Protein Kinases and Transcription Factors Activation in Response to UV-Radiation of Skin: Implications for Carcinogenesis



Abstract
:1. Introduction
2. Signal Transduction Pathways in the Cellular Response to UV Radiation
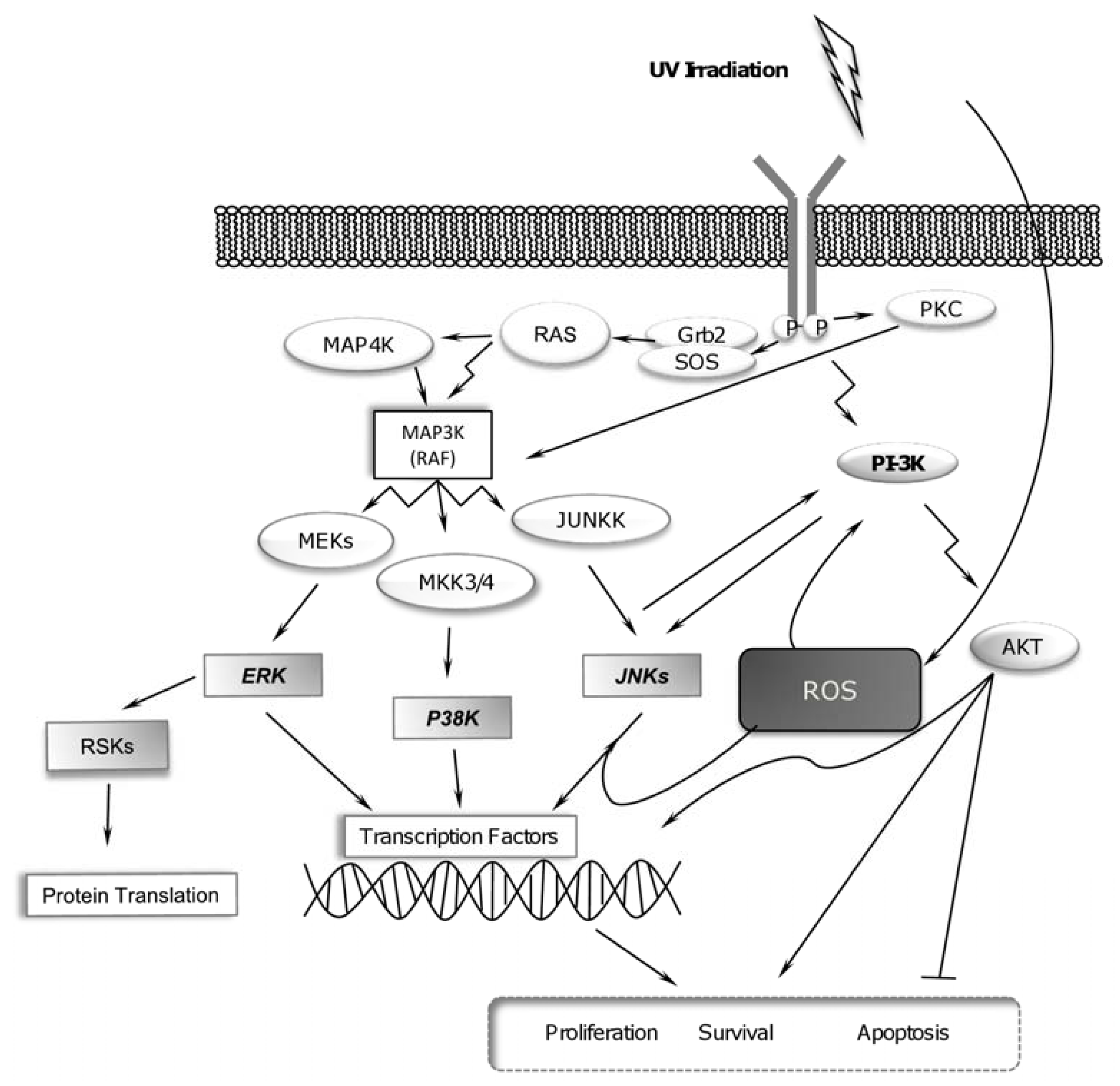
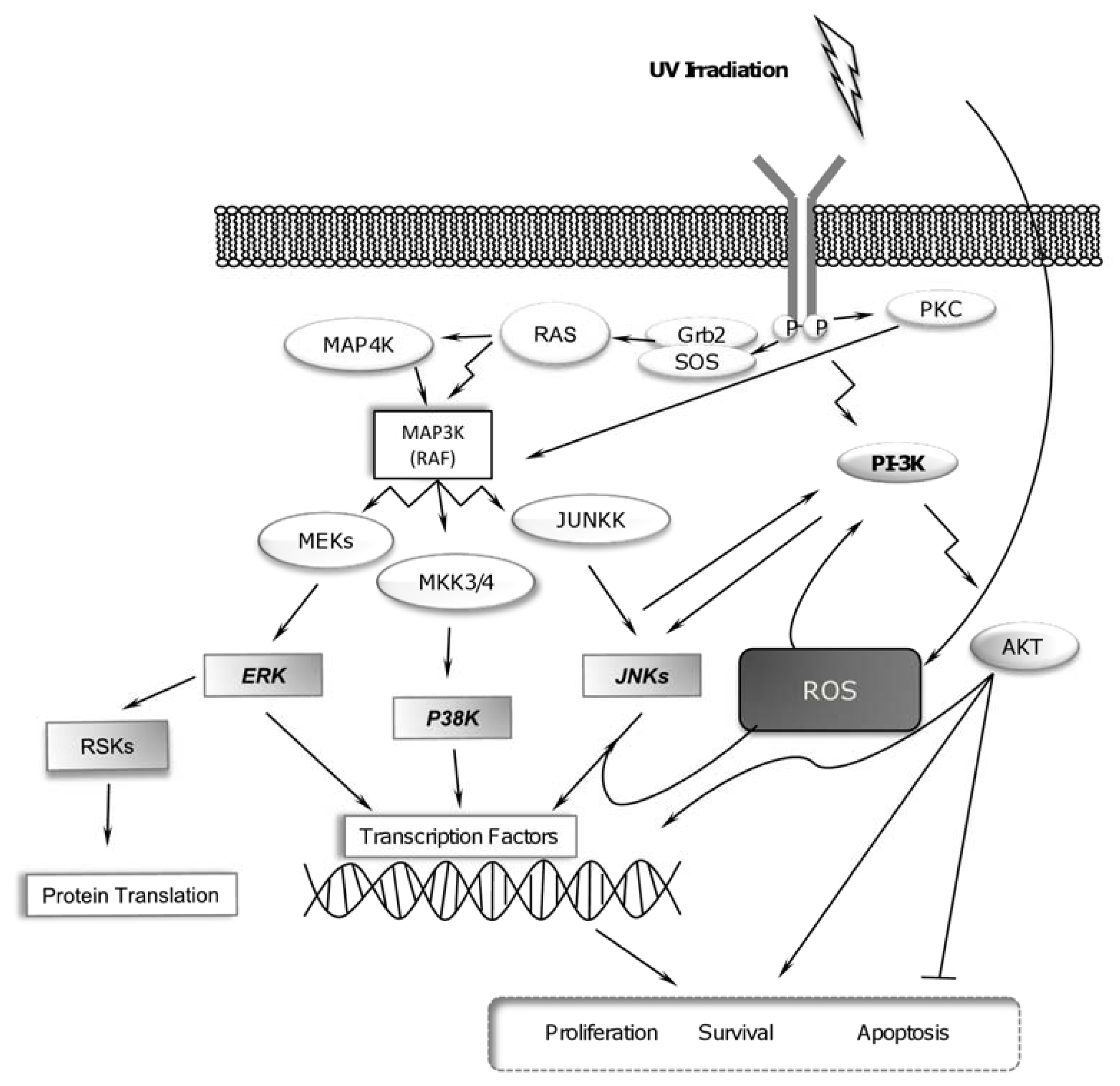
2.1. UV-Radiation Activates MAPK Signaling Pathways
2.2. UV Radiation-Induced Phosphatases
2.3. UV Radiation Induces PI-3K Pathway for Promoting Cell Survival
2.4. RSK Activation and UV Radiation
3. ATM and UV Radiation
4. Role of Transcription Factors in Response to UV Radiation
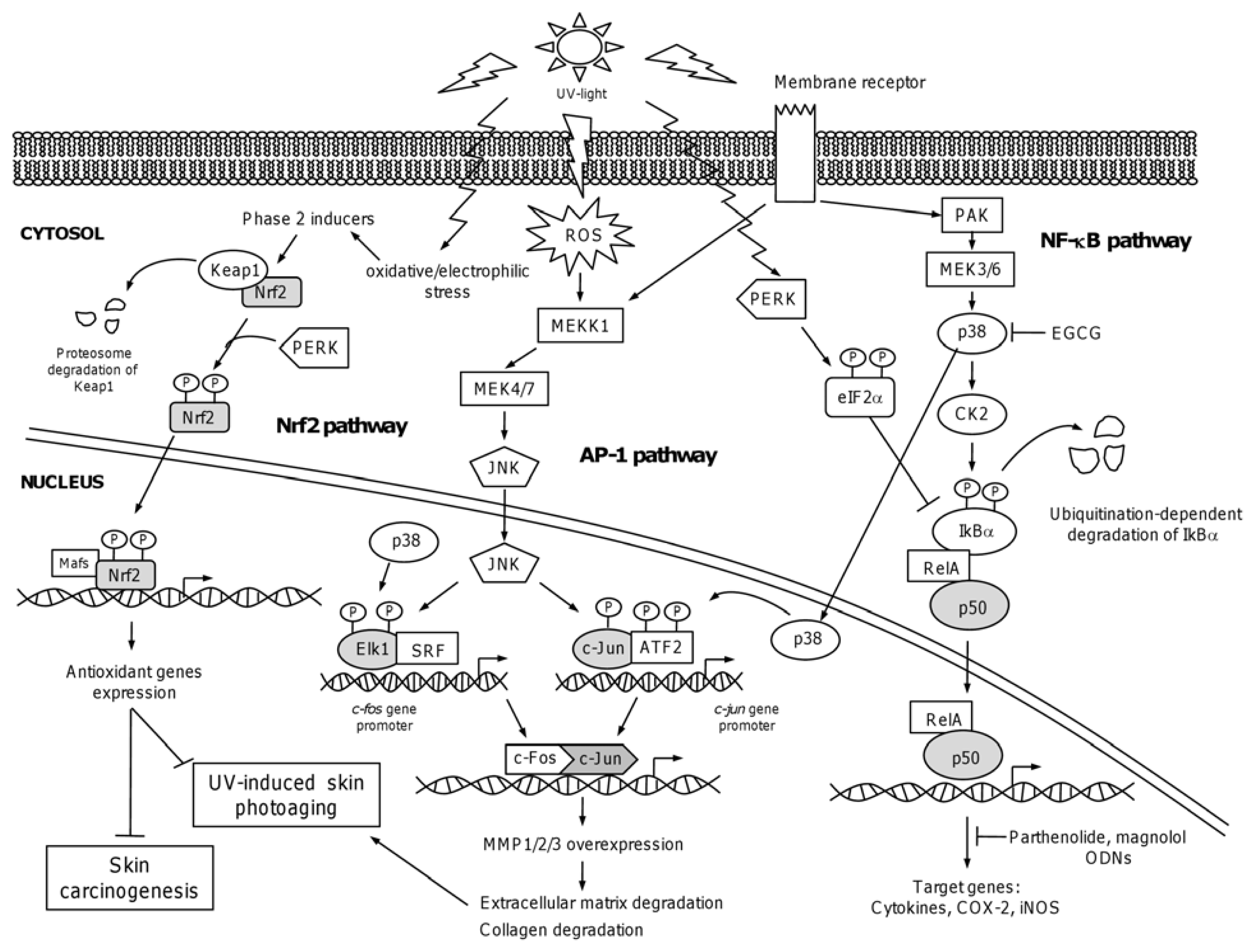
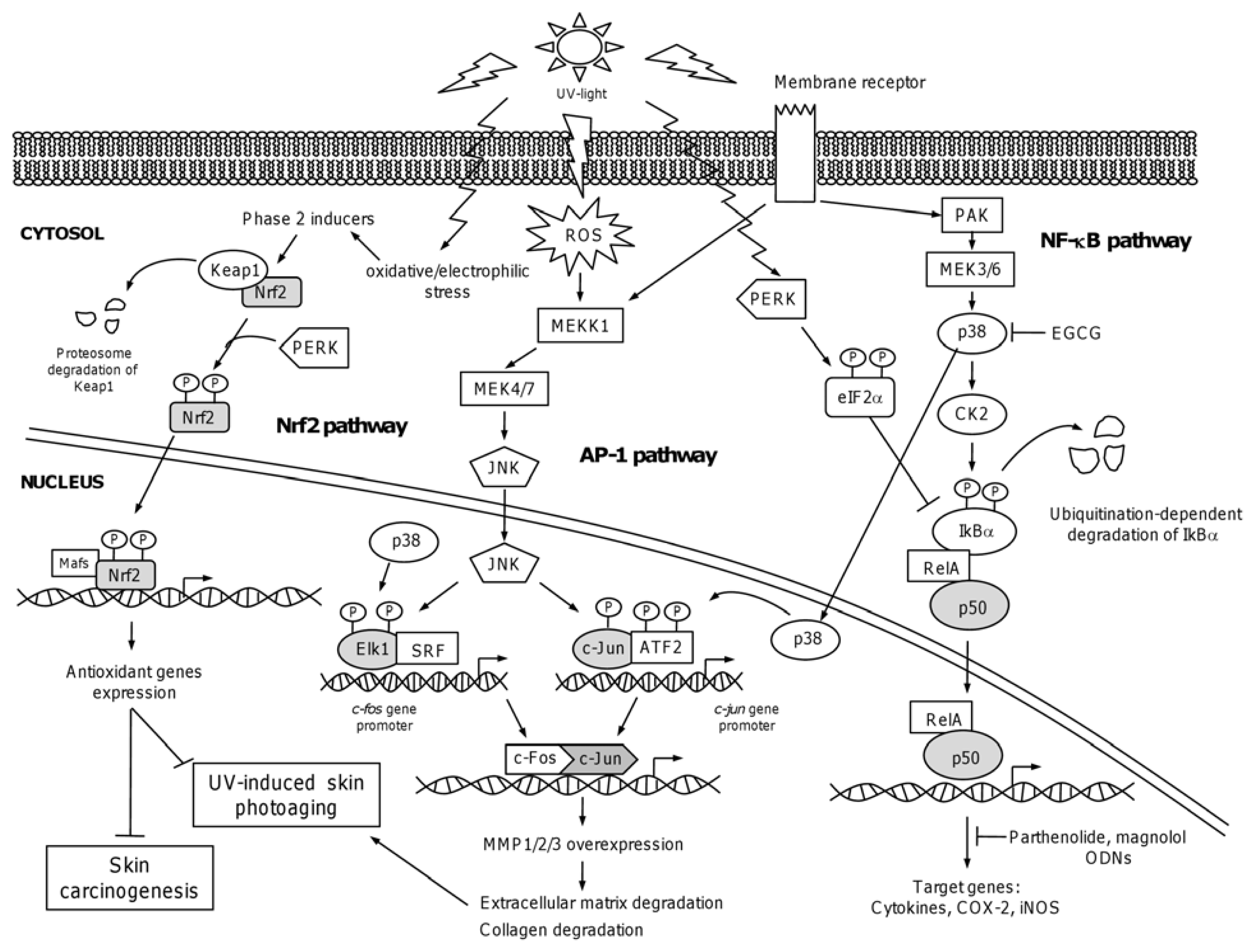
4.1. The Nuclear Factor NF-κB
4.2. Chemopreventive Intervention of NF-κB Pathway
4.3. The Activator Protein-1: AP-1
4.4. The NF-E2-Related Factor 2: Nrf2
4.5. Chemopreventive Intervention of Nfr2-Keap1 Pathway
4.6. Factor of Activated T Cells (NFAT) is Activated in Response to UV Radiation
5. Deciphering Cellular Response to UV Radiation in Skin Using Genomic Tools
6. Concluding Remarks
Acknowledgements
Abbreviations
| UV | Ultraviolet |
| ROS | Reactive Oxygen Species |
| TNFR | Tumor Necrosis Factor Receptor |
| MAPK | Mitogen-Activated Protein Kinases |
| PI-3K | Phosphoinositide 3-Kinases |
| EGFR | Epidermal Growth Factor Receptor |
| PKC | Protein Kinase C |
| ATM | Ataxia Telangiectasia Mutated |
| ATR | ATM-Related |
| JNK | c-Jun NH2-Terminal Kinases |
| RSK2 | p90 Ribosomal S6 Kinase 2 |
| MSK1 | Mitogen-and Stress-Activated Protein Kinase |
| MKPs | Mitogen-Activated Protein Kinase Phosphatases |
| PP2A | Phosphatase 2A |
| Wip1 | Wild-Type p53-Induced Phosphatase |
| PTP | Protein-Tyrosine Phosphatase |
| PIP2 | Phosphatidylinositol-(4,5)-Bisphosphate |
| AP-1 | Activator Protein-1 |
| TLRs | Toll-Like Receptors |
| NF-κB | Nuclear Factor kappa B |
| NIK | NF-κB Inducing Kinase |
| IKK | IκB Kinase |
| NEMO | NF-κB Essential Modulator |
| CK2 | Casein Kinase II |
| LTR | Long Terminal Repeat |
| HIV-1 | Human Immunodeficiency Virus-1 |
| eIF2α | Eukaryotic Translation Initiation Factor 2α |
| PERK | Protein Kinase Like Endoplasmic Reticulum Kinase |
| EGCG | (−)-Epigallocatechin 3-gallate |
| bFGF | Basic Fibroblast Growth Factor |
| MMP-1 | Matrix Metalloproteinase-1 |
| ODNs | Oligodeoxynucleotides |
| TPA | Phorbol Ester 12-O-Tetradecanoylphorbol-13-Acetate |
| MAF | Musculo-Aponeurotic Fibrosarcoma |
| Nrf2 | NF-E2-Related Factor 2 |
| ARE | Antioxidant Responsive Element |
| KGF | Keratinocyte growth factor |
| NFAT | Factor of Activated T Cells |
| COX-2 | Cyclooxygenase 2 |
References
- Epstein, J.H. Photocarcinogenesis, skin cancer, and aging. J. Am. Acad. Dermatol 1983, 9, 487–502. [Google Scholar]
- de Gruijl, F.R.; van Kranen, H.J.; Mullenders, L.H. UV-induced DNA damage, repair, mutations and oncogenic pathways in skin cancer. J. Photochem. Photobiol 2001, B63, 19–27. [Google Scholar]
- Armstrong, B.K.; Kricker, A. The epidemiology of UV induced skin cancer. J. Photochem. Photobiol 2001, B63, 8–18. [Google Scholar]
- Boukamp, P. UV-induced skin cancer: Similarities—variations. J. Dtsch. Dermatol. Ges 2005, 3, 493–503. [Google Scholar]
- Setlow, R.B. The wavelengths in sunlight effective in producing skin cancer: A theoretical analysis. Proc. Natl. Acad. Sci. USA 1974, 71, 3363–3366. [Google Scholar]
- Godar, D.E. UV doses worldwide. Photochem. Photobiol 2005, 81, 736–749. [Google Scholar]
- Kielbassa, C.; Roza, L.; Epe, B. Wavelength dependence of oxidative DNA damage induced by UV and visible light. Carcinogenesis 1997, 18, 811–816. [Google Scholar]
- Ravanat, J.L.; Douki, T.; Cadet, J. Direct and indirect effects of UV radiation on DNA and its components. J. Photochem. Photobiol 2001, B63, 88–102. [Google Scholar]
- Cooper, S.J.; Bowden, G.T. Ultraviolet B regulation of transcription factor families: Roles of nuclear factor-kappa B (NF-κB) and activator protein-1 (AP-1) in UVB-induced skin carcinogenesis. Curr. Cancer Drug Targets 2007, 7, 325–334. [Google Scholar]
- Cole, C.A.; Forbes, P.D.; Davies, R.E. An action spectrum for UV photocarcinogenesis. Photochem. Photobiol 1986, 43, 275–284. [Google Scholar]
- de Gruijl, F.R. Photocarcinogenesis: UVA vs. UVB. Methods Enzymol 2000, 319, 359–366. [Google Scholar]
- Gange, W.R.; Rosen, C.F. UVA effects on mammalian skin and cells. Photochem. Photobiol 1986, 43, 701–705. [Google Scholar]
- Nomura, T.; Nakajima, M.; Hongyo, T.; Taniguchi, E.; Fukuda, K.; Li, L.; Kurooka, M.; Sutoh, K.; Hande, P.; Kawaguchi, T.; et al. Induction of cancer, acidic keratosis, and specific p53 mutations by UVB light in human skin maintained in severe combined immunodeficient mice. Cancer Res 1997, 57, 2081–2084. [Google Scholar]
- Kabuyama, Y.; Homma, M.K.; Kurosaki, T.; Homma, Y. Early signaling events induced by 280-nm UV irradiation. Eur. J. Biochem 2002, 269, 664–670. [Google Scholar]
- Runger, T.M.; Kappes, U.P. Mechanisms of mutation formation with long-wave ultraviolet light (UVA). Photodermatol. Photoimmunol. Photomed 2008, 24, 2–10. [Google Scholar]
- Scharffetter-Kochanek, K.; Wlaschek, M.; Brenneisen, P.; Schauen, M.; Blaudschun, R.; Wenk, J. UV-induced reactive oxygen species in photocarcinogenesis and photoaging. Biol. Chem 1997, 378, 1247–1257. [Google Scholar]
- Bender, K.; Blattner, C.; Knebel, A.; Iordanov, M.; Herrlich, P.; Rahmsdorf, H.J. UV-induced signal transduction. J. Photochem. Photobiol 1997, B37, 1–17. [Google Scholar]
- Tyrrell, R.M. Activation of mammalian gene expression by the UV component of sunlight—from models to reality. Bioessays 1996, 18, 139–148. [Google Scholar]
- Sachsenmaier, C.; Radier-Pohl, A.; Zinck, R.; Nordheim, A.; Herrlich, P.; Rahmsdorf, H.J. Involvement of growth factor receptors in the mammalian UVC response. Cell 1994, 78, 963–972. [Google Scholar]
- Zhang, Y.; Dong, Z.; Bode, A.M.; Ma, W.Y.; Chen, N.; Dong, Z. Induction of EGFR-dependent and EGFR-independent signaling pathways by ultraviolet A irradiation. DNA Cell Biol 2001, 20, 769–779. [Google Scholar]
- Rosette, C.; Karin, M. Ultraviolet light and osmotic stress: Activation of the JNK cascade through multiple growth factor and cytokine receptors. Science 1996, 274, 1194–1197. [Google Scholar]
- Devary, Y.; Gottlieb, R.A.; Smeal, T.; Karin, M. The mammalian ultraviolet response is triggered by activation of Src tyrosine kinases. Cell 1992, 71, 1081–1091. [Google Scholar]
- Coso, O.A.; Chiariello, M.; Yu, J.C.; Teramoto, H.; Crespo, P.; Xu, N.; Miki, T.; Gutkind, J.S. The small GTP-binding proteins Rac1 and Cdc42 regulate the activity of the JNK/SAPK signalling pathway. Cell 1995, 81, 1137–1146. [Google Scholar]
- Kabuyama, Y.; Hamaya, M.; Homma, Y. Wavelength specific activation of PI-3kinase by UVB irradiation. FEBS Lett 1998, 441, 297–301. [Google Scholar]
- Bode, A.M.; Dong, Z. Mitogen-activated protein kinase activation in UV induced signal transduction. Sci. STKE 2003, 2003, RE2. [Google Scholar]
- Zhang, Y.; Ma, W.Y.; Kaji, A.; Bode, A.M.; Dong, Z. Requirement of ATM in UVA-induced signaling and apoptosis. J. Biol. Chem 2002, 277, 3124–3131. [Google Scholar]
- Zhang, Y.; Zhong, S.; Dong, Z.; Chen, N.; Bode, A.M.; Ma, W.; Dong, Z. UVA induces Ser381 phosphorylation of p90RSK/MAPKAP-K1 via ERK and JNK pathways. J. Biol. Chem 2001, 276, 14572–14580. [Google Scholar]
- Ibuki, Y.; Goto, R. Antiapoptotic effects induced by different wavelengths of ultraviolet light. Photochem. Photobiol 2002, 75, 495–502. [Google Scholar]
- Pearson, G.; Robinson, F.; Beers Gibson, T.; Xu, B.E.; Karandikar, M.; Berman, K.; Cobb, M.H. Mitogen-activated protein (MAP) kinase pathways: Regulation and physiological functions. Endocr. Rev 2001, 22, 153–183. [Google Scholar]
- Minden, A.; Lin, A.; McMahon, M.; Lange, C.C.; Derijard, B.; Davis, R.J.; Johnson, L.; Karin, M. Differential activation of ERK and JNK mitogen-activated protein kinases by Raf-1 and MEKK. Science 1994, 266, 1719–1723. [Google Scholar]
- Weston, C.R.; Davis, R.J. The JNK signal transduction pathway. Curr. Opin. Genet. Dev 2002, 12, 14–21. [Google Scholar]
- Roux, P.P.; Blenis, J. ERK and p38 MAPK activated protein kinases: A family of protein kinases with diverse biological functions. Microbiol. Mol. Biol. Rev 2004, 68, 320–344. [Google Scholar]
- Zhong, J.L.; Yang, L.; Lü, F.; Xiao, H.; Xu, R.; Wang, L.; Zhu, F.; Zhang, Y. UVA, UVB and UVC Induce Differential Response Signaling Pathways Converged on the eIF2α phosphorylation. Photochem. Photobiol 2011, 87, 1092–1104. [Google Scholar]
- Nakamura, S.; Takahashi, H.; Kinouchi, M.; Manabe, A.; Ishida-Yamamoto, A.; Hashimoto, Y.; Iizuka, H. Differential phosphorylation of mitogen-activated protein kinase families by epidermal growth factor and ultraviolet B irradiation in SV40-transformed human keratinocytes. J. Dermatol. Sci 2001, 25, 139–149. [Google Scholar]
- Lee, E.R.; Kim, J.H.; Kang, Y.J.; Cho, S.G. The anti-apoptotic and anti-oxidant effect of eriodictyol on UV-induced apoptosis in keratinocytes. Biol. Pharm. Bull 2007, 30, 32–37. [Google Scholar]
- Xu, Y.; Shao, Y.; Zhou, J.; Voorhees, J.J.; Fisher, G.J. Ultraviolet irradiation-induces epidermal growth factor receptor (EGFR) nuclear translocation in human keratinocytes. J. Cell. Biochem 2009, 107, 873–880. [Google Scholar]
- Holt, S.; Alexander, P.; Inman, C.; Davies, D. Epidermal growth factor induced tyrosine phosphorylation of nuclear proteins associated with translocation of epidermal growth factor receptor into the nucleus. Biochem. Pharmacol 1995, 133, 211–222. [Google Scholar]
- Lin, S.; Makino, K.; Xia, W.; Matin, A.; Wen, Y.; Kwong, K.; Bourguignon, L.; Hung, M. Nuclear localization of EGF receptor and its potential new role as a transcription factor. Nat. Cell. Biol 2001, 3, 802–808. [Google Scholar]
- Kyriakis, J.M.; Avruch, J. Mammalian mitogen-activated protein kinase signal transduction pathways activated by stress and inflammation. Physiol. Rev 2001, 81, 807–869. [Google Scholar]
- Raingeaud, J.; Gupta, S.; Rogers, J.S.; Dickens, M.; Han, J.; Ulevitch, R.J.; Davis, R.J. Pro-inflammatory cytokines and environmental stress cause p38 mitogen-activated protein kinase activation by dual phosphorylation on tyrosine and threonine. J. Biol. Chem 1995, 270, 7420–7426. [Google Scholar]
- Huang, C.; Li, J.; Chen, N.; Ma, W.; Bowden, G.T.; Dong, Z. Inhibition of atypical PKC blocks ultraviolet-induced AP-1 activation by specifically inhibiting ERKs activation. Mol. Carcinog 2000, 27, 65–75. [Google Scholar]
- Englaro, W.; Derijard, B.; Ortonne, J.P.; Ballotti, R. Solar ultraviolet light activates extracellular signal-regulated kinases and the ternary complex factor in human normal keratinocytes. Oncogene 1998, 16, 661–664. [Google Scholar]
- Djavaheri-Mergny, M.; Dubertret, L. UV-A-induced AP-1 activation requires the Raf/ERK pathway in human NCTC 2544 keratinocytes. Exp. Dermatol 2001, 10, 204–210. [Google Scholar]
- Li, D.; Turi, T.G.; Schuck, A.; Freedberg, I.M.; Khitrov, G.; Blumenberg, M. Rays and arrays: The transcriptional program in the response of human epidermal keratinocytes to UVB illumination. FASEB J 2001, 15, 2533–2535. [Google Scholar]
- Yanase, H.; Ando, H.; Horikawa, M.; Watanabe, M.; Mori, T.; Matsuda, N. Possible involvement of ERK 1/2 in UVA-induced melanogenesis in cultured normal human epidermal melanocytes. Pigment Cell Res 2001, 14, 103–109. [Google Scholar]
- Mumby, M.C.; Walter, G. Protein serine/threonine phosphatases: Structure, regulation, and functions in cell growth. Physiol. Rev 1993, 73, 673–699. [Google Scholar]
- Lee, E.R.; Kim, J.H.; Choi, H.Y.; Jeon, K.; Cho, S.G. Cytoprotective Effect of Eriodictyol in UV-irradiated Keratinocytes via Phosphatase-dependent Modulation of both the p38 MAPK and Akt Signaling Pathways. Cell. Physiol. Biochem 2011, 27, 513–524. [Google Scholar]
- Song, J.Y.; Han, H.S.; Sabapathy, K.; Lee, B.M.; Yu, E.; Choi, J. Expression of a homeostatic regulator, Wip1 (wild-type p53-induced phosphatase), is temporally induced by c-Jun and p53 in response to UV irradiation. J. Biol. Chem 2010, 285, 9067–9076. [Google Scholar]
- Denu, J.; Tanner, K. Specific and reversible inactivation of protein tyrosine phosphatases by hydrogen peroxide: Evidence for a sulfenic acid intermediate and implications for redox regulation. Biochemistry 1998, 37, 5633–5642. [Google Scholar]
- Knebel, A.; Rahmsdorf, H.; Ullrich, A.; Herrlich, P. Dephosphorylation of receptor tyrosine kinases as target of regulation by radiation, oxidants or alkylating agents. EMBO. J 1996, 15, 5314–5325. [Google Scholar]
- Xu, Y.; Shao, Y.; Voorhees, J.J.; Fisher, G.J. Oxidative inhibition of receptor-type protein-tyrosine phosphatase kappa by ultraviolet irradiation activates epidermal growth factor receptor in human keratinocytes. J. Biol. Chem 2006, 281, 27389–27397. [Google Scholar]
- Cantley, L.C. The phosphoinositide 3-kinase pathway. Science 2002, 296, 1655–1657. [Google Scholar]
- Franke, T.F.; Cantley, L.C. A Bad kinase makes good. Nature 1997, 390, 116–117. [Google Scholar]
- Datta, S.R.; Dudek, H.; Tao, X.; Masters, S.; Fu, H.; Gotoh, Y.; Greenberg, M.E. Akt phosphorylation of bad couples survival signals to the cell-intrinsic death machinery. Cell 1997, 91, 231–241. [Google Scholar]
- Ibuki, Y.; Goto, R. Suppression of apoptosis by UVB irradiation: Survival signaling via PI3-kinase/Akt pathway. Biochem. Biophys. Res. Commun 2000, 279, 872–878. [Google Scholar]
- Wan, Y.S.; Wang, Z.Q.; Shao, Y.; Voorhees, J.J.; Fisher, G.J. Ultraviolet irradiation activates PI 3-kinase/Akt survival pathway via EGF receptors in human skin in vivo. Int. J. Oncol 2001, 18, 461–466. [Google Scholar]
- Dufner, G.T. Ribosomal S6 kinase signaling and the control of translation. Exp. Cell Res 1999, 253, 100–109. [Google Scholar]
- Frodin, M.; Gammeltoft, S. Role and regulation of 90 kDa ribosomal S6 kinase (RSK) in signal transduction. Mol. Cell Endocrinol 1999, 151, 65–77. [Google Scholar]
- Zhang, Y.; Dong, Z.; Nomura, M.; Zhong, S.; Chen, N.; Bode, A.M.; Dong, Z. Signal Transduction Pathways Involved in Phosphorylation and Activation of p70S6K Following Exposure to UVA Irradiation. J. Biol. Chem 2001, 276, 20913–20923. [Google Scholar]
- Watts, R.G.; Huang, C.; Young, M.R.; Li, J.J.; Pennie, W.D.; Colburn, N.H. Expression of dominant negative Erk2 inhibits AP-1 transactivation and neoplastic transformation. Oncogene 1998, 17, 3493–3498. [Google Scholar]
- Rotman, G.; Shiloh, Y. ATM: A mediator of multiple responses to genotoxic stress. Oncogene 1999, 18, 6135–6144. [Google Scholar]
- Kastan, M.B.; Lim, D.S. The many substrates and functions of ATM. Nat. Rev. Mol. Cell Biol 2000, 1, 179–186. [Google Scholar]
- Banin, S.; Moyal, L.; Shieh, S.; Taya, Y.; Anderson, C.W.; Chessa, L.; Smorodinsky, N.I.; Prives, C.; Reiss, Y.; Shiloh, Y.; et al. Enhanced phosphorylation of p53 by ATM in response to DNA damage. Science 1998, 281, 1674–1677. [Google Scholar]
- Westphal, C.H.; Rowan, S.; Schmaltz, C.; Elson, A.; Fisher, D.E.; Leder, P. Atm and p53 cooperate in apoptosis and suppression of tumorigenesis, but not in resistance to acute radiation toxicity. Nat. Genet 1997, 16, 397–401. [Google Scholar]
- Shiloh, Y. ATM (ataxia telangiectasia mutated): Expanding roles in the DNA damage response and cellular homeostasis. Biochem. Soc. Trans 2001, 29, 661–666. [Google Scholar]
- Canman, C.E.; Lim, D.S.; Cimprich, K.A.; Taya, Y.; Tamai, K.; Sakaguchi, K.; Appella, E.; Kastan, M.B.; Siliciano, J.D. Activation of the ATM kinase by ionizing radiation and phosphorylation of p53. Science 1998, 281, 1677–1679. [Google Scholar]
- Shiloh, Y. ATM and ATR: Networking cellular responses to DNA damage. Curr. Opin. Genet. Dev 2001, 11, 71–77. [Google Scholar]
- Devary, Y.; Rosette, C.; DiDonato, J.A.; Karin, M. NF-κB activation by ultraviolet light not dependent on a nuclear signal. Science 1993, 261, 1442–1445. [Google Scholar]
- Nishigori, C. UV-induced DNA damage in carcinogenesis and its repair. J. Dermatol. Sci 2000, 23, S41–S44. [Google Scholar]
- O’Dea, E.; Hoffmann, A. NF-κB signaling. Wiley Interdiscip. Rev. Syst. Biol. Med 2009, 1, 107–115. [Google Scholar]
- O’Dea, E.; Hoffmann, A. The regulatory logic of the NF-κB signaling system. Cold Spring Harb. Perspect. Biol 2010, 2, a000216. [Google Scholar]
- Gilmore, T.D. Introduction to NF-κB: Players, pathways, perspectives. Oncogene 2006, 25, 6680–6684. [Google Scholar]
- Neumann, M.; Naumann, M. Beyond IκBs: Alternative regulation of NF-κB activity. FASEB J 2007, 21, 2642–2654. [Google Scholar]
- Matthew, S.H.; Ghosh, S. Signaling to NF-κB. Genes Dev 2004, 18, 2195–2224. [Google Scholar]
- Muthusamy, V.; Piva, T.J. The UV response of the skin: A review of the MAPK, NF-κB and TNFα signal transduction pathways. Arch. Dermatol. Res 2010, 302, 5–17. [Google Scholar]
- Stein, B.; Rahmsdorf, H.J.; Steffen, A.; Litfin, M.; Herrlich, P. UV-induced DNA damage is an intermediate step in UV-induced expression of human immunodeficiency virus type 1, collagenase, c-fos, and metallothionein. Mol. Cell Biol 1989, 9, 5169–5181. [Google Scholar]
- Li, N.; Karin, M. Ionizing radiation and short wavelength UV activate NF-κB through two distinct mechanisms. Proc. Natl. Acad. Sci. USA 1998, 95, 13012–13017. [Google Scholar]
- Kato, T., Jr; Delhase, M.; Hoffmann, A.; Karin, M. CK2 is a C-terminal IκB kinase responsible for NF-kB activation during UV response. Mol. Cell 2003, 12, 829–839. [Google Scholar]
- Bender, K.; Gottlicher, M.; Whiteside, S.; Rahmsdorf, H.J.; Herrlich, P. Sequential DNA damage-independent and -dependent activation of NF-κB by UV. EMBO J 1998, 17, 5170–5181. [Google Scholar]
- Simon, M.M.; Aragane, Y.; Schwarz, A.; Luger, T.A.; Schwarz, T. UVB light induces nuclear factor kB (NFkB) activity independently from chromosomal DNA damage in cell-free cytosolic extracts. J. Invest. Dermatol 1994, 102, 422–427. [Google Scholar]
- Wu, S.; Tan, M.; Hu, Y.; Wang, J.L.; Scheuner, D.; Kaufman, R.J. Ultraviolet light activates NF-κB through translational inhibition of IκBα synthesis. J. Biol. Chem 2004, 279, 34898–34902. [Google Scholar]
- Lewis, D.A.; Spandau, D.F. UVB activation of NF-κB in normal human keratinocytes occurs via a unique mechanism. Arch. Dermatol. Res 2007, 299, 93–101. [Google Scholar]
- Tanaka, K.; Asamitsu, K.; Uranishi, H.; Iddamalgoda, A.; Ito, K.; Kojima, H.; Okamoto, T. Protecting skin photoaging by NF-κB inhibitor. Curr. Drug Metab 2010, 11, 431–435. [Google Scholar]
- Tanaka, K.; Hasegawa, J.; Asamitsu, K.; Okamoto, T. Prevention of the ultraviolet B-mediated skin photoaging by a nuclear factor kappaB inhibitor, parthenolide. J. Pharmacol. Exp. Ther 2005, 315, 624–630. [Google Scholar]
- Kishida, Y.; Yoshikawa, H.; Myoui, A. Parthenolide, a natural inhibitor of Nuclear Factor-kappaB, inhibits lung colonization of murine osteosarcoma cells. Clin. Cancer Res 2007, 13, 59–67. [Google Scholar]
- Tanaka, K.; Hasegawa, J.; Asamitsu, K.; Okamoto, T. Magnolia ovovata extract and its active component magnolol prevent skin photoaging via inhibition of nuclear factor kappaB. Eur. J. Pharmacol 2007, 565, 212–219. [Google Scholar]
- Mantena, S.K.; Katiyar, S.K. Grape seed proanthocyanidins inhibit UV-radiation-induced oxidative stress and activation of MAPK and NF-κB signaling in human epidermal keratinocytes. Free Radic. Biol. Med 2006, 40, 1603–1614. [Google Scholar]
- Abeyama, K.; Eng, W.; Jester, J.V.; Vink, A.A.; Edelbaum, D.; Cockerell, C.J.; Bergstresser, P.R.; Takashima, A. A role for NF-κB-dependent gene transactivation in sunburn. J. Clin. Invest 2000, 105, 1751–1759. [Google Scholar]
- Yokoyama, S.; Nakano, H.; Yamazaki, T.; Tamai, K.; Hanada, K.; Takahashi, G. Enhancement of ultraviolet-induced apoptosis by NF-κB decoy oligonucleotides. Br. J. Dermatol 2005, 153, S47–S51. [Google Scholar]
- Eferl, R.; Wagner, E.F. AP-1: A double-edged sword in tumorigenesis. Nat. Rev. Cancer 2003, 3, 859–868. [Google Scholar]
- Shaulian, E. AP-1 The Jun proteins: Oncogenes or tumor suppressors in disguise? Cell Signal 2010, 22, 894–899. [Google Scholar]
- Shaulian, E.; Karin, M. AP-1 as a regulator of cell life and death. Nat. Cell Biol 2002, 4, E131–E136. [Google Scholar]
- Bernstein, L.R.; Colburn, N.H. AP1/Jun function is differentially induced in promotion-sensitive and resistant JB6 cells. Science 1989, 244, 566–569. [Google Scholar]
- Domann, F.E., Jr; Levy, J.P.; Finch, J.S.; Bowden, G.T. Constitutive AP-1 DNA binding and transactivating ability of malignant but not benign mouse epidermal cells. Mol. Carcinog 1994, 9, 61–66. [Google Scholar]
- Domann, F.E.; Levy, J.P.; Birrer, M.J.; Bowden, G.T. Stable expression of a c-JUN deletion mutant in two malignant mouse epidermal cell lines blocks tumor formation in nude mice. Cell Growth Differ 1994, 5, 9–16. [Google Scholar]
- Li, J.J.; Rhim, J.S.; Schlegel, R.; Vousden, K.H.; Colburn, N.H. Expression of dominant negative Jun inhibits elevated AP-1 and NF-κB transactivation and suppresses anchorage independent growth of HPV immortalized human keratinocytes. Oncogene 1998, 16, 2711–2721. [Google Scholar]
- Li, J.J.; Westergaard, C.; Ghosh, P.; Colburn, N.H. Inhibitors of both nuclear factor-kappaB and activator protein-1 activation block the neoplastic transformation response. Cancer Res 1997, 57, 3569–3576. [Google Scholar]
- Thompson, E.J.; MacGowan, J.; Young, M.R.; Colburn, N.; Bowden, G.T. A dominant negative c-jun specifically blocks okadaic acid-induced skin tumor promotion. Cancer Res 2002, 62, 3044–3047. [Google Scholar]
- Matthews, C.P.; Birkholz, A.M.; Baker, A.R.; Perella, C.M.; Beck, G.R., Jr; Young, M.R.; Colburn, N.H. Dominant-negative activator protein 1 (TAM67) targets cyclooxygenase-2 and osteopontin under conditions in which it specifically inhibits tumorigenesis. Cancer Res 2007, 67, 2430–2438. [Google Scholar]
- Cooper, S.J.; MacGowan, J.; Ranger-Moore, J.; Young, M.R.; Colburn, N.H.; Bowden, G.T. Expression of dominant negative c-jun inhibits ultraviolet B-induced squamous cell carcinoma number and size in an SKH-1 hairless mouse model. Mol. Cancer Res 2003, 1, 848–854. [Google Scholar]
- Zenz, R.; Wagner, E.F. Jun signalling in the epidermis: From developmental defects to psoriasis and skin tumors. Int. J. Biochem. Cell. Biol 2006, 38, 1043–1049. [Google Scholar]
- Chen, W.; Bowden, G.T. Activation of p38 MAP kinase and ERK are required for ultraviolet-B induced c-fos gene expression in human keratinocytes. Oncogene 1999, 18, 7469–7476. [Google Scholar]
- Ramos, M.C.; Steinbrenner, H.; Stuhlmann, D.; Sies, H.; Brenneisen, P. Induction of MMP-10 and MMP-1 in a squamous cell carcinoma cell line by ultraviolet radiation. Biol. Chem 2004, 385, 75–86. [Google Scholar]
- Fisher, G.J.; Datta, S.C.; Talwar, H.S.; Wang, Z.Q.; Varani, J.; Kang, S.; Voorhees, J.J. Molecular basis of sun- induced premature skin ageing and retinoid antagonism. Nature 1996, 379, 335–339. [Google Scholar]
- Itoh, K.; Wakabayashi, N.; Katoh, Y.; Ishii, T.; O’Connor, T.; Yamamoto, M. Keap1 regulates both cytoplasmic-nuclear shuttling and degradation of Nrf2 in response to electrophiles. Genes Cells 2003, 8, 379–391. [Google Scholar]
- Itoh, K.; Tong, K.I.; Yamamoto, M. Molecular mechanism activating Nrf2-Keap1 pathway in regulation of adaptive response to electrophiles. Free Radic. Biol. Med 2004, 36, 1208–1213. [Google Scholar]
- Motohashi, H.; O’Connor, T.; Katsuoka, F.; Engel, J.D.; Yamamoto, M. Integration and diversity of the regulatory network composed of Maf and CNC families of transcription factors. Gene 2002, 294, 1–12. [Google Scholar]
- Slocum, S.L.; Kensler, T.W. Nrf2: Control of sensitivity to carcinogens. Arch. Toxicol 2011, 85, 273–284. [Google Scholar]
- Braun, S.; Hanselmann, C.; Gassmann, M.G.; auf dem Keller, U.; Born-Berclaz, C.; Chan, K.; Kan, Y.W.; Werner, S. Nrf2 transcription factor, a novel target of keratinocytes growth factor action which regulates gene expression and inflammation in the healing skin wound. Mol. Cell. Biol 2002, 22, 5492–5505. [Google Scholar]
- Hirota, A.; Kawachi, Y.; Itoh, K.; Nakamura, Y.; Xu, X.; Banno, T.; Takahashi, T.; Yamamoto, M.; Otsuka, F. Ultraviolet A irradiation induces NF-E2-related factor 2 activation in dermal fibroblasts: Protective role in UVA-induced apoptosis. J. Invest. Dermatol 2005, 124, 825–832. [Google Scholar]
- Kannan, S.; Jaiswal, A.K. Low and high dose UVB regulation of transcription factor NF-E2-related factor 2. Cancer Res 2006, 66, 8421–8429. [Google Scholar]
- Durchdewald, M.; Beyer, T.A.; Johnson, D.A.; Johnson, J.A.; Werner, S.; auf dem Keller, U. Electrophilic chemicals but not UV irradiation or reactive oxygen species activate Nrf2 in keratinocytes in vitro and in vivo. J. Invest. Dermatol 2007, 127, 646–653. [Google Scholar]
- Faraonio, R.; Vergara, P.; Di Marzo, D.; Pierantoni, M.G.; Napolitano, M.; Russo, T.; Cimino, F. p53 suppresses the Nrf2-dependent transcription of antioxidant response genes. J. Biol. Chem 2006, 281, 39776–39784. [Google Scholar]
- Schäfer, M.; Dütsch, S.; auf dem Keller, U.; Navid, F.; Schwarz, A.; Johnson, D.A.; Johnson, J.A.; Werner, S. Nrf2 establishes a glutathione-mediated gradient of UVB cytoprotection in the epidermis. Genes Dev 2010, 24, 1045–1058. [Google Scholar]
- auf dem Keller, U.; Huber, M.; Beyer, T.A.; Kümin, A.; Siemes, C.; Braun, S.; Bugnon, P.; Mitropoulos, V.; Johnson, D.A.; Johnson, J.A.; et al. Nrf transcription factors in keratinocytes are essential for skin tumor prevention but not for wound healing. Mol. Cell. Biol 2006, 26, 3773–3784. [Google Scholar]
- Kawachi, Y.; Xu, X.; Taguchi, S.; Sakurai, H.; Nakamura, Y.; Ishii, Y.; Fujisawa, Y.; Furuta, J.; Takahashi, T.; Itoh, K.; et al. Attenuation of UVB-induced sunburn reaction and oxidative DNA damage with no alterations in UVB-induced skin carcinogenesis in Nrf2 gene-deficient mice. J. Invest. Dermatol 2008, 128, 1773–1779. [Google Scholar]
- Cullinan, S.B.; Diehl, J.A. PERK-dependent activation of Nrf2 contributes to redox homeostasis and cell survival following endoplasmic reticulum stress. J. Biol. Chem 2004, 279, 20108–20117. [Google Scholar]
- Huang, H.C.; Nguyen, T.; Pickett, C.B. Phosphorylation of Nrf2 at Ser-40 by protein kinase C regulates antioxidant response element-mediated transcription. J. Biol. Chem 2002, 277, 42769–42774. [Google Scholar]
- Yu, R.; Chen, C.; Mo, Y.Y.; Hebbar, V.; Owuor, E.D.; Tan, T.H.; Kong, A.N. Activation of mitogen-activated protein kinase pathways induces antioxidant response element-mediated gene expression via a Nrf2-dependent mechanism. J. Biol. Chem 2000, 275, 39907–39913. [Google Scholar]
- Yu, R.; Mandlekar, S.; Lei, W.; Fahl, W.E.; Tan, T.H.; Kong, A.N. p38 mitogen-activated protein kinase negatively regulates the induction of phase II drug-metabolizing enzymes that detoxify carcinogens. J. Biol. Chem 2000, 275, 2322–2327. [Google Scholar]
- Hu, R.; Saw, C.L.; Yu, R.; Kong, A.N. Regulation of NF-E2-related factor 2 signaling for cancer chemoprevention: Antioxidant coupled with antiinflammatory. Antioxid. Redox Signal 2010, 13, 1679–1698. [Google Scholar]
- Dinkova-Kostova, A.T.; Jenkins, S.N.; Fahey, J.W.; Ye, L.; Wehage, S.L.; Liby, K.T.; Stephenson, K.K.; Wade, K.L.; Talalay, P. Protection against UV-light-induced skin carcinogenesis in SKH-1 high-risk mice by sulforaphane-containing broccoli sprout extracts. Cancer Lett 2006, 240, 243–252. [Google Scholar]
- Kimura, S.; Warabi, E.; Yanagawa, T.; Ma, D.; Itoh, K.; Ishii, Y.; Kawachi, Y.; Ishii, T. Essential role of Nrf2 in keratinocyte protection from UVA by quercetin. Biochem. Biophys. Res. Commun 2009, 387, 109–114. [Google Scholar]
- Rao, A.; Luo, C.; Hogan, P.G. Transcription factors of the NFAT family: Regulation and function. Annu. Rev. Immunol 1997, 15, 707–747. [Google Scholar]
- Hogan, P.G.; Chen, L.; Nardone, J.; Rao, A. Transcriptional regulation by calcium, calcineurin, and NFAT. Genes. Dev 2003, 17, 2205–2232. [Google Scholar]
- Maziere, C.; Morliere, P.; Louandre, C.; Conte, M.A.; Gomilla, C.; Santus, R.; Antonicelli, F.; Hornebeck, W.; Maziere, J.C. Low UVA doses activate the transcription factor NFAT in human fibroblasts by a calcium-calcineurin pathway. Free. Radic. Biol. Med 2005, 39, 1629–1637. [Google Scholar]
- Akunda, J.K.; Chun, K.S.; Sessoms, A.R.; Lao, H.C.; Fischer, S.M.; Langenbach, R. Cyclooxygenase-2 deficiency increases epidermal apoptosis and impairs recovery following acute UVB exposure. Mol. Carcinog 2007, 46, 354–362. [Google Scholar]
- Flockhart, R.J.; Diffey, B.L.; Farr, P.M.; Lloyd, J.; Reynolds, N.J. NFAT regulates induction of COX-2 and apoptosis of keratinocytes in response to ultraviolet radiation exposure. FASEB J 2008, 22, 4218–4227. [Google Scholar]
- Slominski, A.; Pawelek, J. Animals under the sun: Effects of ultraviolet radiation on mammalian skin. Clin. Dermatol 1998, 16, 503–515. [Google Scholar]
- Nickoloff, B.J.; Qin, J.Z.; Chaturvedi, V.; Bacon, P.; Panella, J.; Denning, M.F. Life and death signaling pathways contributing to skin cancer. J. Investig. Dermatol. Symp. Proc 2002, 7, 27–35. [Google Scholar]
- Scott, M.C.; Suzuki, I.; Abdel-Malek, Z.A. Regulation of the human melanocortin 1 receptor expression in epidermal melanocytes by paracrine and endocrine factors and by ultraviolet radiation. Pigment Cell Res 2002, 15, 433–439. [Google Scholar]
- Corre, S.; Primot, A.; Sviderskaya, E.; Bennett, D.C.; Vaulont, S.; Goding, C.R.; Galibert, M.D. UV-induced expression of key component of the tanning process, the POMC and MC1R genes, is dependent on the p-38-activated upstream stimulating factor-1 (USF-1). J. Biol. Chem 2004, 279, 51226–51233. [Google Scholar]
- Pillai, S.; Oresajo, C.; Hayward, J. Ultraviolet radiation and skin aging: Roles of reactive oxygen species, inflammation and protease activation, and strategies for prevention of inflammation-induced matrix degradation—a review. Int. J. Cosmet. Sci 2005, 27, 17–34. [Google Scholar]
- Assefa, Z.; Garmyn, M.; Bouillon, R.; Merlevede, W.; Vandenheede, J.R.; Agostinis, P. Differential stimulation of ERK and JNK activities by ultraviolet B irradiation and epidermal growth factor in human keratinocytes. J. Invest. Dermatol 1997, 108, 886–891. [Google Scholar]
- Derijard, B.; Hibi, M.; Wu, I.H.; Barrett, T.; Su, B.; Deng, T.; Karin, M.; Davis, R.J. JNK1: A protein kinase stimulated by UV light and Ha-Ras that binds and phosphorylates the c-Jun activation domain. Cell 1994, 76, 1025–1037. [Google Scholar]
- Garmyn, M.; Degreef, H.; Gilchrest, B.A. The effect of acute and chronic photodamage on gene expression in human keratinocytes. Dermatology 1995, 190, 305–308. [Google Scholar]
- Delva, E.; Tucker, D.K.; Kowalczyk, A.P. The desmosome. Cold Spring Harb. Perspect. Biol 2009, 1, a002543. [Google Scholar]
- Fujisawa, H.; Wang, B.; Sauder, D.N.; Kondo, S. Effects of interferons on the production of interleukin-6 and interleukin-8 in human keratinocytes. J. Interferon Cytokine Res 1997, 17, 347–353. [Google Scholar]
- Aragane, Y.; Kulms, D.; Luger, T.A.; Schwarz, T. Down-regulation of interferon gamma-activated STAT1 by UV light. Proc. Natl. Acad. Sci. USA 1997, 94, 11490–11495. [Google Scholar]
- Freedberg, I.M.; Tomic-Canic, M.; Komine, M.; Blumenberg, M. Keratins and the keratinocyte activation cycle. J. Invest Dermatol 2001, 116, 633–640. [Google Scholar]
- Sesto, A.; Navarro, M.; Burslem, F.; Jorcano, J.L. Analysis of the ultraviolet B response in primary human keratinocytes using oligonucleotide microarrays. Proc. Natl. Acad. Sci. USA 2002, 99, 2965–2970. [Google Scholar]
- Takao, J.; Ariizumi, K.; Dougherty, I.I.; Cruz, P.D., Jr. Genomic scale analysis of the human keratinocyte response to broad-band ultraviolet-B irradiation. Photodermatol. Photoimmunol. Photomed 2002, 18, 5–13. [Google Scholar]
- Howell, B.G.; Wang, B.; Freed, I.; Mamelak, A.J.; Watanabe, H.; Sauder, D.N. Microarray analysis of UVB-regulated genes in keratinocytes: Downregulation of angiogenesis inhibitor thrombospondin-1. J. Dermatol. Sci 2004, 34, 185–194. [Google Scholar]
- Serre, C.; Lebleu, A.; Bergeron, L.; Plantivaux, A.; Botto, J.M.; Dal, F.C.; Domloge, N. Microarray profiling of gene expression in human keratinocytes suggests a new protective activity against UV-induced DNA damage for a compound previously known to interact with SCF-KIT signalling pathway. Int. J. Cosmet. Sci 2011, 33, 398–407. [Google Scholar]
- Pisarchik, A.; Wortsman, J.; Slominski, A. A novel microarray to evaluate stress-related genes in skin: Effect of ultraviolet light radiation. Gene 2004, 341, 199–207. [Google Scholar]
- Lee, K.M.; Lee, J.G.; Seo, E.Y.; Lee, W.H.; Nam, Y.H.; Yang, J.M.; Kee, S.H.; Seo, Y.J.; Park, J.K.; Kim, C.D.; et al. Analysis of genes responding to ultraviolet B irradiation of HaCaT keratinocytes using a cDNA microarray. Br. J. Dermatol 2005, 152, 52–59. [Google Scholar]
- Murakami, T.; Fujimoto, M.; Ohtsuki, M.; Nakagawa, H. Expression profiling of cancer-related genes in human keratinocytes following non-lethal ultraviolet B irradiation. J. Dermatol. Sci 2001, 27, 121–129. [Google Scholar]
- Dazard, J.E.; Gal, H.; Amariglio, N.; Rechavi, G.; Domany, E.; Givol, D. Genome-wide comparison of human keratinocyte and squamous cell carcinoma responses to UVB irradiation: Implications for skin and epithelial cancer. Oncogene 2003, 22, 2993–3006. [Google Scholar]
- Nordlund, J.J. The melanocyte and the epidermal melanin unit: An expanded concept. Dermatol. Clin 2007, 25, 271–281. [Google Scholar]
- Duval, C.; Regnier, M.; Schmidt, R. Distinct melanogenic response of human melanocytes in mono-culture, in co-culture with keratinocytes and in reconstructed epidermis, to UV exposure. Pigment Cell Res 2001, 14, 348–355. [Google Scholar]
- Duval, C.; Smit, N.P.; Kolb, A.M.; Regnier, M.; Pavel, S.; Schmidt, R. Keratinocytes control the pheo/eumelanin ratio in cultured normal human melanocytes. Pigment Cell Res 2002, 15, 440–446. [Google Scholar]
- Regnier, M.; Duval, C.; Galey, J.B.; Philippe, M.; Lagrange, A.; Tuloup, R.; Schmidt, R. Keratinocyte-melanocyte co-cultures and pigmented reconstructed human epidermis: Models to study modulation of melanogenesis. Cell Mol. Biol 1999, 45, 969–980. [Google Scholar]
- Rees, J.L. Genetics of hair and skin color. Annu. Rev. Genet 2003, 37, 67–90. [Google Scholar]
- Barker, D.; Dixon, K.; Medrano, E.E.; Smalara, D.; Im, S.; Mitchell, D.; Babcock, G.; Abdel-Malek, Z.A. Comparison of the responses of human melanocytes with different melanin contents to ultraviolet B irradiation. Cancer Res 1995, 55, 4041–4046. [Google Scholar]
- Pavey, S.; Conroy, S.; Russell, T.; Gabrielli, B. Ultraviolet radiation induces p16CDKN2A expression in human skin. Cancer Res 1999, 59, 4185–4189. [Google Scholar]
- Sheen, I.S.; Jeng, K.S.; Wu, J.Y. Is p53 gene mutation an indicatior of the biological behaviors of recurrence of hepatocellular carcinoma? World J. Gastroenterol 2003, 9, 1202–1207. [Google Scholar]
- Chouinard, N.; Valerie, K.; Rouabhia, M.; Huot, J. UVB-mediated activation of p38 mitogen-activated protein kinase enhances resistance of normal human keratinocytes to apoptosis by stabilizing cytoplasmic p53. Biochem. J 2002, 365, 133–145. [Google Scholar]
- McGill, G.G.; Horstmann, M.; Widlund, H.R.; Du, J.; Motyckova, G.; Nishimura, E.K.; Lin, Y.L.; Ramaswamy, S.; Avery, W.; Ding, H.F.; et al. Bcl2 regulation by the melanocyte master regulator Mitf modulates lineage survival and melanoma cell viability. Cell 2002, 109, 707–718. [Google Scholar]
- Tada, A.; Pereira, E.; Beitner-Johnson, D.; Kavanagh, R.; Abdel-Malek, Z.A. Mitogen- and ultraviolet-B-induced signaling pathways in normal human melanocytes. J. Invest. Dermatol 2002, 118, 316–322. [Google Scholar]
- Kadekaro, A.L.; Kanto, H.; Kavanagh, R.; Abdel-Malek, Z.A. Significance of the melanocortin 1 receptor in regulating human melanocyte pigmentation, proliferation, and survival. Ann. N.Y. Acad. Sci 2003, 994, 359–365. [Google Scholar]
- Zhang, H.; Rosdahl, I. Ultraviolet A and B differently induce intracellular protein expression in human skin melanocytes—a speculation of separate pathways in initiation of melanoma. Carcinogenesis 2003, 24, 1929–1934. [Google Scholar]
- Valery, C.; Grob, J.J.; Verrando, P. Identification by cDNA microarray technology of genes modulated by artificial ultraviolet radiation in normal human melanocytes: Relation to melanocarcinogenesis. J. Invest. Dermatol 2001, 117, 1471–1482. [Google Scholar]
- Hoek, K.; Rimm, D.L.; Williams, K.R.; Zhao, H.; Ariyan, S.; Lin, A.; Kluger, H.M.; Berger, A.J.; Cheng, E.; Trombetta, E.S.; et al. Expression profiling reveals novel pathways in the transformation of melanocytes to melanomas. Cancer Res 2004, 64, 5270–5282. [Google Scholar]
- Winnepenninckx, V.; Lazar, V.; Michiels, S.; Dessen, P.; Stas, M.; Alonso, S.R.; Avril, M.F.; Ortiz Romero, P.L.; Robert, T.; Balacescu, O.; et al. Gene expression profiling of primary cutaneous melanoma and clinical outcome. J. Natl. Cancer Inst 2006, 98, 472–482. [Google Scholar]
- Yang, G.; Zhang, G.; Pittelkow, M.R.; Ramoni, M.; Tsao, H. Expression profiling of UVB response in melanocytes identifies a set of p53-target genes. J. Invest. Dermatol 2006, 126, 2490–2506. [Google Scholar]
- Runger, T.M. Role of UVA in the pathogenesis of melanoma and non-melanoma skin cancer. A short review. Photodermatol. Photoimmunol. Photomed 1999, 15, 212–216. [Google Scholar]
- Duval, C.; Schmidt, R.; Regnier, M.; Facy, V.; Asselineau, D.; Bernerd, F. The use of reconstructed human skin to evaluate UV-induced modifications and sunscreen efficacy. Exp. Dermatol 2003, 12, 64–70. [Google Scholar]
- Halliday, G.M.; Bestak, R.; Yuen, K.S.; Cavanagh, L.L.; Barnetson, R.S. UVA-induced immunosuppression. Mutat. Res 1998, 422, 139–145. [Google Scholar]
- Agar, N.S.; Halliday, G.M.; Barnetson, R.S.; Ananthaswamy, H.N.; Wheeler, M.; Jones, A.M. The basal layer in human squamous tumors harbors more UVA than UVB fingerprint mutations: A role for UVA in human skin carcinogenesis. Proc. Natl. Acad. Sci. USA 2004, 101, 4954–4959. [Google Scholar]
- Choi, W.; Miyamura, Y.; Wolber, R.; Smuda, C.; Reinhold, W.; Liu, H.; Kolbe, L.; Hearing, V.J. Regulation of human skin pigmentation in situ by repetitive UV exposure: Molecular characterization of responses to UVA and/or UVB. J. Invest. Dermatol 2010, 130, 1685–1696. [Google Scholar]
- Marionnet, C.; Grether-Beck, S.; Seite, S.; Marini, A.; Jaenicke, T.; Lejeune, F.; Bastien, P.; Rougier, A.; Bernerd, F.; Krutmann, J. A broad-spectrum sunscreen prevents UVA radiation-induced gene expression in reconstructed skin in vitro and in human skin in vivo. Exp. Dermatol 2011, 20, 477–482. [Google Scholar]
- Marionnet, C.; Pierrard, C.; Lejeune, F.; Sok, J.; Thomas, M.; Bernerd, F. Different oxidative stress response in keratinocytes and fibroblasts of reconstructed skin exposed to non extreme daily-ultraviolet radiation. PLoS One 2010, 5, e12059. [Google Scholar]

{kind=link}
{kind=link}
| MAPK pathway | PI-3K pathway | Transcription factors | |
|---|---|---|---|
| UV-A | EGFR, SRC, RAS, RAF, ERK1/2, p70s6k and p90RSK | ATM | c-JUN, Nrf2, NFAT |
| UV-B | PKC, JNK, p38 kinase, RSK2, MSK1 | EGFR, PI-3K, Akt, p70s6k and p90RSK | c-FOS, NF-κB NFAT |
| UV-C | EGFR, SRC, ERK, p38 kinase JNK1/2 | ATR | AP-1 |
© 2012 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
López-Camarillo, C.; Aréchaga Ocampo, E.; López Casamichana, M.; Pérez-Plasencia, C.; Álvarez-Sánchez, E.; Marchat, L.A. Protein Kinases and Transcription Factors Activation in Response to UV-Radiation of Skin: Implications for Carcinogenesis. Int. J. Mol. Sci. 2012, 13, 142-172. https://doi.org/10.3390/ijms13010142
López-Camarillo C, Aréchaga Ocampo E, López Casamichana M, Pérez-Plasencia C, Álvarez-Sánchez E, Marchat LA. Protein Kinases and Transcription Factors Activation in Response to UV-Radiation of Skin: Implications for Carcinogenesis. International Journal of Molecular Sciences. 2012; 13(1):142-172. https://doi.org/10.3390/ijms13010142
Chicago/Turabian StyleLópez-Camarillo, César, Elena Aréchaga Ocampo, Mavil López Casamichana, Carlos Pérez-Plasencia, Elizbeth Álvarez-Sánchez, and Laurence A. Marchat. 2012. "Protein Kinases and Transcription Factors Activation in Response to UV-Radiation of Skin: Implications for Carcinogenesis" International Journal of Molecular Sciences 13, no. 1: 142-172. https://doi.org/10.3390/ijms13010142
APA StyleLópez-Camarillo, C., Aréchaga Ocampo, E., López Casamichana, M., Pérez-Plasencia, C., Álvarez-Sánchez, E., & Marchat, L. A. (2012). Protein Kinases and Transcription Factors Activation in Response to UV-Radiation of Skin: Implications for Carcinogenesis. International Journal of Molecular Sciences, 13(1), 142-172. https://doi.org/10.3390/ijms13010142