The mTOR Signalling Pathway in Human Cancer




Abstract
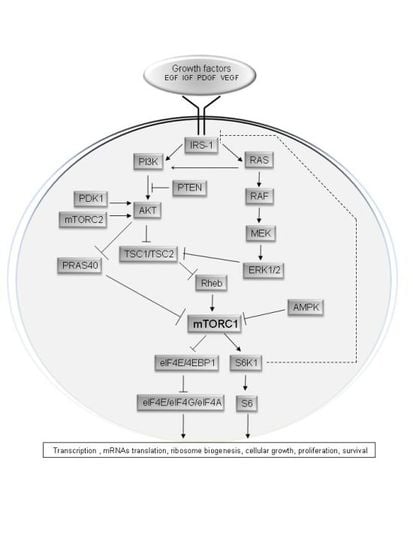
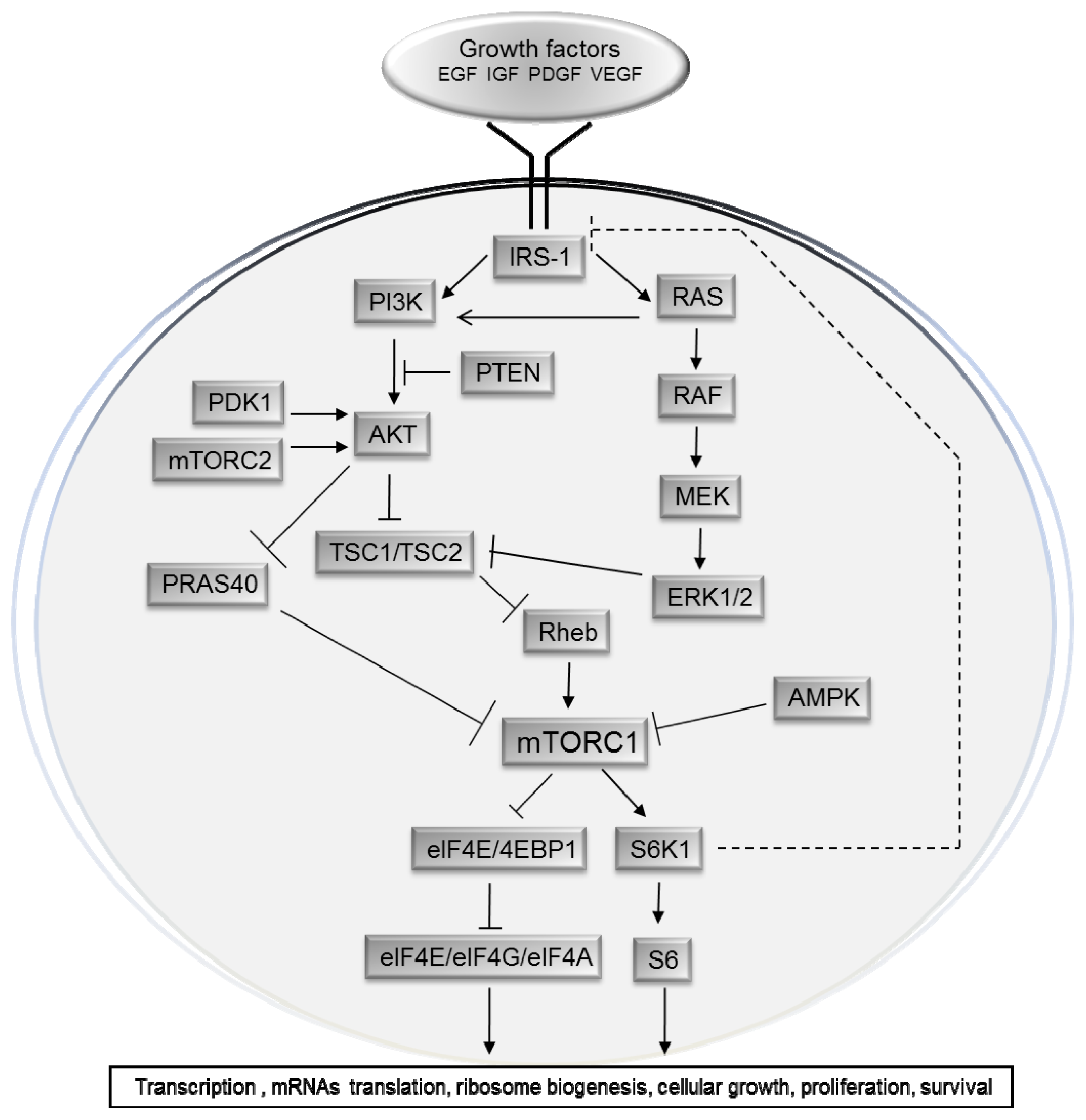
:
1. Introduction
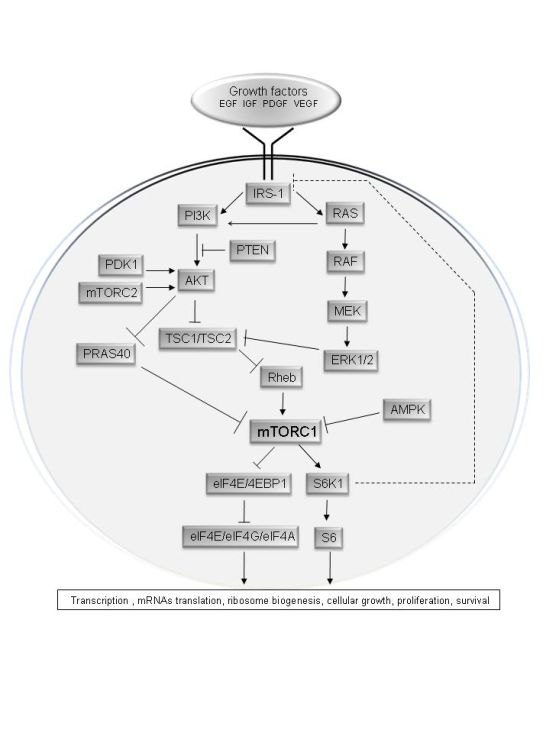
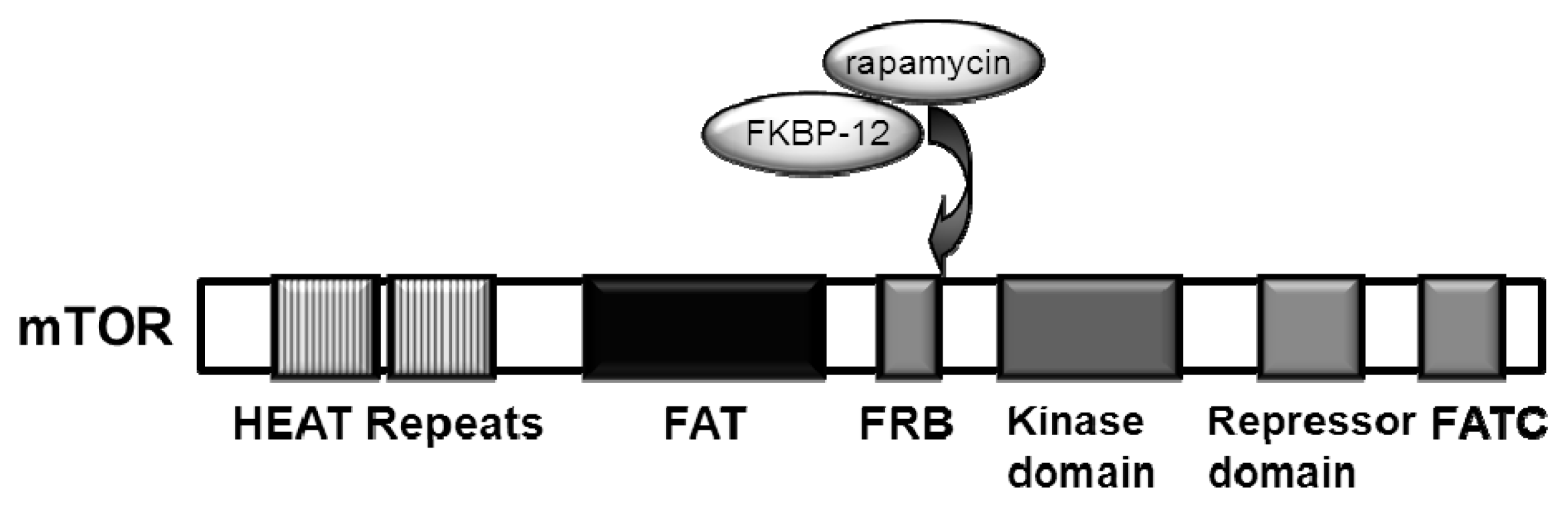
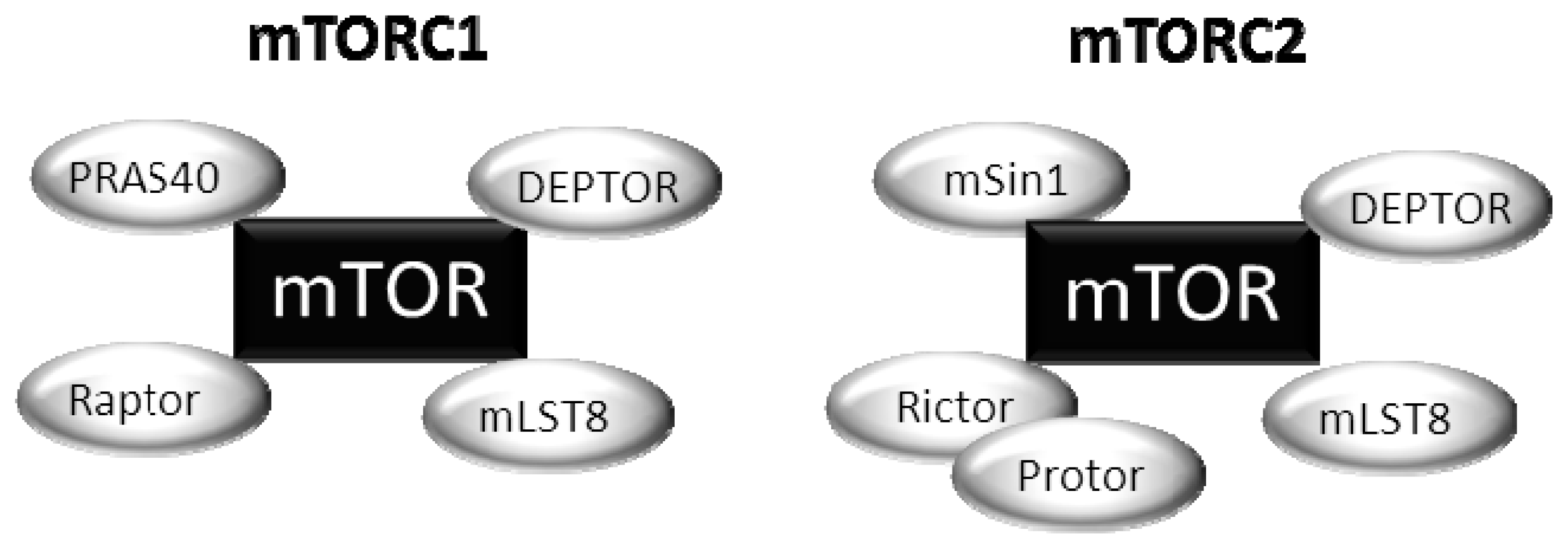
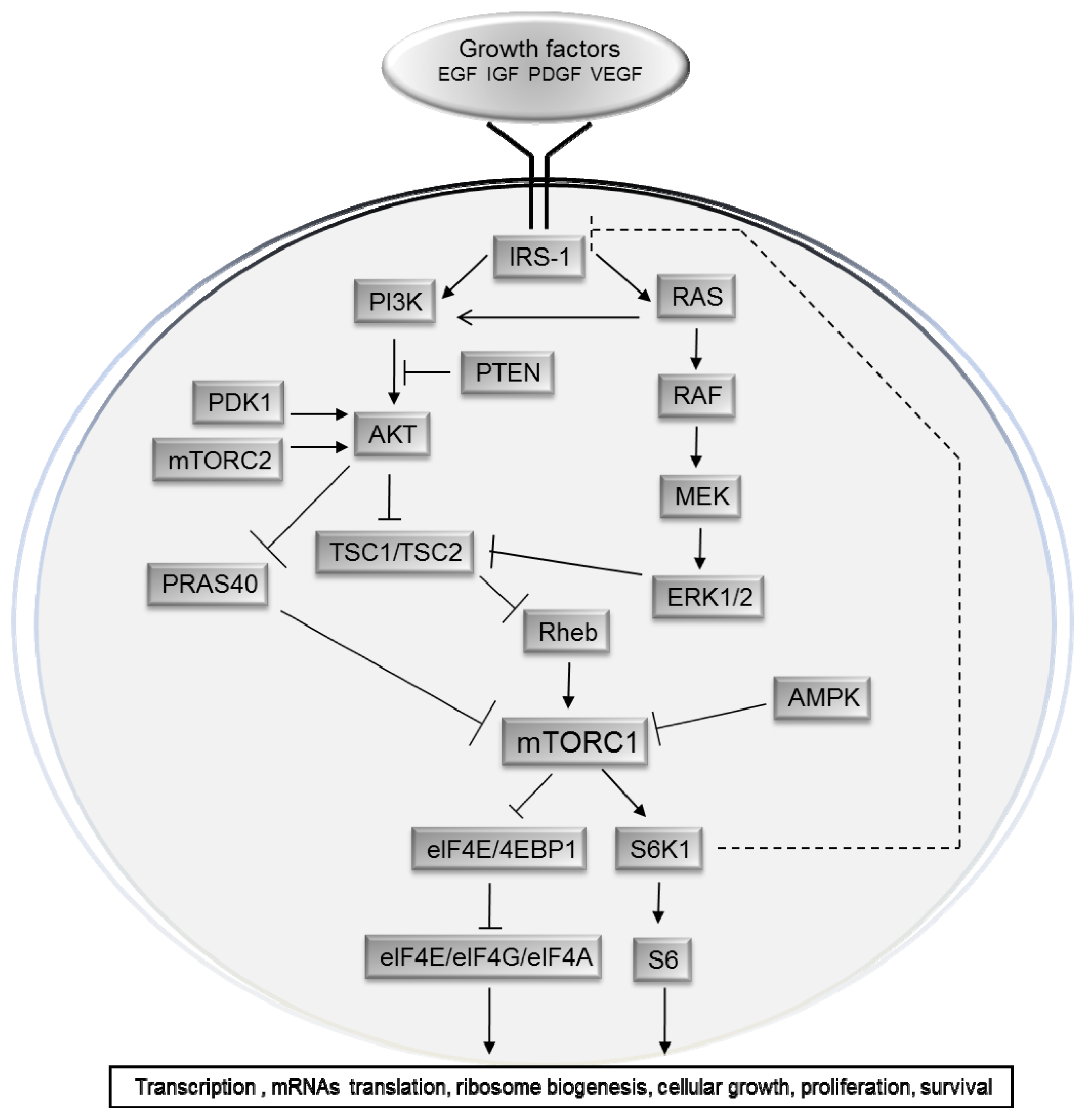
2. Upstream Regulation of the mTOR Pathway
3. Physiological Roles of the mTOR Pathway in Control of Growth and Lifespan
4. The mTOR Pathway in Cancer
5. The mTOR Pathway in Melanoma
6. mTOR Pathway Inhibitors in Cancer Therapy
7. mTOR Therapy Predictive Biomarkers
8. Conclusions
Acknowledgments
- Conflict of InterestThe authors declare no conflict of interest.
Non-Standard Abbreviations
| 4EBP1 | Eukaryotic translation initiation factor 4E-binding protein 1 |
| AMPK | 5′ Adenosine monophosphate-activated protein kinase |
| AKT/PKB | Protein kinase B |
| Bcl-2 | B-cell lymphoma 2 |
| BRAF | v-RAF murine sarcoma viral oncogene homolog B |
| DEPTOR | DEP domain-containing mTOR-interacting protein |
| eEF2K | Eukaryotic elongation factor-2 kinase |
| EGFR | Epidermal growth factor receptor |
| eIF4E | Eucaryotic translation initiation factor 4E |
| ERK | Extracellular regulated MAP kinase |
| FDA | Food and drug administration |
| FOXO | Forkhead transcriptor factor |
| GAP | Gtpase-activating protein |
| GNAQ | Guanine nucleotide-binding protein G(q) subunit alpha |
| GSK3 | Glycogen synthase kinase 3 |
| HIF-1 | Hypoxia-inducible factor 1 |
| Hsp70 | Heat shock protein 70-alpha |
| IRS-1 | Insulin receptor substrate 1 |
| KIT | v-kit Hardy-Zuckerman 4 feline sarcoma viral oncogene homolog |
| MAPK | Mitogen-activated protein kinase |
| MEK | Mitogen activated protein kinase kinase |
| MITF | Microphthalmia-associated transcription factor |
| mLST8 | Mammalian lethal with SEC13 protein 8 |
| mSin1 | Mammalian stress activated protein kinase interacting protein 1 |
| mTOR | Mammalian target of rapamycin |
| mTORC1/2 | mTOR complex 1/2 |
| MYC | Myelocytomatosis viral oncogene |
| NRAS | Neuroblastoma RAS viral (v-ras) oncogene homolog |
| PARP | Poly (ADP-ribose) polymerase |
| PDK 1 | Phosphoinositide-dependent kinase 1 |
| PI3K | Phosphatidylinositol 3-kinase |
| PIP2/ PtdIns(4,5)P2 | Phosphatidylinositol-4,5-bisphosphate |
| PIP3/PtdIns(3,4,5)P3 | Phosphatidylinositol-3,4,5-triphosphate |
| PKC | Protein kinase C |
| PRAS40 | Proline rich Akt substrate 40 |
| PRR5/Protor | Proline rich protein 5 / protein observed with rictor |
| PTEN | Phosphatase and tensin homologue deleted on chromosome ten |
| Raptor | Regulatory associated protein of mTOR |
| RAS | Rat sarcoma virus oncogene |
| REDD1 | Protein regulated in development and DNA damage response 1 |
| Rheb | Ras homolog enriched in brain |
| Rictor | Rapamycin insensitive companion of mTOR |
| RSK | p90 ribosomal S6 kinase |
| RTK | Receptor tyrosine kinase |
| S6K | S6 kinase |
| STAT | Signal transducer and activator of transcription |
| TSC 1/2 | Tuberous sclerosis complex 1/2 |
| VEGFR | Vascular Endothelial Growth Factor Receptor |
References
- Huang, S.; Houghton, P.J. Targeting mTOR signaling for cancer therapy. Curr. Opin. Pharmacol 2003, 3, 371–377. [Google Scholar]
- Wullschleger, S.; Loewith, R.; Hall, M.N. TOR signaling in growth and metabolism. Cell 2006, 124, 471–484. [Google Scholar]
- Guertin, D.A.; Sabatini, D.M. Defining the role of mTOR in cancer. Cancer Cell 2007, 12, 9–22. [Google Scholar]
- Heitman, J.; Movva, N.R.; Hall, M.N. Targets for cell cycle arrest by the immunosuppressant rapamycin in yeast. Science 1991, 253, 905–909. [Google Scholar]
- Sabers, C.J.; Martin, M.M.; Brunn, G.J.; Williams, J.M.; Dumont, F.J.; Wiederrecht, G.; Abraham, R.T. Isolation of a protein target of the FKBP12-rapamycin complex in mammalian cells. J. Biol. Chem 1995, 270, 815–822. [Google Scholar]
- Sabatini, D.M.; Erdjument-Bromage, H.; Lui, M.; Tempst, P.; Snyder, S.H. RAFT1: A mammalian protein that binds to FKBP12 in a rapamycin-dependent fashion and is homologous to yeast TORs. Cell 1994, 78, 35–43. [Google Scholar]
- Brown, E.J.; Albers, M.W.; Shin, T.B.; Ichikawa, K.; Keith, C.T.; Lane, W.S.; Schreiber, S.L. A mammalian protein targeted by G1-arresting rapamycin-receptor complex. Nature 1994, 369, 756–758. [Google Scholar]
- Keith, C.T.; Schreiber, S.L. PIK-related kinases: DNA repair, recombination, and cell cycle checkpoints. Science 1995, 270, 50–51. [Google Scholar]
- Kunz, J.; Henriquez, R.; Schneider, U.; Deuter-Reinhard, M.; Movva, N.R.; Hall, M.N. Target of rapamycin in yeast, TOR2, is an essential phosphatidylinositol kinase homolog required for G1 progression. Cell 1993, 73, 585–596. [Google Scholar]
- Zhou, H.; Huang, S. The complexes of mammalian target of rapamycin. Curr. Protein Pept. Sci 2010, 11, 409–424. [Google Scholar]
- Loewith, R.; Jacinto, E.; Wullschleger, S.; Lorberg, A.; Crespo, J.L.; Bonenfant, D.; Oppliger, W.; Jenoe, P.; Hall, M.N. Two TOR complexes, only one of which is rapamycin sensitive, have distinct roles in cell growth control. Mol. Cell 2002, 10, 457–468. [Google Scholar]
- Sarbassov, D.D.; Ali, S.M.; Kim, D.H.; Guertin, D.A.; Latek, R.R.; Erdjument-Bromage, H.; Tempst, P.; Sabatini, D.M. Rictor, a novel binding partner of mTOR, defines a rapamycin-insensitive and raptor-independent pathway that regulates the cytoskeleton. Curr. Biol 2004, 14, 1296–1302. [Google Scholar]
- Hara, K.; Maruki, Y.; Long, X.; Yoshino, K.; Oshiro, N.; Hidayat, S.; Tokunaga, C.; Avruch, J.; Yonezawa, K. Raptor, a binding partner of target of rapamycin (TOR), mediates TOR action. Cell 2002, 110, 177–189. [Google Scholar]
- Kim, D.H.; Sarbassov, D.D.; Ali, S.M.; King, J.E.; Latek, R.R.; Erdjument-Bromage, H.; Tempst, P.; Sabatini, D.M. mTOR interacts with raptor to form a nutrient-sensitive complex that signals to the cell growth machinery. Cell 2002, 110, 163–175. [Google Scholar]
- Sancak, Y.; Thoreen, C.C.; Peterson, T.R.; Lindquist, R.A.; Kang, S.A.; Spooner, E.; Carr, S.A.; Sabatini, D.M. PRAS40 is an insulin-regulated inhibitor of the mTORC1 protein kinase. Mol. Cell 2007, 25, 903–915. [Google Scholar]
- Peterson, T.R.; Laplante, M.; Thoreen, C.C.; Sancak, Y.; Kang, S.A.; Kuehl, W.M.; Gray, N.S.; Sabatini, D.M. DEPTOR is an mTOR inhibitor frequently overexpressed in multiple myeloma cells and required for their survival. Cell 2009, 137, 873–886. [Google Scholar]
- Gwinn, D.M.; Shackelford, D.B.; Egan, D.F.; Mihaylova, M.M.; Mery, A.; Vasquez, D.S.; Turk, B.E.; Shaw, R.J. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol Cell 2008, 30, 214–226. [Google Scholar]
- Kim, D.H.; Sarbassov, D.D.; Ali, S.M.; Latek, R.R.; Guntur, K.V.; Erdjument-Bromage, H.; Tempst, P.; Sabatini, D.M. GβL, a positive regulator of the rapamycin-sensitive pathway required for the nutrient-sensitive interaction between raptor and mTOR. Mol. Cell 2003, 11, 895–904. [Google Scholar]
- Guertin, D.A.; Stevens, D.M.; Thoreen, C.C.; Burds, A.A.; Kalaany, N.Y.; Moffat, J.; Brown, M.; Fitzgerald, K.J.; Sabatini, D.M. Ablation in mice of the mTORC components raptor, rictor, or mLST8 reveals that mTORC2 is required for signaling to Akt-FOXO and PKCα, but not S6K1. Dev. Cell 2006, 11, 859–871. [Google Scholar]
- Zeng, Z.; Sarbassov dos, D.; Samudio, I.J.; Yee, K.W.; Munsell, M.F.; Ellen Jackson, C.; Giles, F.J.; Sabatini, D.M.; Andreeff, M.; Konopleva, M. Rapamycin derivatives reduce mTORC2 signaling and inhibit AKT activation in AML. Blood 2007, 109, 3509–3512. [Google Scholar]
- Hay, N.; Sonenberg, N. Upstream and downstream of mTOR. Genes Dev 2004, 18, 1926–1945. [Google Scholar]
- Choi, J.H.; Bertram, P.G.; Drenan, R.; Carvalho, J.; Zhou, H.H.; Zheng, X.F. The FKBP12-rapamycin-associated protein (FRAP) is a CLIP-170 kinase. EMBO Rep 2002, 3, 988–994. [Google Scholar]
- Redpath, N.T.; Foulstone, E.J.; Proud, C.G. Regulation of translation elongation factor-2 by insulin via a rapamycin-sensitive signalling pathway. EMBO J 1996, 15, 2291–2297. [Google Scholar]
- Seidel, E.R.; Ragan, V.L. Inhibition by rapamycin of ornithine decarboxylase and epithelial cell proliferation in intestinal IEC-6 cells in culture. Br. J. Pharmacol 1997, 120, 571–574. [Google Scholar]
- Azpiazu, I.; Saltiel, A.R.; DePaoli-Roach, A.A.; Lawrence, J.C. Regulation of both glycogen synthase and PHAS-I by insulin in rat skeletal muscle involves mitogen-activated protein kinase-independent and rapamycin-sensitive pathways. J. Biol. Chem 1996, 271, 5033–5039. [Google Scholar]
- Hudson, C.C.; Liu, M.; Chiang, G.G.; Otterness, D.M.; Loomis, D.C.; Kaper, F.; Giaccia, A.J.; Abraham, R.T. Regulation of hypoxia-inducible factor 1α expression and function by the mammalian target of rapamycin. Mol. Cell Biol 2002, 22, 7004–7014. [Google Scholar]
- Huffman, T.A.; Mothe-Satney, I.; Lawrence, J.C., Jr. Insulin-stimulated phosphorylation of lipin mediated by the mammalian target of rapamycin. Proc. Natl. Acad. Sci. USA 2002, 99, 1047–1052. [Google Scholar]
- Parekh, D.; Ziegler, W.; Yonezawa, K.; Hara, K.; Parker, P.J. Mammalian TOR controls one of two kinase pathways acting upon nPKCδ and nPKCɛ. J. Biol. Chem 1999, 274, 34758–34764. [Google Scholar]
- Peterson, R.T.; Desai, B.N.; Hardwick, J.S.; Schreiber, S.L. Protein phosphatase 2A interacts with the 70-kDa S6 kinase and is activated by inhibition of FKBP12-rapamycinassociated protein. Proc. Natl. Acad. Sci. USA 1999, 96, 4438–4442. [Google Scholar]
- Huang, S.; Liu, L.N.; Hosoi, H.; Dilling, M.B.; Shikata, T.; Houghton, P.J. p53/p21(CIP1) cooperate in enforcing rapamycin-induced G(1) arrest and determine the cellular response to rapamycin. Cancer Res 2001, 61, 3373–3381. [Google Scholar]
- Nourse, J.; Firpo, E.; Flanagan, W.M.; Coats, S.; Polyak, K.; Lee, M.H.; Massague, J.; Crabtree, G.R.; Roberts, J.M. Interleukin-2-mediated elimination of the p27Kip1 cyclin-dependent kinase inhibitor prevented by rapamycin. Nature 1994, 372, 570–573. [Google Scholar]
- Usui, I.; Haruta, T.; Iwata, M.; Takano, A.; Uno, T.; Kawahara, J.; Ueno, E.; Sasaoka, T.; Kobayashi, M. Retinoblastoma protein phosphorylation via PI 3-kinase and mTOR pathway regulates adipocyte differentiation. Biochem. Biophys. Res. Commun 2000, 275, 115–120. [Google Scholar]
- Yokogami, K.; Wakisaka, S.; Avruch, J.; Reeves, S.A. Serine phosphorylation and maximal activation of STAT3 during CNTF signaling is mediated by the rapamycin target mTOR. Curr. Biol 2000, 10, 47–50. [Google Scholar]
- Jacinto, E.; Loewith, R.; Schmidt, A.; Lin, S.; Ruegg, M.A.; Hall, A.; Hall, M.N. Mammalian TOR complex 2 controls the actin cytoskeleton and is rapamycin insensitive. Nat. Cell Biol 2004, 6, 1122–1128. [Google Scholar]
- Frias, M.A.; Thoreen, C.C.; Jaffe, J.D.; Schroder, W.; Sculley, T.; Carr, S.A.; Sabatini, D.M. mSin1 is necessary for Akt/PKB phosphorylation, and its isoforms define three distinct mTORC2s. Curr. Biol 2006, 16, 1865–1870. [Google Scholar]
- Pearce, L.R.; Huang, X.; Boudeau, J.; Pawlowski, R.; Wullschleger, S.; Deak, M.; Ibrahim, A.F.; Gourlay, R.; Magnuson, M.A.; Alessi, D.R. Identification of Protor as a novel Rictor-binding component of mTOR complex-2. Biochem. J 2007, 405, 513–522. [Google Scholar]
- Martin, J.; Masri, J.; Bernath, A.; Nishimura, R.N.; Gera, J. Hsp70 associates with Rictor and is required for mTORC2 formation and activity. Biochem. Biophys. Res. Commun 2008, 372, 578–583. [Google Scholar]
- Hresko, R.C.; Mueckler, M. mTOR.RICTOR is the Ser473 kinase for Akt/protein kinase B in 3T3-L1 adipocytes. J. Biol. Chem 2005, 280, 40406–40416. [Google Scholar]
- Sarbassov, D.D.; Ali, S.M.; Sengupta, S.; Sheen, J.H.; Hsu, P.P.; Bagley, A.F.; Markhard, A.L.; Sabatini, D.M. Prolonged rapamycin treatment inhibits mTORC2 assembly and Akt/PKB. Mol. Cell 2006, 22, 159–168. [Google Scholar]
- Um, S.H.; D’Alessio, D.; Thomas, G. Nutrient overload, insulin resistance, and ribosomal protein S6 kinase 1, S6K1. Cell Metab 2006, 3, 393–402. [Google Scholar]
- Alessi, D.R.; James, S.R.; Downes, C.P.; Holmes, A.B.; Gaffney, P.R.; Reese, C.B.; Cohen, P. Characterization of a 3-phosphoinositide-dependent protein kinase which phosphorylates and activates protein kinase Bα. Curr. Biol 1997, 7, 261–269. [Google Scholar]
- Stokoe, D.; Stephens, L.R.; Copeland, T.; Gaffney, P.R.; Reese, C.B.; Painter, G.F.; Holmes, A.B.; McCormick, F.; Hawkins, P.T. Dual role of phosphatidylinositol-3,4,5-trisphosphate in the activation of protein kinase B. Science 1997, 277, 567–570. [Google Scholar]
- Sarbassov, D.D.; Guertin, D.A.; Ali, S.M.; Sabatini, D.M. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science 2005, 307, 1098–1101. [Google Scholar]
- Stambolic, V.; Suzuki, A.; de la Pompa, J.L.; Brothers, G.M.; Mirtsos, C.; Sasaki, T.; Ruland, J.; Penninger, J.M.; Siderovski, D.P.; Mak, T.W. Negative regulation of PKB/Akt-dependent cell survival by the tumor suppressor PTEN. Cell 1998, 95, 29–39. [Google Scholar]
- Inoki, K.; Li, Y.; Zhu, T.; Wu, J.; Guan, K.L. TSC2 is phosphorylated and inhibited by Akt and suppresses mTOR signalling. Nat. Cell Biol 2002, 4, 648–657. [Google Scholar]
- Inoki, K.; Li, Y.; Xu, T.; Guan, K.L. Rheb GTPase is a direct target of TSC2 GAP activity and regulates mTOR signaling. Genes Dev 2003, 17, 1829–1834. [Google Scholar]
- Long, X.; Lin, Y.; Ortiz-Vega, S.; Yonezawa, K.; Avruch, J. Rheb binds and regulates the mTOR kinase. Curr. Biol 2005, 15, 702–713. [Google Scholar]
- Takahashi, K.; Nakagawa, M.; Young, S.G.; Yamanaka, S. Differential membrane localization of ERas and Rheb, two Ras-related proteins involved in the phosphatidylinositol 3-kinase/mTOR pathway. J. Biol. Chem 2005, 280, 32768–32774. [Google Scholar]
- Buerger, C.; DeVries, B.; Stambolic, V. Localization of Rheb to the endomembrane is critical for its signaling function. Biochem. Biophys. Res. Commun 2006, 344, 869–880. [Google Scholar]
- Kovacina, K.S.; Park, G.Y.; Bae, S.S.; Guzzetta, A.W.; Schaefer, E.; Birnbaum, M.J.; Roth, R.A. Identification of a proline-rich Akt substrate as a 14-3-3 binding partner. J Biol Chem 2003, 278, 10189–10194. [Google Scholar]
- Lim, H.K.; Choi, Y.A.; Park, W.; Lee, T.; Ryu, S.H.; Kim, S.Y.; Kim, J.R.; Kim, J.H.; Baek, S.H. Phosphatidic acid regulates systemic inflammatory responses by modulating the Akt-mammalian target of rapamycin-p70 S6 kinase 1 pathway. J. Biol. Chem 2003, 278, 45117–45127. [Google Scholar]
- Nojima, H.; Tokunaga, C.; Eguchi, S.; Oshiro, N.; Hidayat, S.; Yoshino, K.; Hara, K.; Tanaka, N.; Avruch, J.; Yonezawa, K. The mammalian target of rapamycin (mTOR) partner, raptor, binds the mTOR substrates p70 S6 kinase and 4E-BP1 through their TOR signaling (TOS) motif. J. Biol. Chem 2003, 278, 15461–15464. [Google Scholar]
- Schalm, S.S.; Fingar, D.C.; Sabatini, D.M.; Blenis, J. TOS motif-mediated raptor binding regulates 4E-BP1 multisite phosphorylation and function. Curr. Biol 2003, 13, 797–806. [Google Scholar]
- Schalm, S.S.; Blenis, J. Identification of a conserved motif required for mTOR signaling. Curr. Biol 2002, 12, 632–639. [Google Scholar]
- Dennis, P.B.; Pullen, N.; Kozma, S.C.; Thomas, G. The principal rapamycin-sensitive p70(s6k) phosphorylation sites, T-229 and T-389, are differentially regulated by rapamycin-insensitive kinase kinases. Mol. Cell Biol 1996, 16, 6242–6251. [Google Scholar]
- Faivre, S.; Kroemer, G.; Raymond, E. Current development of mTOR inhibitors as anticancer agents. Nat. Rev. Drug Discov 2006, 5, 671–688. [Google Scholar]
- Sonenberg, N.; Gingras, A.C. The mRNA 5′ cap-binding protein eIF4E and control of cell growth. Curr. Opin. Cell Biol 1998, 10, 268–275. [Google Scholar]
- Pause, A.; Belsham, G.J.; Gingras, A.C.; Donze, O.; Lin, T.A.; Lawrence, J.C., Jr; Sonenberg, N. Insulin-dependent stimulation of protein synthesis by phosphorylation of a regulator of 5′-cap function. Nature 1994, 371, 762–767. [Google Scholar]
- Jastrzebski, K.; Hannan, K.M.; Tchoubrieva, E.B.; Hannan, R.D.; Pearson, R.B. Coordinate regulation of ribosome biogenesis and function by the ribosomal protein S6 kinase, a key mediator of mTOR function. Growth Factors 2007, 25, 209–226. [Google Scholar]
- Harrington, L.S.; Findlay, G.M.; Gray, A.; Tolkacheva, T.; Wigfield, S.; Rebholz, H.; Barnett, J.; Leslie, N.R.; Cheng, S.; Shepherd, P.R.; et al. The TSC1-2 tumor suppressor controls insulin-PI3K signaling via regulation of IRS proteins. J. Cell Biol 2004, 166, 213–223. [Google Scholar]
- Shah, O.J.; Wang, Z.; Hunter, T. Inappropriate activation of the TSC/Rheb/mTOR/S6K cassette induces IRS1/2 depletion, insulin resistance, and cell survival deficiencies. Curr. Biol 2004, 14, 1650–1656. [Google Scholar]
- Manning, B.D.; Cantley, L.C. Rheb fills a GAP between TSC and TOR. Trends Biochem. Sci 2003, 28, 573–576. [Google Scholar]
- Carracedo, A.; Ma, L.; Teruya-Feldstein, J.; Rojo, F.; Salmena, L.; Alimonti, A.; Egia, A.; Sasaki, A.T.; Thomas, G.; Kozma, S.C.; et al. Inhibition of mTORC1 leads to MAPK pathway activation through a PI3K-dependent feedback loop in human cancer. J. Clin. Invest 2008, 118, 3065–3074. [Google Scholar]
- Vander Haar, E.; Lee, S.I.; Bandhakavi, S.; Griffin, T.J.; Kim, D.H. Insulin signalling to mTOR mediated by the Akt/PKB substrate PRAS40. Nat. Cell Biol 2007, 9, 316–323. [Google Scholar]
- Engelman, J.A.; Luo, J.; Cantley, L.C. The evolution of phosphatidylinositol 3-kinases as regulators of growth and metabolism. Nat. Rev. Genet 2006, 7, 606–619. [Google Scholar]
- Dann, S.G.; Thomas, G. The amino acid sensitive TOR pathway from yeast to mammals. FEBS Lett 2006, 580, 2821–2829. [Google Scholar]
- Kimball, S.R.; Jefferson, L.S. Signaling pathways and molecular mechanisms through which branched-chain amino acids mediate translational control of protein synthesis. J. Nutr 2006, 136, 227S–231S. [Google Scholar]
- Nobukuni, T.; Joaquin, M.; Roccio, M.; Dann, S.G.; Kim, S.Y.; Gulati, P.; Byfield, M.P.; Backer, J.M.; Natt, F.; Bos, J.L.; et al. Amino acids mediate mTOR/raptor signaling through activation of class 3 phosphatidylinositol 3OH-kinase. Proc. Natl. Acad. Sci. USA 2005, 102, 14238–14243. [Google Scholar]
- Byfield, M.P.; Murray, J.T.; Backer, J.M. hVps34 is a nutrient-regulated lipid kinase required for activation of p70 S6 kinase. J. Biol. Chem 2005, 280, 33076–33082. [Google Scholar]
- Inoki, K.; Zhu, T.; Guan, K.L. TSC2 mediates cellular energy response to control cell growth and survival. Cell 2003, 115, 577–590. [Google Scholar]
- Hahn-Windgassen, A.; Nogueira, V.; Chen, C.C.; Skeen, J.E.; Sonenberg, N.; Hay, N. Akt activates the mammalian target of rapamycin by regulating cellular ATP level and AMPK activity. J. Biol. Chem 2005, 280, 32081–32089. [Google Scholar]
- Sofer, A.; Lei, K.; Johannessen, C.M.; Ellisen, L.W. Regulation of mTOR and cell growth in response to energy stress by REDD1. Mol. Cell Biol 2005, 25, 5834–5845. [Google Scholar]
- Brugarolas, J.; Lei, K.; Hurley, R.L.; Manning, B.D.; Reiling, J.H.; Hafen, E.; Witters, L.A.; Ellisen, L.W.; Kaelin, W.G., Jr. Regulation of mTOR function in response to hypoxia by REDD1 and the TSC1/TSC2 tumor suppressor complex. Genes Dev. 2004, 18, 2893–2904. [Google Scholar] [Green Version]
- Shoshani, T.; Faerman, A.; Mett, I.; Zelin, E.; Tenne, T.; Gorodin, S.; Moshel, Y.; Elbaz, S.; Budanov, A.; Chajut, A.; et al. Identification of a novel hypoxia-inducible factor 1-responsive gene, RTP801, involved in apoptosis. Mol. Cell Biol 2002, 22, 2283–2293. [Google Scholar]
- Liu, L.; Cash, T.P.; Jones, R.G.; Keith, B.; Thompson, C.B.; Simon, M.C. Hypoxia-induced energy stress regulates mRNA translation and cell growth. Mol. Cell 2006, 21, 521–531. [Google Scholar]
- Wolthuis, R.M.; Bos, J.L. Ras caught in another affair: The exchange factors for Ral. Curr. Opin. Genet. Dev 1999, 9, 112–117. [Google Scholar]
- Repasky, G.A.; Chenette, E.J.; Der, C.J. Renewing the conspiracy theory debate: Does Raf function alone to mediate Ras oncogenesis? Trends Cell Biol 2004, 14, 639–647. [Google Scholar]
- Roberts, P.J.; Der, C.J. Targeting the Raf-MEK-ERK mitogen-activated protein kinase cascade for the treatment of cancer. Oncogene 2007, 26, 3291–3310. [Google Scholar]
- Schubbert, S.; Bollag, G.; Shannon, K. Deregulated Ras signaling in developmental disorders: New tricks for an old dog. Curr. Opin. Genet. Dev 2007, 17, 15–22. [Google Scholar]
- Ballif, B.A.; Roux, P.P.; Gerber, S.A.; MacKeigan, J.P.; Blenis, J.; Gygi, S.P. Quantitative phosphorylation profiling of the ERK/p90 ribosomal S6 kinase-signaling cassette and its targets, the tuberous sclerosis tumor suppressors. Proc. Natl. Acad. Sci. USA 2005, 102, 667–672. [Google Scholar]
- Ma, L.; Chen, Z.; Erdjument-Bromage, H.; Tempst, P.; Pandolfi, P.P. Phosphorylation and functional inactivation of TSC2 by Erk implications for tuberous sclerosis and cancer pathogenesis. Cell 2005, 121, 179–193. [Google Scholar]
- Ma, L.; Teruya-Feldstein, J.; Bonner, P.; Bernardi, R.; Franz, D.N.; Witte, D.; Cordon-Cardo, C.; Pandolfi, P.P. Identification of S664 TSC2 phosphorylation as a marker for extracellular signal-regulated kinase mediated mTOR activation in tuberous sclerosis and human cancer. Cancer Res 2007, 67, 7106–7112. [Google Scholar]
- Carriere, A.; Ray, H.; Blenis, J.; Roux, P.P. The RSK factors of activating the Ras/MAPK signaling cascade. Front. Biosci 2008, 13, 4258–4275. [Google Scholar]
- Pyronnet, S.; Imataka, H.; Gingras, A.C.; Fukunaga, R.; Hunter, T.; Sonenberg, N. Human eukaryotic translation initiation factor 4G (eIF4G) recruits mnk1 to phosphorylate eIF4E. EMBO J 1999, 18, 270–279. [Google Scholar]
- Scheper, G.C.; Morrice, N.A.; Kleijn, M.; Proud, C.G. The mitogen-activated protein kinase signal-integrating kinase Mnk2 is a eukaryotic initiation factor 4E kinase with high levels of basal activity in mammalian cells. Mol. Cell Biol 2001, 21, 743–754. [Google Scholar]
- Ozes, O.N.; Akca, H.; Mayo, L.D.; Gustin, J.A.; Maehama, T.; Dixon, J.E.; Donner, D.B. A phosphatidylinositol 3-kinase/Akt/mTOR pathway mediates and PTEN antagonizes tumor necrosis factor inhibition of insulin signaling through insulin receptor substrate-1. Proc. Natl. Acad. Sci. USA 2001, 98, 4640–4645. [Google Scholar]
- Glantschnig, H.; Fisher, J.E.; Wesolowski, G.; Rodan, G.A.; Reszka, A.A. M-CSF, TNFα and RANK ligand promote osteoclast survival by signaling through mTOR/S6 kinase. Cell Death Differ 2003, 10, 1165–1177. [Google Scholar]
- Lee, D.F.; Kuo, H.P.; Chen, C.T.; Hsu, J.M.; Chou, C.K.; Wei, Y.; Sun, H.L.; Li, L.Y.; Ping, B.; Huang, W.C.; et al. IKK β suppression of TSC1 links inflammation and tumor angiogenesis via the mTOR pathway. Cell 2007, 130, 440–455. [Google Scholar]
- Magnusson, C.; Vaux, D.L. Signalling by CD95 and TNF receptors: Not only life and death. Immunol. Cell Biol 1999, 77, 41–46. [Google Scholar]
- Karin, M. The IκB kinase—a bridge between inflammation and cancer. Cell Res 2008, 18, 334–342. [Google Scholar]
- Dan, H.C.; Baldwin, A.S. Differential involvement of IκB kinases α and β in cytokine- and insulin-induced mammalian target of rapamycin activation determined by Akt. J. Immunol 2008, 180, 7582–7589. [Google Scholar]
- Jacinto, E.; Facchinetti, V.; Liu, D.; Soto, N.; Wei, S.; Jung, S.Y.; Huang, Q.; Qin, J.; Su, B. SIN1/MIP1 maintains rictor-mTOR complex integrity and regulates Akt phosphorylation and substrate specificity. Cell 2006, 127, 125–137. [Google Scholar]
- Huang, J.; Dibble, C.C.; Matsuzaki, M.; Manning, B.D. The TSC1-TSC2 complex is required for proper activation of mTOR complex 2. Mol. Cell Biol 2008, 28, 4104–4115. [Google Scholar]
- Tremblay, F.; Brule, S.; Hee Um, S.; Li, Y.; Masuda, K.; Roden, M.; Sun, X.J.; Krebs, M.; Polakiewicz, R.D.; Thomas, G.; Marette, A. Identification of IRS-1 Ser-1101 as a target of S6K1 in nutrient- and obesity-induced insulin resistance. Proc. Natl. Acad. Sci. USA 2007, 104, 14056–14061. [Google Scholar]
- Oldham, S.; Montagne, J.; Radimerski, T.; Thomas, G.; Hafen, E. Genetic and biochemical characterization of dTOR, the Drosophila homolog of the target of rapamycin. Genes Dev 2000, 14, 2689–2694. [Google Scholar] [Green Version]
- Fingar, D.C.; Salama, S.; Tsou, C.; Harlow, E.; Blenis, J. Mammalian cell size is controlled by mTOR and its downstream targets S6K1 and 4EBP1/eIF4E. Genes Dev 2002, 16, 1472–1487. [Google Scholar]
- Audhya, A.; Loewith, R.; Parsons, A.B.; Gao, L.; Tabuchi, M.; Zhou, H.; Boone, C.; Hall, M.N.; Emr, S.D. Genome-wide lethality screen identifies new PI4,5P2 effectors that regulate the actin cytoskeleton. EMBO J 2004, 23, 3747–3757. [Google Scholar]
- Fadri, M.; Daquinag, A.; Wang, S.; Xue, T.; Kunz, J. The pleckstrin homology domain proteins Slm1 and Slm2 are required for actin cytoskeleton organization in yeast and bind phosphatidylinositol-4,5-bisphosphate and TORC2. Mol. Biol. Cell 2005, 16, 1883–1900. [Google Scholar]
- Peng, T.; Golub, T.R.; Sabatini, D.M. The immunosuppressant rapamycin mimics a starvation-like signal distinct from amino acid and glucose deprivation. Mol. Cell Biol 2002, 22, 5575–5584. [Google Scholar]
- Hannan, K.M.; Brandenburger, Y.; Jenkins, A.; Sharkey, K.; Cavanaugh, A.; Rothblum, L.; Moss, T.; Poortinga, G.; McArthur, G.A.; Pearson, R.B.; et al. mTOR-dependent regulation of ribosomal gene transcription requires S6K1 and is mediated by phosphorylation of the carboxy-terminal activation domain of the nucleolar transcription factor UBF. Mol. Cell Biol 2003, 23, 8862–8877. [Google Scholar]
- Mayer, C.; Zhao, J.; Yuan, X.; Grummt, I. mTOR-dependent activation of the transcription factor TIF-IA links rRNA synthesis to nutrient availability. Genes Dev 2004, 18, 423–434. [Google Scholar]
- Kristof, A.S.; Marks-Konczalik, J.; Billings, E.; Moss, J. Stimulation of signal transducer and activator of transcription-1 (STAT1)-dependent gene transcription by lipopolysaccharide and interferon-gamma is regulated by mammalian target of rapamycin. J. Biol. Chem 2003, 278, 33637–33644. [Google Scholar]
- Cardenas, M.E.; Cutler, N.S.; Lorenz, M.C.; Di Como, C.J.; Heitman, J. The TOR signaling cascade regulates gene expression in response to nutrients. Genes Dev 1999, 13, 3271–3279. [Google Scholar]
- Hardwick, J.S.; Kuruvilla, F.G.; Tong, J.K.; Shamji, A.F.; Schreiber, S.L. Rapamycin-modulated transcription defines the subset of nutrient-sensitive signaling pathways directly controlled by the Tor proteins. Proc. Natl. Acad. Sci. USA 1999, 96, 14866–14870. [Google Scholar]
- Powers, T.; Walter, P. Regulation of ribosome biogenesis by the rapamycin-sensitive TOR-signaling pathway in Saccharomyces cerevisiae. Mol. Biol. Cell 1999, 10, 987–1000. [Google Scholar]
- Levine, B.; Kroemer, G. Autophagy in the pathogenesis of disease. Cell 2008, 132, 27–42. [Google Scholar]
- Mizushima, N.; Levine, B.; Cuervo, A.M.; Klionsky, D.J. Autophagy fights disease through cellular self-digestion. Nature 2008, 451, 1069–1075. [Google Scholar]
- Levine, B.; Klionsky, D.J. Development by self-digestion: Molecular mechanisms and biological functions of autophagy. Dev. Cell 2004, 6, 463–477. [Google Scholar]
- Wu, H.; Yang, J.M.; Jin, S.; Zhang, H.; Hait, W.N. Elongation factor-2 kinase regulates autophagy in human glioblastoma cells. Cancer Res 2006, 66, 3015–3023. [Google Scholar]
- Yu, L.; McPhee, C.K.; Zheng, L.; Mardones, G.A.; Rong, Y.; Peng, J.; Mi, N.; Zhao, Y.; Liu, Z.; Wan, F.; et al. Termination of autophagy and reformation of lysosomes regulated by mTOR. Nature 2010, 465, 942–946. [Google Scholar]
- Tee, A.R.; Blenis, J. mTOR, translational control and human disease. Semin. Cell Dev. Biol 2005, 16, 29–37. [Google Scholar]
- Thomas, G.V.; Horvath, S.; Smith, B.L.; Crosby, K.; Lebel, L.A.; Schrage, M.; Said, J.; de Kernion, J.; Reiter, R.E.; Sawyers, C.L. Antibody-based profiling of the phosphoinositide 3-kinase pathway in clinical prostate cancer. Clin. Cancer Res 2004, 10, 8351–8356. [Google Scholar]
- Kim, J.E.; Chen, J. regulation of peroxisome proliferator-activated receptor-gamma activity by mammalian target of rapamycin and amino acids in adipogenesis. Diabetes 2004, 53, 2748–2756. [Google Scholar]
- Lazar, M.A. PPAR gamma, 10 years later. Biochimie 2005, 87, 9–13. [Google Scholar]
- Um, S.H.; Frigerio, F.; Watanabe, M.; Picard, F.; Joaquin, M.; Sticker, M.; Fumagalli, S.; Allegrini, P.R.; Kozma, S.C.; Auwerx, J.; Thomas, G. Absence of S6K1 protects against age- and diet-induced obesity while enhancing insulin sensitivity. Nature 2004, 431, 200–205. [Google Scholar]
- Martin, D.E.; Hall, M.N. The expanding TOR signaling network. Curr. Opin. Cell Biol 2005, 17, 158–166. [Google Scholar]
- Colman, R.J.; Anderson, R.M.; Johnson, S.C.; Kastman, E.K.; Kosmatka, K.J.; Beasley, T.M.; Allison, D.B.; Cruzen, C.; Simmons, H.A.; Kemnitz, J.W.; et al. Caloric restriction delays disease onset and mortality in rhesus monkeys. Science 2009, 325, 201–204. [Google Scholar]
- Harrison, D.E.; Strong, R.; Sharp, Z.D.; Nelson, J.F.; Astle, C.M.; Flurkey, K.; Nadon, N.L.; Wilkinson, J.E.; Frenkel, K.; Carter, C.S.; et al. Rapamycin fed late in life extends lifespan in genetically heterogeneous mice. Nature 2009, 460, 392–395. [Google Scholar]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar]
- Sato, T.; Nakashima, A.; Guo, L.; Coffman, K.; Tamanoi, F. Single amino-acid changes that confer constitutive activation of mTOR are discovered in human cancer. Oncogene 2010, 29, 2746–2752. [Google Scholar]
- Zhou, B.P.; Hu, M.C.; Miller, S.A.; Yu, Z.; Xia, W.; Lin, S.Y.; Hung, M.C. HER-2/neu blocks tumor necrosis factor-induced apoptosis via the Akt/NF-κB pathway. J. Biol. Chem 2000, 275, 8027–8031. [Google Scholar]
- Chung, J.; Bachelder, R.E.; Lipscomb, E.A.; Shaw, L.M.; Mercurio, A.M. Integrin (α6β4) regulation of eIF-4E activity and VEGF translation: A survival mechanism for carcinoma cells. J. Cell Biol 2002, 158, 165–174. [Google Scholar]
- Stemke-Hale, K.; Gonzalez-Angulo, A.M.; Lluch, A.; Neve, R.M.; Kuo, W.L.; Davies, M.; Carey, M.; Hu, Z.; Guan, Y.; Sahin, A.; et al. An integrative genomic and proteomic analysis of PIK3CA, PTEN, and AKT mutations in breast cancer. Cancer Res 2008, 68, 6084–6091. [Google Scholar]
- Sansal, I.; Sellers, W.R. The biology and clinical relevance of the PTEN tumor suppressor pathway. J. Clin. Oncol 2004, 22, 2954–2963. [Google Scholar]
- Tamguney, T.; Stokoe, D. New insights into PTEN. J. Cell Sci 2007, 120, 4071–4079. [Google Scholar]
- Vivanco, I.; Sawyers, C.L. The phosphatidylinositol 3-Kinase AKT pathway in human cancer. Nat. Rev. Cancer 2002, 2, 489–501. [Google Scholar]
- Armengol, G.; Rojo, F.; Castellvi, J.; Iglesias, C.; Cuatrecasas, M.; Pons, B.; Baselga, J.; Ramon y Cajal, S. 4E-binding protein 1: A key molecular “funnel factor” in human cancer with clinical implications. Cancer Res 2007, 67, 7551–7555. [Google Scholar]
- Coleman, L.J.; Peter, M.B.; Teall, T.J.; Brannan, R.A.; Hanby, A.M.; Honarpisheh, H.; Shaaban, A.M.; Smith, L.; Speirs, V.; Verghese, E.T.; et al. Combined analysis of eIF4E and 4E-binding protein expression predicts breast cancer survival and estimates eIF4E activity. Br. J. Cancer 2009, 100, 1393–1399. [Google Scholar]
- No, J.H.; Jeon, Y.T.; Park, I.A.; Kim, Y.B.; Kim, J.W.; Park, N.H.; Kang, S.B.; Han, J.Y.; Lim, J.M.; Song, Y.S. Activation of mTOR signaling pathway associated with adverse prognostic factors of epithelial ovarian cancer. Gynecol. Oncol 2011, 121, 8–12. [Google Scholar]
- Bjornsti, M.A.; Houghton, P.J. Lost in translation: Dysregulation of cap-dependent translation and cancer. Cancer Cell 2004, 5, 519–523. [Google Scholar]
- Ruggero, D.; Montanaro, L.; Ma, L.; Xu, W.; Londei, P.; Cordon-Cardo, C.; Pandolfi, P.P. The translation factor eIF-4E promotes tumor formation and cooperates with c-Myc in lymphomagenesis. Nat. Med 2004, 10, 484–486. [Google Scholar]
- Li, J.; Yen, C.; Liaw, D.; Podsypanina, K.; Bose, S.; Wang, S.I.; Puc, J.; Miliaresis, C.; Rodgers, L.; McCombie, R.; et al. PTEN, a putative protein tyrosine phosphatase gene mutated in human brain, breast, and prostate cancer. Science 1997, 275, 1943–1947. [Google Scholar]
- Campbell, I.G.; Russell, S.E.; Choong, D.Y.; Montgomery, K.G.; Ciavarella, M.L.; Hooi, C.S.; Cristiano, B.E.; Pearson, R.B.; Phillips, W.A. Mutation of the PIK3CA gene in ovarian and breast cancer. Cancer Res 2004, 64, 7678–7681. [Google Scholar]
- Shaw, R.J.; Cantley, L.C. Ras, PI(3)K and mTOR signalling controls tumour cell growth. Nature 2006, 441, 424–430. [Google Scholar]
- Basso, A.D.; Mirza, A.; Liu, G.; Long, B.J.; Bishop, W.R.; Kirschmeier, P. The farnesyl transferase inhibitor (FTI) SCH66336 (lonafarnib) inhibits Rheb farnesylation and mTOR signaling. Role in FTI enhancement of taxane and tamoxifen anti-tumor activity. J. Biol. Chem 2005, 280, 31101–31108. [Google Scholar]
- Lu, Z.H.; Shvartsman, M.B.; Lee, A.Y.; Shao, J.M.; Murray, M.M.; Kladney, R.D.; Fan, D.; Krajewski, S.; Chiang, G.G.; Mills, G.B.; et al. Mammalian target of rapamycin activator RHEB is frequently overexpressed in human carcinomas and is critical and sufficient for skin epithelial carcinogenesis. Cancer Res 2010, 70, 3287–3298. [Google Scholar]
- Barlund, M.; Forozan, F.; Kononen, J.; Bubendorf, L.; Chen, Y.; Bittner, M.L.; Torhorst, J.; Haas, P.; Bucher, C.; Sauter, G.; et al. Detecting activation of ribosomal protein S6 kinase by complementary DNA and tissue microarray analysis. J. Natl. Cancer Inst 2000, 92, 1252–1259. [Google Scholar]
- Populo, H.; Soares, P.; Faustino, A.; Rocha, A.S.; Silva, P.; Azevedo, F.; Lopes, J.M. mTOR pathway activation in cutaneous melanoma is associated with poorer prognosis characteristics. Pigment Cell Melanoma Res 2011, 24, 254–257. [Google Scholar]
- Hiramatsu, M.; Ninomiya, H.; Inamura, K.; Nomura, K.; Takeuchi, K.; Satoh, Y.; Okumura, S.; Nakagawa, K.; Yamori, T.; Matsuura, M.; et al. Activation status of receptor tyrosine kinase downstream pathways in primary lung adenocarcinoma with reference of KRAS and EGFR mutations. Lung Cancer 2010, 70, 94–102. [Google Scholar]
- Noh, W.C.; Kim, Y.H.; Kim, M.S.; Koh, J.S.; Kim, H.A.; Moon, N.M.; Paik, N.S. Activation of the mTOR signaling pathway in breast cancer and its correlation with the clinicopathologic variables. Breast Cancer Res. Treat 2008, 110, 477–483. [Google Scholar]
- Pantuck, A.J.; Seligson, D.B.; Klatte, T.; Yu, H.; Leppert, J.T.; Moore, L.; O’Toole, T.; Gibbons, J.; Belldegrun, A.S.; Figlin, R.A. Prognostic relevance of the mTOR pathway in renal cell carcinoma: Implications for molecular patient selection for targeted therapy. Cancer 2007, 109, 2257–2267. [Google Scholar]
- Zhou, L.; Huang, Y.; Li, J.; Wang, Z. The mTOR pathway is associated with the poor prognosis of human hepatocellular carcinoma. Med. Oncol 2010, 27, 255–261. [Google Scholar]
- Inoki, K.; Corradetti, M.N.; Guan, K.L. Dysregulation of the TSC-mTOR pathway in human disease. Nat. Genet 2005, 37, 19–24. [Google Scholar]
- Vignot, S.; Faivre, S.; Aguirre, D.; Raymond, E. mTOR-targeted therapy of cancer with rapamycin derivatives. Ann. Oncol 2005, 16, 525–537. [Google Scholar]
- Hager, M.; Haufe, H.; Kemmerling, R.; Mikuz, G.; Kolbitsch, C.; Moser, P.L. PTEN expression in renal cell carcinoma and oncocytoma and prognosis. Pathology 2007, 39, 482–485. [Google Scholar]
- Madhunapantula, S.V.; Robertson, G.P. The PTEN-AKT3 signaling cascade as a therapeutic target in melanoma. Pigment Cell Melanoma Res 2009, 22, 400–419. [Google Scholar]
- Johnson, S.R.; Tattersfield, A.E. Lymphangioleiomyomatosis. Semin. Respir. Crit. Care Med 2002, 23, 85–92. [Google Scholar]
- Kwiatkowski, D.J. Tuberous sclerosis: From tubers to mTOR. Ann. Hum. Genet 2003, 67, 87–96. [Google Scholar]
- Manning, B.D.; Cantley, L.C. United at last: The tuberous sclerosis complex gene products connect the phosphoinositide 3-kinase/Akt pathway to mammalian target of rapamycin (mTOR) signalling. Biochem. Soc. Trans 2003, 31, 573–578. [Google Scholar]
- De Benedetti, A.; Graff, J.R. eIF-4E expression and its role in malignancies and metastases. Oncogene 2004, 23, 3189–3199. [Google Scholar]
- Nakamura, J.L.; Garcia, E.; Pieper, R.O. S6K1 plays a key role in glial transformation. Cancer Res 2008, 68, 6516–6523. [Google Scholar]
- Liaw, D.; Marsh, D.J.; Li, J.; Dahia, P.L.; Wang, S.I.; Zheng, Z.; Bose, S.; Call, K.M.; Tsou, H.C.; Peacocke, M.; et al. Germline mutations of the PTEN gene in Cowden disease, an inherited breast and thyroid cancer syndrome. Nat. Genet 1997, 16, 64–67. [Google Scholar]
- Shaw, R.J.; Bardeesy, N.; Manning, B.D.; Lopez, L.; Kosmatka, M.; DePinho, R.A.; Cantley, L.C. The LKB1 tumor suppressor negatively regulates mTOR signaling. Cancer Cell 2004, 6, 91–99. [Google Scholar]
- Johannessen, C.M.; Reczek, E.E.; James, M.F.; Brems, H.; Legius, E.; Cichowski, K. The NF1 tumor suppressor critically regulates TSC2 and mTOR. Proc. Natl. Acad. Sci. USA 2005, 102, 8573–8578. [Google Scholar]
- Dahl, C.; Guldberg, P. The genome and epigenome of malignant melanoma. Apmis 2007, 115, 1161–1176. [Google Scholar]
- Dai, D.L.; Martinka, M.; Li, G. Prognostic significance of activated Akt expression in melanoma: A clinicopathologic study of 292 cases. J. Clin. Oncol 2005, 23, 1473–1482. [Google Scholar]
- Wu, H.; Goel, V.; Haluska, F.G. PTEN signaling pathways in melanoma. Oncogene 2003, 22, 3113–3122. [Google Scholar]
- Reifenberger, J.; Wolter, M.; Bostrom, J.; Buschges, R.; Schulte, K.W.; Megahed, M.; Ruzicka, T.; Reifenberger, G. Allelic losses on chromosome arm 10q and mutation of the PTEN (MMAC1) tumour suppressor gene in primary and metastatic malignant melanomas. Virchows Arch 2000, 436, 487–493. [Google Scholar]
- Celebi, J.T.; Shendrik, I.; Silvers, D.N.; Peacocke, M. Identification of PTEN mutations in metastatic melanoma specimens. J. Med. Genet 2000, 37, 653–657. [Google Scholar]
- Tsao, H.; Zhang, X.; Fowlkes, K.; Haluska, F.G. Relative reciprocity of NRAS and PTEN/MMAC1 alterations in cutaneous melanoma cell lines. Cancer Res 2000, 60, 1800–1804. [Google Scholar]
- Tsao, H.; Goel, V.; Wu, H.; Yang, G.; Haluska, F.G. Genetic interaction between NRAS and BRAF mutations and PTEN/MMAC1 inactivation in melanoma. J. Invest. Dermatol 2004, 122, 337–341. [Google Scholar]
- Davies, M.A.; Stemke-Hale, K.; Tellez, C.; Calderone, T.L.; Deng, W.; Prieto, V.G.; Lazar, A.J.; Gershenwald, J.E.; Mills, G.B. A novel AKT3 mutation in melanoma tumours and cell lines. Br. J. Cancer 2008, 99, 1265–1268. [Google Scholar]
- Dhawan, P.; Singh, A.B.; Ellis, D.L.; Richmond, A. Constitutive activation of Akt/protein kinase B in melanoma leads to up-regulation of nuclear factor-κB and tumor progression. Cancer Res 2002, 62, 7335–7342. [Google Scholar]
- Stahl, J.M.; Sharma, A.; Cheung, M.; Zimmerman, M.; Cheng, J.Q.; Bosenberg, M.W.; Kester, M.; Sandirasegarane, L.; Robertson, G.P. Deregulated Akt3 activity promotes development of malignant melanoma. Cancer Res 2004, 64, 7002–7010. [Google Scholar]
- Meier, F.; Schittek, B.; Busch, S.; Garbe, C.; Smalley, K.; Satyamoorthy, K.; Li, G.; Herlyn, M. The RAS/RAF/MEK/ERK and PI3K/AKT signaling pathways present molecular targets for the effective treatment of advanced melanoma. Front. Biosci 2005, 10, 2986–3001. [Google Scholar]
- Cheung, M.; Sharma, A.; Madhunapantula, S.V.; Robertson, G.P. Akt3 and mutant V600EB-Raf cooperate to promote early melanoma development. Cancer Res 2008, 68, 3429–3439. [Google Scholar]
- Madhunapantula, S.V.; Sharma, A.; Robertson, G.P. PRAS40 deregulates apoptosis in malignant melanoma. Cancer Res 2007, 67, 3626–3636. [Google Scholar]
- Saraiva, V.S.; Caissie, A.L.; Segal, L.; Edelstein, C.; Burnier, M.N., Jr. Immunohistochemical expression of phospho-Akt in uveal melanoma. Melanoma Res. 2005, 15, 245–250. [Google Scholar]
- Populo, H.; Soares, P.; Rocha, A.S.; Silva, P.; Lopes, J.M. Evaluation of the mTOR pathway in ocular (uvea and conjunctiva) melanoma. Melanoma Res 2010, 20, 107–117. [Google Scholar]
- Abdel-Rahman, M.H.; Yang, Y.; Zhou, X.P.; Craig, E.L.; Davidorf, F.H.; Eng, C. High frequency of submicroscopic hemizygous deletion is a major mechanism of loss of expression of PTEN in uveal melanoma. J. Clin. Oncol 2006, 24, 288–295. [Google Scholar]
- Smalley, K.S.; Eisen, T.G. Farnesyl transferase inhibitor SCH66336 is cytostatic, pro-apoptotic and enhances chemosensitivity to cisplatin in melanoma cells. Int. J. Cancer 2003, 105, 165–175. [Google Scholar]
- Easton, J.B.; Houghton, P.J. mTOR and cancer therapy. Oncogene 2006, 25, 6436–6446. [Google Scholar]
- Vezina, C.; Kudelski, A.; Sehgal, S.N. Rapamycin (AY-22,989), a new antifungal antibiotic. I. Taxonomy of the producing streptomycete and isolation of the active principle. J. Antibiot. (Tokyo) 1975, 28, 721–726. [Google Scholar]
- Eng, C.P.; Sehgal, S.N.; Vezina, C. Activity of rapamycin (AY-22,989) against transplanted tumors. J. Antibiot. (Tokyo) 1984, 37, 1231–1237. [Google Scholar]
- Linhares, M.M.; Gonzalez, A.M.; Trivino, T.; Melaragno, C.; Moura, R.M.; Garcez, M.H.; Sa, J.R.; Aguiar, W.F.; Succi, T.; Barbosa, C.S.; et al. Simultaneous pancreas-kidney transplantation initial experience. Transplant. Proc 2003, 35, 1109. [Google Scholar]
- Huang, S.; Houghton, P.J. Resistance to rapamycin: A novel anticancer drug. Cancer Metastasis Rev 2001, 20, 69–78. [Google Scholar]
- Dilling, M.B.; Dias, P.; Shapiro, D.N.; Germain, G.S.; Johnson, R.K.; Houghton, P.J. Rapamycin selectively inhibits the growth of childhood rhabdomyosarcoma cells through inhibition of signaling via the type I insulin-like growth factor receptor. Cancer Res 1994, 54, 903–907. [Google Scholar]
- Geoerger, B.; Kerr, K.; Tang, C.B.; Fung, K.M.; Powell, B.; Sutton, L.N.; Phillips, P.C.; Janss, A.J. Antitumor activity of the rapamycin analog CCI-779 in human primitive neuroectodermal tumor/medulloblastoma models as single agent and in combination chemotherapy. Cancer Res 2001, 61, 1527–1532. [Google Scholar]
- Seufferlein, T.; Rozengurt, E. Rapamycin inhibits constitutive p70s6k phosphorylation, cell proliferation, and colony formation in small cell lung cancer cells. Cancer Res 1996, 56, 3895–3897. [Google Scholar]
- Ogawa, T.; Tokuda, M.; Tomizawa, K.; Matsui, H.; Itano, T.; Konishi, R.; Nagahata, S.; Hatase, O. Osteoblastic differentiation is enhanced by rapamycin in rat osteoblast-like osteosarcoma (ROS 17/2.8) cells. Biochem. Biophys. Res. Commun 1998, 249, 226–230. [Google Scholar]
- Grewe, M.; Gansauge, F.; Schmid, R.M.; Adler, G.; Seufferlein, T. Regulation of cell growth and cyclin D1 expression by the constitutively active FRAP-p70s6K pathway in human pancreatic cancer cells. Cancer Res 1999, 59, 3581–3587. [Google Scholar]
- Pang, H.; Faber, L.E. Estrogen and rapamycin effects on cell cycle progression in T47D breast cancer cells. Breast Cancer Res. Treat 2001, 70, 21–26. [Google Scholar]
- van der Poel, H.G.; Hanrahan, C.; Zhong, H.; Simons, J.W. Rapamycin induces Smad activity in prostate cancer cell lines. Urol. Res 2003, 30, 380–386. [Google Scholar]
- Muthukkumar, S.; Ramesh, T.M.; Bondada, S. Rapamycin, a potent immunosuppressive drug, causes programmed cell death in B lymphoma cells. Transplantation 1995, 60, 264–270. [Google Scholar]
- Phung, T.L.; Ziv, K.; Dabydeen, D.; Eyiah-Mensah, G.; Riveros, M.; Perruzzi, C.; Sun, J.; Monahan-Earley, R.A.; Shiojima, I.; Nagy, J.A.; et al. Pathological angiogenesis is induced by sustained Akt signaling and inhibited by rapamycin. Cancer Cell 2006, 10, 159–170. [Google Scholar]
- Thomas, G.V.; Tran, C.; Mellinghoff, I.K.; Welsbie, D.S.; Chan, E.; Fueger, B.; Czernin, J.; Sawyers, C.L. Hypoxia-inducible factor determines sensitivity to inhibitors of mTOR in kidney cancer. Nat. Med 2006, 12, 122–127. [Google Scholar]
- Gingras, A.C.; Raught, B.; Gygi, S.P.; Niedzwiecka, A.; Miron, M.; Burley, S.K.; Polakiewicz, R.D.; Wyslouch-Cieszynska, A.; Aebersold, R.; Sonenberg, N. Hierarchical phosphorylation of the translation inhibitor 4E-BP1. Genes Dev 2001, 15, 2852–2864. [Google Scholar]
- Dumont, F.J.; Su, Q. Mechanism of action of the immunosuppressant rapamycin. Life Sci 1996, 58, 373–395. [Google Scholar]
- Dancey, J.E. Therapeutic targets: MTOR and related pathways. Cancer Biol. Ther 2006, 5, 1065–1073. [Google Scholar]
- Rini, B.I. Temsirolimus, an inhibitor of mammalian target of rapamycin. Clin. Cancer Res 2008, 14, 1286–1290. [Google Scholar]
- Rizzieri, D.A.; Feldman, E.; Dipersio, J.F.; Gabrail, N.; Stock, W.; Strair, R.; Rivera, V.M.; Albitar, M.; Bedrosian, C.L.; Giles, F.J. A phase 2 clinical trial of deforolimus (AP23573, MK-8669), a novel mammalian target of rapamycin inhibitor, in patients with relapsed or refractory hematologic malignancies. Clin. Cancer Res 2008, 14, 2756–2762. [Google Scholar]
- Wolpin, B.M.; Hezel, A.F.; Abrams, T.; Blaszkowsky, L.S.; Meyerhardt, J.A.; Chan, J.A.; Enzinger, P.C.; Allen, B.; Clark, J.W.; Ryan, D.P.; et al. Oral mTOR inhibitor everolimus in patients with gemcitabine-refractory metastatic pancreatic cancer. J. Clin. Oncol 2009, 27, 193–198. [Google Scholar]
- Johnston, P.B.; Inwards, D.J.; Colgan, J.P.; Laplant, B.R.; Kabat, B.F.; Habermann, T.M.; Micallef, I.N.; Porrata, L.F.; Ansell, S.M.; Reeder, C.B.; et al. A Phase II trial of the oral mTOR inhibitor everolimus in relapsed Hodgkin lymphoma. Am. J. Hematol 2010, 85, 320–324. [Google Scholar]
- Witzig, T.E.; Reeder, C.B.; LaPlant, B.R.; Gupta, M.; Johnston, P.B.; Micallef, I.N.; Porrata, L.F.; Ansell, S.M.; Colgan, J.P.; Jacobsen, E.D.; et al. A phase II trial of the oral mTOR inhibitor everolimus in relapsed aggressive lymphoma. Leukemia 2011, 25, 341–347. [Google Scholar]
- Ellard, S.L.; Clemons, M.; Gelmon, K.A.; Norris, B.; Kennecke, H.; Chia, S.; Pritchard, K.; Eisen, A.; Vandenberg, T.; Taylor, M.; et al. Randomized phase II study comparing two schedules of everolimus in patients with recurrent/metastatic breast cancer: NCIC Clinical Trials Group IND.163. J. Clin. Oncol 2009, 27, 4536–4541. [Google Scholar]
- Oza, A.M.; Elit, L.; Tsao, M.S.; Kamel-Reid, S.; Biagi, J.; Provencher, D.M.; Gotlieb, W.H.; Hoskins, P.J.; Ghatage, P.; Tonkin, K.S.; et al. Phase II study of temsirolimus in women with recurrent or metastatic endometrial cancer: A trial of the NCIC Clinical Trials Group. J. Clin. Oncol 2011, 29, 3278–3285. [Google Scholar]
- Hess, G.; Herbrecht, R.; Romaguera, J.; Verhoef, G.; Crump, M.; Gisselbrecht, C.; Laurell, A.; Offner, F.; Strahs, A.; Berkenblit, A.; et al. Phase III study to evaluate temsirolimus compared with investigator’s choice therapy for the treatment of relapsed or refractory mantle cell lymphoma. J. Clin. Oncol 2009, 27, 3822–389. [Google Scholar]
- O’Reilly, K.E.; Rojo, F.; She, Q.B.; Solit, D.; Mills, G.B.; Smith, D.; Lane, H.; Hofmann, F.; Hicklin, D.J.; Ludwig, D.L.; et al. mTOR inhibition induces upstream receptor tyrosine kinase signaling and activates Akt. Cancer Res 2006, 66, 1500–1508. [Google Scholar]
- Del Bufalo, D.; Ciuffreda, L.; Trisciuoglio, D.; Desideri, M.; Cognetti, F.; Zupi, G.; Milella, M. Antiangiogenic potential of the Mammalian target of rapamycin inhibitor temsirolimus. Cancer Res 2006, 66, 5549–5554. [Google Scholar]
- Gossage, L.; Eisen, T. Alterations in VHL as potential biomarkers in renal-cell carcinoma. Nat. Rev. Clin. Oncol 2010, 7, 277–288. [Google Scholar]
- Feldman, M.E.; Apsel, B.; Uotila, A.; Loewith, R.; Knight, Z.A.; Ruggero, D.; Shokat, K.M. Active-site inhibitors of mTOR target rapamycin-resistant outputs of mTORC1 and mTORC2. PLoS Biol 2009, 7, e38. [Google Scholar]
- Thoreen, C.C.; Kang, S.A.; Chang, J.W.; Liu, Q.; Zhang, J.; Gao, Y.; Reichling, L.J.; Sim, T.; Sabatini, D.M.; Gray, N.S. An ATP-competitive mammalian target of rapamycin inhibitor reveals rapamycin-resistant functions of mTORC1. J. Biol. Chem 2009, 284, 8023–8032. [Google Scholar]
- Molckovsky, A.; Siu, L.L. First-in-class, first-in-human phase I results of targeted agents: Highlights of the 2008 American society of clinical oncology meeting. J. Hematol. Oncol 2008, 1, 20. [Google Scholar]
- Yap, T.A.; Garrett, M.D.; Walton, M.I.; Raynaud, F.; de Bono, J.S.; Workman, P. Targeting the PI3K-AKT-mTOR pathway: Progress, pitfalls, and promises. Curr. Opin. Pharmacol 2008, 8, 393–412. [Google Scholar]
- Liu, T.J.; Koul, D.; LaFortune, T.; Tiao, N.; Shen, R.J.; Maira, S.M.; Garcia-Echevrria, C.; Yung, W.K. NVP-BEZ235, a novel dual phosphatidylinositol 3-kinase/mammalian target of rapamycin inhibitor, elicits multifaceted antitumor activities in human gliomas. Mol. Cancer Ther 2009, 8, 2204–2210. [Google Scholar]
- Ballou, L.M.; Lin, R.Z. Rapamycin and mTOR kinase inhibitors. J. Chem. Biol 2008, 1, 27–36. [Google Scholar]
- Chresta, C.M.; Davies, B.R.; Hickson, I.; Harding, T.; Cosulich, S.; Critchlow, S.E.; Vincent, J.P.; Ellston, R.; Jones, D.; Sini, P.; et al. AZD8055 is a potent, selective, and orally bioavailable ATP-competitive mammalian target of rapamycin kinase inhibitor with in vitro and in vivo antitumor activity. Cancer Res 2010, 70, 288–298. [Google Scholar]
- Garcia-Martinez, J.M.; Moran, J.; Clarke, R.G.; Gray, A.; Cosulich, S.C.; Chresta, C.M.; Alessi, D.R. Ku-0063794 is a specific inhibitor of the mammalian target of rapamycin (mTOR). Biochem. J 2009, 421, 29–42. [Google Scholar]
- Yu, K.; Toral-Barza, L.; Shi, C.; Zhang, W.G.; Lucas, J.; Shor, B.; Kim, J.; Verheijen, J.; Curran, K.; Malwitz, D.J.; et al. Biochemical, cellular, and in vivo activity of novel ATP-competitive and selective inhibitors of the mammalian target of rapamycin. Cancer Res 2009, 69, 6232–6240. [Google Scholar]
- Raynaud, F.I.; Eccles, S.; Clarke, P.A.; Hayes, A.; Nutley, B.; Alix, S.; Henley, A.; Di-Stefano, F.; Ahmad, Z.; Guillard, S.; et al. Pharmacologic characterization of a potent inhibitor of class I phosphatidylinositide 3-kinases. Cancer Res 2007, 67, 5840–5850. [Google Scholar]
- Venkatesan, A.M.; Chen, Z.; dos Santos, O.; Dehnhardt, C.; Santos, E.D.; Ayral-Kaloustian, S.; Mallon, R.; Hollander, I.; Feldberg, L.; Lucas, J.; et al. PKI-179: An orally efficacious dual phosphatidylinositol-3-kinase (PI3K)/mammalian target of rapamycin (mTOR) inhibitor. Bioorg. Med. Chem. Lett 2010, 20, 5869–5873. [Google Scholar]
- Venkatesan, A.M.; Dehnhardt, C.M.; Delos Santos, E.; Chen, Z.; Dos Santos, O.; Ayral-Kaloustian, S.; Khafizova, G.; Brooijmans, N.; Mallon, R.; Hollander, I.; et al. Bis(morpholino-1,3,5-triazine) derivatives: Potent adenosine 5′-triphosphate competitive phosphatidylinositol-3-kinase/mammalian target of rapamycin inhibitors: Discovery of compound 26 (PKI-587), a highly efficacious dual inhibitor. J. Med. Chem 2010, 53, 2636–2645. [Google Scholar]
- Grunwald, V.; DeGraffenried, L.; Russel, D.; Friedrichs, W.E.; Ray, R.B.; Hidalgo, M. Inhibitors of mTOR reverse doxorubicin resistance conferred by PTEN status in prostate cancer cells. Cancer Res 2002, 62, 6141–6145. [Google Scholar]
- Mondesire, W.H.; Jian, W.; Zhang, H.; Ensor, J.; Hung, M.C.; Mills, G.B.; Meric-Bernstam, F. Targeting mammalian target of rapamycin synergistically enhances chemotherapy-induced cytotoxicity in breast cancer cells. Clin. Cancer Res 2004, 10, 7031–7042. [Google Scholar]
- Steelman, L.S.; Navolanic, P.M.; Sokolosky, M.L.; Taylor, J.R.; Lehmann, B.D.; Chappell, W.H.; Abrams, S.L.; Wong, E.W.; Stadelman, K.M.; Terrian, D.M.; et al. Suppression of PTEN function increases breast cancer chemotherapeutic drug resistance while conferring sensitivity to mTOR inhibitors. Oncogene 2008, 27, 4086–4095. [Google Scholar]
- Kreisl, T.N.; Lassman, A.B.; Mischel, P.S.; Rosen, N.; Scher, H.I.; Teruya-Feldstein, J.; Shaffer, D.; Lis, E.; Abrey, L.E. A pilot study of everolimus and gefitinib in the treatment of recurrent glioblastoma (GBM). J. Neurooncol 2009, 92, 99–105. [Google Scholar]
- Reardon, D.A.; Desjardins, A.; Vredenburgh, J.J.; Gururangan, S.; Friedman, A.H.; Herndon, J.E., 2nd; Marcello, J.; Norfleet, J.A.; McLendon, R.E.; Sampson, J.H.; et al. Phase 2 trial of erlotinib plus sirolimus in adults with recurrent glioblastoma. J. Neurooncol 2010, 96, 219–230. [Google Scholar]
- Johnston, S.R. New strategies in estrogen receptor-positive breast cancer. Clin. Cancer Res 2010, 16, 1979–1987. [Google Scholar]
- Morrow, P.K.; Wulf, G.M.; Ensor, J.; Booser, D.J.; Moore, J.A.; Flores, P.R.; Xiong, Y.; Zhang, S.; Krop, I.E.; Winer, E.P.; et al. Phase I/II study of trastuzumab in combination with everolimus (RAD001) in patients with HER2-overexpressing metastatic breast cancer who progressed on trastuzumab-based therapy. J. Clin. Oncol 2011, 29, 3126–3132. [Google Scholar]
- Merchan, J.R.; Pitot, H.C.; Qin, R.; Liu, G.; Fitch, T.R.; Picus, J.; Maples, W.J.; Erlichman, C. Phase I/II trial of CCI 779 and bevacizumab in advanced renal cell carcinoma (RCC): Safety and activity in RTKI refractory RCC patients. J. Clin. Oncol 2009, 27. abstract 5039. [Google Scholar]
- Patnaik, A.; Ricart, A.; Cooper, J.; Papadopoulos, K.; Beeram, M.; Mita, C.; Mita, M.M.; Hufnagel, D.; Izbicka, E.; Tolcher, A.W. A phase I, pharmacokinetic and pharmacodynamic study of sorafenib (S), a multi-targeted kinase inhibitor in combination with temsirolimus (T), an mTOR inhibitor in patients with advanced solid malignancies. J. Clin. Oncol 2007, 25. abstract 3512. [Google Scholar]
- Patel, P.H.; Senico, P.L.; Curiel, R.E.; Motzer, R.J. Phase I study combining treatment with temsirolimus and sunitinib malate in patients with advanced renal cell carcinoma. Clin. Genitourin. Cancer 2009, 7, 24–27. [Google Scholar]
- Houghton, P. Targeting the IGF-1/mTOR pathway. Second AACR Centennial Conference on Translational Cancer Medicine, Monterey, CA, USA, 20–23 July 2008; AACR: Washington DC, Philadelphia, USA, 2008. Abstract number PL05-03. [Google Scholar]
- Kurmasheva, R.T.; Easton, J.B.; Houghton, P.J. Combined targeting of mTOR and the insulin-like growth factor pathway. ASCO Educ. Book 2008 2008, 460–464. [Google Scholar]
- Lasithiotakis, K.G.; Sinnberg, T.W.; Schittek, B.; Flaherty, K.T.; Kulms, D.; Maczey, E.; Garbe, C.; Meier, F.E. Combined inhibition of MAPK and mTOR signaling inhibits growth, induces cell death, and abrogates invasive growth of melanoma cells. J. Invest. Dermatol 2008, 128, 2013–2023. [Google Scholar]
- McDaid, H.M.; Legrier, M.; Yang, C.H.; Yan, H.G.; Lopez-Barcons, L.; Keller, S.M.; Horwitz, S.B. Combined MEK and mTOR suppression is synergistic in human NSCLC and is mediated via inhibition of protein translation. J. Clin. Oncol 2007, 25. abstract 10615. [Google Scholar]
- Molhoek, K.R.; Brautigan, D.L.; Slingluff, C.L., Jr. Synergistic inhibition of human melanoma proliferation by combination treatment with B-Raf inhibitor BAY43-9006 and mTOR inhibitor Rapamycin. J. Transl. Med 2005, 3. [Google Scholar] [CrossRef]
- Romano, M.F.; Avellino, R.; Petrella, A.; Bisogni, R.; Romano, S.; Venuta, S. Rapamycin inhibits doxorubicin-induced NF-κB/Rel nuclear activity and enhances the apoptosis of melanoma cells. Eur. J. Cancer 2004, 40, 2829–2836. [Google Scholar]
- Werzowa, J.; Cejka, D.; Fuereder, T.; Dekrout, B.; Thallinger, C.; Pehamberger, H.; Wacheck, V.; Pratscher, B. Suppression of mTOR complex 2-dependent AKT phosphorylation in melanoma cells by combined treatment with rapamycin and LY294002. Br. J. Dermatol 2009, 160, 955–964. [Google Scholar]
- Thallinger, C.; Poeppl, W.; Pratscher, B.; Mayerhofer, M.; Valent, P.; Tappeiner, G.; Joukhadar, C. CCI-779 plus cisplatin is highly effective against human melanoma in a SCID mouse xenotranplantation model. Pharmacology 2007, 79, 207–213. [Google Scholar]
- Eberle, J.; Kurbanov, B.M.; Hossini, A.M.; Trefzer, U.; Fecker, L.F. Overcoming apoptosis deficiency of melanoma-hope for new therapeutic approaches. Drug Resist. Updat 2007, 10, 218–234. [Google Scholar]
- Noh, W.C.; Mondesire, W.H.; Peng, J.; Jian, W.; Zhang, H.; Dong, J.; Mills, G.B.; Hung, M.C.; Meric-Bernstam, F. Determinants of rapamycin sensitivity in breast cancer cells. Clin. Cancer Res 2004, 10, 1013–1023. [Google Scholar]
- Neshat, M.S.; Mellinghoff, I.K.; Tran, C.; Stiles, B.; Thomas, G.; Petersen, R.; Frost, P.; Gibbons, J.J.; Wu, H.; Sawyers, C.L. Enhanced sensitivity of PTEN-deficient tumors to inhibition of FRAP/mTOR. Proc. Natl. Acad. Sci. USA 2001, 98, 10314–10319. [Google Scholar]
- Yu, K.; Toral-Barza, L.; Discafani, C.; Zhang, W.G.; Skotnicki, J.; Frost, P.; Gibbons, J.J. mTOR, a novel target in breast cancer: The effect of CCI-779, an mTOR inhibitor, in preclinical models of breast cancer. Endocr. Relat. Cancer 2001, 8, 249–258. [Google Scholar]
- Cloughesy, T.F.; Yoshimoto, K.; Nghiemphu, P.; Brown, K.; Dang, J.; Zhu, S.; Hsueh, T.; Chen, Y.; Wang, W.; Youngkin, D.; et al. Antitumor activity of rapamycin in a Phase I trial for patients with recurrent PTEN-deficient glioblastoma. PLoS Med 2008, 5. [Google Scholar] [CrossRef]
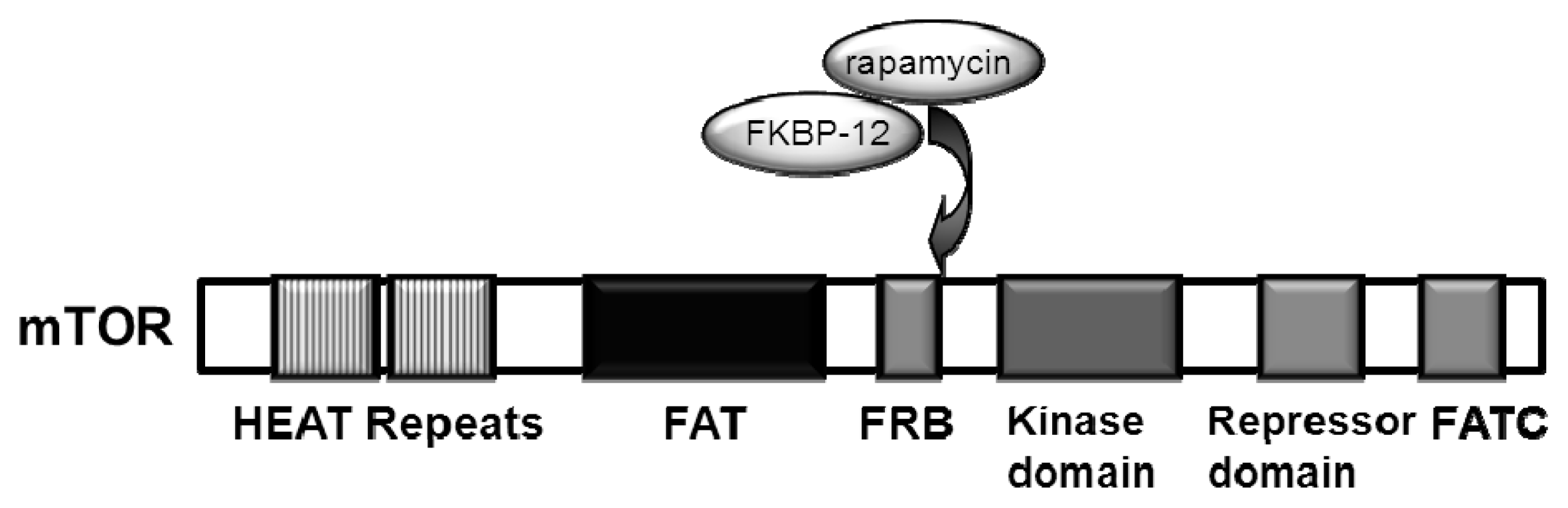
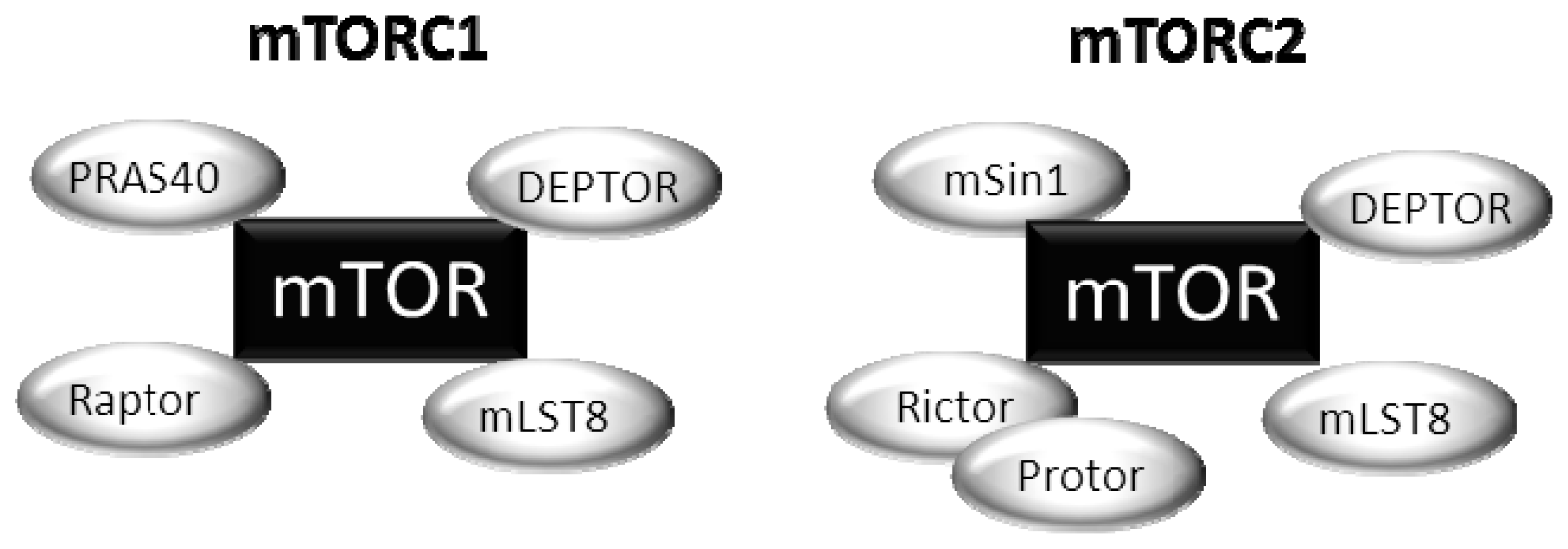

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Proto-oncogenes | Alterations described | References |
|---|---|---|
| AKT | AKT is amplified in a subset of human cancers, such as breast and ovarian cancers. | [126] |
| 4EBP1 | 4EBP1 expression was found to be associated with poor prognosis in several human tumours, such as breast, colon, ovarian and prostate cancers. The phosphorylation of 4EBP1 was also found to be associated with chemoresistance in ovarian cancer. | [127–129] |
| eIF4E | Ectopic overexpression of eIF4E can transform cells ex vivo and in vivo. eIF4E is overexpressed in many human tumours, such as breast, colon, and head and neck cancers, non-Hodgkin’s lymphomas, and chronic and acute myelogenous leukemias. | [130,131] |
| PI3K | High PI3K activity was implicated in cell transformation and tumour progression and described in several human cancers, such as ovarian, gastrointestinal, breast and prostate cancers. | [126, 132–134] |
| Rheb | Rheb overexpression is described in many tumour cells, and Rheb upregulation is critical for squamous carcinoma and associates with poor prognosis in breast and head and neck cancers. | [135,136] |
| S6K1 | S6K1 is overexpressed in in lung and ovary cancers and its expression correlates with poor prognosis in breast, kidney and hepatocellular carcinomas. | [137–142] |
| Tumour suppressor genes | ||
| LKB1 | Individuals with mutations in LKB1 develop Peutz-Jeghers syndrome, which includes the occurrence of gastrointestinal tract hamartomas. | [111,143] |
| PTEN | Loss of PTEN function has been described in a large proportion of advanced human cancers, such as melanoma, breast, prostate and renal cancers. Individuals with inherited mutations in PTEN develop hamartoma tumour syndromes (Cowden disease, Bannayan-Riley-Ruvalcaba syndrome, Proteus syndrome, Lhermitte-Duclos disease) and are at higher risk for developing several cancers. | [124,132, 143–146] |
| TSC1/TSC2 | Patients with mutations in TSC1 or TSC2 develop tuberous sclerosis complex (TSC), a syndrome that includes the development of hamartomas in many organs. Mutations in TSC2 may also lead to the development of Lymphangioleiomyomatosis (LAM). | [147–149] |
| mTOR inhibitors | Mechanism of action | References |
|---|---|---|
| Rapamycin and analogues | ||
| Deforolimus | Binding to the immunophilin FKBP12 Partial mTORC1 inhibitor Cell-type specific mTORC2 inhibitor | [206] |
| Everolimus | Binding to the immunophilin FKBP12 Partial mTORC1 inhibitor Cell-type specific mTORC2 inhibitor | [206] |
| Sirolimus | Binding to the immunophilin FKBP12 Partial mTORC1 inhibitor Cell-type specific mTORC2 inhibitor | [206] |
| Temsirolimus | Binding to the immunophilin FKBP12 Partial mTORC1 inhibitor Cell-type specific mTORC2 inhibitor | [206] |
| Small molecule inhibitors of kinases | ||
| AZD8055 | ATP competitive inhibitor of mTOR | [207] |
| Ku-0063794 | Specific mTORC1 and mTORC2 inhibitor | [208] |
| PP242 | mTOR kinase inhibitor | [201] |
| PP30 | mTOR kinase inhibitor | [201] |
| Torin1 | mTOR kinase inhibitor | [202] |
| WYE-354 | ATP competitive inhibitor of mTOR | [209] |
| mTOR and PI3K dual-specificity inhibitors | ||
| NVP-BEZ235 | ATP-competitive inhibitor of PI3K and mTOR | [205] |
| PI-103 | ATP competitive inhibitor of DNA-PK, PI3K and mTOR | [210] |
| PKI-179, PKI-587 | ATP competitive inhibitor of DNA-PK, PI3K and mTOR | [211,212] |
| XL765 | ATP-competitive inhibitor of DNA-PK, PI3K and mTOR | [203] |
© 2012 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Pópulo, H.; Lopes, J.M.; Soares, P. The mTOR Signalling Pathway in Human Cancer. Int. J. Mol. Sci. 2012, 13, 1886-1918. https://doi.org/10.3390/ijms13021886
Pópulo H, Lopes JM, Soares P. The mTOR Signalling Pathway in Human Cancer. International Journal of Molecular Sciences. 2012; 13(2):1886-1918. https://doi.org/10.3390/ijms13021886
Chicago/Turabian StylePópulo, Helena, José Manuel Lopes, and Paula Soares. 2012. "The mTOR Signalling Pathway in Human Cancer" International Journal of Molecular Sciences 13, no. 2: 1886-1918. https://doi.org/10.3390/ijms13021886
APA StylePópulo, H., Lopes, J. M., & Soares, P. (2012). The mTOR Signalling Pathway in Human Cancer. International Journal of Molecular Sciences, 13(2), 1886-1918. https://doi.org/10.3390/ijms13021886