Function Prediction and Analysis of Mycobacterium tuberculosis Hypothetical Proteins

Abstract
:1. Introduction
2. Results and Discussion
2.1. General Topological Parameters of the MTB Functional Network
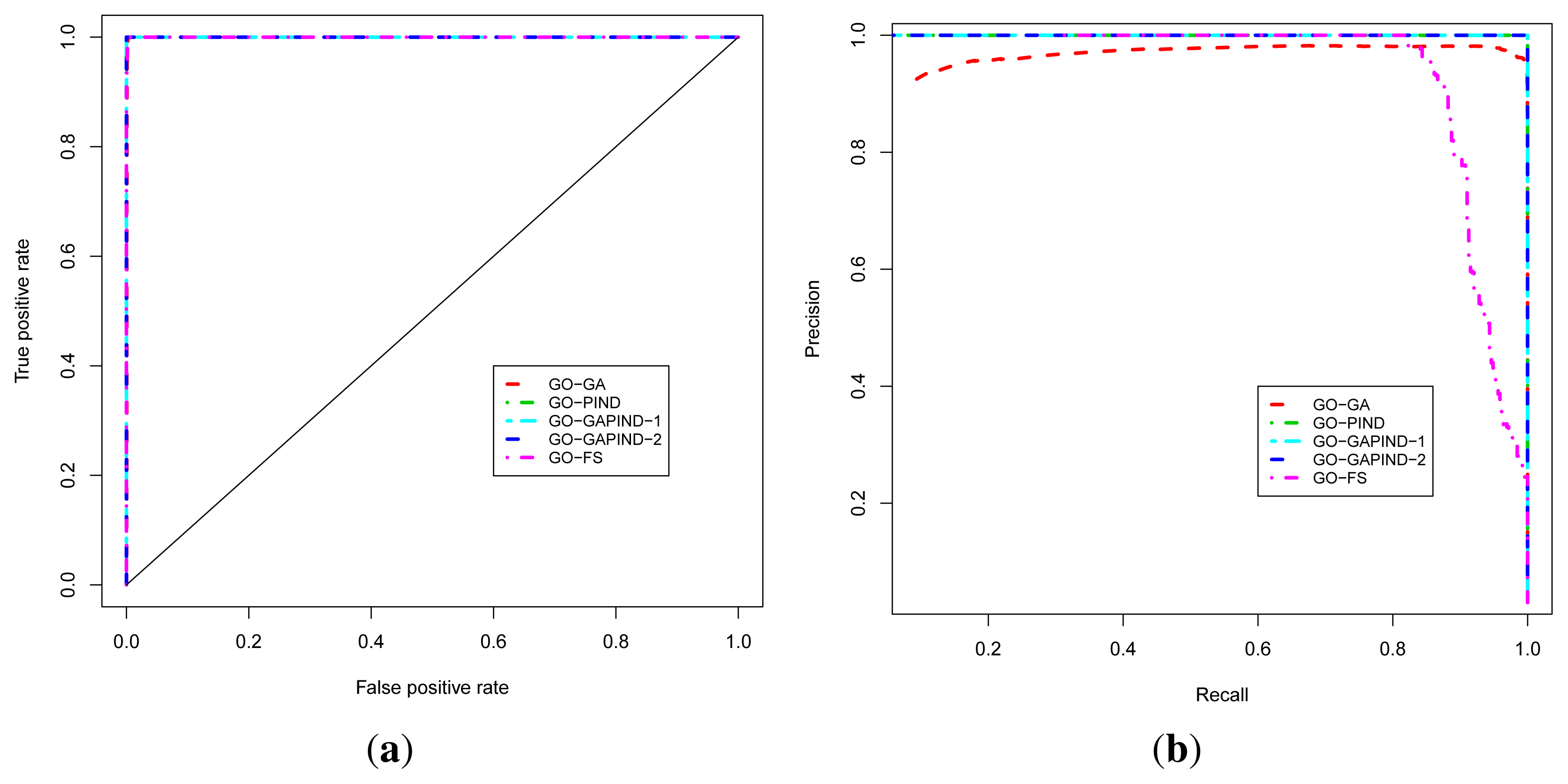
2.2. Topological Analysis of MTB Hypothetical Proteins
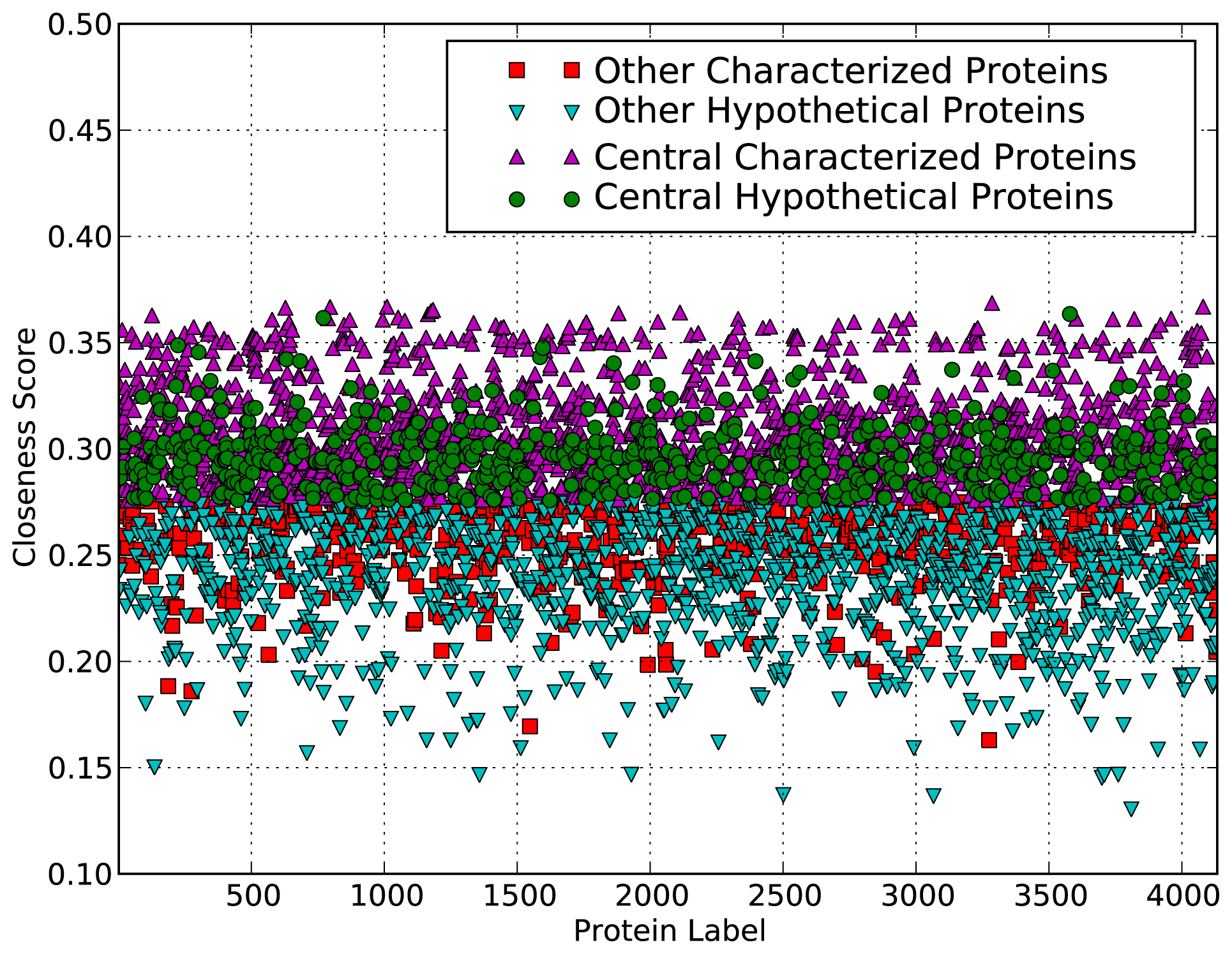
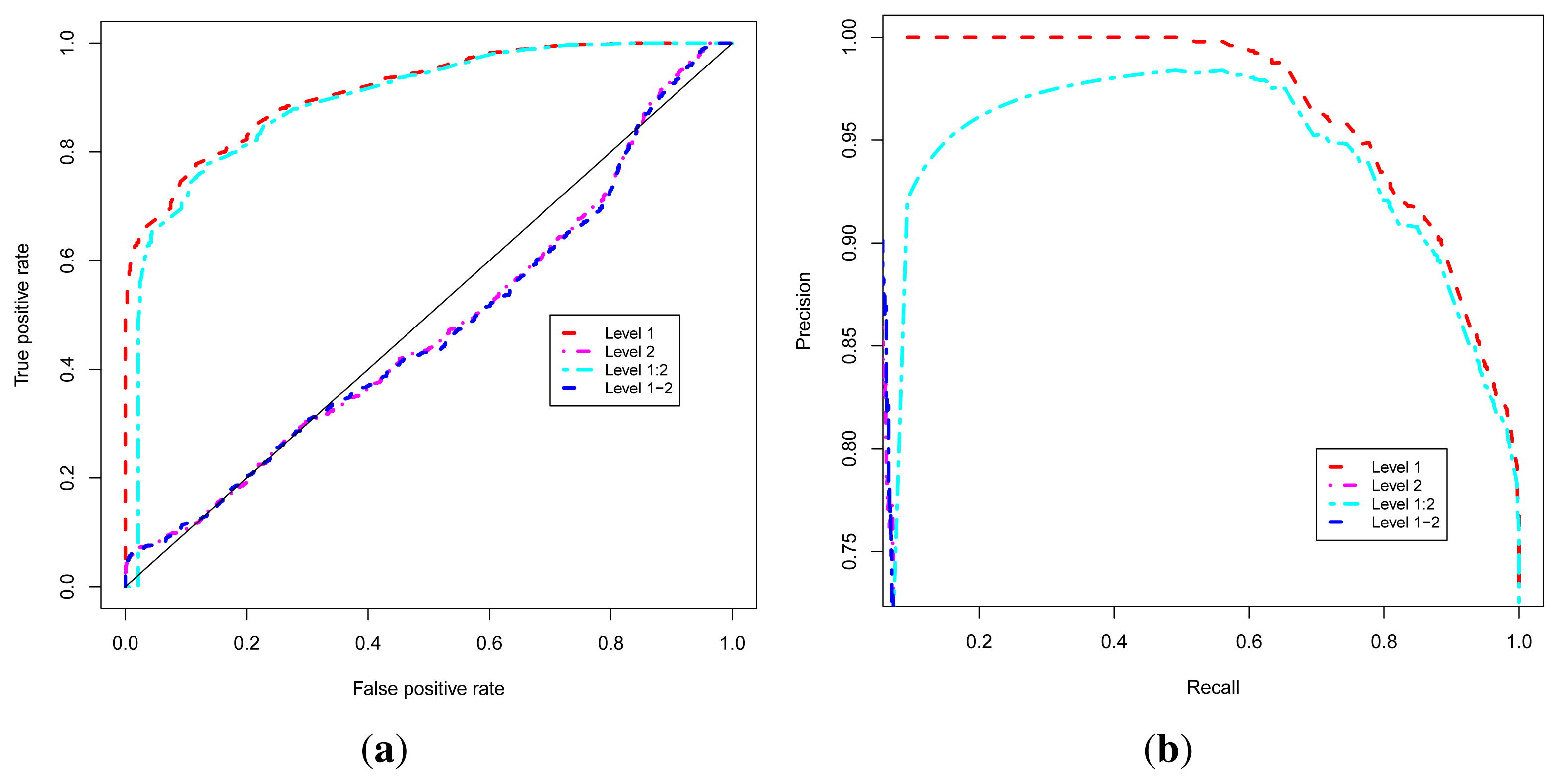
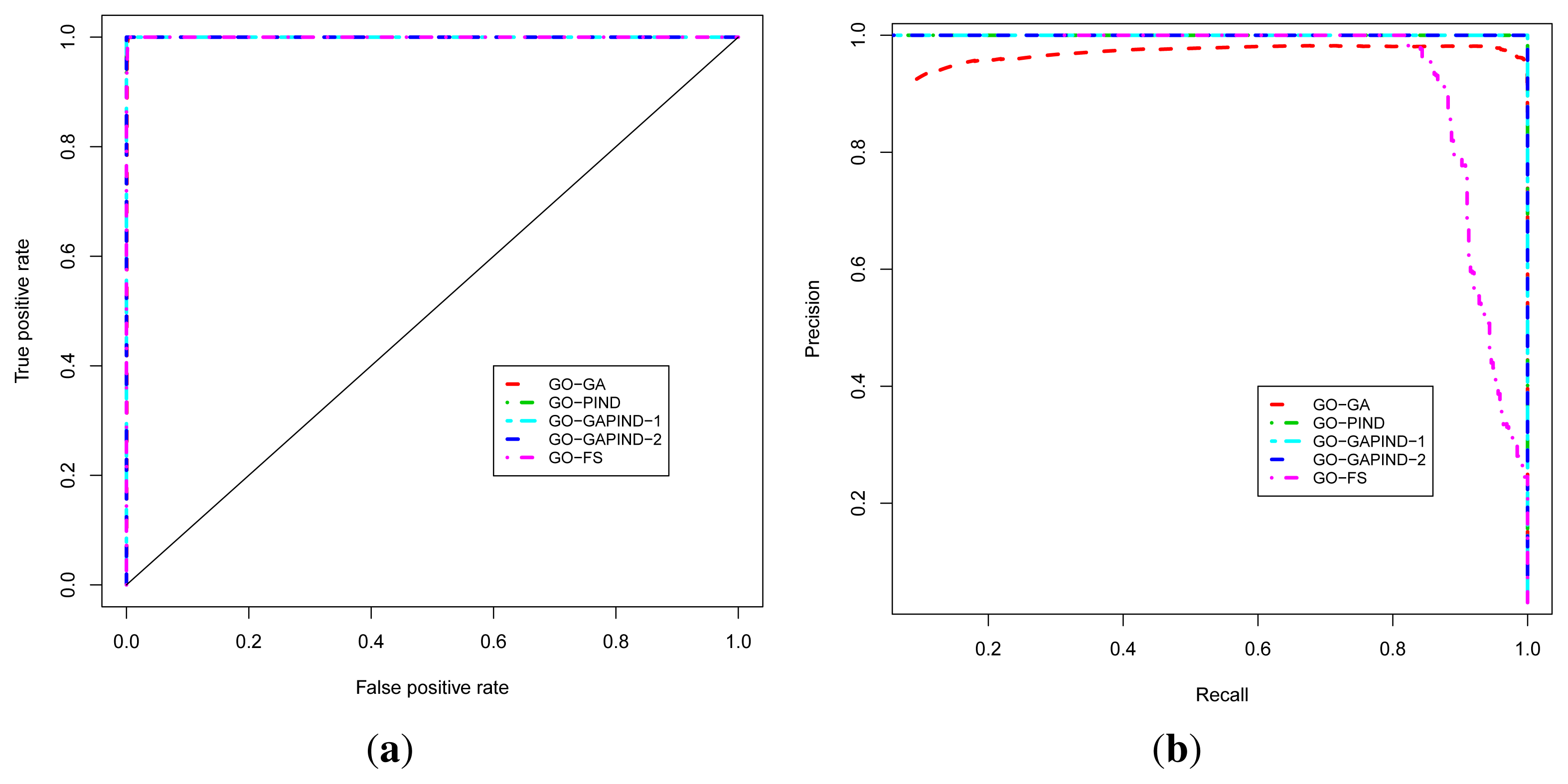
2.3. Evaluation of Function Predictions
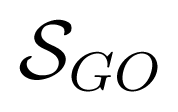 (t, Ip) = max{
(t, s) : s ∈ Ip} with
(t, s) the GO-universal semantic similarity score between GO terms t and s [36], and |Ip| stands for the number of GO BP terms in the set
, with IPRp the InterPro terms of the protein p and mapGO(t) the set of GO BP terms mapping the InterPro term t.
(t, Ip) = max{
(t, s) : s ∈ Ip} with
(t, s) the GO-universal semantic similarity score between GO terms t and s [36], and |Ip| stands for the number of GO BP terms in the set
, with IPRp the InterPro terms of the protein p and mapGO(t) the set of GO BP terms mapping the InterPro term t.2.4. Functional Analysis of MTB Hypothetical Proteins and Adaptability
3. Materials and Methods
3.1. Topological Features of the MTB Hypothetical Proteins
3.2. Protein Annotation Prediction
3.3. Annotation Enrichment Analysis
4. Conclusions
Acknowledgements
References
- Enault, F.; Suhre, K.; Claverie, J.M. Phydbac “Gene Function Predictor”: A gene annotation tool based on genomic context analysis. BMC Bioinforma 2005, 6. [Google Scholar] [CrossRef]
- Mazandu, G.K.; Mulder, N.J. Scoring protein relationships in functional interaction networks predicted from sequence data. PLoS One 2011, 6. [Google Scholar] [CrossRef] [Green Version]
- Lord, P.W.; Stevens, P.W.; Brass, A.; Goble, C.A. Investigating semantic similarity measures across the Gene Ontology: The relationship between sequence and annotation. emphBioinformatics 2003, 19, 1275–1283. [Google Scholar]
- Gruber, T.R. Toward principles for the design of ontologies used for knowledge sharing. Int. J. Hum.-Comput. Stud 1995, 43, 907–928. [Google Scholar]
- Gruber, T.R. A translation approach to portable ontology specifications. Knowl. Acquis 1993, 5, 199–220. [Google Scholar]
- Stevens, R.; Goble, C.A.; Bechhofer, S. Ontology-based knowledge representation for bioinformatics. Brief. Bioinforma 2000, 1, 398–414. [Google Scholar]
- Ciocoiu, M.; Gruninger, M.; Nau, D. Ontologies for integrating engineering applications. J. Comput. Inf. Sci. Eng 2001, 1, 45–60. [Google Scholar]
- Uschold, M.; Gruninger, M. Ontologies and semantics for seamless connectivity. SIGMOD Rec 2004, 33, 58–64. [Google Scholar]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene Ontology: Tool for the unification of biology. Nat. Genet 2000, 25, 25–29. [Google Scholar]
- GO-Consortium. The Gene Ontology in 2010: Extensions and refinements. Nucleic Acids Res 2009, 38, D331–D335.
- Camon, E.; Magrane, M.; Barrell, D.; Binns, D.; Fleischmann, W.; Kersey, P.; Mulder, N.; Oinn, T.; Maslen, J.; Cox, A.; et al. The Gene Ontology Annotation (GOA) project: Implementation of GO in SWISS-PROT, TrEMBL, and InterPro. Genome Res 2003, 13, 662–672. [Google Scholar]
- Camon, E.; Barrell, D.; Lee, V.; Dimmer, E.; Apweiler, R. The Gene Ontology Annotation (GOA) Database—An integrated resource of GO annotations to the UniProt Knowledgebase. Silico Biol 2004, 4, 5–6. [Google Scholar]
- Camon, E.; Magrane, M.; Barrell, D.; Lee, V.; Dimmer, E.; Maslen, J.; Binns, D.; Harte, N.; Lopez, R.; Apweiler, R. The Gene Ontology Annotation (GOA) Database: Sharing knowledge in Uniprot with Gene Ontology. Nucleic Acids Res 2004, 32, D262–D266. [Google Scholar]
- Barrell, D.; Dimmer, E.; Huntley, R.P.; Binns, D.; O’Donovan, C.; Apweiler, R. The GOA database in 2009—An integrated Gene Ontology Annotation resource. Nucleic Acids Res 2009, 37, D396–D403. [Google Scholar]
- Dimmer, E.C.; Huntley, R.P.; Barrell, D.G.; Binns, D.; Draghici, S.; Camon, E.B.; Hubank, M.; Talmud, P.J.; Apweiler, R.; Lovering, R.C. The Gene Ontology—Providing a functional role in proteomic studies. Proteomics 2008, 8(Suppl), 2–11. [Google Scholar]
- Camon, E.B.; Barrell, D.G.; Dimmer, E.C.; Lee, V.; Magrane, M.; Maslen, J.; Binns, D.; Apweiler, R. An evaluation of GO annotation retrieval for BioCreAtIve and GOA. BMC Bioinforma 2005, 6. [Google Scholar] [CrossRef]
- Mazandu, G.K.; Mulder, N.J. Using the underlying biological organization of the MTB functional network for protein function prediction. Infect. Genet. Evol 2011, 12, 922–932. [Google Scholar]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. A basic local alignment search tool. J. Mol. Biolol 1990, 215, 403–410. [Google Scholar]
- Altschul, S.F.; Madden, T.L.; Shaffer, A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res 1997, 25, 3389–3402. [Google Scholar]
- Browne, F.; Zheng, H.; Wang, H.; Azuaje, F. An integrative bayesian approach to supporting the prediction of protein-protein interactions: A case study in human heart failure. World Acad. Sci. Eng. Technol 2009, 53, 457–463. [Google Scholar]
- Persener, J. Bioinformatics and Functional Genomics; John Wiley & Sons: Hoboken, NJ, USA, 2003. [Google Scholar]
- Brosch, R.; Gordon, V.; Eiglmeier, K.; Garnier, T.; Tekala, F.; Yeramian, E.; Cole, S.T. Genomics, Biology and Evolution of the Mycobacterium tuberculosis Complex. In Molecular Genetics of Mycobacteria; ASM Press: Washington DC, USA, 2000; pp. 19–36. [Google Scholar]
- Abdallah, A.M.; Verboom, T.; Weerdenburg, E.M.; Gey van Pittius, N.C.; Mahasha, P.W.; Jiménez, C.; Parra, M.; Cadieux, N.; Brennan, M.J.; Appelmelk, B.J.; et al. PPE and PE PGRS proteins of Mycobacterium marinum are transported via the type VII secretion system ESX-5. Mol. Microbiol 2009, 73, 329–340. [Google Scholar]
- Delogu, G.; Brennan, M. Comparative immune response to PE and PE PGRS antigens of Mycobacterium tuberculosis. Infect. Immun 2001, 69, 5606–5611. [Google Scholar]
- Brennan, M.J.; Delogu, G.; Chen, Y.; Bardarov, S.; Kriakov, J.; Alavi, M.; Jacobs, W.R., Jr. Evidence that Mycobacterial PE PGRS Proteins are cell surface constituents that influence interactions with other cells. Infect. Immun 2001, 69, 7326–7333. [Google Scholar]
- Banu, S.; Honore, N.; Saint-Joanis, B.; Philpott, D.; Prevost, M.C.; Cole, S.T. Are the PE-PGRS proteins of Mycobacterium tuberculosis variable surface antigens? Mol. Microbiol 2002, 44, 9–19. [Google Scholar]
- Huang, Y.; Wang, Y.; Bai, Y.; Wang, Z.G.; Yang, L.; Zhao, D. Expression of PE PGRS 62 protein in Mycobacterium smegmatis decrease mRNA expression of proinflammatory cytokines IL-1β, IL-6 in macrophages. Mol. Cell Biochem 2010, 340, 223–229. [Google Scholar]
- Mazandu, G.K.; Opap, K.; Mulder, N.J. Contribution of microarray data to the advancement of knowledge on the Mycobacterium tuberculosis interactome: Use of the random partial least squares approach. Infect. Genet. Evol 2011, 11, 181–189. [Google Scholar]
- Mazandu, G.K.; Mulder, N.J. Generation and analysis of large-scale data driven Mycobacterium tuberculosis functional networks for drug target identification. Adv. Bioinforma 2011, 2011. [Google Scholar] [CrossRef]
- Tsoka, S.; Ouzounis, C.A. Recent developments and future directions in computational genomics. FEBS Lett 2000, 480, 42–48. [Google Scholar]
- Mason, O.; Verwoerd, M. Graph theory and networks in biology. IET Syst. Biol 2007, 1, 89–119. [Google Scholar]
- Gursoy, A.; Keskin, O.; Nussinov, R. Topological properties of protein interaction networks from structural perspective. Biochem. Soc. Trans 2008, 36, 1398–1403. [Google Scholar]
- Sassetti, C.M.; Boyd, D.H.; Rubin, E.J. Genes required for mycobacterial growth defined by high density mutagenesis. Mol. Microbiol 2003, 48, 77–84. [Google Scholar]
- Sassetti, C.M.; Rubin, E.J. Genetic requirements for mycobacterial survival during infection. PNAS 2003, 100, 12989–12994. [Google Scholar]
- InterPro Database. Available online: http://www.ebi.ac.uk/interpro accessed on 20 April 2012.
- Mazandu, G.K.; Mulder, N.J. A topology-based metric for measuring term similarity in the gene ontology. Adv. Bioinforma 2012, 2012. [Google Scholar] [CrossRef]
- Brennan, P.J. Structure, function, and biogenesis of the cell wall of Mycobacterium tuberculosis. Tuberculosis 2003, 83, 91–97. [Google Scholar]
- Brennan, P.J.; Crick, D.C. The cell-wall core of Mycobacterium tuberculosis in the context of drug discovery. Curr. Top. Med. Chem 2007, 7, 475–488. [Google Scholar]
- Wolfe, L.M.; Mahaffey, S.B.; Kruh, N.A.; Dobos, K.M. Proteomic definition of the cell wall of Mycobacterium tuberculosis. J. Proteome Res 2010, 9, 5816–5826. [Google Scholar]
- Brennan, P.J.; Nikaido, H. The envelope of mycobacteria. Annu. Rev. Biochem 1995, 64, 29–63. [Google Scholar]
- Gu, S.; Chen, J.; Dobos, K.M.; Bradbury, E.M.; Belisle, J.T.; Chen, X. Comprehensive proteomic profiling of the membrane constituents of a Mycobacterium tuberculosis strain. Mol. Cell Proteomics 2003, 2, 1284–1296. [Google Scholar]
- Ng, S.K.; Zhang, Z.; Tan, S.H. Integrative approach for computationally inferring protein domain interactions. Bioinformatics 2003, 19, 923–929. [Google Scholar]
- Walhout, A.J.; Sordella, R.; Lu, X.; Hartley, J.L.; Temple, G.F.; Brasch, M.A.; Thierry-Mieg, N.; Vidal, M. Protein interaction mapping in C. elegans using proteins involved in vulval development. Science 2000, 287, 116–122. [Google Scholar]
- IntAct Database. http://www.ebi.ac.uk/intact/main.xhtml accessed on 6 October 2011.
- Aranda, B.; Achuthan, P.; Alam-Faruque, Y.; Armean, I.; Bridge, A.; Derow, C.; Feuermann, M.; Ghanbarian, A.; Kerrien, S.; Khadake, J.; et al. The IntAct molecular interaction database in 2010. Nucleic Acids Res 2010, 38(Database issue), D525–D531. [Google Scholar]
- Kerrien, S.; Aranda, B.; Breuza, L.; Bridge, A.; Broackes-Carter, F.; Chen, C.; Duesbury, M.; Dumousseau, M.; Feuermann, M.; Hinz, U.; et al. The IntAct molecular interaction database in 2012. Nucleic Acids Res 2012, 40, D841–D846. [Google Scholar]
- Integr8 Project. Available online: http://www.ebi.ac.uk/integr8 accessed on 28 October 2011.
- Pruess, M.; Kersey, P.; Apweiler, R. The Integr8 project—A resource for genomic and proteomic data. Silico Biol 2004, 5, 179–185. [Google Scholar]
- Ng, S.K.; Zhang, Z.; Tan, S.H.; Lin, K. InterDom: A database of putative interacting protein domains for validating predicted protein interactions and complexes. Nucleic Acids Res 2003, 31, 251–254. [Google Scholar]
- Pagel, P.; Oesterheld, M.; Tovstukhina, O.; Strack, N.; Stümpflen, V.; Frishman, D. DIMA 2.0—predicted and known domain interactions. Nucleic Acids Res 2008, 36, D651–D655. [Google Scholar]
- TubercuList Database. Available online: http://genolist.pasteur.fr/Tuberculist accessed on 28 October 2011.
- Swets, J. Measuring the accuracy of diagnostic systems. Science 1988, 240, 1285–1293. [Google Scholar]
- Swets, J.; Dawes, R.; Monahan, J. Better decisions through science. Sci. Am 2000, 283, 82–87. [Google Scholar]
- Buckland, M.; Gey, F. The relationship between recall and precision. J. Am. Soc. Inf. Sci 1994, 45, 12–19. [Google Scholar]
- Sing, T.; Sander, O.; Beerenwinkel, N.; Lengauer, T. ROCR: Visualizing classifier performance in R. Bioinformatics 2005, 21, 3940–3941. [Google Scholar]
- R Development Core Team, R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2010.
- R Development Core Team, R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2011.
- Apweiler, R.; Bairoch, A.; Wu, C.H.; Barker, W.C.; Boeckmann, B.; Ferro, S.; Gasteiger, E.; Huang, H.; Lopez, R.; Magrane, M.; et al. UniProt: The Universal Protein knowledgebase. Nucleic Acids Res 2004, 32, D115–D119. [Google Scholar]
- UniProt-Consortium. The Universal Protein Resource (UniProt) in 2010. Nucleic Acids Res 2010, 38, D142–D148.
- Jain, E.; Bairoch, A.; Duvaud, S.; Phan, I.; Redaschi, N.; Suzek, B.E.; Martin, M.J.; McGarvey, P.; Gasteiger, E. Infrastructure for the life sciences: Design and implementation of the UniProt website. BMC Bioinforma 2009, 10, 136. [Google Scholar]
- Martin, D.; Brun, C.; Remy, E.; Mouren, P.; Thieffry, D.; Jacq, B. GOToolBox: Functional analysis of gene datasets based on Gene Ontology. Genome Biol 2004, 5. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Chen, H.J. A Note on the determination of sample sizes for hypergeometric distributions. Commun. Stat. Theory Methods 1999, 28, 1749–1757. [Google Scholar]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameters | Value |
|---|---|
| Number of Proteins (Nodes) | 4136 |
| Number of Functional Interactions (Edges) | 59,919 |
| Average Degree (in and out) | 28.974 |
| Average Shortest Path Length | 3.6274 |
| Maximum Path Length | 11 |
| Number of Connected Components | 23 |
| % of Nodes in Largest Component | 98.7% |
| Number of Hubs | 201 |
| Metric | Average values | p-values | |||
|---|---|---|---|---|---|
| Hypothetical | Other proteins | Expected value | Other proteins | Expected value | |
| Degree | 16.16892 | 37.77267 | 28.09381 | < 2.2 × 10−16 | < 2.2 × 10−16 |
| Closeness | 0.277673 | 0.293277 | 0.27568 | 5.562 × 10−08 | 0.9792 |
| Betweenness | 6948.217 | 13913 | 15003 | < 2.2 × 10−16 | < 2.2 × 10−16 |
| Eigenvector | 0.000314 | 0.00591 | 0.00340 | < 2.2 × 10−16 | < 2.2 × 10−16 |
| UniProt-ID | InterPro-ID | GO mapping | GO Prediction | Power |
|---|---|---|---|---|
| P67745 | IPR000835 | GO:0006355 [8] | GO:0006355 [8] | 1.00000 |
| Q8VKE6 | IPR002514 | GO:0006313 [8] | GO:0006313 [8] | 1.00000 |
| Q7D8E8 | IPR001845 | GO:0006355 [8] | GO:0006355 [8] | 1.00000 |
| Q7D5V1 | IPR000836 | GO:0009116 [6] | GO:0009116 [6] | 1.00000 |
| Q7D8W2 | IPR001087 | GO:0006629 [3] | GO:0006629 [3] | 1.00000 |
| Q8VJC1 | IPR006059 | GO:0006810 [3] | GO:0006810 [3] | 1.00000 |
| Q7D9M5 | IPR020946 | GO:0055114 [2] | GO:0055114 [2] | 1.00000 |
| P64725 | IPR002539 | GO:0008152 [1] | GO:0008152 [1] | 1.00000 |
| P71788 | IPR013216 | GO:0008152 [1] | GO:0008152 [1] | 1.00000 |
| Q10777 | IPR000873 | GO:0008152 [1] | GO:0008152 [1] | 1.00000 |
| O05796 | IPR013216 | GO:0008152 [1] | GO:0008152 [1] | 1.00000 |
| O07197 | IPR013094 | GO:0008152 [1] | GO:0008152 [1] | 1.00000 |
| P0A5F5 | IPR005674 | GO:0008152 [1] | GO:0008152 [1] | 0.66604 |
| IPR000383 | GO:0006508 [4] | GO:0044238 [2] | ||
| O06547 | IPR013216 | GO:0008152 [1] | GO:0044237 [2] | 0.24819 |
| Q7D8C2 | IPR013216 | GO:0008152 [1] | GO:0042158 [5] | 0.03033 |
| O05294 | IPR012908 | GO:0006886 [6] | GO:0044237 [2] | 0.01435 |
| GO:0006505 [8] |
| Functional Class | # Proteins before | Prediction | PE/PPE- | p-values | # Proteins after | % change |
|---|---|---|---|---|---|---|
| 1 virulence, detoxification, adaptation | 176 | 85 | 1 | 3.33067 × 10−16 | 261 | 32.6 |
| 2 lipid metabolism | 230 | 80 | 6 | 9.76996 × 10−15 | 310 | 25.8 |
| 3 information pathways | 245 | 93 | 3 | 9.76996 × 10−15 | 338 | 27.5 |
| 4 cell wall and cell processes | 618 | 418 | 62 | 3.33067 × 10−16 | 1036 | 40.3 |
| 5 insertion seqs and phages | 82 | 65 | 4 | 1.11022 × 10−16 | 147 | 44.2 |
| 6 PE/PPE | 147 | −115 | - | - | 32 | - |
| 7 intermediary metabolism and respiration | 884 | 637 | 38 | 4.95160 × 10−14 | 1521 | 41.9 |
| 8 unknown | 1637 | −1351 | - | - | 286 | |
| 9 regulatory proteins | 176 | 88 | 1 | 9.54792 × 10−15 | 264 | 33.3 |
| Total | 4195 | 1466 | 115 | - | 4195 | 37.8 |
| GO ID | GO name | Frequency | p-Value |
|---|---|---|---|
| GO:0006730 | one-carbon metabolic process | 364 | 0.00000 |
| GO:0009132 | nucleoside diphosphate metabolic process | 74 | 0.00000 |
| GO:0009123 | nucleoside monophosphate metabolic process | 72 | 0.00000 |
| GO:0009141 | nucleoside triphosphate metabolic process | 71 | 0.00000 |
| GO:0006353 | transcription termination, DNA-dependent | 88 | 1.11022 × 10−16 |
| GO:0019538 | protein metabolic process | 87 | 1.11022 × 10−16 |
| GO:0022900 | electron transport chain | 354 | 2.22045 × 10−16 |
| GO:0006793 | phosphorus metabolic process | 324 | 2.22045 × 10−16 |
| GO:0009061 | anaerobic respiration | 135 | 2.22045 × 10−16 |
| GO:0009307 | DNA restriction-modification system | 73 | 2.22045 × 10−16 |
| GO:0006662 | glycerol ether metabolic process | 277 | 3.33067 × 10−16 |
| GO:0015074 | DNA integration | 157 | 3.33067 × 10−16 |
| GO:0019419 | sulfate reduction | 86 | 3.33067 × 10−16 |
| GO:0051090 | regulation of sequence-specific DNA binding transcription factor activity | 64 | 3.33067 × 10−16 |
| GO:0006139 | nucleobase-containing compound metabolic process | 336 | 4.44089 × 10−16 |
| GO:0006259 | DNA metabolic process | 169 | 4.44089 × 10−16 |
| GO:0006313 | transposition, DNA-mediated | 113 | 4.44089 × 10−16 |
| GO:0006797 | polyphosphate metabolic process | 672 | 5.55112 × 10−16 |
| GO:0006796 | phosphate-containing compound metabolic process | 643 | 5.55112 × 10−16 |
| GO:0000103 | sulfate assimilation | 603 | 5.55112 × 10−16 |
| GO:0044238 | primary metabolic process | 548 | 5.55112 × 10−16 |
| GO:0006281 | DNA repair | 246 | 5.55112 × 10−16 |
| GO:0006413 | translational initiation | 156 | 5.55112 × 10−16 |
| GO:0009116 | nucleoside metabolic process | 107 | 5.55112 × 10−16 |
| GO:0044255 | cellular lipid metabolic process | 444 | 6.66134 × 10−16 |
| GO:0044262 | cellular carbohydrate metabolic process | 317 | 6.66134 × 10−16 |
| GO:0001121 | transcription from bacterial-type RNA polymerase promoter | 273 | 6.66134 × 10−16 |
| GO:0006302 | double-strand break repair | 200 | 6.66134 × 10−16 |
| GO:0009225 | nucleotide-sugar metabolic process | 177 | 6.66134 × 10−16 |
| GO:0006310 | DNA recombination | 170 | 6.66134 × 10−16 |
| GO:0006260 | DNA replication | 156 | 6.66134 × 10−16 |
| GO:0006396 | RNA processing | 109 | 6.66134 × 10−16 |
| GO:0015977 | carbon fixation | 534 | 7.77161 × 10−16 |
| GO:0042126 | nitrate metabolic process | 477 | 7.77156 × 10−16 |
| GO:0006082 | organic acid metabolic process | 244 | 7.77156 × 10−16 |
| GO:0006266 | DNA ligation | 150 | 7.77156 × 10−16 |
| GO:0006104 | succinyl-CoA metabolic process | 144 | 7.77156 × 10−16 |
| GO:0009060 | aerobic respiration | 118 | 7.77156 × 10−16 |
| GO:0006352 | transcription initiation, DNA-dependent | 112 | 7.77156 × 10−16 |
| GO:0044249 | cellular biosynthetic process | 618 | 8.88178 × 10−16 |
| GO:0006351 | transcription, DNA-dependent | 333 | 8.88178 × 10−16 |
| GO:0009399 | nitrogen fixation | 271 | 8.88178 × 10−16 |
| GO:0016042 | lipid catabolic process | 107 | 8.88178 × 10−16 |
| GO:0009117 | nucleotide metabolic process | 101 | 8.88178 × 10−16 |
| GO:0043620 | regulation of DNA-dependent transcription in response to stress | 96 | 8.88178 × 10−16 |
| GO:0001522 | pseudouridine synthesis | 92 | 8.88178 × 10−16 |
| GO:0006314 | intron homing | 137 | 9.99201 × 10−16 |
| GO:0006066 | alcohol metabolic process | 635 | 1.11022 × 10−15 |
| GO:0090305 | nucleic acid phosphodiester bond hydrolysis | 222 | 1.11022 × 10−15 |
| GO:0006284 | base-excision repair | 212 | 1.11022 × 10−15 |
| GO:0006289 | nucleotide-excision repair | 210 | 1.33227 × 10−15 |
| GO:0009451 | RNA modification | 101 | 1.33227 × 10−15 |
| GO:0008610 | lipid biosynthetic process | 184 | 1.66534 × 10−15 |
| GO:0044267 | cellular protein metabolic process | 58 | 3.66374 × 10−15 |
| GO:0006268 | DNA unwinding involved in replication | 53 | 7.32747 × 10−15 |
| GO:0006270 | DNA-dependent DNA replication initiation | 52 | 1.36557 × 10−14 |
| GO:0006412 | translation | 207 | 4.56302 × 10−14 |
| GO:0031554 | regulation of transcription termination, DNA-dependent | 50 | 5.19584 × 10−14 |
| GO:0030261 | chromosome condensation | 56 | 1.21125 × 10−13 |
| GO:0006801 | superoxide metabolic process | 693 | 2.91767 × 10−13 |
| GO:0000725 | recombinational repair | 46 | 8.03135 × 10−13 |
| Association Evidence by Type | Low Confidence | Medium Confidence | High Confidence |
|---|---|---|---|
| Previous Functional Network | 6850 | 32488 | 25605 |
| Domain-domain | 0 | 5082 | 864 |
| Interologs | 0 | 0 | 1701 |
| Combined Score | 6844 | 30142 | 29776 |
© 2012 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Mazandu, G.K.; Mulder, N.J. Function Prediction and Analysis of Mycobacterium tuberculosis Hypothetical Proteins. Int. J. Mol. Sci. 2012, 13, 7283-7302. https://doi.org/10.3390/ijms13067283
Mazandu GK, Mulder NJ. Function Prediction and Analysis of Mycobacterium tuberculosis Hypothetical Proteins. International Journal of Molecular Sciences. 2012; 13(6):7283-7302. https://doi.org/10.3390/ijms13067283
Chicago/Turabian StyleMazandu, Gaston K., and Nicola J. Mulder. 2012. "Function Prediction and Analysis of Mycobacterium tuberculosis Hypothetical Proteins" International Journal of Molecular Sciences 13, no. 6: 7283-7302. https://doi.org/10.3390/ijms13067283
APA StyleMazandu, G. K., & Mulder, N. J. (2012). Function Prediction and Analysis of Mycobacterium tuberculosis Hypothetical Proteins. International Journal of Molecular Sciences, 13(6), 7283-7302. https://doi.org/10.3390/ijms13067283