, M.G.; Nava, P.; Koch, S.; Lee, W.Y.; Capaldo, C.T.; Peatman, E.; Severson, E.A.; Flower, R.J.; Perretti, M.; et al. Annexin A1 regulates intestinal mucosal injury, inflammation, and repair. J. Immunol 2008, 181, 5035–5044. [Google Scholar]
- Martin, G.R.; Perretti, M.; Flower, R.J.; Wallace, J.L. Annexin-1 modulates repair of gastric mucosal injury. Am. J. Physiol. Gastrointest Liver Physiol 2008, 294, G764–G769. [Google Scholar]
- Bouter, A.; Gounou, C.; Berat, R.; Tan, S.; Gallois, B.; Granier, T.; d’Estaintot, B.L.; Poschl, E.; Brachvogel, B.; Brisson, A.R. Annexin-A5 assembled into two-dimensional arrays promotes cell membrane repair. Nat. Commun 2011, 2. [Google Scholar] [CrossRef] [Green Version]
- Polk, D.B.; Peek, R.M., Jr. Helicobacter pylori: Gastric cancer and beyond. Nat. Rev. Cancer 2010, 10, 403–414. [Google Scholar]
- Blaser, M.J. Linking Helicobacter pylori to gastric cancer. Nat. Med 2000, 6, 376–377. [Google Scholar]
- Yamaoka, Y. Mechanisms of disease: Helicobacter pylori virulence factors. Nat. Rev. Gastroenterol. Hepatol 2010, 7, 629–641. [Google Scholar]
- Lai, Y.P.; Yang, J.C.; Lin, T.Z.; Lin, J.T.; Wang, J.T. Helicobacter pylori infection and CagA protein translocation in human primary gastric epithelial cell culture. Helicobacter 2006, 11, 451–459. [Google Scholar]
- Beatty, W.L. Lysosome repair enables host cell survival and bacterial persistence following Chlamydia trachomatis infection. Cell. Microbiol 2007, 9, 2141–2152. [Google Scholar]
- McNeil, P.L.; Ito, S. Gastrointestinal cell plasma membrane wounding and resealing in vivo. Gastroenterology 1989, 96, 1238–1248. [Google Scholar]
- Theodoropoulos, P.A.; Gravanis, A.; Tsapara, A.; Margioris, A.N.; Papadogiorgaki, E.; Galanopoulos, V.; Stournaras, C. Cytochalasin B may shorten actin filaments by a mechanism independent of barbed end capping. Biochem. Pharmacol 1994, 47, 1875–1881. [Google Scholar]
- Papini, E.; de Bernard, M.; Milia, E.; Bugnoli, M.; Zerial, M.; Rappuoli, R.; Montecucco, C. Cellular vacuoles induced by Helicobacter pylorioriginate from late endosomal compartments. Proc. Natl. Acad. Sci. USA 1994, 91, 9720–9724. [Google Scholar]
- Marlink, K.L.; Bacon, K.D.; Sheppard, B.C.; Ashktorab, H.; Smoot, D.T.; Cover, T.L.; Deveney, C.W.; Rutten, M.J. Effects of Helicobacter pylori on intracellular Ca2+ signaling in normal human gastric mucous epithelial cells. Am. J. Physiol. Gastrointest. Liver Physiol 2003, 285, G163–G176. [Google Scholar]
- Gerke, V.; Creutz, C.E.; Moss, S.E. Annexins: Linking Ca2+ signalling to membrane dynamics. Nat. Rev. Mol. Cell Biol 2005, 6, 449–461. [Google Scholar]
- Draeger, A.; Monastyrskaya, K.; Babiychuk, E.B. Plasma membrane repair and cellular damage control: The annexin survival kit. Biochem. Pharmacol 2011, 81, 703–712. [Google Scholar]
- Lennon, N.J.; Kho, A.; Bacskai, B.J.; Perlmutter, S.L.; Hyman, B.T.; Brown, R.H., Jr. Dysferlin interacts with annexins A1 and A2 and mediates sarcolemmal wound-healing. J. Biol. Chem. 2003, 278, 50466–50473. [Google Scholar]
- Lin, L.L.; Chen, C.N.; Lin, W.C.; Lee, P.H.; Chang, K.J.; Lai, Y.P.; Wang, J.T.; Juan, H.F. Annexin A4: A novel molecular marker for gastric cancer with Helicobacter pylori infection using proteomics approach. Proteomics Clin. Appl 2008, 2, 619–634. [Google Scholar]
- Togo, T.; Alderton, J.M.; Bi, G.Q.; Steinhardt, R.A. The mechanism of facilitated cell membrane resealing. J. Cell Sci 1999, 112, 719–731. [Google Scholar]
- Rodriguez, A.; Webster, P.; Ortego, J.; Andrews, N.W. Lysosomes behave as Ca2+-regulated exocytic vesicles in fibroblasts and epithelial cells. J. Cell Biol 1997, 137, 93–104. [Google Scholar]
- Watanabe, T.; Tada, M.; Nagai, H.; Sasaki, S.; Nakao, M. Helicobacter pylori infection induces gastric cancer in mongolian gerbils. Gastroenterology 1998, 115, 642–648. [Google Scholar]
- Huynh, C.; Roth, D.; Ward, D.M.; Kaplan, J.; Andrews, N.W. Defective lysosomal exocytosis and plasma membrane repair in Chediak-Higashi/beige cells. Proc. Natl. Acad. Sci. USA 2004, 101, 16795–16800. [Google Scholar]
- McNeil, P.L.; Warder, E. Glass beads load macromolecules into living cells. J. Cell Sci 1987, 88, 669–678. [Google Scholar]
- Mellgren, R.L.; Zhang, W.; Miyake, K.; McNeil, P.L. Calpain is required for the rapid, calcium-dependent repair of wounded plasma membrane. J. Biol. Chem 2007, 282, 2567–2575. [Google Scholar]
- McNeil, P.L.; Vogel, S.S.; Miyake, K.; Terasaki, M. Patching plasma membrane disruptions with cytoplasmic membrane. J. Cell Sci 2000, 113, 1891–1902. [Google Scholar]
- Liu, R.; Li, Z.; Bai, S.; Zhang, H.; Tang, M.; Lei, Y.; Chen, L.; Liang, S.; Zhao, Y.L.; Wei, Y.; Huang, C. Mechanism of cancer cell adaptation to metabolic stress: Proteomics identification of a novel thyroid hormone-mediated gastric carcinogenic signaling pathway. Mol. Cell. Proteomics 2009, 8, 70–85. [Google Scholar]
- Amieva, M.R.; Vogelmann, R.; Covacci, A.; Tompkins, L.S.; Nelson, W.J.; Falkow, S. Disruption of the epithelial apical-junctional complex by Helicobacter pylori CagA. Science 2003, 300, 1430–1434. [Google Scholar]
- Lytton, S.D.; Fischer, W.; Nagel, W.; Haas, R.; Beck, F.X. Production of ammonium by Helicobacter pylori mediates occludin processing and disruption of tight junctions in Caco-2 cells. Microbiology 2005, 151, 3267–3276. [Google Scholar]
- Idone, V.; Tam, C.; Goss, J.W.; Toomre, D.; Pypaert, M.; Andrews, N.W. Repair of injured plasma membrane by rapid Ca2+-dependent endocytosis. J. Cell Biol 2008, 180, 905–914. [Google Scholar]
- Monastyrskaya, K.; Babiychuk, E.B.; Draeger, A. The annexins: Spatial and temporal coordination of signaling events during cellular stress. Cell. Mol. Life Sci 2009, 66, 2623–2642. [Google Scholar]
- Duncan, R.; Carpenter, B.; Main, L.C.; Telfer, C.; Murray, G.I. Characterisation and protein expression profiling of annexins in colorectal cancer. Br. J. Cancer 2008, 98, 426–433. [Google Scholar]
- Jeon, Y.J.; Kim, D.H.; Jung, H.; Chung, S.J.; Chi, S.W.; Cho, S.; Lee, S.C.; Park, B.C.; Park, S.G.; Bae, K.H. Annexin A4 interacts with the NF-kappaB p50 subunit and modulates NF-kappaB transcriptional activity in a Ca2+-dependent manner. Cell. Mol. Life Sci 2010, 67, 2271–2281. [Google Scholar]
- Xin, W.; Rhodes, D.R.; Ingold, C.; Chinnaiyan, A.M.; Rubin, M.A. Dysregulation of the annexin family protein family is associated with prostate cancer progression. Am. J. Pathol 2003, 162, 255–261. [Google Scholar]
- Zimmermann, U.; Balabanov, S.; Giebel, J.; Teller, S.; Junker, H.; Schmoll, D.; Protzel, C.; Scharf, C.; Kleist, B.; Walther, R. Increased expression and altered location of annexin IV in renal clear cell carcinoma: A possible role in tumour dissemination. Cancer Lett 2004, 209, 111–118. [Google Scholar]
- Shen, J.; Person, M.D.; Zhu, J.; Abbruzzese, J.L.; Li, D. Protein expression profiles in pancreatic adenocarcinoma compared with normal pancreatic tissue and tissue affected by pancreatitis as detected by two-dimensional gel electrophoresis and mass spectrometry. Cancer Res 2004, 64, 9018–9026. [Google Scholar]
- Miao, Y.; Cai, B.; Liu, L.; Yang, Y.; Wan, X. Annexin IV is differentially expressed in clear cell carcinoma of the ovary. Int. J. Gynecol. Cancer Off. J. Int. Gynecol. Cancer Soc 2009, 19, 1545–1549. [Google Scholar]
- Toyama, A.; Suzuki, A.; Shimada, T.; Aoki, C.; Aoki, Y.; Umino, Y.; Nakamura, Y.; Aoki, D.; Sato, T.A. Proteomic characterization of ovarian cancers identifying annexin-A4, phosphoserine aminotransferase, cellular retinoic acid-binding protein 2, and serpin B5 as histology-specific biomarkers. Cancer Sci 2012, 103, 747–755. [Google Scholar]
- Lai, Y.P.; Yang, J.C.; Lin, T.Z.; Wang, J.T.; Lin, J.T. CagA tyrosine phosphorylation in gastric epithelial cells caused by Helicobacter pylori in patients with gastric adenocarcinoma. Helicobacter 2003, 8, 235–243. [Google Scholar]
- Tzeng, C.C.; Meng, C.L.; Jin, L.; Hsieh, H.F. Cytogenetic studies of gastric adenocarcinoma. Cancer Genet. Cytogenet 1991, 55, 67–71. [Google Scholar]
© 2012 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Lin, L.-L.; Huang, H.-C.; Ogihara, S.; Wang, J.-T.; Wu, M.-C.; McNeil, P.L.; Chen, C.-N.; Juan, H.-F. Helicobacter pylori Disrupts Host Cell Membranes, Initiating a Repair Response and Cell Proliferation. Int. J. Mol. Sci. 2012, 13, 10176-10192. https://doi.org/10.3390/ijms130810176
Lin L-L, Huang H-C, Ogihara S, Wang J-T, Wu M-C, McNeil PL, Chen C-N, Juan H-F. Helicobacter pylori Disrupts Host Cell Membranes, Initiating a Repair Response and Cell Proliferation. International Journal of Molecular Sciences. 2012; 13(8):10176-10192. https://doi.org/10.3390/ijms130810176
Chicago/Turabian StyleLin, Li-Ling, Hsuan-Cheng Huang, Satoshi Ogihara, Jin-Town Wang, Meng-Chuan Wu, Paul L. McNeil, Chiung-Nien Chen, and Hsueh-Fen Juan. 2012. "Helicobacter pylori Disrupts Host Cell Membranes, Initiating a Repair Response and Cell Proliferation" International Journal of Molecular Sciences 13, no. 8: 10176-10192. https://doi.org/10.3390/ijms130810176
APA StyleLin, L.-L., Huang, H.-C., Ogihara, S., Wang, J.-T., Wu, M.-C., McNeil, P. L., Chen, C.-N., & Juan, H.-F. (2012). Helicobacter pylori Disrupts Host Cell Membranes, Initiating a Repair Response and Cell Proliferation. International Journal of Molecular Sciences, 13(8), 10176-10192. https://doi.org/10.3390/ijms130810176