Asymmetric Dimethyarginine as Marker and Mediator in Ischemic Stroke

{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
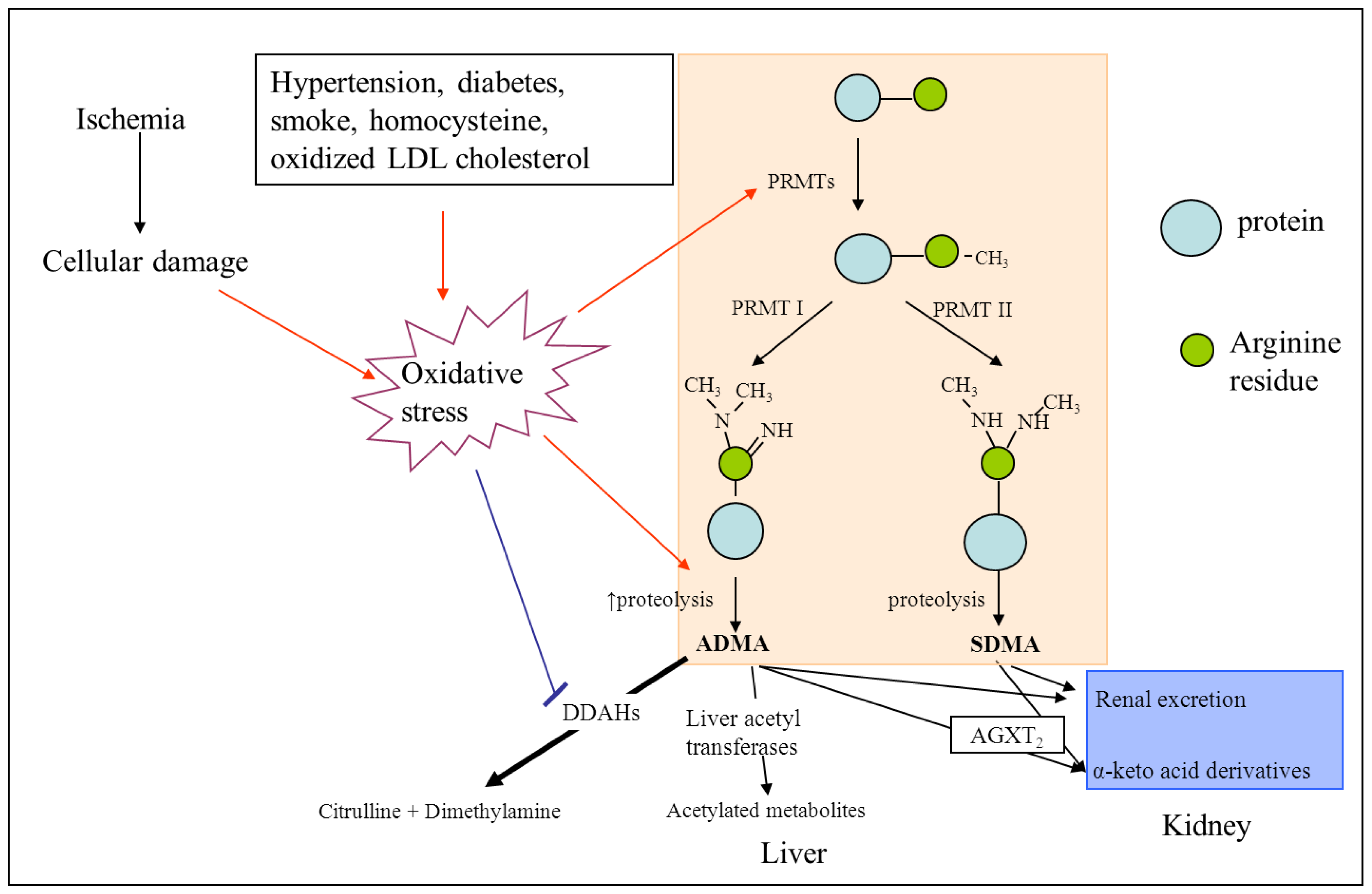
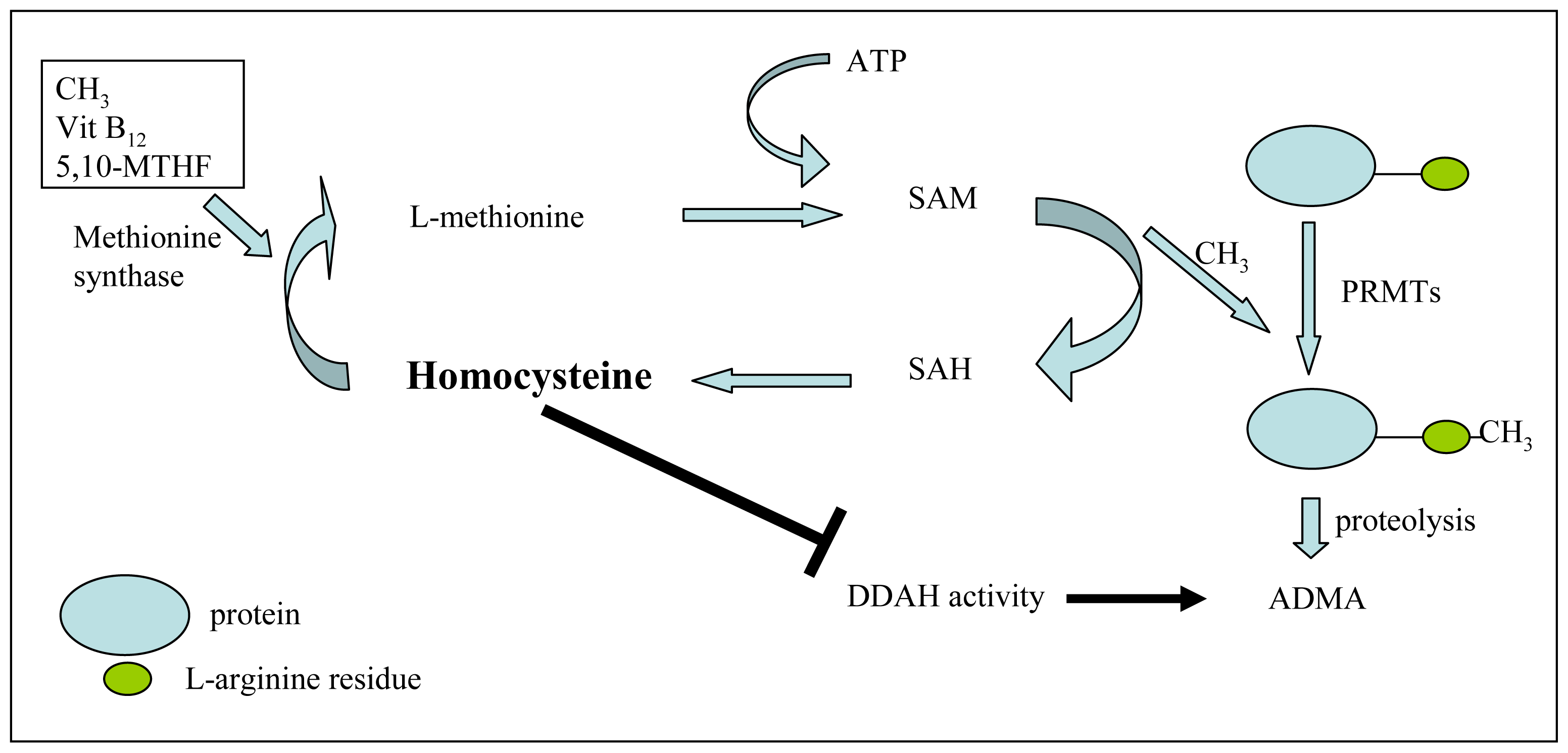
2. ADMA and NOS
3. ADMA and Cerebrovascular Risk
3.1. ADMA as a Mediator of Endothelial Dysfunction and Atherosclerosis
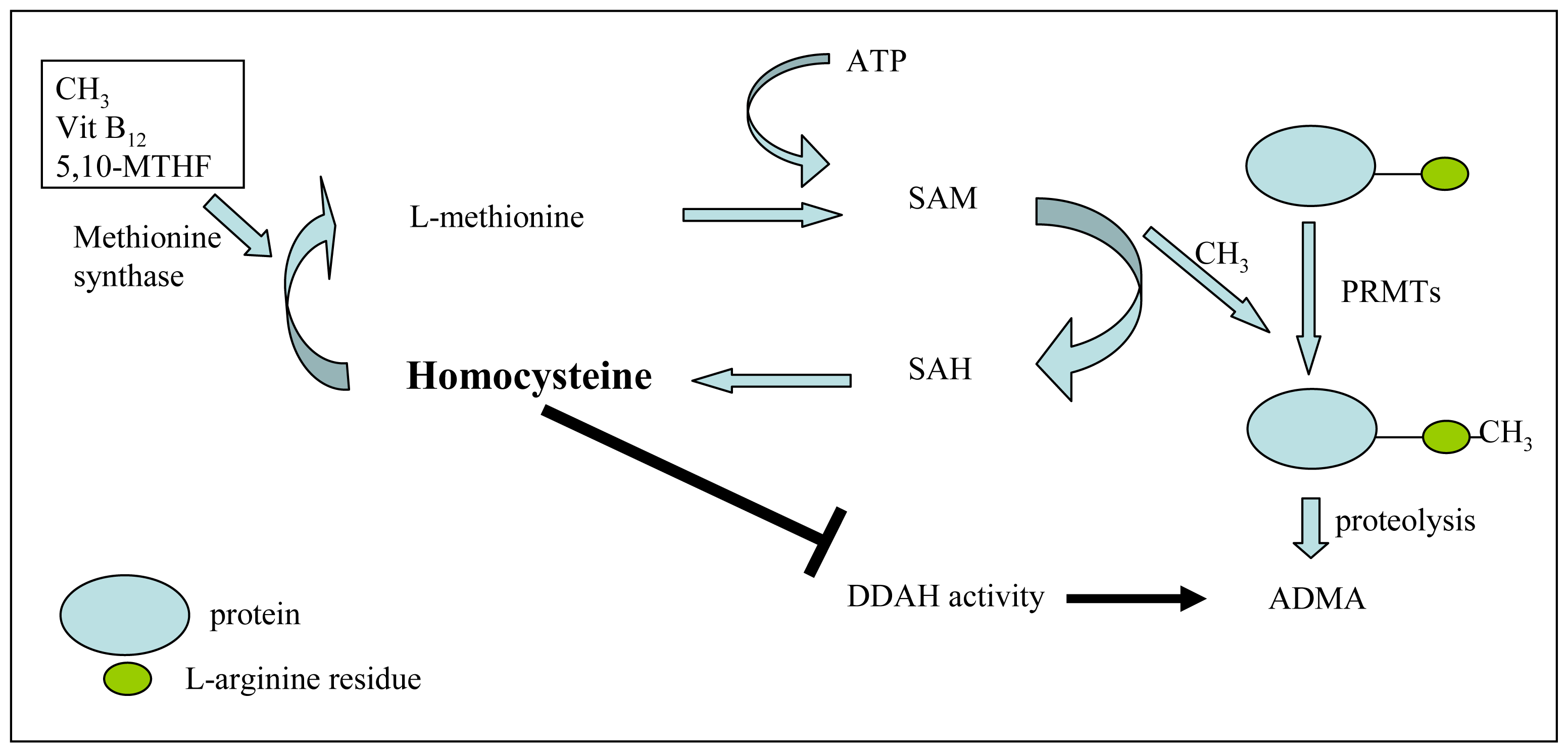
3.2. ADMA and Its Association with Vascular Risk Factors
3.3. ADMA and Its Association with Carotid Intima-Media Thickness
3.4. ADMA and Its Association with Cardiovascular and Cerebrovascular Events
3.5. ADMA as Pharmacological Target for Treatment of Cardiovascular Risk
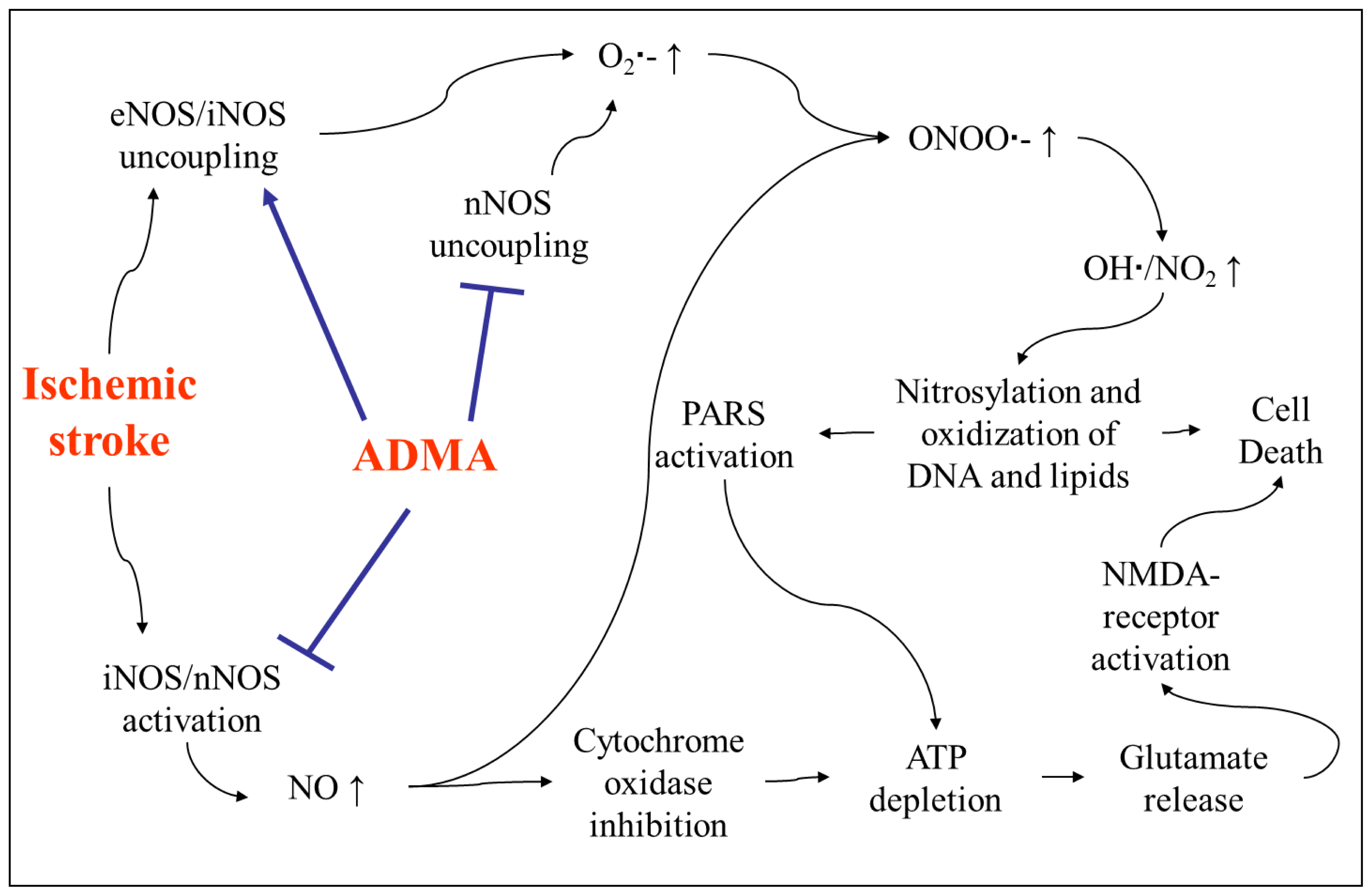
4. The Role of ADMA in Acute Ischemic Stroke
4.1. Increase of ADMA after Acute Ischemic Stroke and Its Association with Outcome
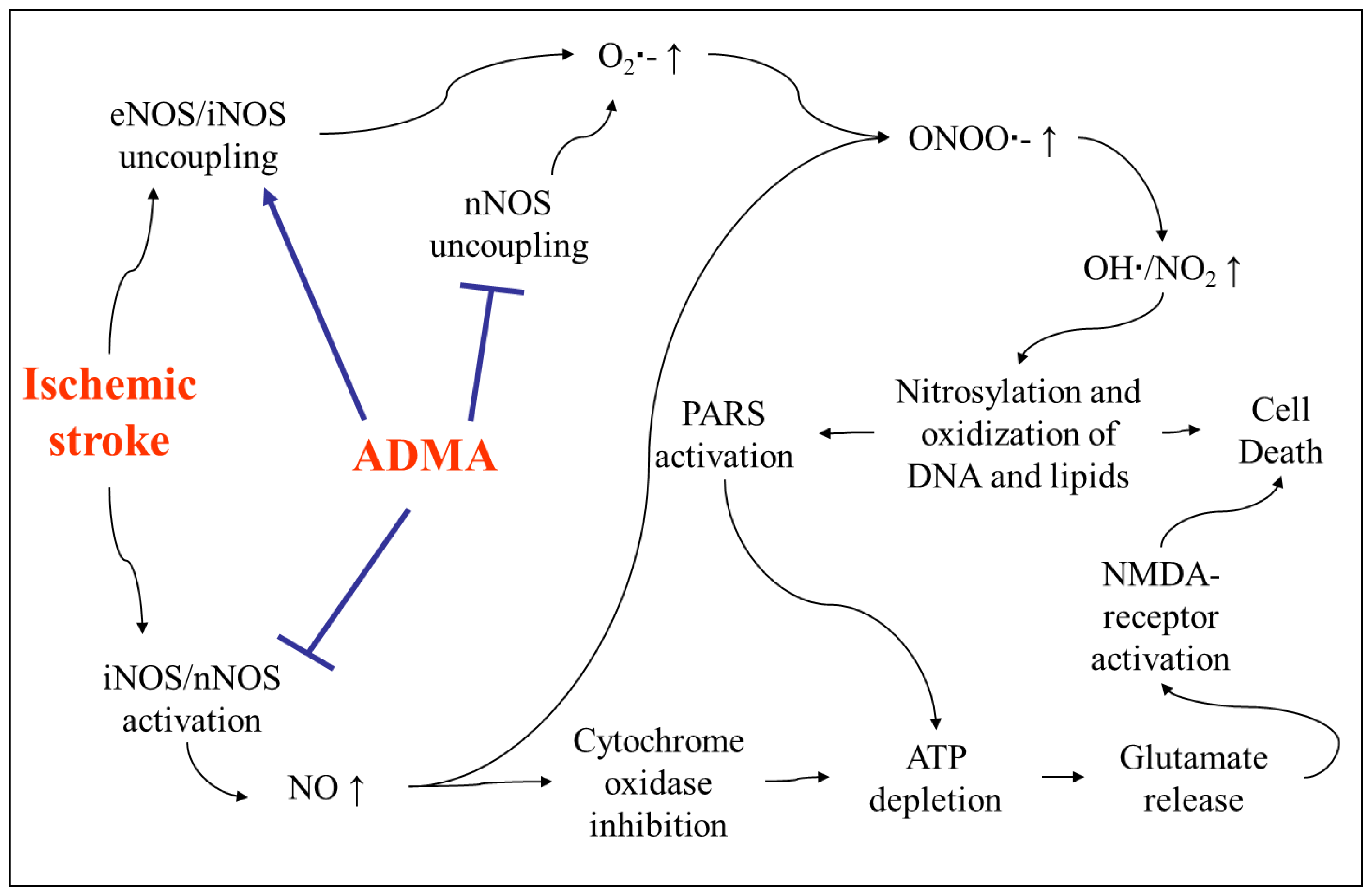
4.2. Involvement of ADMA in Brain Injury after Stroke
5. Conclusions
- Conflict of InterestJ.T. Kielstein runs and hosts the website www.adma.com. S. Chen was funded by a grant from Boehringer-Ingelheim, Germany. This work was supported by a grant from the HiLF program of Hannover Medical School awarded to H. Worthmann.
References
- World Health Statistics 2008; World Health Organization: Geneva, Switzerland; p. 2008.
- Rosamond, W.; Flegal, K.; Furie, K.; Go, A.; Greenlund, K.; Haase, N.; Hailpern, S.M.; Ho, M.; Howard, V.; Kissela, B.; et al. Heart disease and stroke statistics—2008 update: A report from the American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Circulation 2008, 117, e25–e146. [Google Scholar]
- Vladimirova-Kitova, L.; Terzieva, D.; Marinov, B. Intima-media thickness and flow-mediated vasodilation in asymptomatic subjects with newly detected severe hypercholesterolemia. Echocardiography 2009, 26, 1060–1068. [Google Scholar]
- Kielstein, J.T.; Bode-Boger, S.M.; Frolich, J.C.; Ritz, E.; Haller, H.; Fliser, D. Asymmetric dimethylarginine, blood pressure, and renal perfusion in elderly subjects. Circulation 2003, 107, 1891–1895. [Google Scholar]
- Abbasi, F.; Asagmi, T.; Cooke, J.P.; Lamendola, C.; McLaughlin, T.; Reaven, G.M.; Stuehlinger, M.; Tsao, P.S. Plasma concentrations of asymmetric dimethylarginine are increased in patients with type 2 diabetes mellitus. Am. J. Cardiol 2001, 88, 1201–1203. [Google Scholar]
- Boger, R.H.; Lentz, S.R.; Bode-Boger, S.M.; Knapp, H.R.; Haynes, W.G. Elevation of asymmetrical dimethylarginine may mediate endothelial dysfunction during experimental hyperhomocyst(e)inaemia in humans. Clin. Sci. (Lond.) 2001, 100, 161–167. [Google Scholar]
- Stuhlinger, M.C.; Stanger, O. Asymmetric dimethyl-L-arginine (ADMA): A possible link between homocyst(e)ine and endothelial dysfunction. Curr. Drug Metab 2005, 6, 3–14. [Google Scholar]
- Sobczak, A.; Goniewicz, M.L.; Szoltysek-Boldys, I. ADMA and SDMA levels in healthy men exposed to tobacco smoke. Atherosclerosis 2009, 205, 357–359. [Google Scholar]
- Nishiyama, Y.; Ueda, M.; Katsura, K.I.; Otsuka, T.; Abe, A.; Nagayama, H.; Katayama, Y. Asymmetric dimethylarginine (ADMA) as a possible risk marker for ischemic stroke. J. Neurol. Sci 2010, 290, 12–15. [Google Scholar]
- Pikula, A.; Boger, R.H.; Beiser, A.S.; Maas, R.; DeCarli, C.; Schwedhelm, E.; Himali, J.J.; Schulze, F.; Au, R.; Kelly-Hayes, M.; et al. Association of plasma ADMA levels with MRI markers of vascular brain injury: Framingham offspring study. Stroke 2009, 40, 2959–2964. [Google Scholar]
- Worthmann, H.; Chen, S.; Martens-Lobenhoffer, J.; Li, N.; Deb, M.; Tryc, A.B.; Goldbecker, A.; Dong, Q.; Kielstein, J.T.; Bode-Boger, S.M.; et al. High plasma dimethylarginine levels are associated with adverse clinical outcome after stroke. J. Atheroscler. Thromb 2011, 18, 753–761. [Google Scholar]
- McDermott, J.R. Studies on the catabolism of Ng-methylarginine, Ng, Ng-dimethylarginine and Ng, Ng-dimethylarginine in the rabbit. Biochem. J 1976, 154, 179–184. [Google Scholar]
- Nakajima, T.; Matsuoka, Y.; Kakimoto, Y. Isolation and identification of N-G-monomethyl, N-G,N-G-dimethyl- and N-G,N′ G-dimethylarginine from the hydrolysate of proteins of bovine brain. Biochim. Biophys. Acta 1971, 230, 212–222. [Google Scholar]
- Lee, D.Y.; Teyssier, C.; Strahl, B.D.; Stallcup, M.R. Role of protein methylation in regulation of transcription. Endocr. Rev 2005, 26, 147–170. [Google Scholar]
- Anthony, S.; Leiper, J.; Vallance, P. Endogenous production of nitric oxide synthase inhibitors. Vasc. Med 2005, 10, S3–S9. [Google Scholar]
- Rodionov, R.N.; Murry, D.J.; Vaulman, S.F.; Stevens, J.W.; Lentz, S.R. Human alanine-glyoxylate aminotransferase 2 lowers asymmetric dimethylarginine and protects from inhibition of nitric oxide production. J. Biol. Chem 2010, 285, 5385–5391. [Google Scholar]
- Caplin, B.; Wang, Z.; Slaviero, A.; Tomlinson, J.; Dowsett, L.; Delahey, M.; Salama, A.; Wheeler, D.C.; Leiper, J. Alanine-Glyoxylate Aminotransferase-2 Metabolizes Endogenous Methylarginines, Regulates NO, and Controls Blood Pressure. Arterioscler. Thromb. Vasc. Biol. 2012. [Google Scholar] [CrossRef]
- Cooke, J.P. Does ADMA cause endothelial dysfunction? Arterioscler. Thromb. Vasc. Biol 2000, 20, 2032–2037. [Google Scholar]
- Ogawa, T.; Kimoto, M.; Sasaoka, K. Purification and properties of a new enzyme, NG,NG-dimethylarginine dimethylaminohydrolase, from rat kidney. J. Biol. Chem 1989, 264, 10205–10209. [Google Scholar]
- Leiper, J.M.; Santa Maria, J.; Chubb, A.; MacAllister, R.J.; Charles, I.G.; Whitley, G.S.; Vallance, P. Identification of two human dimethylarginine dimethylaminohydrolases with distinct tissue distributions and homology with microbial arginine deiminases. Biochem. J 1999, 343, 209–214. [Google Scholar]
- Onozato, M.L.; Tojo, A.; Leiper, J.; Fujita, T.; Palm, F.; Wilcox, C.S. Expression of NG,NG-dimethylarginine dimethylaminohydrolase and protein arginine N-methyltransferase isoforms in diabetic rat kidney: Effects of angiotensin II receptor blockers. Diabetes 2008, 57, 172–180. [Google Scholar]
- Tran, C.T.; Fox, M.F.; Vallance, P.; Leiper, J.M. Chromosomal localization, gene structure, and expression pattern of DDAH1: Comparison with DDAH2 and implications for evolutionary origins. Genomics 2000, 68, 101–105. [Google Scholar]
- Hu, X.; Xu, X.; Zhu, G.; Atzler, D.; Kimoto, M.; Chen, J.; Schwedhelm, E.; Luneburg, N.; Boger, R.H.; Zhang, P.; et al. Vascular endothelial-specific dimethylarginine dimethylaminohydrolase-1-deficient mice reveal that vascular endothelium plays an important role in removing asymmetric dimethylarginine. Circulation 2009, 120, 2222–2229. [Google Scholar]
- Hu, X.; Atzler, D.; Xu, X.; Zhang, P.; Guo, H.; Lu, Z.; Fassett, J.; Schwedhelm, E.; Boger, R.H.; Bache, R.J.; et al. Dimethylarginine dimethylaminohydrolase-1 is the critical enzyme for degrading the cardiovascular risk factor asymmetrical dimethylarginine. Arterioscler. Thromb. Vasc. Biol 2011, 31, 1540–1546. [Google Scholar]
- Leiper, J.; Murray-Rust, J.; McDonald, N.; Vallance, P. S-nitrosylation of dimethylarginine dimethylaminohydrolase regulates enzyme activity: further interactions between nitric oxide synthase and dimethylarginine dimethylaminohydrolase. Proc. Natl. Acad. Sci. USA 2002, 99, 13527–13532. [Google Scholar]
- Vallance, P.; Leiper, J. Cardiovascular biology of the asymmetric dimethylarginine:dimethylarginine dimethylaminohydrolase pathway. Arterioscler. Thromb. Vasc. Biol 2004, 24, 1023–1030. [Google Scholar]
- Ogawa, T.; Kimoto, M.; Watanabe, H.; Sasaoka, K. Metabolism of NG,NG-and NG,N’G-dimethylarginine in rats. Arch. Biochem. Biophys 1987, 252, 526–537. [Google Scholar]
- Caplin, B.; Leiper, J. Endogenous nitric oxide synthase inhibitors in the biology of disease: Markers, mediators, and regulators? Arterioscler. Thromb. Vasc. Biol 2012, 32, 1343–1353. [Google Scholar]
- Marescau, B.; Nagels, G.; Possemiers, I.; De Broe, M.E.; Becaus, I.; Billiouw, J.M.; Lornoy, W.; De Deyn, P.P. Guanidino compounds in serum and urine of nondialyzed patients with chronic renal insufficiency. Metabolism 1997, 46, 1024–1031. [Google Scholar]
- Napoli, C.; Ignarro, L.J. Nitric oxide and pathogenic mechanisms involved in the development of vascular diseases. Arch. Pharm. Res 2009, 32, 1103–1108. [Google Scholar]
- Samdani, A.F.; Dawson, T.M.; Dawson, V.L. Nitric oxide synthase in models of focal ischemia. Stroke 1997, 28, 1283–1288. [Google Scholar]
- Vallance, P.; Leone, A.; Calver, A.; Collier, J.; Moncada, S. Endogenous dimethylarginine as an inhibitor of nitric oxide synthesis. J. Cardiovasc. Pharmacol 1992, 20, S60–S62. [Google Scholar]
- Closs, E.I.; Basha, F.Z.; Habermeier, A.; Forstermann, U. Interference of l-arginine analogues with L-arginine transport mediated by the y+ carrier hCAT-2B. Nitric Oxide 1997, 1, 65–73. [Google Scholar]
- Bode-Boger, S.M.; Scalera, F.; Kielstein, J.T.; Martens-Lobenhoffer, J.; Breithardt, G.; Fobker, M.; Reinecke, H. Symmetrical dimethylarginine: a new combined parameter for renal function and extent of coronary artery disease. J. Am. Soc. Nephrol 2006, 17, 1128–1134. [Google Scholar]
- Schepers, E.; Barreto, D.V.; Liabeuf, S.; Glorieux, G.; Eloot, S.; Barreto, F.C.; Massy, Z.; Vanholder, R. Symmetric dimethylarginine as a proinflammatory agent in chronic kidney disease. Clin. J. Am. Soc. Nephrol 2011, 6, 2374–2383. [Google Scholar]
- Kwasny-Krochin, B.; Gluszko, P.; Undas, A. Plasma asymmetric dimethylarginine in active rheumatoid arthritis: links with oxidative stress and inflammation. Pol. Arch. Med. Wewn 2012, 122, 270–276. [Google Scholar]
- Schulze, F.; Carter, A.M.; Schwedhelm, E.; Ajjan, R.; Maas, R.; von Holten, R.A.; Atzler, D.; Grant, P.J.; Boger, R.H. Symmetric dimethylarginine predicts all-cause mortality following ischemic stroke. Atherosclerosis 2010, 208, 518–523. [Google Scholar]
- Landim, M.B.; Casella Filho, A.; Chagas, A.C. Asymmetric dimethylarginine (ADMA) and endothelial dysfunction: Implications for atherogenesis. Clinics (Sao Paulo) 2009, 64, 471–478. [Google Scholar]
- Jia, S.J.; Zhou, Z.; Zhang, B.K.; Hu, Z.W.; Deng, H.W.; Li, Y.J. Asymmetric dimethylarginine damages connexin43-mediated endothelial gap junction intercellular communication. Biochem. Cell. Biol 2009, 87, 867–874. [Google Scholar]
- Chen, M.; Li, Y.; Yang, T.; Wang, Y.; Bai, Y.; Xie, X. ADMA induces monocyte adhesion via activation of chemokine receptors in cultured THP-1 cells. Cytokine 2008, 43, 149–159. [Google Scholar]
- Chen, M.F.; Li, Y.J.; Yang, T.L.; Lou, B.; Xie, X.M. Losartan inhibits monocytic adhesion induced by ADMA via downregulation of chemokine receptors in monocytes. Eur. J. Clin. Pharmacol 2009, 65, 457–464. [Google Scholar]
- Woollard, K.J.; Geissmann, F. Monocytes in atherosclerosis: subsets and functions. Nat. Rev. Cardiol 2010, 7, 77–86. [Google Scholar]
- Boisvert, W.A.; Curtiss, L.K.; Terkeltaub, R.A. Interleukin-8 and its receptor CXCR2 in atherosclerosis. Immunol. Res 2000, 21, 129–137. [Google Scholar]
- Sun, L.; Zhang, T.; Yu, X.; Xin, W.; Lan, X.; Zhang, D.; Huang, C.; Du, G. Asymmetric dimethylarginine confers the communication between endothelial and smooth muscle cells and leads to VSMC migration through p38 and ERK1/2 signaling cascade. FEBS Lett 2011, 585, 2727–2734. [Google Scholar]
- Smirnova, I.V.; Kajstura, M.; Sawamura, T.; Goligorsky, M.S. Asymmetric dimethylarginine upregulates LOX-1 in activated macrophages: Role in foam cell formation. Am. J. Physiol. Heart Circ. Physiol 2004, 287, H782–H790. [Google Scholar]
- Jiang, D.J.; Jia, S.J.; Dai, Z.; Li, Y.J. Asymmetric dimethylarginine induces apoptosis via p38 MAPK/caspase-3-dependent signaling pathway in endothelial cells. J. Mol. Cell. Cardiol 2006, 40, 529–539. [Google Scholar]
- Yuan, Q.; Jiang, D.J.; Chen, Q.Q.; Wang, S.; Xin, H.Y.; Deng, H.W.; Li, Y.J. Role of asymmetric dimethylarginine in homocysteine-induced apoptosis of vascular smooth muscle cells. Biochem. Biophys. Res. Commun 2007, 356, 880–885. [Google Scholar]
- Thum, T.; Tsikas, D.; Stein, S.; Schultheiss, M.; Eigenthaler, M.; Anker, S.D.; Poole-Wilson, P.A.; Ertl, G.; Bauersachs, J. Suppression of endothelial progenitor cells in human coronary artery disease by the endogenous nitric oxide synthase inhibitor asymmetric dimethylarginine. J. Am. Coll. Cardiol 2005, 46, 1693–1701. [Google Scholar]
- Jacobi, J.; Maas, R.; Cardounel, A.J.; Arend, M.; Pope, A.J.; Cordasic, N.; Heusinger-Ribeiro, J.; Atzler, D.; Strobel, J.; Schwedhelm, E.; et al. Dimethylarginine dimethylaminohydrolase overexpression ameliorates atherosclerosis in apolipoprotein E-deficient mice by lowering asymmetric dimethylarginine. Am. J. Pathol 2010, 176, 2559–2570. [Google Scholar]
- Boger, R.H.; Bode-Boger, S.M.; Szuba, A.; Tsao, P.S.; Chan, J.R.; Tangphao, O.; Blaschke, T.F.; Cooke, J.P. Asymmetric dimethylarginine (ADMA): A novel risk factor for endothelial dysfunction: its role in hypercholesterolemia. Circulation 1998, 98, 1842–1847. [Google Scholar]
- Gao, H.W.; Xie, C.; Wang, H.N.; Lin, Y.J.; Hong, T.P. Beneficial metabolic effects of nateglinide versus acarbose in patients with newly-diagnosed type 2 diabetes. Acta Pharmacol. Sin 2007, 28, 534–539. [Google Scholar]
- Juonala, M.; Viikari, J.S.; Alfthan, G.; Marniemi, J.; Kahonen, M.; Taittonen, L.; Laitinen, T.; Raitakari, O.T. Brachial artery flow-mediated dilation and asymmetrical dimethylarginine in the cardiovascular risk in young Finns study. Circulation 2007, 116, 1367–1373. [Google Scholar]
- Stuhlinger, M.C.; Abbasi, F.; Chu, J.W.; Lamendola, C.; McLaughlin, T.L.; Cooke, J.P.; Reaven, G.M.; Tsao, P.S. Relationship between insulin resistance and an endogenous nitric oxide synthase inhibitor. JAMA 2002, 287, 1420–1426. [Google Scholar]
- Yoo, J.H.; Lee, S.C. Elevated levels of plasma homocyst(e)ine and asymmetric dimethylarginine in elderly patients with stroke. Atherosclerosis 2001, 158, 425–430. [Google Scholar]
- Notsu, Y.; Nabika, T.; Bokura, H.; Suyama, Y.; Kobayashi, S.; Yamaguchi, S.; Masuda, J. Evaluation of asymmetric dimethylarginine and homocysteine in microangiopathy-related cerebral damage. Am. J. Hypertens 2009, 22, 257–262. [Google Scholar]
- Khan, U.; Hassan, A.; Vallance, P.; Markus, H.S. Asymmetric dimethylarginine in cerebral small vessel disease. Stroke 2007, 38, 411–413. [Google Scholar]
- Yin, Q.F.; Xiong, Y. Pravastatin restores DDAH activity and endothelium-dependent relaxation of rat aorta after exposure to glycated protein. J. Cardiovasc. Pharmacol 2005, 45, 525–532. [Google Scholar]
- Ito, A.; Tsao, P.S.; Adimoolam, S.; Kimoto, M.; Ogawa, T.; Cooke, J.P. Novel mechanism for endothelial dysfunction: dysregulation of dimethylarginine dimethylaminohydrolase. Circulation 1999, 99, 3092–3095. [Google Scholar]
- Boger, R.H.; Sydow, K.; Borlak, J.; Thum, T.; Lenzen, H.; Schubert, B.; Tsikas, D.; Bode-Boger, S.M. LDL cholesterol upregulates synthesis of asymmetrical dimethylarginine in human endothelial cells: Involvement of S-adenosylmethionine-dependent methyltransferases. Circ. Res 2000, 87, 99–105. [Google Scholar]
- Lentz, S.R.; Rodionov, R.N.; Dayal, S. Hyperhomocysteinemia, endothelial dysfunction, and cardiovascular risk: the potential role of ADMA. Atheroscler. Suppl 2003, 4, 61–65. [Google Scholar]
- Stuhlinger, M.C.; Tsao, P.S.; Her, J.H.; Kimoto, M.; Balint, R.F.; Cooke, J.P. Homocysteine impairs the nitric oxide synthase pathway: Role of asymmetric dimethylarginine. Circulation 2001, 104, 2569–2575. [Google Scholar]
- Boger, R.H.; Bode-Boger, S.M.; Sydow, K.; Heistad, D.D.; Lentz, S.R. Plasma concentration of asymmetric dimethylarginine, an endogenous inhibitor of nitric oxide synthase, is elevated in monkeys with hyperhomocyst(e)inemia or hypercholesterolemia. Arterioscler. Thromb. Vasc. Biol 2000, 20, 1557–1564. [Google Scholar]
- Selley, M.L. Homocysteine increases the production of asymmetric dimethylarginine in cultured neurons. J. Neurosci. Res 2004, 77, 90–93. [Google Scholar]
- Nanayakkara, P.W.; Teerlink, T.; Stehouwer, C.D.; Allajar, D.; Spijkerman, A.; Schalkwijk, C.; ter Wee, P.M.; van Guldener, C. Plasma asymmetric dimethylarginine (ADMA) concentration is independently associated with carotid intima-media thickness and plasma soluble vascular cell adhesion molecule-1 (sVCAM-1) concentration in patients with mild-to-moderate renal failure. Kidney Int 2005, 68, 2230–2236. [Google Scholar]
- Maas, R.; Xanthakis, V.; Polak, J.F.; Schwedhelm, E.; Sullivan, L.M.; Benndorf, R.; Schulze, F.; Vasan, R.S.; Wolf, P.A.; Boger, R.H.; et al. Association of the endogenous nitric oxide synthase inhibitor ADMA with carotid artery intimal media thickness in the Framingham Heart Study offspring cohort. Stroke 2009, 40, 2715–2719. [Google Scholar]
- Riccioni, G.; Bucciarelli, V.; Scotti, L.; Aceto, A.; Orazio, N.D.; Di Ilio, E.; Bucciarelli, T. Relationship between asymmetric dimethylarginine and asymptomatic carotid atherosclerosis. J. Biol. Regul. Homeost. Agents 2010, 24, 351–358. [Google Scholar]
- Furuki, K.; Adachi, H.; Enomoto, M.; Otsuka, M.; Fukami, A.; Kumagae, S.; Matsuoka, H.; Nanjo, Y.; Kakuma, T.; Imaizumi, T. Plasma level of asymmetric dimethylarginine (ADMA) as a predictor of carotid intima-media thickness progression: Six-year prospective study using carotid ultrasonography. Hypertens. Res 2008, 31, 1185–1189. [Google Scholar]
- Zsuga, J.; Torok, J.; Magyar, M.T.; Valikovics, A.; Gesztelyi, R.; Keki, S.; Csiba, L.; Zsuga, M.; Bereczki, D. Serum asymmetric dimethylarginine negatively correlates with intima-media thickness in early-onset atherosclerosis. Cerebrovasc. Dis 2007, 23, 388–394. [Google Scholar]
- Bai, Y.; Sun, L.; Du, L.; Zhang, T.; Xin, W.; Lan, X.; Du, G. Association of circulating levels of asymmetric dimethylarginine (ADMA) with carotid intima-media thickness: Evidence from 6168 participants. Ageing Res. Rev. 2012, in press. [Google Scholar]
- Wilson, A.M.; Shin, D.S.; Weatherby, C.; Harada, R.K.; Ng, M.K.; Nair, N.; Kielstein, J.; Cooke, J.P. Asymmetric dimethylarginine correlates with measures of disease severity, major adverse cardiovascular events and all-cause mortality in patients with peripheral arterial disease. Vasc. Med 2010, 15, 267–274. [Google Scholar]
- Tripepi, G.; Mattace, F.R.; Sijbrands, E.; Seck, M.S.; Maas, R.; Boger, R.; Witteman, J.; Rapisarda, F.; Malatino, L.; Mallamaci, F.; et al. Inflammation and asymmetric dimethylarginine for predicting death and cardiovascular events in ESRD patients. Clin. J. Am. Soc. Nephrol 2011, 6, 1714–1721. [Google Scholar]
- Leong, T.; Zylberstein, D.; Graham, I.; Lissner, L.; Ward, D.; Fogarty, J.; Bengtsson, C.; Bjorkelund, C.; Thelle, D. Asymmetric dimethylarginine independently predicts fatal and nonfatal myocardial infarction and stroke in women: 24-year follow-up of the population study of women in Gothenburg. Arterioscler. Thromb. Vasc. Biol 2008, 28, 961–967. [Google Scholar]
- Krzyzanowska, K.; Mittermayer, F.; Wolzt, M.; Schernthaner, G. Asymmetric dimethylarginine predicts cardiovascular events in patients with type 2 diabetes. Diabetes Care 2007, 30, 1834–1839. [Google Scholar]
- Zoccali, C.; Bode-Boger, S.; Mallamaci, F.; Benedetto, F.; Tripepi, G.; Malatino, L.; Cataliotti, A.; Bellanuova, I.; Fermo, I.; Frolich, J.; et al. Plasma concentration of asymmetrical dimethylarginine and mortality in patients with end-stage renal disease: A prospective study. Lancet 2001, 358, 2113–2117. [Google Scholar]
- Wang, Z.; Tang, W.H.; Cho, L.; Brennan, D.M.; Hazen, S.L. Targeted metabolomic evaluation of arginine methylation and cardiovascular risks: potential mechanisms beyond nitric oxide synthase inhibition. Arterioscler. Thromb. Vasc. Biol 2009, 29, 1383–1391. [Google Scholar]
- Boger, R.H.; Sullivan, L.M.; Schwedhelm, E.; Wang, T.J.; Maas, R.; Benjamin, E.J.; Schulze, F.; Xanthakis, V.; Benndorf, R.A.; Vasan, R.S. Plasma asymmetric dimethylarginine and incidence of cardiovascular disease and death in the community. Circulation 2009, 119, 1592–1600. [Google Scholar]
- Siegerink, B.; Maas, R.; Vossen, C.Y.; Schwedhelm, E.; Koenig, W.; Boger, R.; Rothenbacher, D.; Brenner, H.; Breitling, L.P. Asymmetric and symmetric dimethylarginine and risk of secondary cardiovascular disease events and mortality in patients with stable coronary heart disease: The KAROLA follow-up study. Clin. Res. Cardiol. 2012, in press. [Google Scholar]
- Lisabeth, L.D.; Beiser, A.S.; Brown, D.L.; Murabito, J.M.; Kelly-Hayes, M.; Wolf, P.A. Age at natural menopause and risk of ischemic stroke: the Framingham heart study. Stroke 2009, 40, 1044–1049. [Google Scholar]
- Rufa, A.; Blardi, P.; De Lalla, A.; Cevenini, G.; De Stefano, N.; Zicari, E.; Auteri, A.; Federico, A.; Dotti, M.T. Plasma levels of asymmetric dimethylarginine in cerebral autosomal dominant arteriopathy with subcortical infarct and leukoencephalopathy. Cerebrovasc. Dis 2008, 26, 636–640. [Google Scholar]
- Ding, H.; Wu, B.; Wang, H.; Lu, Z.; Yan, J.; Wang, X.; Shaffer, J.R.; Hui, R.; Wang, D.W. A novel loss-of-function DDAH1 promoter polymorphism is associated with increased susceptibility to thrombosis stroke and coronary heart disease. Circ. Res 2010, 106, 1145–1152. [Google Scholar]
- Lu, T.M.; Lin, S.J.; Lin, M.W.; Hsu, C.P.; Chung, M.Y. The association of dimethylarginine dimethylaminohydrolase 1 gene polymorphism with type 2 diabetes: a cohort study. Cardiovasc. Diabetol 2011, 10, 16. [Google Scholar]
- Maas, R. Pharmacotherapies and their influence on asymmetric dimethylargine (ADMA). Vasc. Med 2005, 10, S49–S57. [Google Scholar]
- Lu, T.M.; Ding, Y.A.; Leu, H.B.; Yin, W.H.; Sheu, W.H.; Chu, K.M. Effect of rosuvastatin on plasma levels of asymmetric dimethylarginine in patients with hypercholesterolemia. Am. J. Cardiol 2004, 94, 157–161. [Google Scholar]
- Nishiyama, Y.; Ueda, M.; Otsuka, T.; Katsura, K.; Abe, A.; Nagayama, H.; Katayama, Y. Statin Treatment Decreased Serum Asymmetric Dimethylarginine (ADMA) Levels in Ischemic Stroke Patients. J. Atheroscler. Thromb 2011, 18, 131–137. [Google Scholar]
- Pereira, E.C.; Bertolami, M.C.; Faludi, A.A.; Salem, M.; Bersch, D.; Abdalla, D.S. Effects of simvastatin and l-arginine on vasodilation, nitric oxide metabolites and endogenous NOS inhibitors in hypercholesterolemic subjects. Free Radic. Res 2003, 37, 529–536. [Google Scholar]
- Eid, H.M.; Eritsland, J.; Larsen, J.; Arnesen, H.; Seljeflot, I. Increased levels of asymmetric dimethylarginine in populations at risk for atherosclerotic disease. Effects of pravastatin. Atherosclerosis 2003, 166, 279–284. [Google Scholar]
- Panichi, V.; Mantuano, E.; Paoletti, S.; Santi, S.; Manca Rizza, G.; Cutrupi, S.; Pizzini, P.; Spoto, B.; Tripepi, G.; Zoccali, C. Effect of simvastatin on plasma asymmetric dimethylarginine concentration in patients with chronic kidney disease. J. Nephrol 2008, 21, 38–44. [Google Scholar]
- Verhoeven, M.O.; Hemelaar, M.; van der Mooren, M.J.; Kenemans, P.; Teerlink, T. Oral, more than transdermal, oestrogen therapy lowers asymmetric dimethylarginine in healthy postmenopausal women: A randomized, placebo-controlled study. J. Int. Med 2006, 259, 199–208. [Google Scholar]
- Monsalve, E.; Oviedo, P.J.; Garcia-Perez, M.A.; Tarin, J.J.; Cano, A.; Hermenegildo, C. Estradiol counteracts oxidized LDL-induced asymmetric dimethylarginine production by cultured human endothelial cells. Cardiovasc. Res 2007, 73, 66–72. [Google Scholar]
- Wadham, C.; Mangoni, A.A. Dimethylarginine dimethylaminohydrolase regulation: a novel therapeutic target in cardiovascular disease. Expert Opin. Drug Metab. Toxicol 2009, 5, 303–319. [Google Scholar]
- Chen, H.; Yoshioka, H.; Kim, G.S.; Jung, J.E.; Okami, N.; Sakata, H.; Maier, C.M.; Narasimhan, P.; Goeders, C.E.; Chan, P.H. Oxidative stress in ischemic brain damage: mechanisms of cell death and potential molecular targets for neuroprotection. Antioxid. Redox Signal 2011, 14, 1505–1517. [Google Scholar]
- Sydow, K.; Munzel, T. ADMA and oxidative stress. Atheroscler. Suppl 2003, 4, 41–51. [Google Scholar]
- Brouns, R.; Marescau, B.; Possemiers, I.; Sheorajpanday, R.; De Deyn, P.P. Dimethylarginine levels in cerebrospinal fluid of hyperacute ischemic stroke patients are associated with stroke severity. Neurochem. Res 2009, 34, 1642–1649. [Google Scholar]
- Adams, H.P., Jr; Bendixen, B.H.; Kappelle, L.J.; Biller, J.; Love, B.B.; Gordon, D.L.; Marsh, E.E., III. Classification of subtype of acute ischemic stroke. Definitions for use in a multicenter clinical trial. TOAST. Trial of Org 10172 in Acute Stroke Treatment. Stroke 1993, 24, 35–41. [Google Scholar]
- Scherbakov, N.; Sandek, A.; Martens-Lobenhoffer, J.; Kung, T.; Turhan, G.; Liman, T.; Ebinger, M.; von Haehling, S.; Bode-Boger, S.M.; Endres, M.; et al. Endothelial dysfunction of the peripheral vascular bed in the acute phase after ischemic stroke. Cerebrovasc. Dis 2012, 33, 37–46. [Google Scholar]
- Wanby, P.; Teerlink, T.; Brudin, L.; Brattstrom, L.; Nilsson, I.; Palmqvist, P.; Carlsson, M. Asymmetric dimethylarginine (ADMA) as a risk marker for stroke and TIA in a Swedish population. Atherosclerosis 2006, 185, 271–277. [Google Scholar]
- Rueda-Clausen, C.F.; Cordoba-Porras, A.; Bedoya, G.; Silva, F.A.; Zarruk, J.G.; Lopez-Jaramillo, P.; Villa, L.A. Increased plasma levels of total homocysteine but not asymmetric dimethylarginine in Hispanic subjects with ischemic stroke FREC-VI sub-study. Eur. J. Neurol 2012, 19, 417–425. [Google Scholar]
- Luneburg, N.; von Holten, R.A.; Topper, R.F.; Schwedhelm, E.; Maas, R.; Boger, R.H. Symmetric dimethylarginine is a marker of detrimental outcome in the acute phase after ischaemic stroke: role of renal function. Clin. Sci. (Lond.) 2012, 122, 105–111. [Google Scholar]
- Iadecola, C.; Pelligrino, D.A.; Moskowitz, M.A.; Lassen, N.A. Nitric oxide synthase inhibition and cerebrovascular regulation. J. Cereb. Blood Flow Metab 1994, 14, 175–192. [Google Scholar]
- Faraci, F.M.; Brian, J.E., Jr; Heistad, D.D. Response of cerebral blood vessels to an endogenous inhibitor of nitric oxide synthase. Am. J. Physiol. 1995, 269, H1522–H1527. [Google Scholar]
- Dayoub, H.; Rodionov, R.N.; Lynch, C.; Cooke, J.P.; Arning, E.; Bottiglieri, T.; Lentz, S.R.; Faraci, F.M. Overexpression of dimethylarginine dimethylaminohydrolase inhibits asymmetric dimethylarginine-induced endothelial dysfunction in the cerebral circulation. Stroke 2008, 39, 180–184. [Google Scholar]
- Segarra, G.; Medina, P.; Ballester, R.M.; Lluch, P.; Aldasoro, M.; Vila, J.M.; Lluch, S.; Pelligrino, D.A. Effects of some guanidino compounds on human cerebral arteries. Stroke 1999, 30, 2206–2210, discussion 2210–2211. [Google Scholar]
- Kielstein, J.T.; Donnerstag, F.; Gasper, S.; Menne, J.; Kielstein, A.; Martens-Lobenhoffer, J.; Scalera, F.; Cooke, J.P.; Fliser, D.; Bode-Boger, S.M. ADMA increases arterial stiffness and decreases cerebral blood flow in humans. Stroke 2006, 37, 2024–2029. [Google Scholar]
- Leypoldt, F.; Choe, C.U.; Gelderblom, M.; von Leitner, E.C.; Atzler, D.; Schwedhelm, E.; Gerloff, C.; Sydow, K.; Boger, R.H.; Magnus, T. Dimethylarginine dimethylaminohydrolase-1 transgenic mice are not protected from ischemic stroke. PLoS One 2009, 4, e7337. [Google Scholar]
- Xu, L.; Wang, B.; Kaur, K.; Kho, M.F.; Cooke, J.P.; Giffard, R.G. NOx and ADMA changes with focal ischemia, amelioration with the chaperonin GroEL. Neurosci. Lett 2007, 418, 201–204. [Google Scholar]
- Cardounel, A.J.; Zweier, J.L. Endogenous methylarginines regulate neuronal nitric-oxide synthase and prevent excitotoxic injury. J. Biol. Chem 2002, 277, 33995–34002. [Google Scholar]
- Wells, S.M.; Holian, A. Asymmetric dimethylarginine induces oxidative and nitrosative stress in murine lung epithelial cells. Am. J. Respir. Cell. Mol. Biol 2007, 36, 520–528. [Google Scholar]
- Rashid, P.A.; Whitehurst, A.; Lawson, N.; Bath, P.M. Plasma nitric oxide (nitrate/nitrite) levels in acute stroke and their relationship with severity and outcome. J. Stroke Cerebrovasc. Dis 2003, 12, 82–87. [Google Scholar]
- Huang, Z.; Huang, P.L.; Panahian, N.; Dalkara, T.; Fishman, M.C.; Moskowitz, M.A. Effects of cerebral ischemia in mice deficient in neuronal nitric oxide synthase. Science 1994, 265, 1883–1885. [Google Scholar]
- Zhao, X.; Haensel, C.; Araki, E.; Ross, M.E.; Iadecola, C. Gene-dosing effect and persistence of reduction in ischemic brain injury in mice lacking inducible nitric oxide synthase. Brain Res 2000, 872, 215–218. [Google Scholar]
- Cardounel, A.J.; Xia, Y.; Zweier, J.L. Endogenous methylarginines modulate superoxide as well as nitric oxide generation from neuronal nitric-oxide synthase: differences in the effects of monomethyl- and dimethylarginines in the presence and absence of tetrahydrobiopterin. J. Biol. Chem 2005, 280, 7540–7549. [Google Scholar]
- Murphy, S.; Gibson, C.L. Nitric oxide, ischaemia and brain inflammation. Biochem. Soc. Trans 2007, 35, 1133–1137. [Google Scholar]
- Fickling, S.A.; Holden, D.P.; Cartwright, J.E.; Nussey, S.S.; Vallance, P.; Whitley, G.S. Regulation of macrophage nitric oxide synthesis by endothelial cells: A role for NG,NG-dimethylarginine. Acta Physiol. Scand 1999, 167, 145–150. [Google Scholar]
- Leiper, J.; Nandi, M.; Torondel, B.; Murray-Rust, J.; Malaki, M.; O’Hara, B.; Rossiter, S.; Anthony, S.; Madhani, M.; Selwood, D.; et al. Disruption of methylarginine metabolism impairs vascular homeostasis. Nat. Med 2007, 13, 198–203. [Google Scholar]
- Zhang, G.G.; Bai, Y.P.; Chen, M.F.; Shi, R.Z.; Jiang, D.J.; Fu, Q.M.; Tan, G.S.; Li, Y.J. Asymmetric dimethylarginine induces TNF-alpha production via ROS/NF-kappaB dependent pathway in human monocytic cells and the inhibitory effect of reinioside C. Vasc. Pharmacol 2008, 48, 115–121. [Google Scholar]
- Jiang, J.L.; Wang, S.; Li, N.S.; Zhang, X.H.; Deng, H.W.; Li, Y.J. The inhibitory effect of simvastatin on the ADMA-induced inflammatory reaction is mediated by MAPK pathways in endothelial cells. Biochem. Cell. Biol 2007, 85, 66–77. [Google Scholar]
- Worthmann, H.; Tryc, A.B.; Goldbecker, A.; Ma, Y.T.; Tountopoulou, A.; Hahn, A.; Dengler, R.; Lichtinghagen, R.; Weissenborn, K. The temporal profile of inflammatory markers and mediators in blood after acute ischemic stroke differs depending on stroke outcome. Cerebrovasc. Dis 2010, 30, 85–92. [Google Scholar]
- Von Leitner, E.C.; Klinke, A.; Atzler, D.; Slocum, J.L.; Lund, N.; Kielstein, J.T.; Maas, R.; Schmidt-Haupt, R.; Pekarova, M.; Hellwinkel, O.; et al. Pathogenic cycle between the endogenous nitric oxide synthase inhibitor asymmetrical dimethylarginine and the leukocyte-derived hemoprotein myeloperoxidase. Circulation 2011, 124, 2735–2745. [Google Scholar]
- Tanaka, M.; Sydow, K.; Gunawan, F.; Jacobi, J.; Tsao, P.S.; Robbins, R.C.; Cooke, J.P. Dimethylarginine dimethylaminohydrolase overexpression suppresses graft coronary artery disease. Circulation 2005, 112, 1549–1556. [Google Scholar]
- Konishi, H.; Sydow, K.; Cooke, J.P. Dimethylarginine dimethylaminohydrolase promotes endothelial repair after vascular injury. J. Am. Coll. Cardiol 2007, 49, 1099–1105. [Google Scholar]
- Stuhlinger, M.C.; Conci, E.; Haubner, B.J.; Stocker, E.M.; Schwaighofer, J.; Cooke, J.P.; Tsao, P.S.; Pachinger, O.; Metzler, B. Asymmetric dimethyl l-arginine (ADMA) is a critical regulator of myocardial reperfusion injury. Cardiovasc. Res 2007, 75, 417–425. [Google Scholar]
- Tojo, A.; Welch, W.J.; Bremer, V.; Kimoto, M.; Kimura, K.; Omata, M.; Ogawa, T.; Vallance, P.; Wilcox, C.S. Colocalization of demethylating enzymes and NOS and functional effects of methylarginines in rat kidney. Kidney Int 1997, 52, 1593–1601. [Google Scholar]
- Schepers, E.; Glorieux, G.; Dhondt, A.; Leybaert, L.; Vanholder, R. Role of symmetric dimethylarginine in vascular damage by increasing ROS via store-operated calcium influx in monocytes. Nephrol. Dial. Transplant 2009, 24, 1429–1435. [Google Scholar]
- Oner-Iyidogan, Y.; Oner, P.; Kocak, H.; Gurdol, F.; Bekpinar, S.; Unlucerci, Y.; Caliskan, Y.; Cetinalp-Demircan, P.; Kocak, T.; Turkmen, A. Dimethylarginines and inflammation markers in patients with chronic kidney disease undergoing dialysis. Clin. Exp. Med 2009, 9, 235–241. [Google Scholar]
- Caglar, K.; Yilmaz, M.I.; Sonmez, A.; Cakir, E.; Kaya, A.; Acikel, C.; Eyileten, T.; Yenicesu, M.; Oguz, Y.; Bilgi, C.; et al. ADMA, proteinuria, and insulin resistance in non-diabetic stage I chronic kidney disease. Kidney Int 2006, 70, 781–787. [Google Scholar]
© 2012 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Chen, S.; Li, N.; Deb-Chatterji, M.; Dong, Q.; Kielstein, J.T.; Weissenborn, K.; Worthmann, H. Asymmetric Dimethyarginine as Marker and Mediator in Ischemic Stroke. Int. J. Mol. Sci. 2012, 13, 15983-16004. https://doi.org/10.3390/ijms131215983
Chen S, Li N, Deb-Chatterji M, Dong Q, Kielstein JT, Weissenborn K, Worthmann H. Asymmetric Dimethyarginine as Marker and Mediator in Ischemic Stroke. International Journal of Molecular Sciences. 2012; 13(12):15983-16004. https://doi.org/10.3390/ijms131215983
Chicago/Turabian StyleChen, Shufen, Na Li, Milani Deb-Chatterji, Qiang Dong, Jan T. Kielstein, Karin Weissenborn, and Hans Worthmann. 2012. "Asymmetric Dimethyarginine as Marker and Mediator in Ischemic Stroke" International Journal of Molecular Sciences 13, no. 12: 15983-16004. https://doi.org/10.3390/ijms131215983
APA StyleChen, S., Li, N., Deb-Chatterji, M., Dong, Q., Kielstein, J. T., Weissenborn, K., & Worthmann, H. (2012). Asymmetric Dimethyarginine as Marker and Mediator in Ischemic Stroke. International Journal of Molecular Sciences, 13(12), 15983-16004. https://doi.org/10.3390/ijms131215983