Neuroprotective Effects of Pre-Treament with l-Carnitine and Acetyl-l-Carnitine on Ischemic Injury In Vivo and In Vitro

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
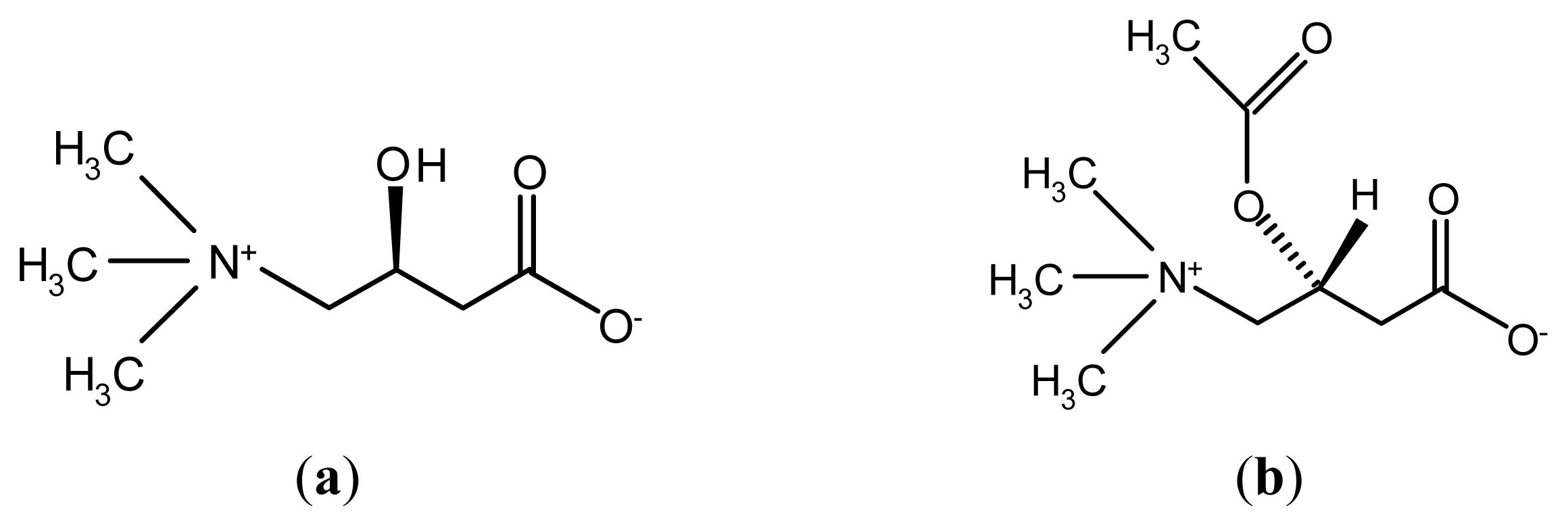
:1. Introduction
2. Results
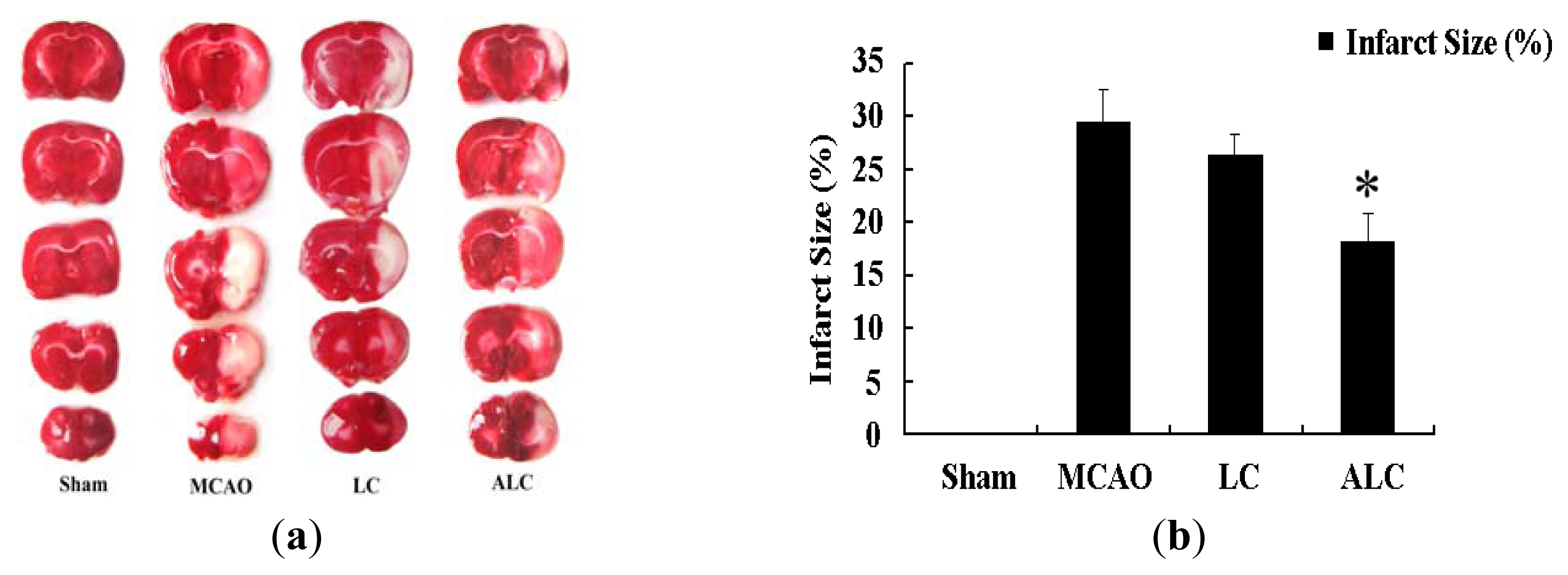
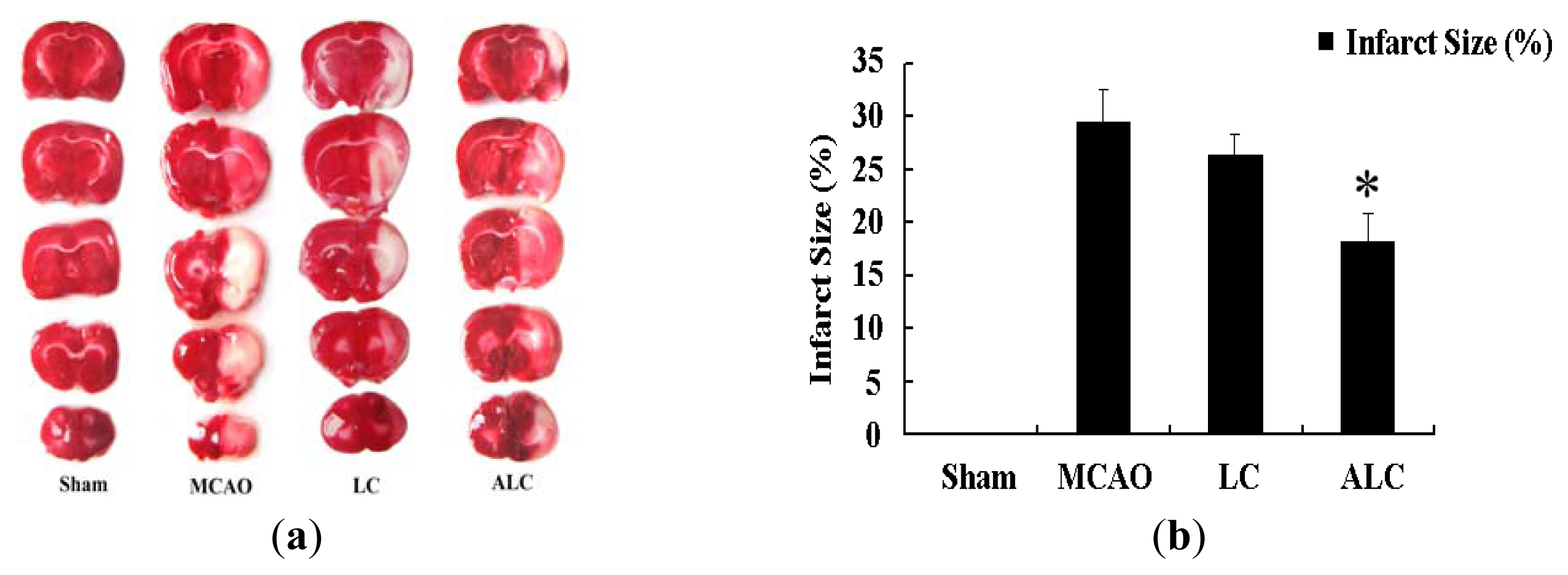
2.1. ALC, But Not LC, Decreases Infarction Size in SD Rats after MCAO
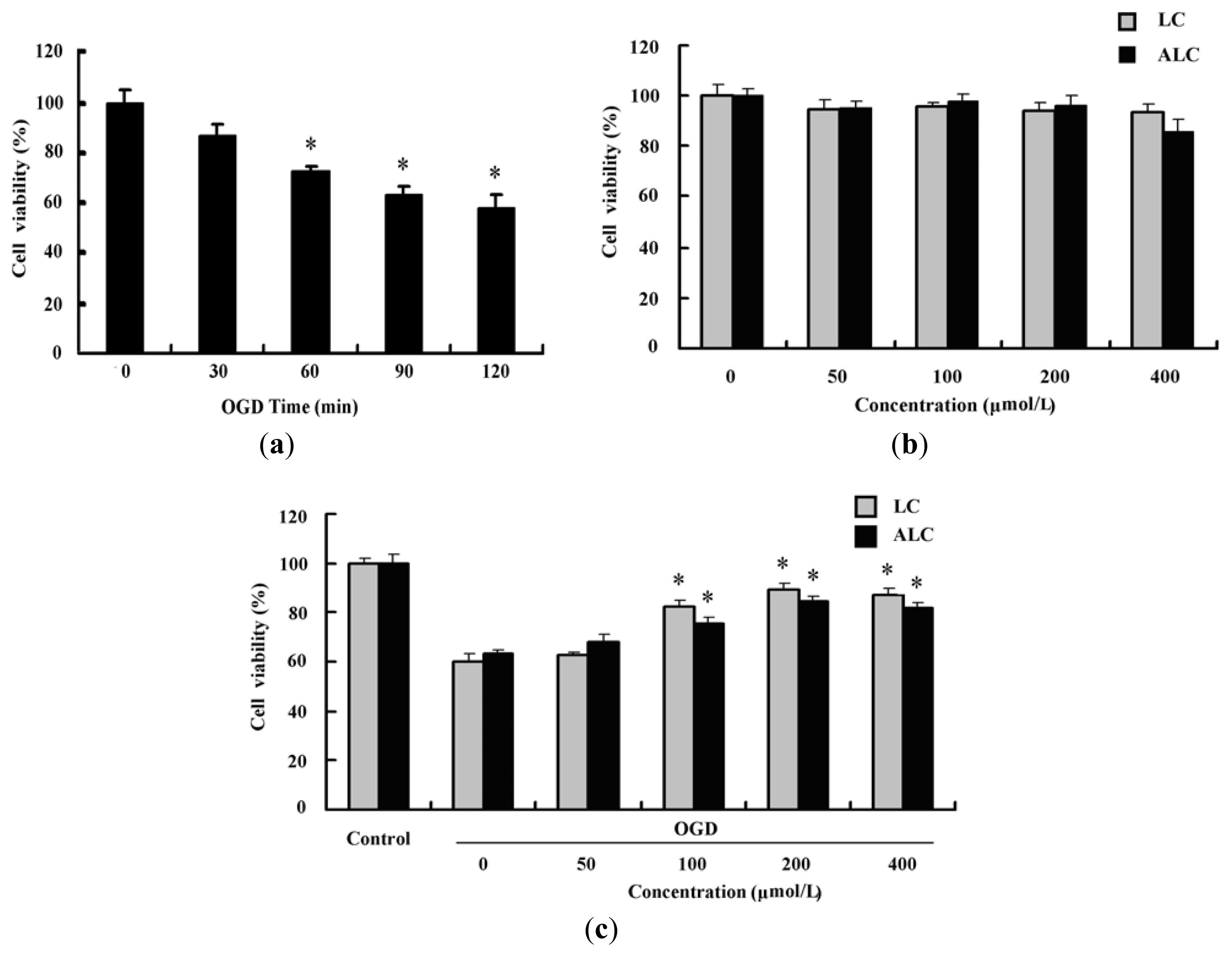
2.2. LC and ALC Pretreatment Reduces Oxygen-Glucose Deprivation-Induced Cell Injury
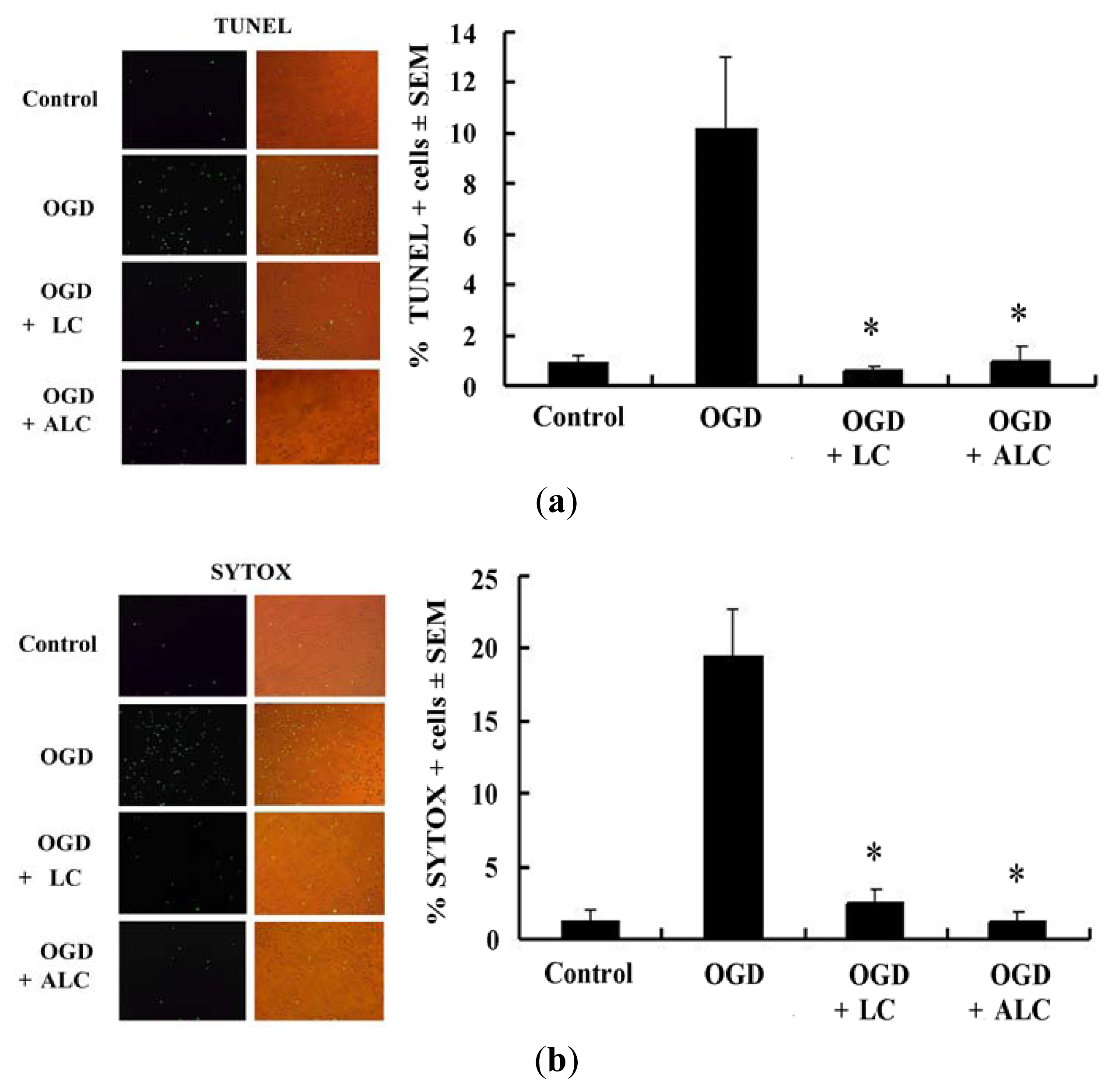
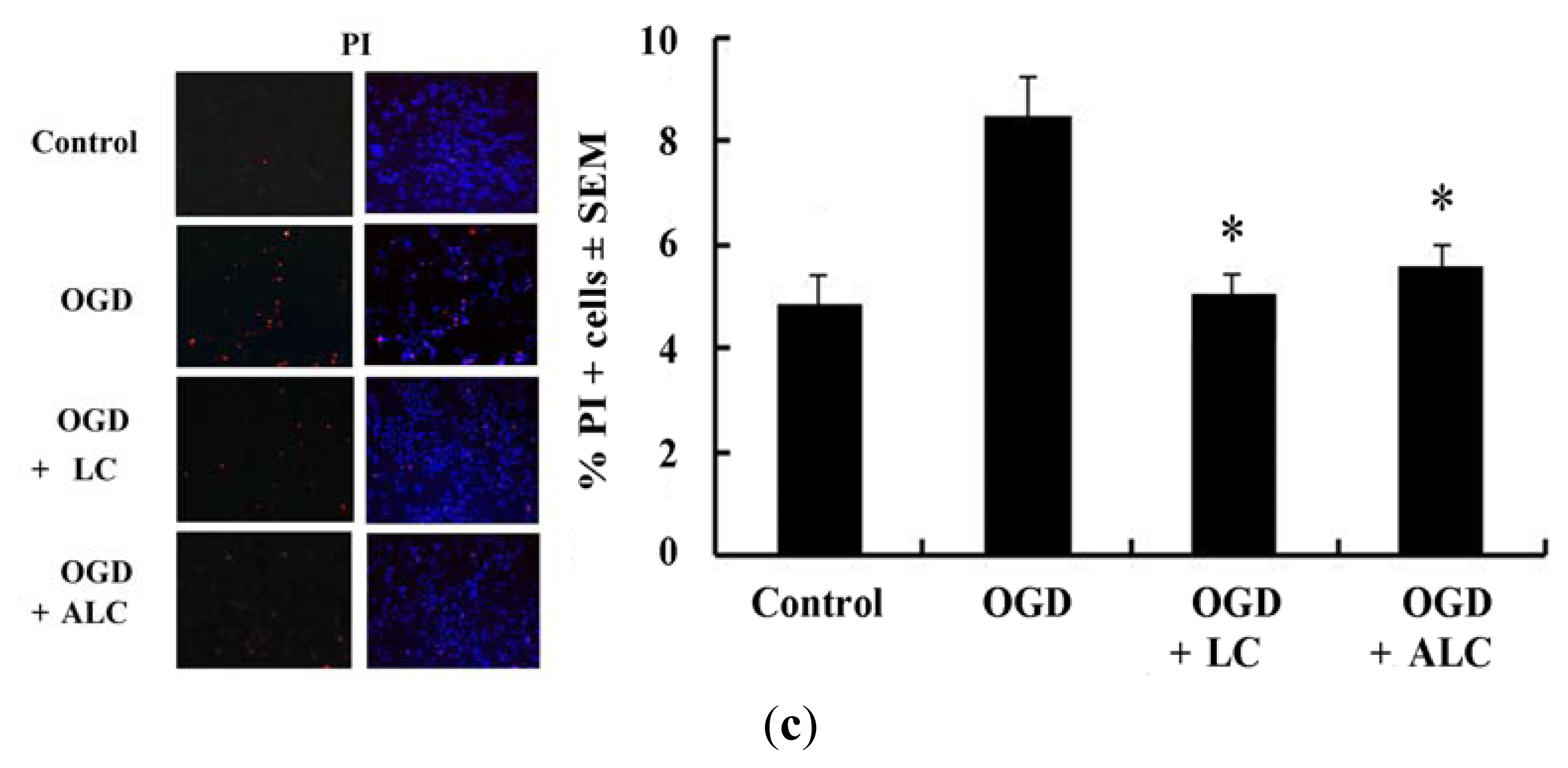
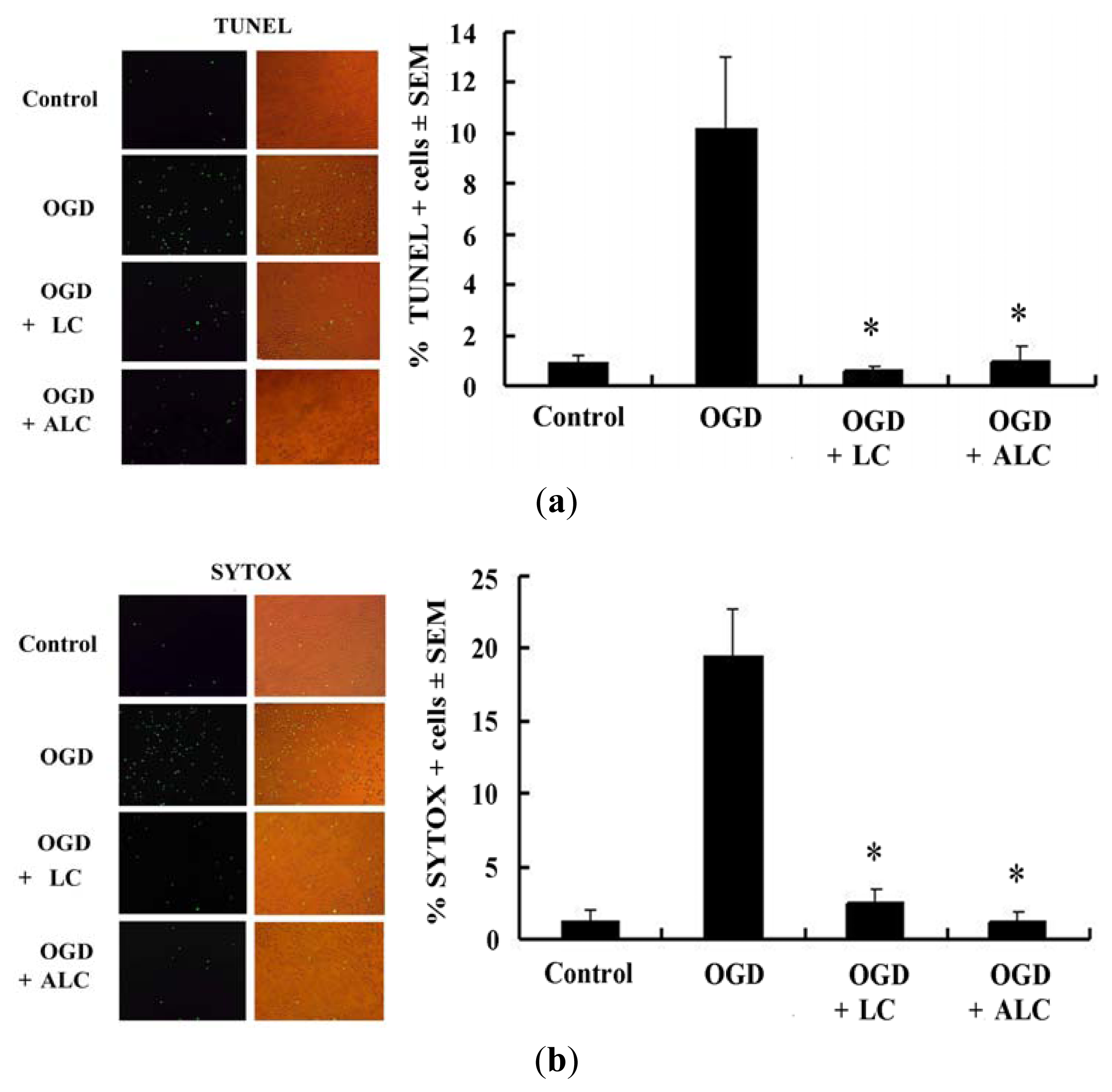
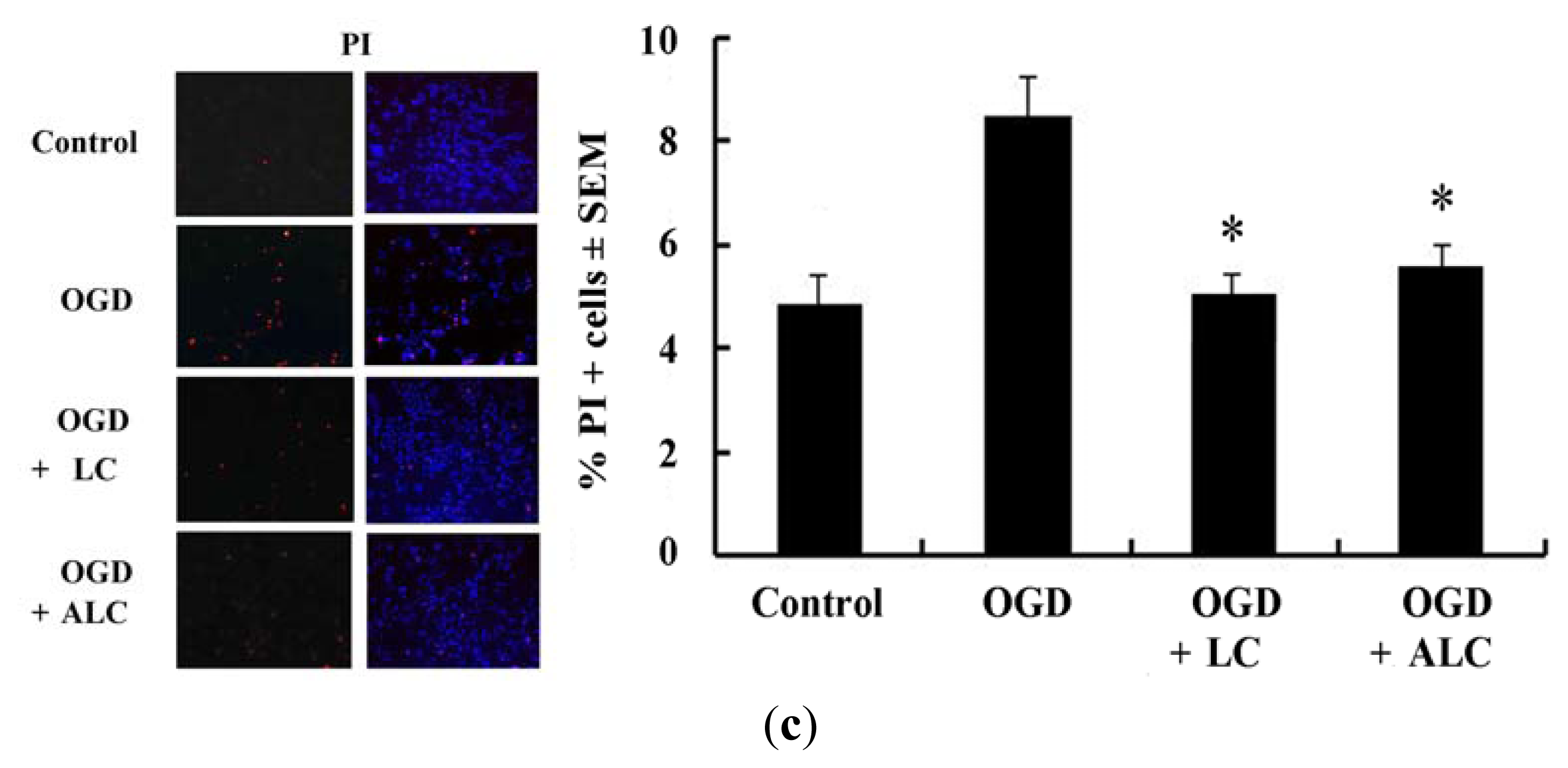
2.3. Pretreatment with LC or ALC Decreases OGD-Induced Cell Apoptosis and Death
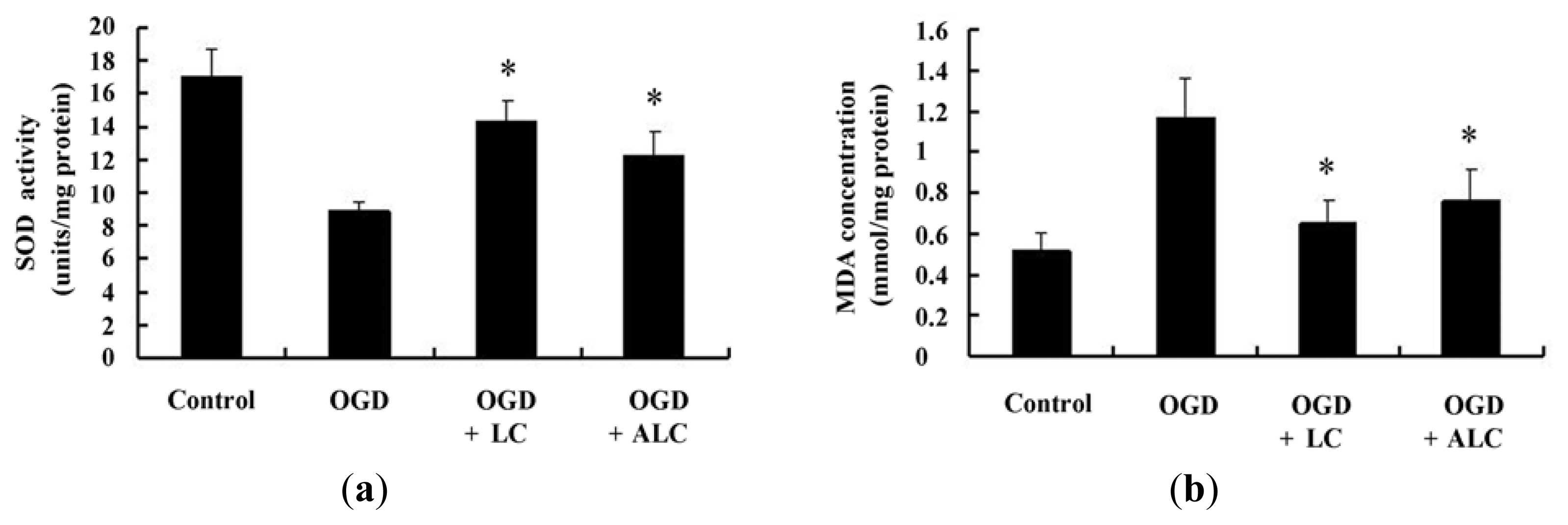
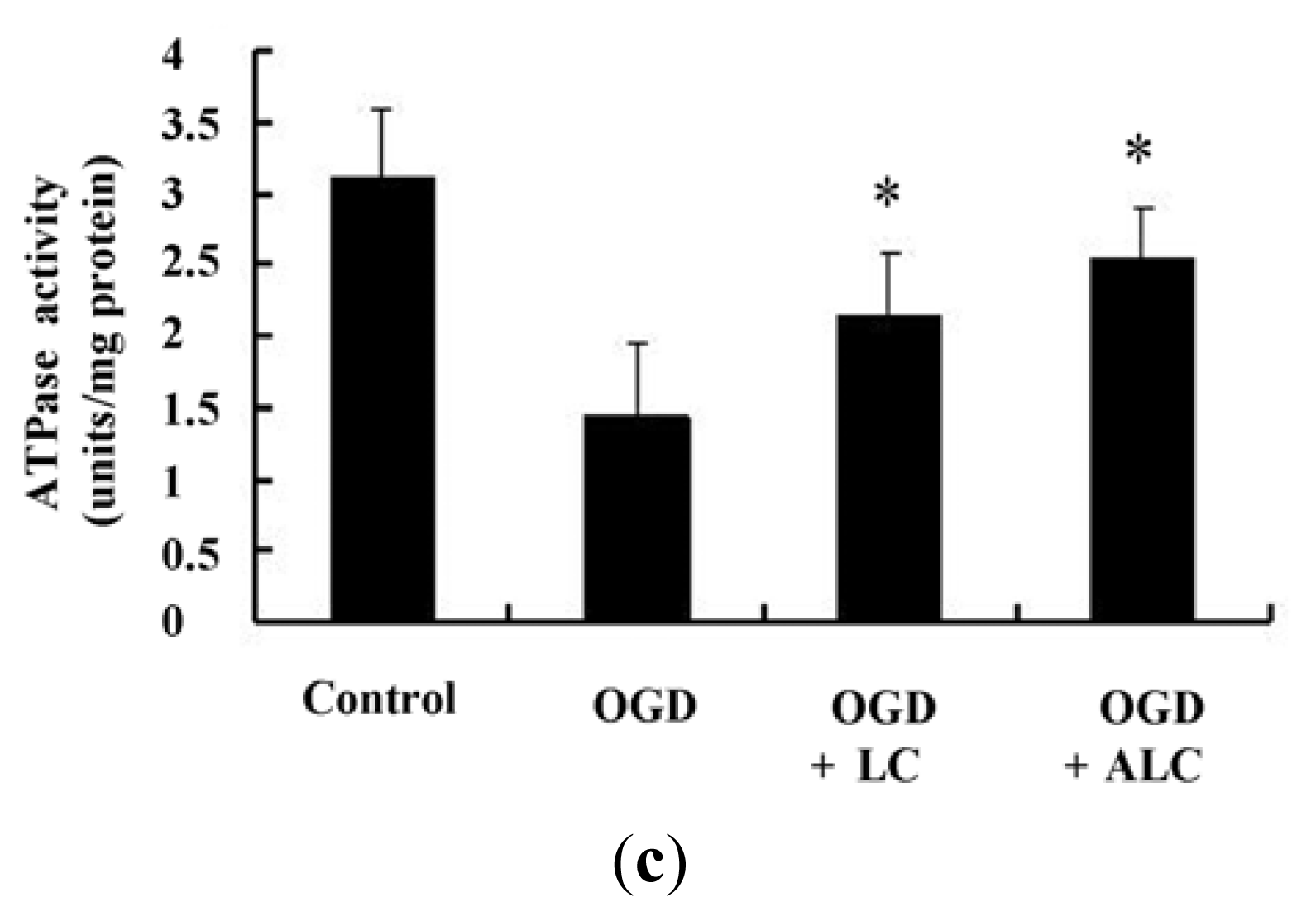
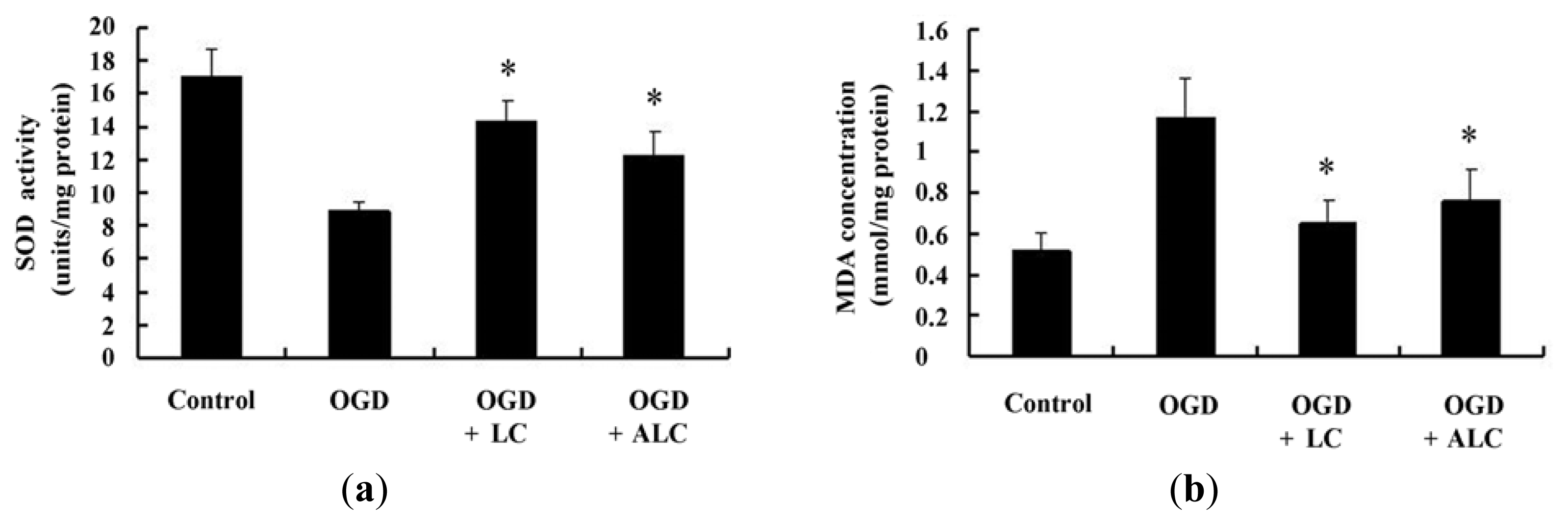
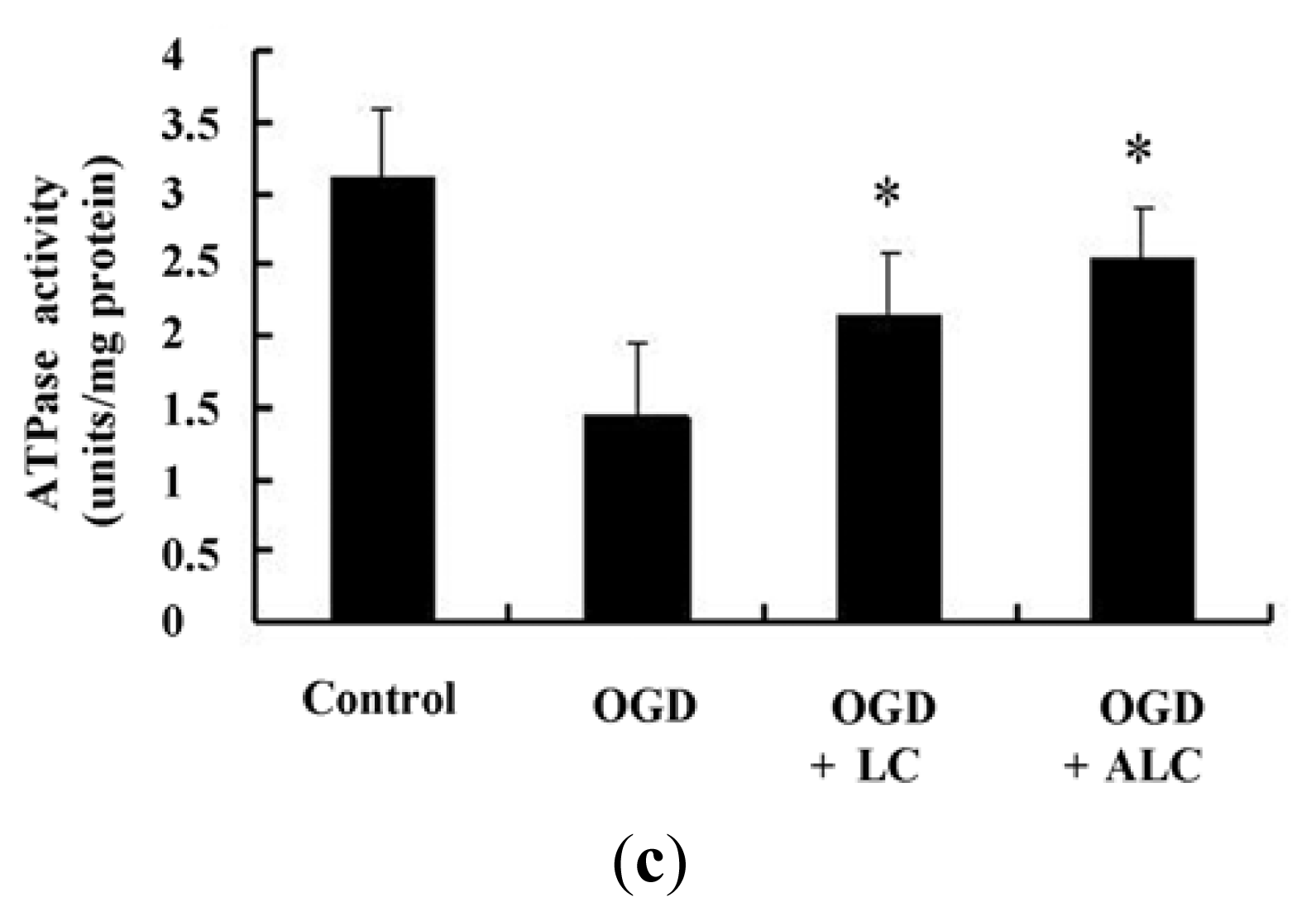
2.4. Effects of LC or ALC on the SOD, MDA, and ATPase in OGD-Induced PC12 Cells
3. Discussion
4. Experimental Section
4.1. Experimental Animals
4.2. Transient Middle Cerebral Artery Occlusion (MCAO)
4.3. Measurement of the Infarct Size
4.4. Cell Culture and Treatments
4.5. Oxygen-Glucose Deprivation (OGD)
4.6. Cell Viability Assay
4.7. Measurement of Cell Death and Apoptosis
4.8. Measurement of SOD, ATPase Activities and MDA Content
4.9. Statistical Analysis
5. Conclusions
Acknowledgments
References
- Murray, C.J.; Lopez, A.D. Mortality by cause for eight regions of the world: Global burden of disease study. Lancet 1997, 349, 1269–1276. [Google Scholar]
- Deshpande, J.K.; Siesjo, B.K.; Wieloch, T. Calcium accumulation and neuronal damage in the rat hippocampus following cerebral ischemia. J. Cereb. Blood Flow Metab 1987, 7, 89–95. [Google Scholar]
- MacManus, J.P.; Buchan, A.M.; Hill, I.E.; Rasquinha, I.; Preston, E. Global ischemia can cause DNA fragmentation indicative of apoptosis in rat brain. Neurosci. Lett 1993, 164, 89–92. [Google Scholar]
- Leist, M.; Jaattela, M. Four deaths and a funeral: From caspases to alternative mechanisms. Nat. Rev. Mol. Cell Biol 2001, 2, 589–598. [Google Scholar]
- Picconi, B.; Barone, I.; Pisani, A.; Nicolai, R.; Benatti, P.; Bernardi, G.; Calvani, M.; Calabresi, P. Acetyl-l-carnitine protects striatal neurons against in vitro ischemia: The role of endogenous acetylcholine. Neuropharmacology 2006, 50, 917–923. [Google Scholar]
- Zivin, J.A. Neuroprotective therapies in stroke. Drugs 1997, 54, 83–88, discussion 88–89. [Google Scholar]
- Flanagan, J.L.; Simmons, P.A.; Vehige, J.; Willcox, M.D.; Garrett, Q. Role of carnitine in disease. Nutr. Metab. (Lond.) 2010, 7, 30–43. [Google Scholar]
- Virmani, A.; Binienda, Z. Role of carnitine esters in brain neuropathology. Mol. Aspects Med 2004, 25, 533–549. [Google Scholar]
- Wainwright, M.S.; Kohli, R.; Whitington, P.F.; Chace, D.H. Carnitine treatment inhibits increases in cerebral carnitine esters and glutamate detected by mass spectrometry after hypoxia-ischemia in newborn rats. Stroke 2006, 37, 524–530. [Google Scholar]
- Zanelli, S.A.; Solenski, N.J.; Rosenthal, R.E.; Fiskum, G. Mechanisms of ischemic neuroprotection by acetyl-l-carnitine. Ann. N.Y. Acad. Sci 2005, 1053, 153–161. [Google Scholar]
- Slivka, A.; Silbersweig, D.; Pulsinelli, W. Carnitine treatment for stroke in rats. Stroke 1990, 21, 808–811. [Google Scholar]
- Turkyilmaz, C.; Turkyilmaz, Z.; Onal, E.; Atalay, Y.; Soylemezoglu, F.; Celasun, B. l-Carnitine pre-treatment reduces apoptotic cell death in seven-day-old rats hypoxia ischemia. Restor. Neurol. Neurosci 2010, 28, 817–824. [Google Scholar]
- Jalal, F.Y.; Bohlke, M.; Maher, T.J. Acetyl-l-carnitine reduces the infarct size and striatal glutamate outflow following focal cerebral ischemia in rats. Ann. N.Y. Acad. Sci 2010, 1199, 95–104. [Google Scholar]
- Sitprija, V. Altered fluid, electrolyte and mineral status in tropical disease, with an emphasis on malaria and leptospirosis. Nat. Clin. Pract. Nephrol 2008, 4, 91–101. [Google Scholar]
- Llansola, M.; Felipo, V. Carnitine prevents NMDA receptor-mediated activation of MAP-kinase and phosphorylation of microtubule-associated protein 2 in cerebellar neurons in culture. Brain Res 2002, 947, 50–56. [Google Scholar]
- Breitkreutz, R.; Babylon, A.; Hack, V.; Schuster, K.; Tokus, M.; Bohles, H.; Hagmuller, E.; Edler, L.; Holm, E.; Droge, W. Effect of carnitine on muscular glutamate uptake and intramuscular glutathione in malignant diseases. Br. J. Cancer 2000, 82, 399–403. [Google Scholar]
- Ferrari-Toninelli, G.; Maccarinelli, G.; Uberti, D.; Buerger, E.; Memo, M. Mitochondria-targeted antioxidant effects of S(−) and R(+) pramipexole. BMC Pharmacol 2010, 10. [Google Scholar] [CrossRef]
- Garrett, Q.; Xu, S.; Simmons, P.A.; Vehige, J.; Flanagan, J.L.; Willcox, M.D. Expression and localization of carnitine/organic cation transporter OCTN1 and OCTN2 in ocular epithelium. Invest. Ophthalmol. Vis. Sci 2008, 49, 4844–4849. [Google Scholar]
- Inazu, M.; Takeda, H.; Maehara, K.; Miyashita, K.; Tomoda, A.; Matsumiya, T. Functional expression of the organic cation/carnitine transporter 2 in rat astrocytes. J. Neurochem 2006, 97, 424–434. [Google Scholar]
- Muneer, P.M.A.; Alikunju, S.; Szlachetka, A.M.; Mercer, A.J.; Haorah, J. Ethanol impairs glucose uptake by human astrocytes and neurons: Protective effects of acetyl-l-carnitine. Int. J. Physiol. Pathophysiol. Pharmacol 2011, 3, 48–56. [Google Scholar]
- Scafidi, S.; Fiskum, G.; Lindauer, S.L.; Bamford, P.; Shi, D.; Hopkins, I.; McKenna, M.C. Metabolism of acetyl-l-carnitine for energy and neurotransmitter synthesis in the immature rat brain. J. Neurochem 2010, 114, 820–831. [Google Scholar]
- Singh, G.; Siddiqui, M.A.; Khanna, V.K.; Kashyap, M.P.; Yadav, S.; Gupta, Y.K.; Pant, K.K.; Pant, A.B. Oxygen glucose deprivation model of cerebral stroke in PC-12 cells: Glucose as a limiting factor. Toxicol. Mech. Methods 2009, 19, 154–160. [Google Scholar]
- Hota, K.B.; Hota, S.K.; Chaurasia, O.P.; Singh, S.B. Acetyl-l-carnitine-mediated neuroprotection during hypoxia is attributed to ERK1/2-Nrf2-regulated mitochondrial biosynthesis. Hippocampus 2011. [Google Scholar] [CrossRef]
- Yuan, J. Neuroprotective strategies targeting apoptotic and necrotic cell death for stroke. Apoptosis 2009, 14, 469–477. [Google Scholar]
- Qi, J.; Hong, Z.Y.; Xin, H.; Zhu, Y.Z. Neuroprotective effects of leonurine on ischemia/reperfusion-induced mitochondrial dysfunctions in rat cerebral cortex. Biol. Pharm. Bull 2010, 33, 1958–1964. [Google Scholar]
- Siktar, E.; Ekinci, D.; Beydemir, S.; Gulcin, I.; Gunay, M. Protective role of l-carnitine supplementation against exhaustive exercise induced oxidative stress in rats. Eur. J. Pharmacol 2011, 668, 407–413. [Google Scholar]
- Kim, W.; Kim, D.W.; Yoo, D.Y.; Chung, J.Y.; Hwang, I.K.; Won, M.H.; Choi, S.Y.; Jeon, S.W.; Jeong, J.H.; Hwang, H.S.; et al. Neuroprotective effects of PEP-1-Cu, Zn-SOD against ischemic neuronal damage in the rabbit spinal cord. Neurochem. Res 2011, 37, 307–313. [Google Scholar]
- Haorah, J.; Floreani, N.A.; Knipe, B.; Persidsky, Y. Stabilization of superoxide dismutase by acetyl-l-carnitine in human brain endothelium during alcohol exposure: Novel protective approach. Free Radic. Biol. Med 2011, 51, 1601–1609. [Google Scholar]
- Derin, N.; Izgut-Uysal, V.N.; Agac, A.; Aliciguzel, Y.; Demir, N. l-Carnitine protects gastric mucosa by decreasing ischemia-reperfusion induced lipid peroxidation. J. Physiol. Pharmacol 2004, 55, 595–606. [Google Scholar]
- Shuaib, A.; Waqaar, T.; Wishart, T.; Kanthan, R.; Howlett, W. Acetyl-l-carnitine attenuates neuronal damage in gerbils with transient forebrain ischemia only when given before the insult. Neurochem. Res 1995, 20, 1021–1025. [Google Scholar]
- Lin, T.N.; He, Y.Y.; Wu, G.; Khan, M.; Hsu, C.Y. Effect of brain edema on infarct volume in a focal cerebral ischemia model in rats. Stroke 1993, 24, 117–121. [Google Scholar]
- SPSS, version 16.0; IBM Corporation: Armonk, NY, USA, 2008.
© 2012 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Zhang, R.; Zhang, H.; Zhang, Z.; Wang, T.; Niu, J.; Cui, D.; Xu, S. Neuroprotective Effects of Pre-Treament with l-Carnitine and Acetyl-l-Carnitine on Ischemic Injury In Vivo and In Vitro. Int. J. Mol. Sci. 2012, 13, 2078-2090. https://doi.org/10.3390/ijms13022078
Zhang R, Zhang H, Zhang Z, Wang T, Niu J, Cui D, Xu S. Neuroprotective Effects of Pre-Treament with l-Carnitine and Acetyl-l-Carnitine on Ischemic Injury In Vivo and In Vitro. International Journal of Molecular Sciences. 2012; 13(2):2078-2090. https://doi.org/10.3390/ijms13022078
Chicago/Turabian StyleZhang, Rui, Hong Zhang, Zhongxia Zhang, Tao Wang, Jingya Niu, Dongsheng Cui, and Shunjiang Xu. 2012. "Neuroprotective Effects of Pre-Treament with l-Carnitine and Acetyl-l-Carnitine on Ischemic Injury In Vivo and In Vitro" International Journal of Molecular Sciences 13, no. 2: 2078-2090. https://doi.org/10.3390/ijms13022078
APA StyleZhang, R., Zhang, H., Zhang, Z., Wang, T., Niu, J., Cui, D., & Xu, S. (2012). Neuroprotective Effects of Pre-Treament with l-Carnitine and Acetyl-l-Carnitine on Ischemic Injury In Vivo and In Vitro. International Journal of Molecular Sciences, 13(2), 2078-2090. https://doi.org/10.3390/ijms13022078