The Proteomics Big Challenge for Biomarkers and New Drug-Targets Discovery


{kind=link}
{kind=link}
Abstract
:1. Introduction
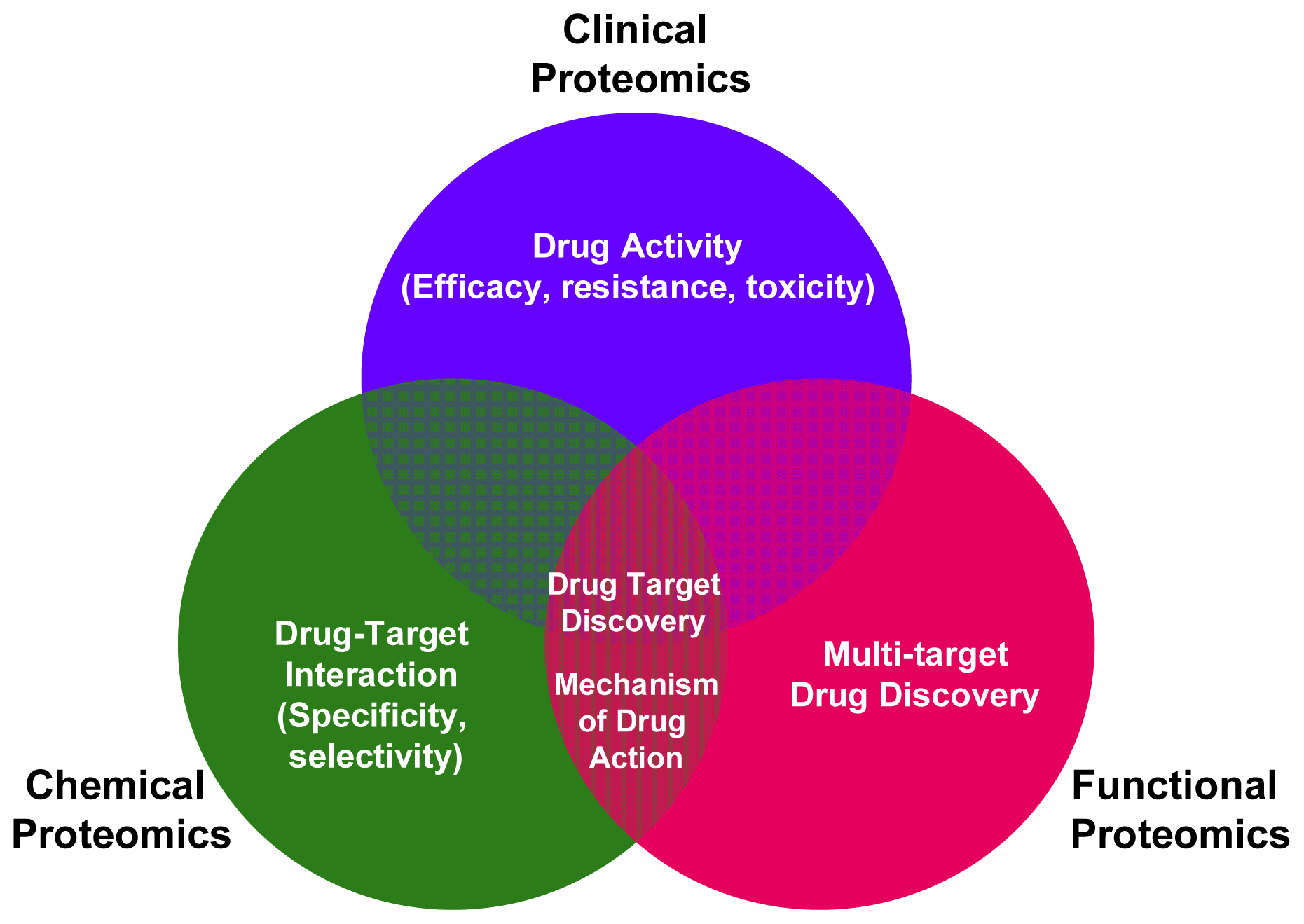
2. Applying Functional Proteomics to Biomarkers and Drug-Targets Discovery
3. Applying Chemical Proteomics to Biomarkers and Drug-Targets Discovery
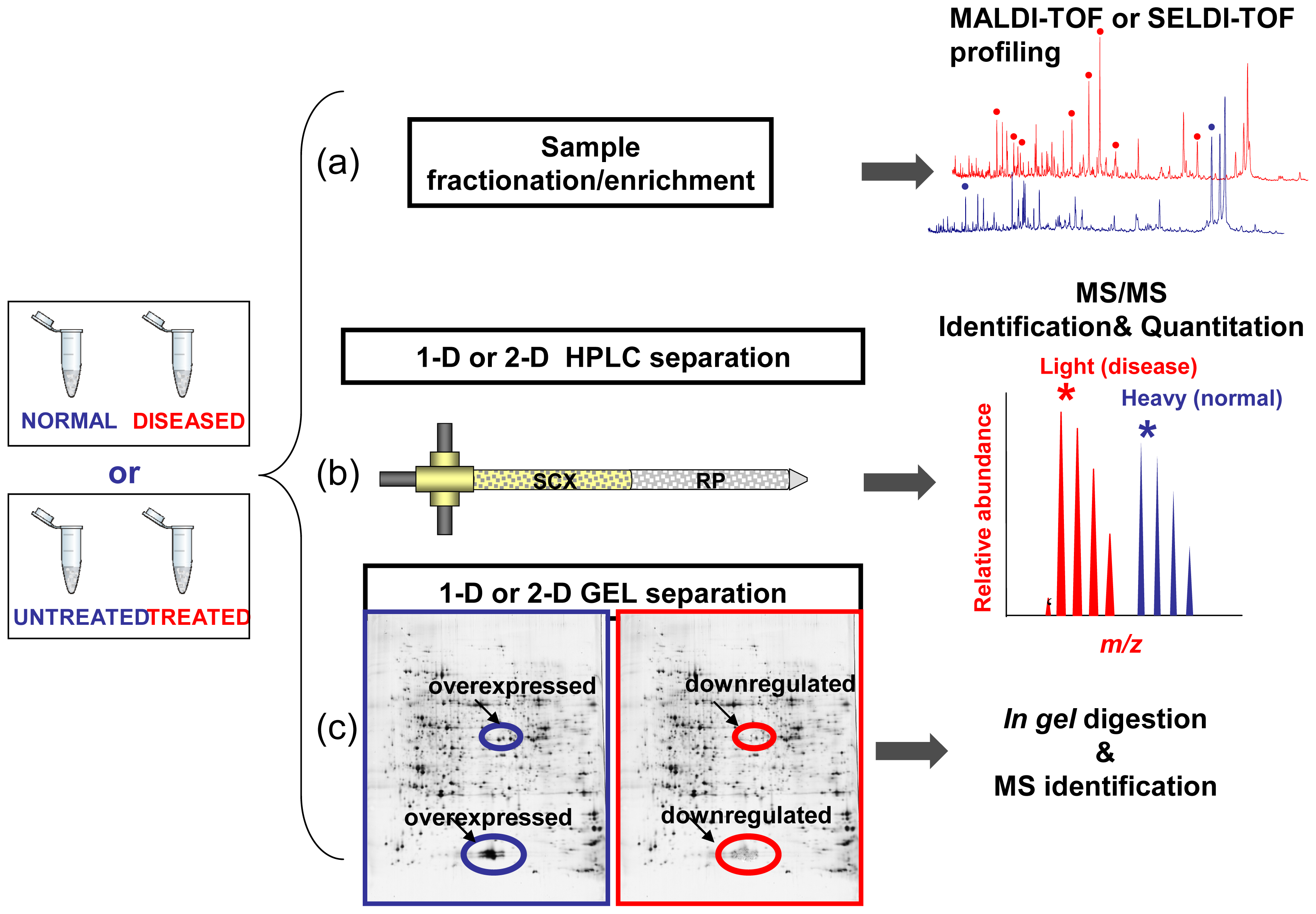
4. Applying Clinical Proteomics to Biomarkers and Drug-Targets Discovery
Limitations of Comparative Profiling Strategies
5. Conclusions
Acknowledgments
- Conflict of InterestThe authors declare no conflict of interest.
References
- Angel, T.E.; Aryal, U.K.; Hengel, S.M.; Baker, E.S.; Kelly, R.T.; Robinson, E.W.; Smith, R.D. Mass spectrometry based proteomics: Existing capabilities and future directions. Chem. Soc. Rev 2012, 41, 3912–3928. [Google Scholar]
- Au, C.E.; Bell, A.W.; Gilchrist, A.; Hiding, J.; Nilsson, T.; Bergeron, J.J. Organellar proteomics to create the cell map. Curr. Opin. Cell Biol 2007, 19, 376–385. [Google Scholar]
- Collins, S.R.; Kemmeren, P.; Zhao, X.C.; Greenblatt, J.F.; Spencer, F.; Holstege, F.C.; Weissman, J.S.; Krogan, N.J. Toward a comprehensive atlas of the physical interactome of Saccharomyces cerevisiae. Mol. Cell. Proteomics 2007, 6, 439–450. [Google Scholar]
- Aebersold, R.; Mann, M. Mass spectrometry-based proteomics. Nature 2003, 422, 198–207. [Google Scholar]
- Pan, S.; Aebersold, R.; Chen, R.; Rush, J.; Goodlett, D.R.; McIntosh, M.W.; Zhang, J.; Brentnall, T.A. Mass spectrometry based targeted protein quantification: Methods and applications. J. Proteome Res 2009, 8, 787–797. [Google Scholar]
- Beretta, L. Proteomics from the clinical perspective: Many hopes and much debate. Nat. Methods 2007, 4, 785–786. [Google Scholar]
- Köcher, T.; Superti-Furga, G. Mass spectrometry-based functional proteomics: From molecular machines to protein networks. Nat. Methods 2007, 4, 807–815. [Google Scholar]
- Major, M.B.; Camp, N.D.; Berndt, J.D.; Yi, X.; Goldenberg, S.J.; Hubbert, C.; Biechele, T.L.; Gingras, A.C.; Zheng, N.; Maccoss, M.J.; et al. Wilms tumor suppressor WTX negatively regulates WNT/β-catenin signaling. Science 2007, 316, 1043–1046. [Google Scholar]
- Tedford, N.C.; Hall, A.B.; Graham, J.R.; Murphy, C.E.; Gordon, N.F.; Radding, J.A. Quantitative analysis of cell signaling and drug action via mass spectrometry-based systems level phosphoproteomics. Proteomics 2009, 9, 1469–1487. [Google Scholar]
- Bantscheff, M.; Scholten, A.; Heck, A.J. Revealing promiscuous drug-target interactions by chemical proteomics. Drug Discov. Today 2009, 14, 1021–1029. [Google Scholar]
- Alberts, B. The cell as a collection of protein machines: Preparing the next generation of molecular biologists. Cell 1998, 92, 291–294. [Google Scholar]
- Wasinger, V.C.; Harris, R.; Williams, K.L.; Humphery-Smith, I. Progress with gene-product mapping of the Mollicutes: Mycoplasma genitalium. Electrophoresis 1995, 16, 1090–1094. [Google Scholar]
- Kolch, W. Meaningful relationships: The regulation of the Ras/Raf/MEK/ERK pathway by protein interactions. Biochem. J 2000, 351, 289–305. [Google Scholar]
- Kolch, W.; Pitt, A. Functional proteomics to dissect tyrosine kinase signalling pathways in cancer. Nat. Rev. Cancer 2010, 10, 618–629. [Google Scholar]
- Neubauer, G.; Gottschalk, A.; Fabrizio, P.; Séraphin, B.; Lührmann, R.; Mann, M. Identification of the proteins of the yeast U1 small nuclear ribonucleoprotein complex by mass spectrometry. Proc. Natl. Acad. Sci. USA 1997, 94, 385–390. [Google Scholar]
- Figeys, D. Proteomics approaches in drug discovery. Anal. Chem 2002, 74, 413A–419A. [Google Scholar]
- Rual, J.F.; Venkatesan, K.; Hao, T.; Hirozane-Kishikawa, T.; Dricot, A.; Li, N.; Berriz, G.F.; Gibbons, F.D.; Dreze, M.; Ayivi-Guedehoussou, N.; et al. Towards a proteome-scale map of the human protein-protein interaction network. Nature 2005, 437, 1173–1178. [Google Scholar]
- Paonessa, G.; Graziani, R.; De Serio, A.; Savino, R.; Ciapponi, L.; Lahm, A.; Salvati, A.L.; Toniatti, C.; Ciliberto, G. Two distinct and independent sites on IL-6 trigger gp 130 dimer formation and signalling. EMBO J 1995, 14, 1942–1951. [Google Scholar]
- Mallick, P.; Kuster, B. Proteomics: A pragmatic perspective. Nat. Biotechnol 2010, 28, 695–709. [Google Scholar]
- Bürckstümmer, T.; Bennett, K.L.; Preradovic, A.; Schütze, G.; Hantschel, O.; Superti-Furga, G.; Bauch, A. An efficient tandem affinity purification procedure for interaction proteomics in mammalian cells. Nat. Methods 2006, 3, 1013–1019. [Google Scholar]
- Gavin, A.C.; Bösche, M.; Krause, R.; Grandi, P.; Marzioch, M.; Bauer, A.; Schultz, J.; Rick, J.M.; Michon, A.M.; Cruciat, C.M.; et al. Functional organization of the yeast proteome by systematic analysis of protein complexes. Nature 2002, 415, 141–147. [Google Scholar]
- Gavin, A.C.; Aloy, P.; Grandi, P.; Krause, R.; Boesche, M.; Marzioch, M.; Rau, C.; Jensen, L.J.; Bastuck, S.; Dümpelfeld, B.; et al. Proteome survey reveals modularity of the yeast cell machinery. Nature 2006, 440, 631–636. [Google Scholar]
- Krogan, N.J.; Cagney, G.; Yu, H.; Zhong, G.; Guo, X.; Ignatchenko, A.; Li, J.; Pu, S.; Datta, N.; Tikuisis, A.P.; et al. Global landscape of protein complexes in the yeast Saccharomyces cerevisiae. Nature 2006, 440, 637–643. [Google Scholar]
- Andersen, J.S.; Lam, Y.W.; Leung, A.K.; Ong, S.E.; Lyon, C.E.; Lamond, A.I.; Mann, M. Nucleolar proteome dynamics. Nature 2005, 433, 77–83. [Google Scholar]
- Bouwmeester, T.; Bauch, A.; Ruffner, H.; Angrand, P.O.; Bergamini, G.; Croughton, K.; Cruciat, C.; Eberhard, D.; Gagneur, J.; Ghidelli, S.; et al. A physical and functional map of the human TNF-α/NF-κB signal transduction pathway. Nat. Cell Biol 2004, 6, 97–105. [Google Scholar]
- Brehme, M.; Hantschel, O.; Colinge, J.; Kaupe, I.; Planyavsky, M.; Köcher, T.; Mechtler, K.; Bennett, K.L.; Superti-Furga, G. Charting the molecular network of the drug target Bcr-Abl. Proc. Natl. Acad. Sci. USA 2009, 106, 7414–7419. [Google Scholar]
- Melo, J.V.; Barnes, D.J. Chronic myeloid leukaemia as a model of disease evolution in human cancer. Nat. Rev. Cancer 2007, 7, 441–453. [Google Scholar]
- Goh, W.W.; Lee, Y.H.; Ramdzan, Z.M.; Chung, M.C.; Wong, L.; Sergot, M.J. A network-based maximum link approach towards MS identifies potentially important roles for undetected ARRB1/2 and ACTB in liver cancer progression. Int. J. Bioinform. Res. Appl 2012, 8, 155–170. [Google Scholar]
- Wang, X.; Zhang, A.; Sun, H.; Wu, G.; Sun, W.; Yan, G. Network generation enhances interpretation of proteomics data sets by a combination of two-dimensional polyacrylamide gel electrophoresis and matrix-assisted laser desorption/ionization-time of flight mass spectrometry. Analyst 2012, 137, 4703–4711. [Google Scholar]
- Cluitmans, J.C.; Hardeman, M.R.; Dinkla, S.; Brock, R.; Bosman, G.J. Red blood cell deformability during storage: Towards functional proteomics and metabolomics in the blood bank. Blood Transfus 2012, 10, s12–s18. [Google Scholar]
- Burkhart, J.M.; Vaudel, M.; Gambaryan, S.; Radau, S.; Walter, U.; Martens, L.; Geiger, J.; Sickmann, A.; Zahedi, R.P. The first comprehensive and quantitative analysis of human platelet protein composition allows the comparative analysis of structural and functional pathways. Blood 2012, 120, e73–e82. [Google Scholar]
- Wishart, T.M.; Rooney, T.M.; Lamont, D.J.; Wright, A.K.; Morton, A.J.; Jackson, M.; Freeman, M.R.; Gillingwater, T.H. Combining comparative proteomics and molecular genetics uncovers regulators of synaptic and axonal stability and degeneration in vivo. PLoS Genet 2012, 8, e1002936. [Google Scholar]
- Zheng, Q.; Ren, Y.; Tzekov, R.; Zhang, Y.; Chen, B.; Hou, J.; Zhao, C.; Zhu, J.; Zhang, Y.; Dai, X.; et al. Differential proteomics and functional research following gene therapy in a mouse model of leber congenital amaurosis. PLoS One 2012, 7, e44855. [Google Scholar]
- Johnson, E.K.; Zhang, L.; Adams, M.E.; Phillips, A.; Freitas, M.A.; Froehner, S.C.; Green-Church, K.B.; Montanaro, F. Proteomic analysis reveals new cardiac-specific dystrophin-associated proteins. PLoS One 2012, 7, e43515. [Google Scholar]
- Becnel, L.B.; McKenna, N.J. Minireview: Progress and challenges in proteomics data management, sharing, and integration. Mol. Endocrinol 2012, 26, 1660–1674. [Google Scholar]
- Chocu, S.; Calvel, P.; Rolland, A.D.; Pineau, C. Spermatogenesis in mammals: Proteomic insights. Syst. Biol. Reprod. Med 2012, 58, 179–190. [Google Scholar]
- Baker, M.A.; Nixon, B.; Naumovski, N.; Aitken, R.J. Proteomic insights into the maturation and capacitation of mammalian spermatozoa. Syst. Biol. Reprod. Med 2012, 58, 211–217. [Google Scholar]
- Dadvar, P.; O’Flaherty, M.; Scholten, A.; Rumpel, K.; Heck, A.J. A chemical proteomics based enrichment technique targeting the interactome of the PDE5 inhibitor PF-4540124. Mol. Biosyst 2009, 5, 472–482. [Google Scholar]
- Bantscheff, M.; Eberhard, D.; Abraham, Y.; Bastuck, S.; Boesche, M.; Hobson, S.; Mathieson, T.; Perrin, J.; Raida, M.; Rau, C.; et al. Quantitative chemical proteomics reveals mechanisms of action of clinical ABL kinase inhibitors. Nat. Biotechnol 2007, 25, 1035–1044. [Google Scholar]
- Trinkle-Mulcahy, L.; Boulon, S.; Lam, Y.W.; Urcia, R.; Boisvert, F.M.; Vandermoere, F.; Morrice, N.A.; Swift, S.; Rothbauer, U.; Leonhardt, H.; et al. Identifying specific protein interaction partners using quantitative mass spectrometry and bead proteomes. J. Cell Biol 2008, 183, 223–239. [Google Scholar] [Green Version]
- Sin, N.; Meng, L.; Auth, H.; Crews, C.M. Eponemycin analogues: Syntheses and use as probes of angiogenesis. Bioorg. Med. Chem 1998, 6, 1209–1217. [Google Scholar]
- Ranish, J.A.; Yi, E.C.; Leslie, D.M.; Purvine, S.O.; Goodlett, D.R.; Eng, J.; Aebersold, R. The study of macromolecular complexes by quantitative proteomics. Nat. Genet 2003, 33, 349–355. [Google Scholar]
- Dadvar, P.; Kovanich, D.; Folkers, G.E.; Rumpel, K.; Raijmakers, R.; Heck, A.J. Phosphatidylethanolamine-binding proteins, including RKIP, exhibit affinity for phosphodiesterase-5 inhibitors. ChemBioChem 2009, 10, 2654–2662. [Google Scholar]
- Rix, U.; Superti-Furga, G. Target profiling of small molecules by chemical proteomics. Nat. Chem. Biol 2009, 5, 616–624. [Google Scholar]
- Brehmer, D.; Godl, K.; Zech, B.; Wissing, J.; Daub, H. Proteome-wide identification of cellular targets affected by bisindolylmaleimide-type protein kinase C inhibitors. Mol. Cell. Proteomics 2004, 3, 490–500. [Google Scholar]
- Bach, S.; Knockaert, M.; Reinhardt, J.; Lozach, O.; Schmitt, S.; Baratte, B.; Koken, M.; Coburn, S.P.; Tang, L.; Jiang, T.; et al. Roscovitine targets, protein kinases and pyridoxal kinase. J. Biol. Chem 2005, 280, 31208–31219. [Google Scholar]
- Rix, U.; Hantschel, O.; Dürnberger, G.; Remsing Rix, L.L.; Planyavsky, M.; Fernbach, N.V.; Kaupe, I.; Bennett, K.L.; Valent, P.; Colinge, J.; et al. Chemical proteomic profiles of the BCR-ABL inhibitors imatinib, nilotinib, and dasatinib reveal novel kinase and nonkinase targets. Blood 2007, 110, 4055–4063. [Google Scholar]
- Moellering, R.E.; Cravatt, B.F. How chemoproteomics can enable drug discovery and development. Chem. Biol 2012, 19, 11–22. [Google Scholar]
- Nomura, D.K.; Dix, M.M.; Cravatt, B.F. Activity-based protein profiling for biochemical pathway discovery in cancer. Nat. Rev. Cancer 2010, 10, 630–638. [Google Scholar]
- Bantscheff, M.; Drewes, G. Chemoproteomic approaches to drug target identification and drug profiling. Bioorg. Med. Chem 2012, 20, 1973–1978. [Google Scholar]
- Rybak, J.N.; Ettorre, A.; Kaissling, B.; Giavazzi, R.; Neri, D.; Elia, G. In vivo protein biotinylation for identification of organ-specific antigens accessible from the vasculature. Nat. Methods 2005, 2, 291–298. [Google Scholar]
- Roesli, C.; Neri, D.; Rybak, J.N. In vivo protein biotinylation and sample preparation for the proteomic identification of organ- and disease-specific antigens accessible from the vasculature. Nat. Protoc 2006, 1, 192–199. [Google Scholar]
- Castronovo, V.; Waltregny, D.; Kischel, P.; Roesli, C.; Elia, G.; Rybak, J.N.; Neri, D. A chemical proteomics approach for the identification of accessible antigens expressed in human kidney cancer. Mol. Cell. Proteomics 2006, 5, 2083–2091. [Google Scholar]
- Conrotto, P.; Roesli, C.; Rybak, J.; Kischel, P.; Waltregny, D.; Neri, D.; Castronovo, V. Identification of new accessible tumor antigens in human colon cancer by ex vivo protein biotinylation and comparative mass spectrometry analysis. Int. J. Cancer 2008, 123, 2856–2864. [Google Scholar]
- Scheurer, S.B.; Rybak, J.N.; Roesli, C.; Brunisholz, R.A.; Potthast, F.; Schlapbach, R.; Neri, D.; Elia, G. Identification and relative quantification of membrane proteins by surface biotinylation and two-dimensional peptide mapping. Proteomics 2005, 5, 2718–2728. [Google Scholar]
- Roesli, C.; Mumprecht, V.; Neri, D.; Detmar, M. Identification of the surface-accessible, lineage-specific vascular proteome by two-dimensional peptide mapping. FASEB J 2008, 22, 1933–1944. [Google Scholar]
- Strassberger, V.; Fugmann, T.; Neri, D.; Roesli, C. Chemical proteomic and bioinformatic strategies for the identification and quantification of vascular antigens in cancer. J. Proteomics 2010, 73, 1954–1973. [Google Scholar]
- Matta, A.; Ralhan, R.; DeSouza, L.V.; Siu, K.W. Mass spectrometry-based clinical proteomics: Head-and-neck cancer biomarkers and drug-targets discovery. Mass Spectrom. Rev 2010, 29, 945–961. [Google Scholar]
- Apweiler, R.; Aslanidis, C.; Deufel, T.; Gerstner, A.; Hansen, J.; Hochstrasser, D.; Kellner, R.; Kubicek, M.; Lottspeich, F.; Maser, E.; et al. Approaching clinical proteomics: Current state and future fields of application in cellular proteomics. Cytometry A 2009, 75, 816–832. [Google Scholar]
- Tammen, H.; Schulte, I.; Hess, R.; Menzel, C.; Kellmann, M.; Mohring, T.; Schulz-Knappe, P. Peptidomic analysis of human blood specimens: Comparison between plasma specimens and serum by differential peptide display. Proteomics 2005, 5, 3414–3422. [Google Scholar]
- Savino, R.; Terracciano, R. Mesopore-assisted profiling strategies in clinical proteomics for drug/target discovery. Drug Discov. Today 2012, 17, 143–152. [Google Scholar]
- Matt, P.; Fu, Z.; Fu, Q.; van Eyk, J.E. Biomarker discovery: Proteome fractionation and separation in biological samples. Physiol. Genomics 2008, 33, 12–17. [Google Scholar]
- Casadonte, F.; Pasqua, L.; Savino, R.; Terracciano, R. Smart trypsin adsorption into N-(2-aminoethyl)-3-aminopropyl-modified mesoporous silica for ultra fast protein digestion. Chemistry 2010, 16, 8998–9001. [Google Scholar]
- Savino, R.; Casadonte, F.; Terracciano, R. In mesopore protein digestion: A new forthcoming strategy in proteomics. Molecules 2011, 16, 5938–5962. [Google Scholar]
- Petricoin, E.F.; Ardekani, A.M.; Hitt, B.A.; Levine, P.J.; Fusaro, V.A.; Steinberg, S.M.; Mills, G.B.; Simone, C.; Fishman, D.A.; Kohn, E.C.; et al. Use of proteomic patterns in serum to identify ovarian cancer. Lancet 2002, 359, 572–577. [Google Scholar]
- Diamandis, E.P. Mass spectrometry as a diagnostic and a cancer biomarker discovery tool: Opportunities and potential limitations. Mol. Cell. Proteomics 2004, 3, 367–378. [Google Scholar]
- Baggerly, K.A.; Morris, J.S.; Coombes, K.R. Reproducibility of SELDI-TOF protein patterns in serum: Comparing datasets from different experiments. Bioinformatics 2004, 20, 777–785. [Google Scholar]
- Lopez, M.F.; Mikulskis, A.; Kuzdzal, S.; Bennett, D.A.; Kelly, J.; Golenko, E.; DiCesare, J.; Denoyer, E.; Patton, W.F.; Ediger, R.; et al. High-resolution serum proteomic profiling of Alzheimer disease samples reveals disease-specific, carrier-protein-bound mass signatures. Clin. Chem 2005, 51, 1946–1954. [Google Scholar]
- Clarke, C.H.; Buckley, J.A.; Fung, E.T. SELDI-TOF-MS proteomics of breast cancer. Clin. Chem. Lab. Med 2005, 43, 1314–1320. [Google Scholar]
- Sauter, E.R.; Shan, S.; Hewett, J.E.; Speckman, P.; Du Bois, G.C. Proteomic analysis of nipple aspirate fluid using SELDI-TOF-MS. Int. J. Cancer 2005, 114, 791–796. [Google Scholar]
- De Seny, D.; Fillet, M.; Meuwis, M.A.; Geurts, P.; Lutteri, L.; Ribbens, C.; Bours, V.; Wehenkel, L.; Piette, J.; Malaise, M.; et al. Discovery of new rheumatoid arthritis biomarkers using the surface-enhanced laser desorption/ionization time-of-flight mass spectrometry ProteinChip approach. Arthritis Rheum 2005, 52, 3801–3812. [Google Scholar]
- He, J.; Gornbein, J.; Shen, D.; Lu, M.; Rovai, L.E.; Shau, H.; Katz, J.; Whitelegge, J.P.; Faull, K.F.; Chang, H.R. Detection of breast cancer biomarkers in nipple aspirate fluid by SELDI-TOF and their identification by combined liquid chromatography-tandem mass spectrometry. Int. J. Oncol 2007, 30, 145–154. [Google Scholar]
- Hodgetts, A.; Levin, M.; Kroll, J.S.; Langford, P.R. Biomarker discovery in infectious diseases using SELDI. Future Microbiol 2007, 2, 35–49. [Google Scholar]
- De Seny, D.; Fillet, M.; Ribbens, C.; Marée, R.; Meuwis, M.A.; Lutteri, L.; Chapelle, J.P.; Wehenkel, L.; Louis, E.; Merville, M.P.; et al. Monomeric calgranulins measured by SELDI-TOF mass spectrometry and calprotectin measured by ELISA as biomarkers in arthritis. Clin. Chem 2008, 54, 1066–1075. [Google Scholar]
- Cazares, L.H.; Diaz, J.; Drake, R.; Semmes, O. MALDI/SELDI Protein Profiling of Serum for the Identification of Cancer Biomarkers. In Methods in Molecular Biology; Vlahou, A., Ed.; Humana Press: Totowa, NJ, USA, 2008; pp. 125–140. [Google Scholar]
- Villanueva, J.; Philip, J.; Chaparro, C.A.; Li, Y.; ToledoCrow, R.; DeNoyer, L.; Fleisher, M.; Robbins, R.J.; Tempst, P. Correcting common errors in identifying cancer-specific serum peptide signatures. J. Prot. Res 2005, 4, 1060–1072. [Google Scholar]
- Zhang, X.; Leung, S.M.; Morris, C.R.; Shigenaga, M.K. Evaluation of a novel, integrated approach using functionalized magnetic beads, bench-top MALDI-TOF-MS with prestructured sample supports, and pattern recognition software for profiling potential biomarkers in human plasma. J. Biomol. Tech 2004, 15, 167–175. [Google Scholar]
- Villanueva, J.; Philip, J.; Entenberg, D.; Chaparro, C.A.; Tanwar, M.K.; Holland, E.C.; Tempst, P. Serum peptide profiling by magnetic particle-assisted, automated sample processing and MALDI-TOF mass spectrometry. Anal. Chem 2004, 76, 1560–1570. [Google Scholar]
- Baumann, S.; Ceglarek, U.; Fiedler, G.M.; Lembcke, J.; Leichtle, A.; Thiery, J. Standardized approach to proteome profiling of human serum based on magnetic bead separation and matrix-assisted laser desorption/ionization time-of-flight mass spectrometry. Clin. Chem 2005, 51, 973–980. [Google Scholar]
- De Noo, M.E.; Tollenaar, R.A.; Ozalp, A.; Kuppen, P.J.; Bladergroen, M.R.; Eilers, P.H.; Deelder, A.M. Reliability of human serum protein profiles generated with C8 magnetic beads assisted MALDI-TOF mass spectrometry. Anal. Chem 2005, 77, 7232–7241. [Google Scholar]
- Callesen, A.K.; Madsen, J.S.; Vach, W.; Kruse, T.A.; Mogensen, O.; Jensen, O.N. Serum protein profiling by solid phase extraction and mass spectrometry: A future diagnostics tool? Proteomics 2009, 9, 1428–1441. [Google Scholar]
- Terracciano, R.; Casadonte, F.; Pasqua, L.; Candeloro, P.; Di Fabrizio, E.; Urbani, A.; Savino, R. Enhancing plasma peptide MALDI-TOF-MS profiling by mesoporous silica assisted crystallization. Talanta 2010, 80, 1532–1538. [Google Scholar]
- Ray, S.; Reddy, P.J.; Jain, R.; Gollapalli, K.; Moiyadi, A.; Srivastava, S. Proteomic technologies for the identification of disease biomarkers in serum: Advances and challenges ahead. Proteomics 2011, 11, 2139–2161. [Google Scholar]
- Ebert, M.P.; Meuer, J.; Wiemer, J.C.; Schulz, H.U.; Reymond, M.A.; Traugott, U.; Malfertheiner, P.; Röcken, C. Identification of gastric cancer patients by serum protein profiling. J. Proteome Res 2004, 3, 1261–1266. [Google Scholar]
- Zhang, Z.; Bast, R.C., Jr; Yu, Y.; Li, J.; Sokoll, L.J.; Rai, A.J.; Rosenzweig, J.M.; Cameron, B.; Wang, Y.Y.; Meng, X.Y.; et al. Three biomarkers identified from serum proteomic analysis for the detection of early stage ovarian cancer. Cancer Res. 2004, 64, 5882–5890. [Google Scholar]
- Chen, Y.D.; Zheng, S.; Yu, J.K.; Hu, X. Artificial neural networks analysis of surface-enhanced laser desorption/ionization mass spectra of serum protein pattern distinguishes colorectal cancer from healthy population. Clin. Cancer Res 2004, 10, 8380–8385. [Google Scholar]
- Carrette, O.; Demalte, I.; Scherl, A.; Yalkinoglu, O.; Corthals, G.; Burkhard, P.; Hochstrasser, D.F.; Sanchez, J.C. A panel of cerebrospinal fluid potential biomarkers for the diagnosis of Alzheimer’s disease. Proteomics 2003, 3, 1486–1494. [Google Scholar]
- Cheng, A.J.; Chen, L.C.; Chien, K.Y.; Chen, Y.J.; Chang, J.T.; Wang, H.M.; Liao, C.T.; Chen, I.H. Oral cancer plasma tumor marker identified with bead-based affinity-fractionated proteomic technology. Clin. Chem 2005, 51, 2236–2244. [Google Scholar]
- Chang, J.T.; Chen, L.C.; Wei, S.Y.; Chen, Y.J.; Wang, H.M.; Liao, C.T.; Chen, I.H.; Cheng, A.J. Increase diagnostic efficacy by combined use of fingerprint markers in mass spectrometry-plasma peptidomes from nasopharyngeal cancer patients for example. Clin. Biochem 2006, 39, 1144–1151. [Google Scholar]
- Zhang, H.; Wu, G.; Tu, H.; Huang, F. Discovery of serum biomarkers in astrocytoma by SELDI-TOF MS and proteinchip technology. J. Neurooncol 2007, 84, 315–323. [Google Scholar]
- Sawai, S.; Humemura, H.; Mori, M.; Satoh, M.; Hayakawa, S.; Kodera, Y.; Tomonaga, T.; Kuwabara, S.; Nomura, F. Serum levels of complement C4 fragments correlate with disease activity in multiple sclerosis: Proteomic analysis. J. Neuroimmunol 2010, 218, 112–115. [Google Scholar]
- Chinello, C.; Gianazza, E.; Zoppis, I.; Mainini, V.; Galbusera, C.; Picozzi, S.; Rocco, F.; Galasso, G.; Bosari, S.; Ferrero, S.; et al. Serum biomarkers of renal cell carcinoma assessed using a protein profiling approach based on clinprot technique. Urology 2010, 75, 842–847. [Google Scholar]
- Mainini, V.; Gianazza, E.; Chinello, C.; Bilo, G.; Revera, M.; Giuliano, A.; Caldara, G.; Lombardi, C.; Piperno, A.; Magni, F.; et al. Modulation of urinary peptidome in humans exposed to high altitude hypoxia. Mol. Biosyst 2012, 8, 959–966. [Google Scholar]
- Merkel, D.; Rist, W.; Seither, P.; Weith, A.; Lenter, M.C. Proteomic study of human bronchoalveolar lavage fluids from smokers with chronic obstructive pulmonary disease by combining surface-enhanced laser desorption/ionization-mass spectrometry profiling with mass spectrometric protein identification. Proteomics 2005, 5, 2972–2980. [Google Scholar]
- Terracciano, R.; Preianò, M.; Palladino, G.P.; Carpagnano, G.E.; Barbaro, M.P.; Pelaia, G.; Savino, R.; Maselli, R. Peptidome profiling of induced sputum by mesoporous silica beads and MALDI-TOF MS for non-invasive biomarker discovery of chronic inflammatory lung diseases. Proteomics 2011, 11, 3402–3414. [Google Scholar]
- Gygi, S.P.; Rist, B.; Gerber, S.A.; Turecek, F.; Gelb, M.H.; Aebersold, R. Quantitative analysis of complex protein mixtures using isotope-coded affinity tags. Nat. Biotechnol 1999, 17, 994–999. [Google Scholar]
- Ross, P.L.; Huang, Y.N.; Marchese, J.N.; Williamson, B.; Parker, K.; Hattan, S.; Khainovski, N.; Pillai, S.; Dey, S.; Daniels, S.; et al. Multiplexed protein quantitation in Saccharomyces cerevisiae using amine-reactive isobaric tagging reagents. Mol. Cell. Proteomics 2004, 3, 1154–1169. [Google Scholar]
- Veenstra, T.D. Global and targeted quantitative proteomics for biomarker discovery. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci 2007, 847, 3–11. [Google Scholar]
- Sui, J.; Zhang, J.; Tan, T.L.; Ching, C.B.; Chen, W.N. Comparative proteomics analysis of vascular smooth muscle cells incubated with S- and R-enantiomers of atenolol using iTRAQ-coupled two-dimensional LC-MS/MS. Mol. Cell. Proteomics 2008, 7, 1007–1018. [Google Scholar]
- Sui, J.; Zhang, J.; Ching, C.B.; Chen, W.N. Expanding proteomics into the analysis of chiral drugs. Mol. Biosyst 2009, 5, 603–608. [Google Scholar]
- Lange, V.; Picotti, P.; Domon, B.; Aebersold, R. Selected reaction monitoring for quantitative proteomics: A tutorial. Mol. Syst. Biol 2008, 4, 222. [Google Scholar]
- Levin, Y.; Schwartz, E.; Wang, L.; Leweke, F.M.; Bahn, S. Label free LC-MS/MS quantitative proteomics for large-scale biomarker discovery in complex samples. J. Sep. Sci 2007, 30, 2198–2203. [Google Scholar]
- Nanni, P.; Levander, F.; Roda, G.; Caponi, A.; James, P.; Roda, A. A label-free nano-liquid chromatography-mass spectrometry approach for quantitative serum peptidomics in Crohn’s disease patients. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci 2009, 877, 3127–3136. [Google Scholar]
- Gámez-Pozo, A.; Sánchez-Navarro, I.; Calvo, E.; Agulló-Ortuño, M.T.; López-Vacas, R.; Díaz, E.; Camafeita, E.; Nistal, M.; Madero, R.; Espinosa, E.; et al. PTRF/cavin-1 and MIF proteins are identified as non-small cell lung cancer biomarkers by label-free proteomics. PLoS One 2012, 7, e33752. [Google Scholar]
- Foss, E.J.; Radulovic, D.; Stirewalt, D.L.; Radich, J.; Sala-Torra, O.; Pogosova-Agadjanyan, E.L.; Hengel, S.M.; Loeb, K.R.; Deeg, H.J.; Meshinchi, S.; et al. Proteomic classification of acute leukemias by alignment-based quantitation of LC-MS/MS data sets. J. Proteome Res 2012, 11, 5005–5010. [Google Scholar]
- Breuker, K.; Jin, M.; Han, X.; Jiang, H.; McLafferty, F.W. Top-down identification and characterization of biomolecules by mass spectrometry. J. Am. Soc. Mass Spectrom 2008, 19, 1045–1053. [Google Scholar]
- Berardi, S.; Caivano, A.; Ria, R.; Nico, B.; Savino, R.; Terracciano, R.; De Tullio, G.; Ferrucci, A.; De Luisi, A.; Moschetta, M.; et al. Four proteins governing overangiogenic endothelial cell phenotype in patients with multiple myeloma are plausible therapeutic targets. Oncogene 2012, 31, 2258–2269. [Google Scholar]
- Descotes, F.; Jézéquel, P.; Spyratos, F.; Campion, L.; Grenot, C.; Lerebours, F.; Campone, M.; Guérin-Charbonnel, C.; Lanoë, D.; Adams, M.; et al. Identification of potential prognostic biomarkers for node-negative breast tumours by proteomic analysis: A multicentric 2004 national PHRC study. Int. J. Oncol 2012, 41, 92–104. [Google Scholar]
- Raimondo, F.; Salemi, C.; Chinello, C.; Fumagalli, D.; Morosi, L.; Rocco, F.; Ferrero, S.; Perego, R.; Bianchi, C.; Sarto, C.; et al. Proteomic analysis in clear cell renal cell carcinoma: Identification of differentially expressed protein by 2-D DIGE. Mol. Biosyst 2012, 8, 1040–1051. [Google Scholar]
- Rai, A.J.; Gelfand, C.A.; Haywood, B.C.; Warunek, D.J.; Yi, J.; Schuchard, M.D.; Mehigh, R.J.; Cockrill, S.L.; Scott, G.B.; Tammen, H.; et al. HUPO plasma proteome project specimen collection and handling: Towards the standardization of parameters for plasma proteome samples. Proteomics 2005, 5, 3262–3277. [Google Scholar]
- Callesen, A.K.; Christensen, R.; Madsen, J.S.; Vach, W.; Zapico, E.; Cold, S.; Jørgensen, P.E.; Mogensen, O.; Kruse, T.A.; Jensen, O.N. Reproducibility of serum protein profiling by systematic assessment using solid-phase extraction and matrix-assisted laser desorption/ionization mass spectrometry. Rapid Commun. Mass Spectrom 2008, 22, 291–300. [Google Scholar]
- Navare, A.; Zhou, M.; McDonald, J.; Noriega, F.G.; Sullards, M.C.; Fernandez, F.M. Serum biomarker profiling by solid-phase extraction with particle-embedded micro tips and matrix-assisted laser desorption/ionization mass spectrometry. Rapid Commun. Mass Spectrom 2008, 22, 997–1008. [Google Scholar]
- Penno, M.A.; Ernst, M.; Hoffman, P. Optimal preparation methods for automated matrix-assisted laser desorption/ionization time-of-flight mass spectrometry profiling of low molecular weight proteins and peptides. Rapid Commun. Mass Spectrom 2009, 23, 2656–2662. [Google Scholar]
- Zolla, L. Proteomics studies reveal important information on small molecule therapeutics: A case study on plasma proteins. Drug Discov. Today 2008, 13, 1042–1051. [Google Scholar]
- D’Alessandro, A.; Zolla, L. Pharmacoproteomics: A chess game on a protein field. Drug Discov. Today 2010, 15, 1015–1023. [Google Scholar]
- Slany, A.; Haudek, V.J.; Gundacker, N.C.; Griss, J.; Mohr, T.; Wimmer, H.; Eisenbauer, M.; Elbling, L.; Gerner, C. Introducing a new parameter for quality control of proteome profiles: Considerations of commonly expressed proteins. Electrophoresis 2009, 30, 1306–1328. [Google Scholar]
- Taylor, C.F.; Paton, N.W.; Lilley, K.S.; Binz, P.A.; Julian, R.K., Jr; Jones, A.R.; Zhu, W.; Apweiler, R.; Aebersold, R.; Deutsch, E.W.; et al. The minimum information about a proteomics experiment (MIAPE). Nat. Biotechnol. 2007, 25, 887–893. [Google Scholar]
- Preianò, M.; Pasqua, L.; Gallelli, L.; Galasso, O.; Gasparini, G.; Savino, R.; Terracciano, R. Simultaneous extraction and rapid visualization of peptidomic & lipidomic body fluids fingerprints by using mesoporous aluminosilicate and MALDI-TOF MS. Proteomics 2012. [Google Scholar] [CrossRef]
- Wegdam, W.; Moerland, P.D.; Meijer, D.; de Jong, S.M.; Hoefsloot, H.C.; Kenter, G.G.; Buist, M.R.; Aerts, J.M. A critical assessment of SELDI-TOF-MS for biomarker discovery in serum and tissue of patients with an ovarian mass. Proteome Sci 2012, 10. [Google Scholar] [CrossRef]
- Zhang, J.; Wang, K.; Zhang, J.; Liu, S.S.; Dai, L.; Zhang, J.Y. Using proteomic approach to identify tumor-associated proteins as biomarkers in human esophageal squamous cell carcinoma. J. Proteome Res 2011, 10, 2863–2872. [Google Scholar]
- Gerszten, R.E.; Asnani, A.; Carr, S.A. Status and prospects for discovery and verification of new biomarkers of cardiovascular disease by proteomics. Circ. Res 2011, 109, 463–474. [Google Scholar]
- Keshishian, H.; Addona, T.; Burgess, M.; Kuhn, E.; Carr, S.A. Quantitative, multiplexed assays for low abundance proteins in plasma by targeted mass spectrometry and stable isotope dilution. Mol. Cell. Proteomics 2007, 6, 2212–2229. [Google Scholar]
- Anderson, N.L.; Anderson, N.G.; Haines, L.R.; Hardie, D.B.; Olafson, R.W.; Pearson, T.W. Mass spectrometric quantitation of peptides and proteins using Stable Isotope Standards and Capture by Anti-Peptide Antibodies (SISCAPA). J. Proteome Res 2004, 3, 235–244. [Google Scholar]
- Picotti, P.; Bodenmiller, B.; Mueller, L.N.; Domon, B.; Aebersold, R. Full dynamic range proteome analysis of S. cerevisiae by targeted proteomics. Cell 2009, 138, 795–806. [Google Scholar]
- Addona, T.A.; Abbatiello, S.E.; Schilling, B.; Skates, S.J.; Mani, D.R.; Bunk, D.M.; Spiegelman, C.H.; Zimmerman, L.J.; Ham, A.J.; Keshishian, H.; et al. Multi-site assessment of the precision and reproducibility of multiple reaction monitoring-based measurements of proteins in plasma. Nat. Biotechnol 2009, 27, 633–641. [Google Scholar]
- Shi, T.; Su, D.; Liu, T.; Tang, K.; Camp, D.G., II; Qian, W.; Smith, R.D. Advancing the sensitivity of selected reaction monitoring-based targeted quantitative proteomics. Proteomics 2012, 12, 1074–1092. [Google Scholar]
- Whiteaker, J.R.; Zhao, L.; Anderson, L.; Paulovich, A.G. An automated and multiplexed method for high throughput peptide immunoaffinity enrichment and multiple reaction monitoring mass spectrometry-based quantification of protein biomarkers. Mol. Cell. Proteomics 2010, 9, 184–196. [Google Scholar]
- Hossain, M.; Kaleta, D.T.; Robinson, E.W.; Liu, T.; Zhao, R.; Page, J.S.; Kelly, R.T.; Moore, R.J.; Tang, K.; Camp, D.G., II; et al. Enhanced sensitivity for selected reaction monitoring mass spectrometry-based targeted proteomics using a dual stage electrodynamic ion funnel interface. Mol. Cell. Proteomics 2011, 10. [Google Scholar] [CrossRef]
- Canterbury, J.D.; Yi, X.; Hoopmann, M.R.; MacCoss, M.J. Assessing the dynamic range and peak capacity of nanoflow LC-FAIMS-MS on an ion trap mass spectrometer for proteomics. Anal. Chem 2008, 80, 6888–6897. [Google Scholar]

© 2012 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Savino, R.; Paduano, S.; Preianò, M.; Terracciano, R. The Proteomics Big Challenge for Biomarkers and New Drug-Targets Discovery. Int. J. Mol. Sci. 2012, 13, 13926-13948. https://doi.org/10.3390/ijms131113926
Savino R, Paduano S, Preianò M, Terracciano R. The Proteomics Big Challenge for Biomarkers and New Drug-Targets Discovery. International Journal of Molecular Sciences. 2012; 13(11):13926-13948. https://doi.org/10.3390/ijms131113926
Chicago/Turabian StyleSavino, Rocco, Sergio Paduano, Mariaimmacolata Preianò, and Rosa Terracciano. 2012. "The Proteomics Big Challenge for Biomarkers and New Drug-Targets Discovery" International Journal of Molecular Sciences 13, no. 11: 13926-13948. https://doi.org/10.3390/ijms131113926
APA StyleSavino, R., Paduano, S., Preianò, M., & Terracciano, R. (2012). The Proteomics Big Challenge for Biomarkers and New Drug-Targets Discovery. International Journal of Molecular Sciences, 13(11), 13926-13948. https://doi.org/10.3390/ijms131113926