A Comparative Reverse Docking Strategy to Identify Potential Antineoplastic Targets of Tea Functional Components and Binding Mode

Abstract
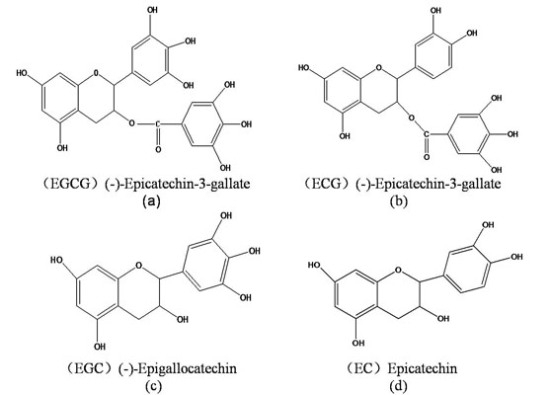
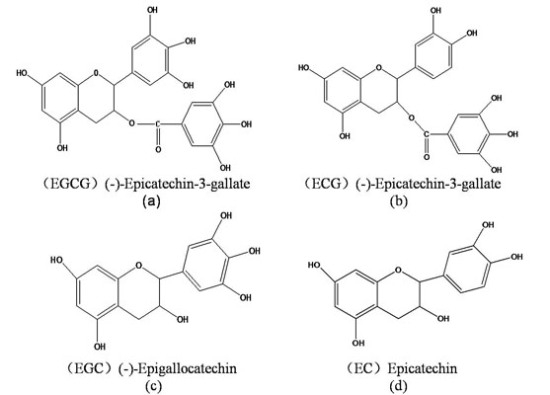
:
1. Introduction
2. Results and Discussion
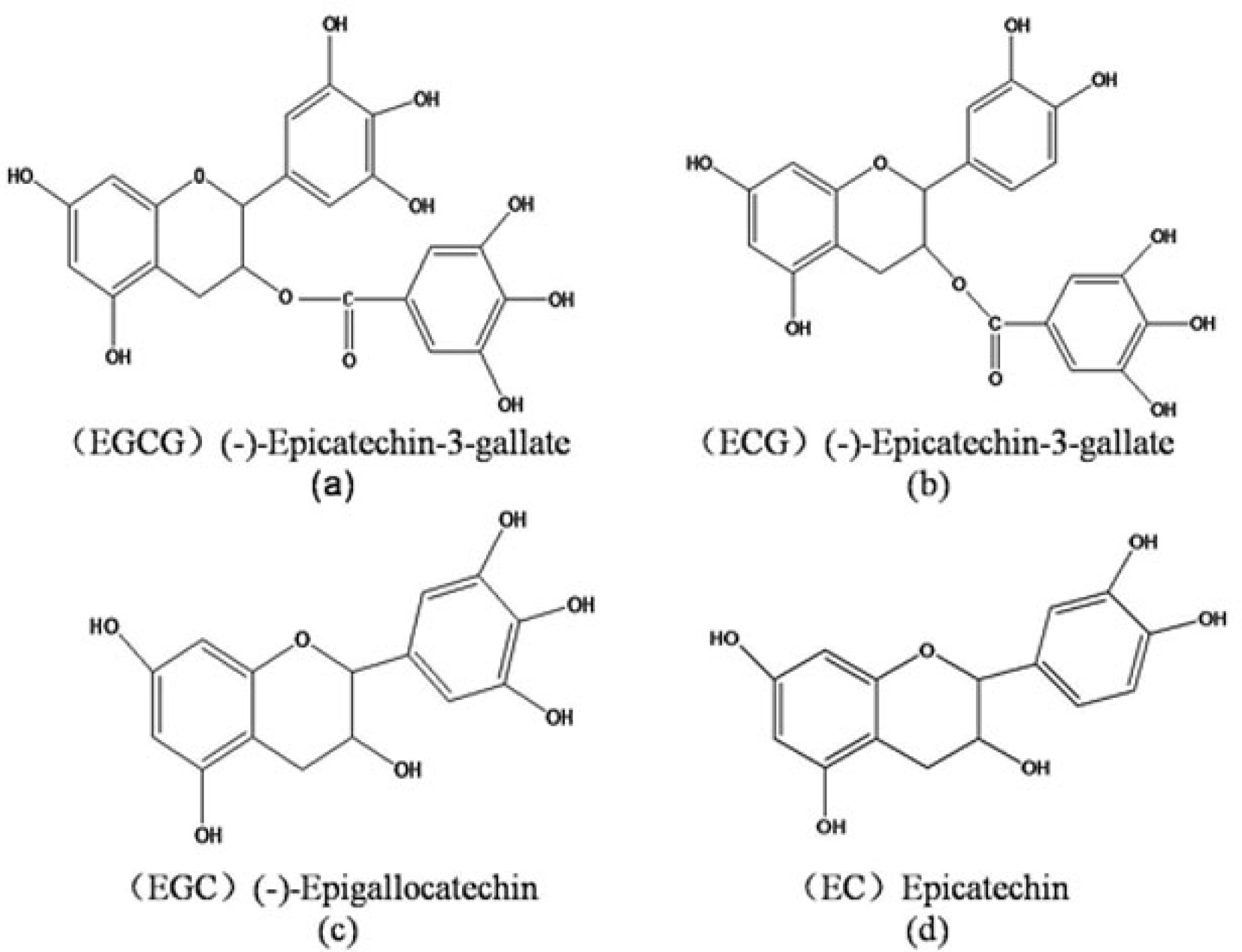
2.1. Potential Protein Targets for EGCG
2.2. Potential Protein Receptors for ECG, EGC and EC
2.3. Comparing Screening Results of Autodock and Tarfisdock
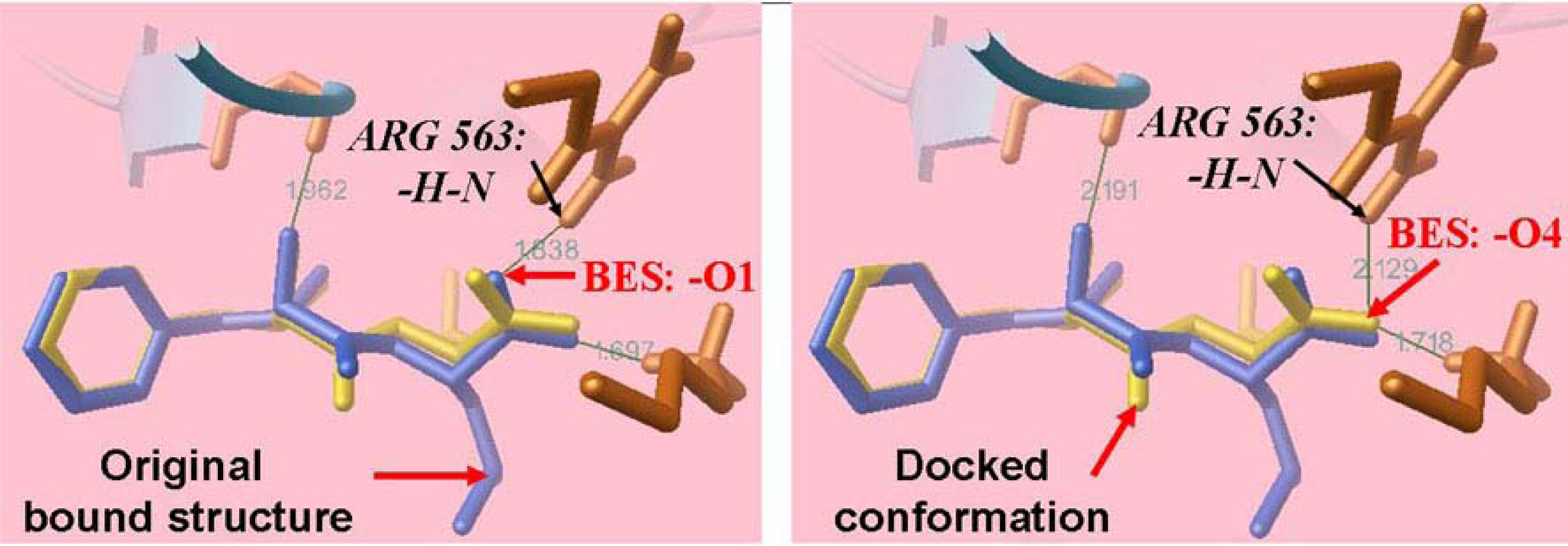
2.4. Docking Results for the Original Ligand BES in Leukotriene A4 hydrolase Crystal Structure
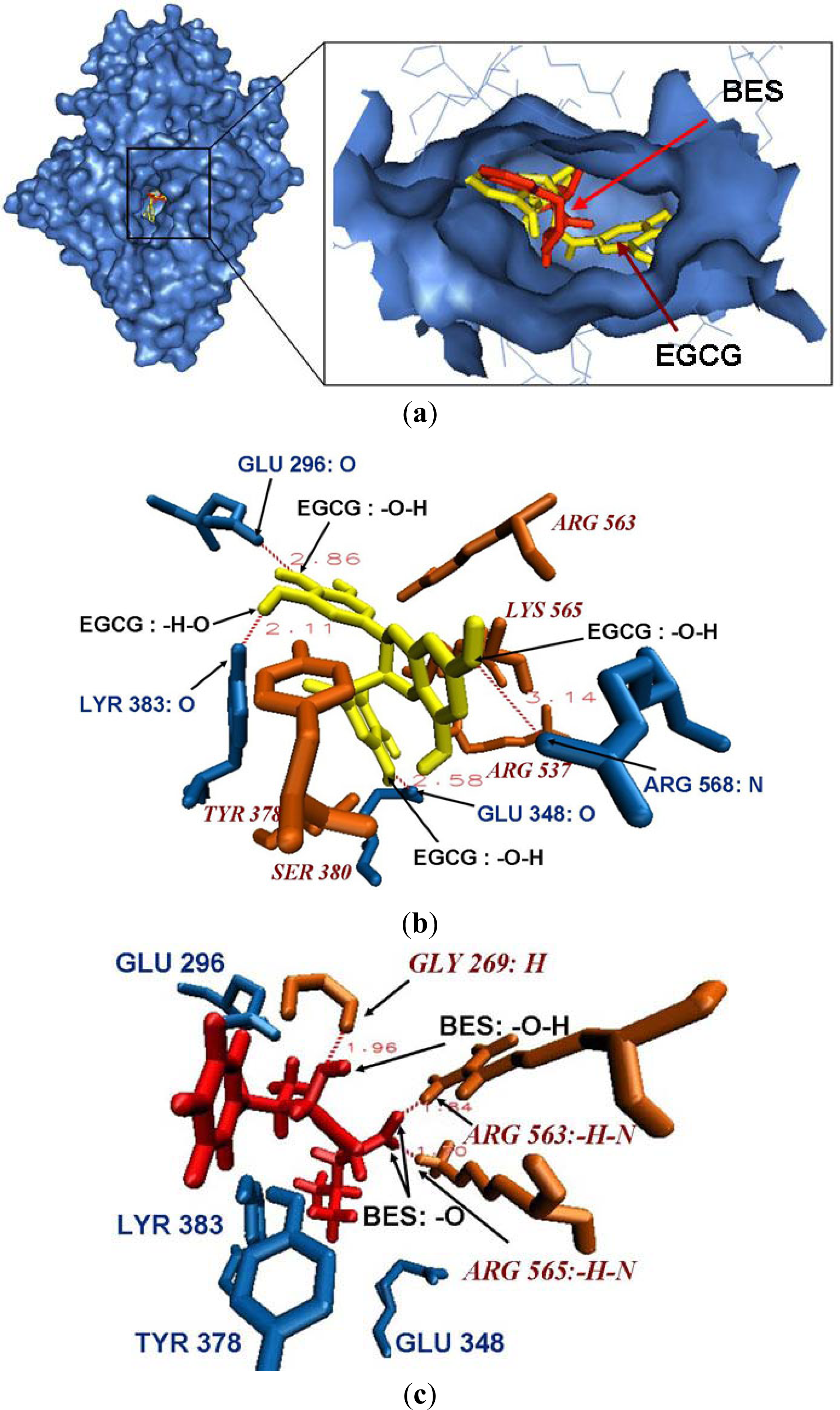
2.5. Binding Mode between Leukotriene A4 Hydrolase and EGCG
3. Experimental Section
3.1. The Three-Dimensional Structures of Tea Polyphenols and Drug Targets
3.2. Reverse Docking Procedure Using Autodock and Tarfisdock
4. Conclusions
Acknowledgments
References
- Wang, HY; Ding, J; Yang, F. Cell signal transduction and development of targeted anti-tumor agent. Chin. J. Cancer Biother 2007, 14, 401–404. [Google Scholar]
- Tachibana, H; Koga, K; Fujimura, Y; Yamada, K. A receptor for green tea polyphenol EGCG. Nat. Struct. Mol. Biol 2004, 11, 380–381. [Google Scholar]
- Umeda, D; Yano, S; Yamada, K; Tachibana, H. Green tea polyphenol epigallocatechin-3-gallate signaling pathway through 67-kDa laminin receptor. J. Biol. Chem 2008, 283, 3050–3058. [Google Scholar]
- Adachi, S; Nagao, T; To, S; Joe, AK; Shimizu, M; Matsushima-Nishiwaki, R; Kozawa, O; Moriwaki, H; Maxfield, FR; Weinstein, IB. (−)-Epigallocatechin gallate causes internalization of the epidermal growth factor receptor in human colon cancer cells. Carcinogenesis 2008, 29, 1986–1993. [Google Scholar]
- Hajduk, PJ; Huth, JR; Tse, C. Predicting protein druggability. Drug Discov. Today 2005, 10, 1675–1682. [Google Scholar]
- Huang, CM; Elmets, CA; Tang, DC; Li, F; Yusuf, N. Proteomics reveals that proteins expressed during the early stage of Bacillus anthracis infection are potential targets for the development of vaccines and drugs. Genomics Proteomics Bioinforma 2004, 3, 143–151. [Google Scholar]
- Persidis, A. Proteomics: An ambitious drug development platform attempts to link gene sequence to expressed phenotype under various physiological states. Nat. Biotechnol 1998, 16, 393–394. [Google Scholar]
- Lin, JH; Lu, AY. Role of pharmacokinetics and metabolism in drug discovery and development. Pharmacol. Rev 1997, 49, 403–449. [Google Scholar]
- Chen, YZ; Ung, CY. Prediction of potential toxicity and side effect protein targets of a small molecule by a ligand-protein inverse docking approach. J. Mol. Graph. Model 2001, 20, 199–218. [Google Scholar]
- Li, H; Gao, Z; Kang, L; Zhang, H; Yang, K; Yu, K; Luo, X; Zhu, W; Chen, K; Shen, J; et al. TarFisDock: A web server for identifying drug targets with docking approach. Nucleic Acids Res 2006, 34, W219–W224. [Google Scholar]
- Ewing, TJA; Kuntz, ID. Critical evaluation of search algorithms for automated molecular docking and database screening. J. Comput. Chem 1997, 18, 1175–1189. [Google Scholar]
- Morris, GM; Goodsell, DS; Halliday, RS; Huey, R; Hart, WE; Belew, RK; Olson, AJ. Automated docking using a lamarckian genetic algorithm and and empirical binding free energy function. J. Comput. Chem 1998, 19, 1639–1662. [Google Scholar]
- Smith, DM; Daniel, KG; Wang, Z; Guida, WC; Chan, TH; Dou, QP. Docking studies and model development of tea polyphenol proteasome inhibitors: Applications to rational drug design. Proteins 2004, 54, 58–70. [Google Scholar]
- Rao, MS; Olson, AJ. Modelling of factor Xa-inhibitor complexes: A computational flexible docking approach. Proteins 1999, 34, 173–183. [Google Scholar]
- Dym, O; Xenarios, I; Ke, H; Colicelli, J. Molecular docking of competitive phosphodiesterase inhibitors. Mol. Pharmacol 2002, 61, 20–25. [Google Scholar]
- Harriman, DJ; Deslongchamps, G. Reverse-docking as a computational tool for the study of asymmetric organocatalysis. J. Comput. Aided Mol. Des 2004, 18, 303–308. [Google Scholar]
- Gao, Z; Li, H; Zhang, H; Liu, X; Kang, L; Luo, X; Zhu, W; Chen, K; Wang, X; Jiang, H. PDTD: A web-accessible protein database for drug target identification. BMC Bioinforma 2008, 9, 104–110. [Google Scholar]
- Yamaguchi, K; Honda, M; Ikigai, H; Hara, Y; Shimamura, T. Inhibitory effects of (−)-epigallocatechin gallate on the life cycle of human immunodeficiency virus type 1 (HIV-1). Antivir. Res 2002, 53, 19–34. [Google Scholar]
- Ramirez-Mares, MV; de Mejia, EG. Comparative study of the antioxidant effect of ardisin and epigallocatechin gallate in rat hepatocytes exposed to benomyl and 1-nitropyrene. Food Chem. Toxicol 2003, 41, 1527–1535. [Google Scholar]
- Ahmed, I; John, A; Vijayasarathy, C; Robin, MA; Raza, H. Differential modulation of growth and glutathione metabolism in cultured rat astrocytes by 4-hydroxynonenal and green tea polyphenol, epigallocatechin-3-gallate. Neurotoxicology 2002, 23, 289–300. [Google Scholar]
- Raederstorff, DG; Schlachter, MF; Elste, V; Weber, P. Effect of EGCG on lipid absorption and plasma lipid levels in rats. J. Nutr. Biochem 2003, 14, 326–332. [Google Scholar]
- Zigman, S. Do tea polyphenols protect dogfish lens (mustelus canis) catalase against UV damage? Biol. Bull 1997, 193, 253–254. [Google Scholar]
- Lee, WJ; Shim, JY; Zhu, BT. Mechanisms for the inhibition of DNA methyltransferases by tea catechins and bioflavonoids. Mol. Pharmacol 2005, 68, 1018–1030. [Google Scholar]
- Shim, JH; Choi, HS; Pugliese, A; Lee, SY; Chae, JI; Choi, BI; Bode, AM; Dong, Z. (−)-Epigallocatechin gallate regulates CD3-mediated T cell receptor signaling in leukemia through the inhibition of ZAP-70 kinase. J. Biol. Chem 2008, 283, 28370–28379. [Google Scholar]
- He, Z; Tang, F; Ermakova, S; Li, M; Zhao, Q; Cho, Y-Y; Ma, W-Y; Choi, H-S; Bode, AM; Yang, CS; et al. Fyn is a novel target of (−)-epigallocatechin gallate in the inhibition of JB6 Cl41 cell transformation. Mol. Carcinog 2008, 47, 172–183. [Google Scholar]
- Okamoto, T. NSAID zaltoprofen improves the decrease in body weight in rodent sickness behavior models: Proposed new applications of NSAIDs (Review). Int. J. Mol. Med 2002, 9, 369–372. [Google Scholar]
- Daniel, L; Lechevallier, E; Giorgi, R; Sichez, H; Zattara-Cannoni, H. Pax-2 expression in adult renal tumors. Hum. Pathol 2001, 32, 282–287. [Google Scholar]
- Huey, R; Morris, GM; Olson, AJ; Goodsell, DS. A semiempirical free energy force field with charge-based desolvation. J. Comput. Chem 2007, 28, 1145–1152. [Google Scholar]
- Hakonarson, H; Thorvaldsson, S; Helgadottir, A; Gudbjartsson, D; Zink, F; Andresdottir, M; Manolescu, A; Arnar, DO; Andersen, K; Sigurdsson, A; et al. Effects of a 5-lipoxygenase-activating protein inhibitor on biomarkers associated with risk of myocardial infarction: a randomized trial. JAMA 2005, 293, 2245–2256. [Google Scholar]
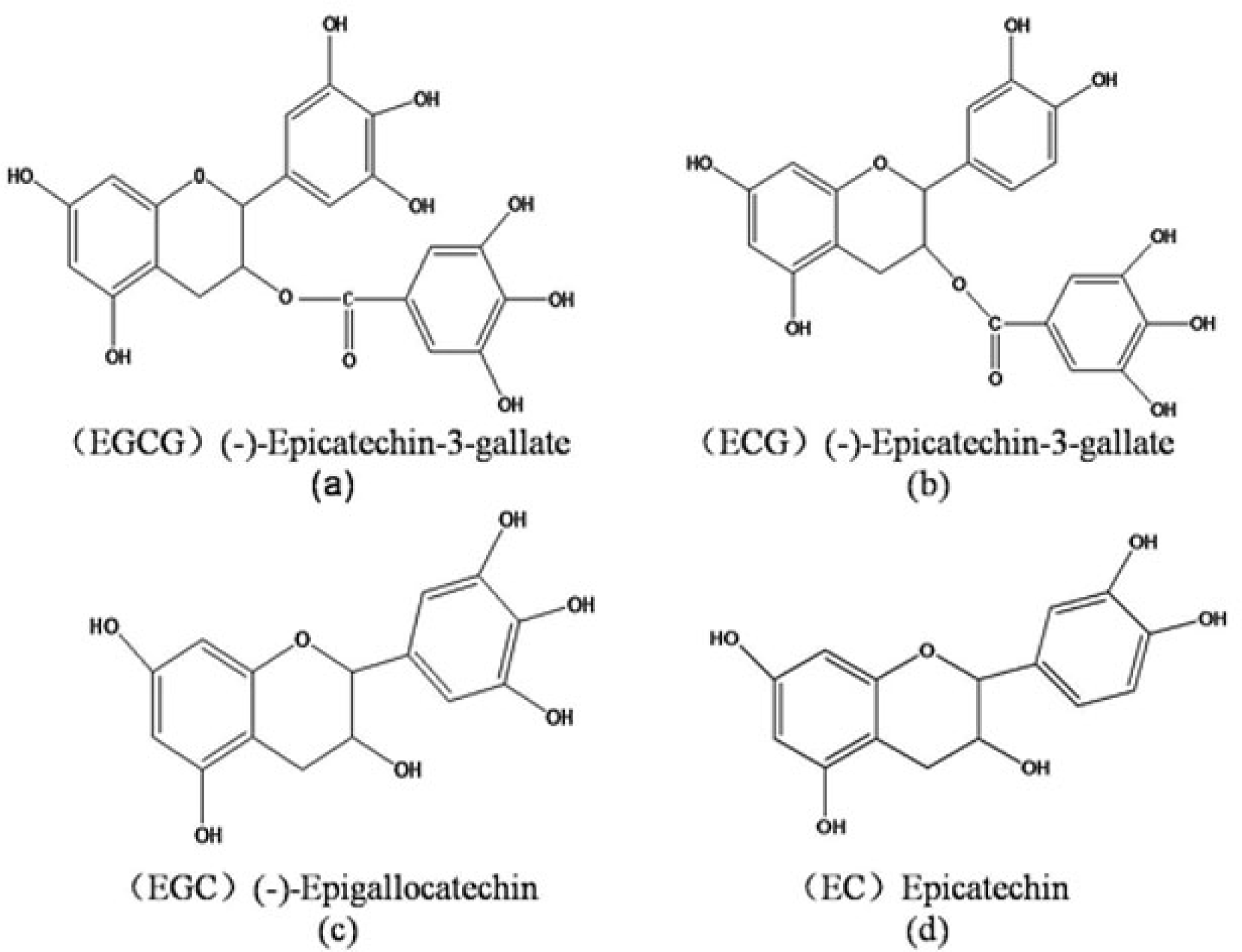

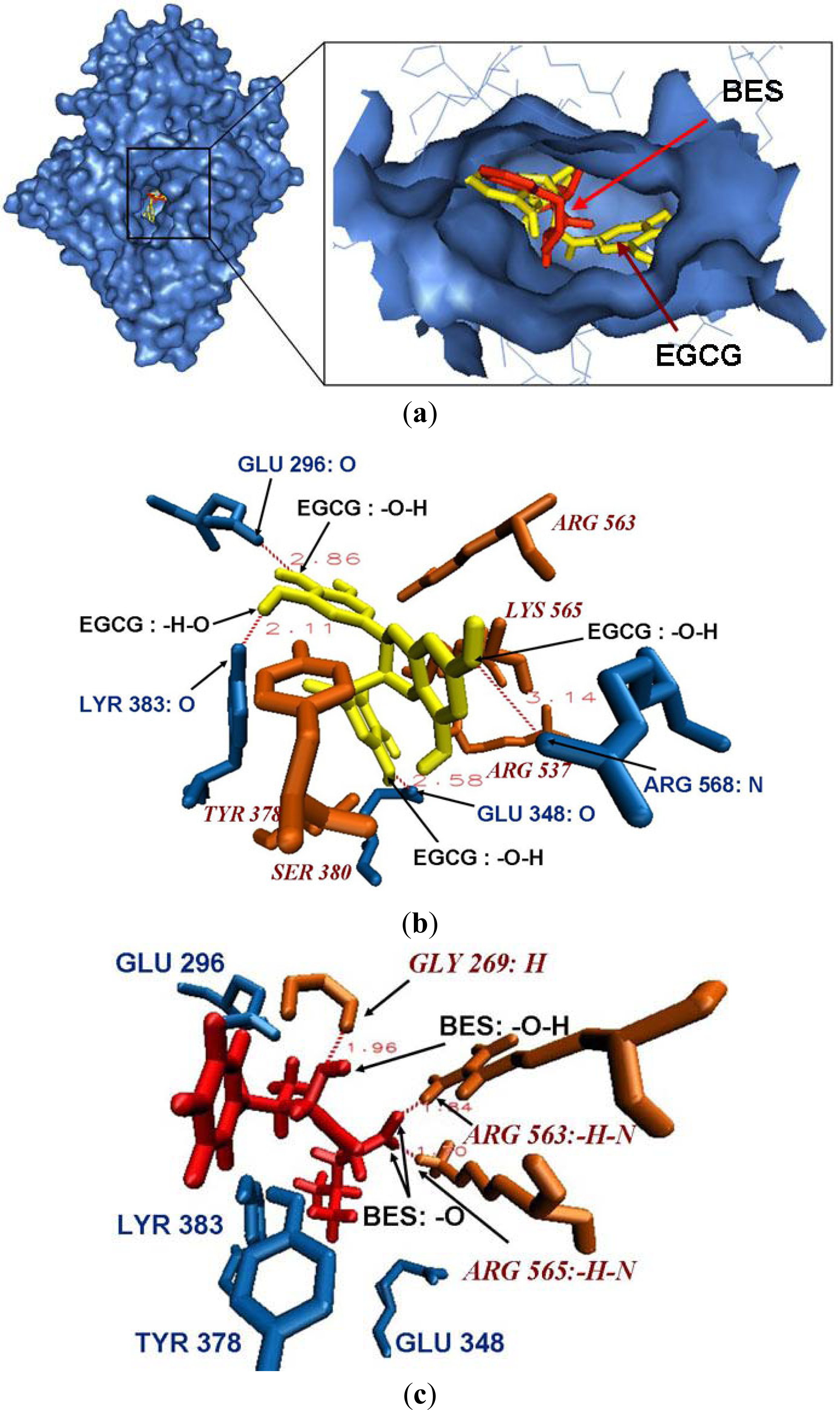

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PDB_ID | Target Name | Predicted by Procedures | Implicated by Experiment | Energy Score (kcal/mol) | Reference or Related Disease |
|---|---|---|---|---|---|
| 3BCH | 67 kD laminin receptor | Autodock | YES | −3.75 | [2] |
| 7HVP | HIV protease | Autodock/Tarfisdock | YES | −5.01/−44.87 | [18] |
| 1GRE | glutathione reductase | Autodock | YES | −6.80 | [19,20] |
| 1IJH | cholesterol oxidase | Autodock | YES | −6.77 | [21] |
| 8CAT | catalase | Autodock | YES | −6.23 | [22] |
| 1JNY | eEF1-α | Autodock | YES | −5.70 | [3] |
| 1BOO | DNA methyltransferase | Autodock | YES | −4.66 | [23] |
| 2OZO | ZAP-70 | Autodock | YES | −3.81 | [24] |
| 2DQ7 | Fyn kinase | Autodock | YES | −4.91 | [25] |
| 1HS6 | Leukotriene A4 | Autodock/Tarfisdock | NO | −5.22/−48.2 | Esophagus cancer |
| 1FT2 | Farnesyl protein transferase | Autodock/Tarfisdock | NO | −4.1/−44.94 | Cancer/Tumour |
| 1UTR | Mammalian PCB-binding protein | Autodock | NO | −7.19 | Lung Cancer |
| 1JVM | Voltage-Gated Potassium Channel | Autodock | NO | −6.58 | Cardiomyopathie |
| 1OG5 | CYP450 | Autodock | NO | −6.49 | Tumour |
| 1VKG | Histone deacetylase | Autodock | NO | −5.63 | Tumour |
| 1OOQ | Dihyrofolate reductase | Tarfisdock | NO | −46.42 | Tumour |
| 1IYH | Hematopoietic Prostagladin Synthase | Tarfisdock | NO | −44.50 | Cancer |
| 1PY5 | TGF-beta receptor type I | Tarfisdock | NO | −44.33 | Renal carcinoma |
| Target Name | 4 Types of Tea-Polyphenols | |||
|---|---|---|---|---|
| EGCG | EGC | ECG | EC | |
| CYP450 | √ | √ | √ | √ |
| Oxidosqualene cyclase | √ | √ | √ | √ |
| Voltage-Gated Potassium Channel | √ | √ | — | √ |
| Phospholipase A2 | √ | √ | √ | √ |
| FK506 binding protein | √ | — | √ | √ |
| Dihyrofolate reductase | — | √ | √ | √ |
| Leukotriene A4 hydrolase | √ | — | √ | — |
| PARP(Poly ADP-Ribose Polymerase) | √ | √ | √ | — |
| Protoporphyrinogen oxidase | √ | √ | √ | — |
| Ornithine Aminotransferase | — | — | — | √ |
| 1,2-Cyclooxygenase | — | — | — | √ |
| histone deacetylases (HDACs) | — | — | √ | √ |
| Amino acid oxidase | — | — | √ | √ |
| Glutamic acid receptor-2 | — | √ | — | — |
| Mammalian PCB-binding protein | √ | — | √ | — |
| Fatty acid-binding protein (FABP) | √ | — | √ | — |
| Catalase | √ | √ | — | — |
| Ligand Name | PDB_ID | Score Tarfisdock (kcal/mol) | Score Autodock (kcal/mol) | Receptor Name |
|---|---|---|---|---|
| EGCG | 1HS6 | −48.21 | −5.22 | Leukotriene A4 hydrolase |
| 1FT2 | −44.94 | −4.10 | Farnesyl protein transferase | |
| 7HVP | −44.87 | −5.01 | HIV protease | |
| 1Y79 | −44.11 | −3.96 | Dipeptidyl peptidase | |
| EGC | 1YTV | −36.44 | −5.66 | Vasopressin V1a receptor |
| 1Q5M | −35.83 | −3.32 | Alpha hydroxysteroid dehydrogenase | |
| 1NR5 | −35.24 | −4.01 | 3-dehydroquinate Synthase | |
| 1Y79 | −35.19 | −3.77 | Peptidyl dipeptidase | |
| 2C6C | −48.27 | −3.31 | Glutamate carboxypeptidase II | |
| 1TCO | −43.82 | −6.07 | FK506 binding protein | |
| ECG | 1W6K | −38.06 | −6.07 | Oxidosqualene cyclase |
| 1HS6 | −46.21 | −5.74 | Leukotriene A4 hydrolase | |
| EC | 1W6K | −38.06 | −6.07 | Oxidosqualene cyclase |
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Zheng, R.; Chen, T.-s.; Lu, T. A Comparative Reverse Docking Strategy to Identify Potential Antineoplastic Targets of Tea Functional Components and Binding Mode. Int. J. Mol. Sci. 2011, 12, 5200-5212. https://doi.org/10.3390/ijms12085200
Zheng R, Chen T-s, Lu T. A Comparative Reverse Docking Strategy to Identify Potential Antineoplastic Targets of Tea Functional Components and Binding Mode. International Journal of Molecular Sciences. 2011; 12(8):5200-5212. https://doi.org/10.3390/ijms12085200
Chicago/Turabian StyleZheng, Rong, Tuan-sheng Chen, and Tun Lu. 2011. "A Comparative Reverse Docking Strategy to Identify Potential Antineoplastic Targets of Tea Functional Components and Binding Mode" International Journal of Molecular Sciences 12, no. 8: 5200-5212. https://doi.org/10.3390/ijms12085200
APA StyleZheng, R., Chen, T.-s., & Lu, T. (2011). A Comparative Reverse Docking Strategy to Identify Potential Antineoplastic Targets of Tea Functional Components and Binding Mode. International Journal of Molecular Sciences, 12(8), 5200-5212. https://doi.org/10.3390/ijms12085200