1. Introduction
Polymeric drug delivery systems are designed to deliver drugs to the local site of action for extended periods of time, so that the therapeutic levels of drugs with short
in vivo half-lives can be maintained [
1]. In addition, different forms of drug delivery devices are used to reduce the fluctuations in plasma drug levels, so a slower and controlled drug release rate can be achieved, which can then provide an effective pharmacological response [
1,
2]. Microencapsulation of a drug within a polymeric device, e.g., microspheres is considered as one of the most common methods of drug delivery [
3]. Drug encapsulated microspheres require less frequent drug administration when compared to the conventional dosage forms. Also, drugs encapsulated within microspheres are kept separate from other microspheres; as a result multiple drug administration in a single injection can be achieved, which would have not been possible otherwise, owing to drug compatibility issues [
1]. Apart from being orally administered, microspheres can also be administered through a number of parenteral pathways such as intraocular, intravenous, intra-arterial, intraspinal and intraosseous [
3]. Often the site of administration influences the performance of drug delivery due to differences in the local tissue environment such as pH and enzyme activity [
3].
The solvent evaporation method is the most frequently used technique to prepare polymeric microspheres for drug delivery. The characteristics of the microspheres produced are largely dependent on the type of polymer (hydrophobic/hydrophilic) and drug used. Other processing parameters such as solvent type and concentration of the emulsifier, drug/polymer ratio and stirring rate are also known to affect the physicochemical properties of the microspheres [
4]. For example, surface morphology and porosity of hydrophobic microspheres are known to be particularly affected by the processing parameters due to which the drug release rate from microspheres is also affected. However, the encapsulation efficiency of drugs within hydrophobic microspheres is largely dependent on the type of microencapsulation procedure and on the solubility of the drug. For example, in the single emulsion method (o/w), a predetermined quantity of the polymer dissolved in the solvent is added to deionized water (inner aqueous phase, w
1) containing polyvinyl alcohol PVA, which is used as a surfactant for the production of microspheres. The solvent is removed from the inner aqueous phase and evaporated through the emulsion-air interface. Polymer precipitation is accelerated leading to the formation of the final microsphere, once the solvent evaporation is initiated. However, the encapsulation efficiency of highly hydrophilic drugs is affected by the fast shrinkage of the microspheres. As a result, the encapsulated drug is drained out of the microspheres during solvent removal. Therefore the o/w method is more suitable for the entrapment of less hydrophilic drugs as they accumulate towards the microsphere surface due to their hydrophobic nature. In the double emulsion method (w
1/o/w
2) the initial emulsion phase (w
1) is added to a second aqueous phase containing PVA (w
2). During solvent evaporation, small microdroplets formed within the microspheres coalesce forming a honeycomb structure. During solvent evaporation, the precipitating polymer wall also forms holes through which the entrapped drug is partly removed. This honeycomb structure is considered most suitable for efficient entrapment of hydrophilic drugs [
5].
The double emulsion method has been used by several researchers to entrap highly hydrophilic drugs such as gentamicin within biodegradable hydrophobic microspheres [
6,
7]. Gentamicin, a highly water soluble drug, is an aminoglycoside that has been used against a wide range of Gram-positive and Gram-negative bacteria [
7]. For example, gentamicin has been used for the treatment of osteomyelitis, an inflammatory bone disease [
7]. Osteomyelitis is the microbial infection of the bone medullary cavity, cortex and periosteum that is known to occur during post-operative sepsis after an orthopaedic procedure. However, for the treatment of osteomyelitis, a prolonged systemic antibiotic treatment such as the use of gentamicin, either oral or parenteral, for a period of 4–6 weeks, is known to cause systemic toxicity and patient discomfort. Therefore, localised drug delivery at the infected site, for example injecting drug loaded microspheres, is being proposed. By this method the risk of high dose administration and possibility of building drug resistance could be avoided [
8]. In a study conducted by Huang
et al. [
8] PLA microspheres containing gentamicin were developed for the treatment of bone infection. PLA microspheres with an average size of 178 μm were incorporated with gentamicin sulphate for the treatment of osteomyelitis, where 80% of gentamicin sulphate was released within 3 weeks of implantation [
8].
Although these biodegradable polymers such as PLA prove to be advantageous since they can circumvent some of the problems faced when long term implants are used, they have a risk of toxicity and immunogenicity due to their acidic by-products. Other properties such as high price, lack of tailorability, presence of chemical catalysts and fast degradation rate has challenged their commercialization [
9]. Thus the need for polyhydroxyalkanoates has arisen due to their tailorable mechanical properties, biocompatibility and biodegradability.
In this paper, P(3HB) microspheres containing gentamicin for the treatment of bone infections such as osteomyelitis were developed. Localised drug delivery at the site of infection using these drug loaded microspheres is being proposed, for example incorporating the microspheres into 3D porous scaffolds [
10]. The main advantage of the P(3HB) microspheres is the lack of risk of toxicity and immunogenicity due to acidic by products, which is a disadvantage when PLA is used [
11,
12]. In this work, the processing conditions that particularly affect the characteristics of the P(3HB) microspheres were investigated so that microspheres with a high gentamicin loading efficiency, controllable porosity and uniform drug distribution could be produced.
2. Materials and Methods
2.1. Materials
Distilled water and high performance liquid chromatography (HPLC) grade water were used where appropriate. The two types of water were obtained from Elga Pure Lab Options distillation units. All chemicals, buffers and drugs used were purchased from Sigma-Aldrich (Dorset UK). Care was taken to make sure that the chemicals used were of the best grade available (analytical or HPLC where appropriate) for the experiment.
P(3HB) was isolated from the Gram-positive bacteria Bacillus cereus SPV which was obtained from the culture collection of University of Westminster, London, UK. All chemicals required for the growth of Bacillus cereus SPV and extraction of polymer from the bacterial cells were obtained from Sigma-Aldrich Company Ltd. and VWR Chemicals (England) except for nutrient broth and yeast extract, which was obtained from DIFCO (BD UK Ltd., Oxford, UK). Antifoam (FG-10) was purchased from Dow corning (Edison, NJ, USA) for fermentation study.
2.2. P(3HB) Microsphere Production
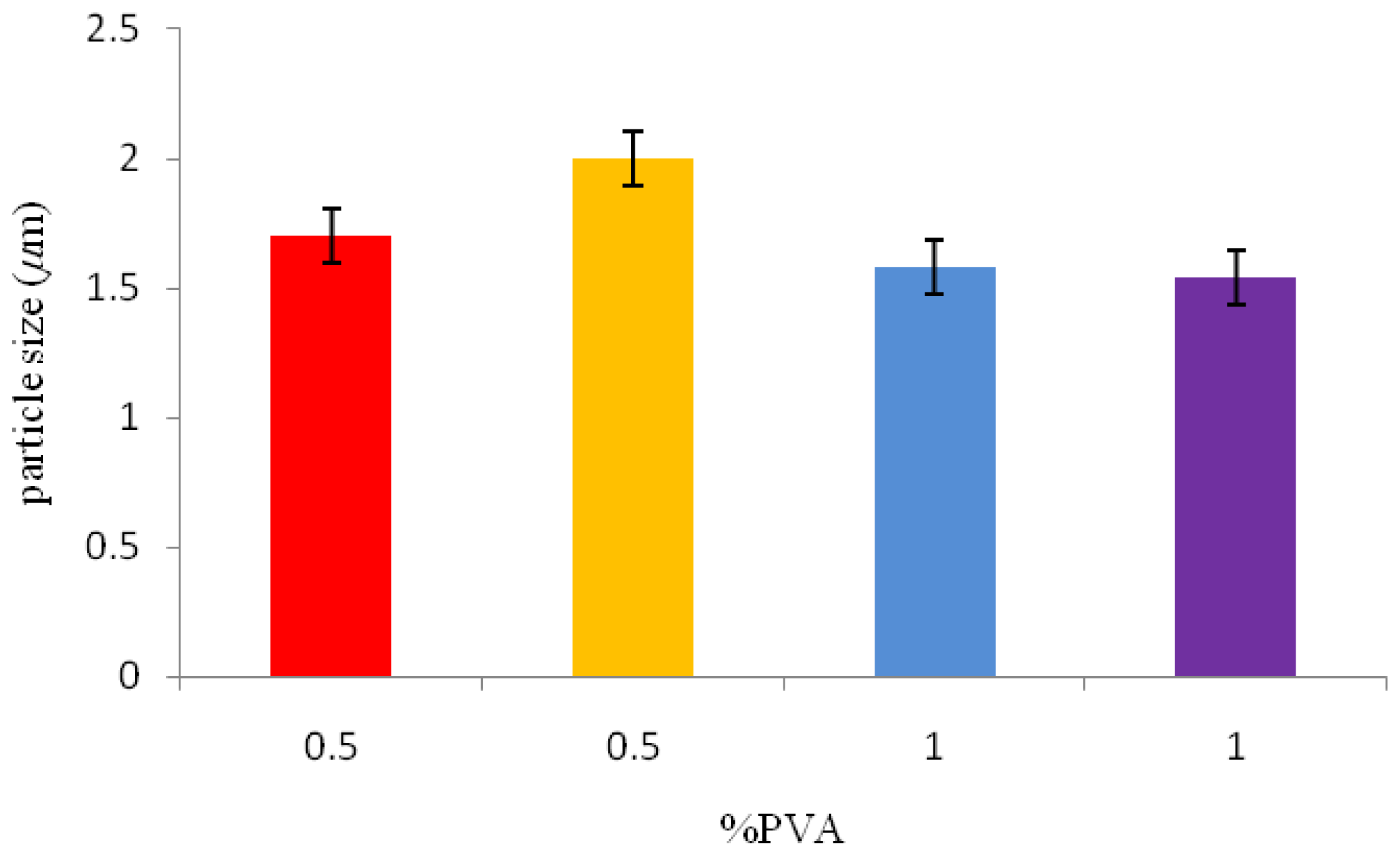
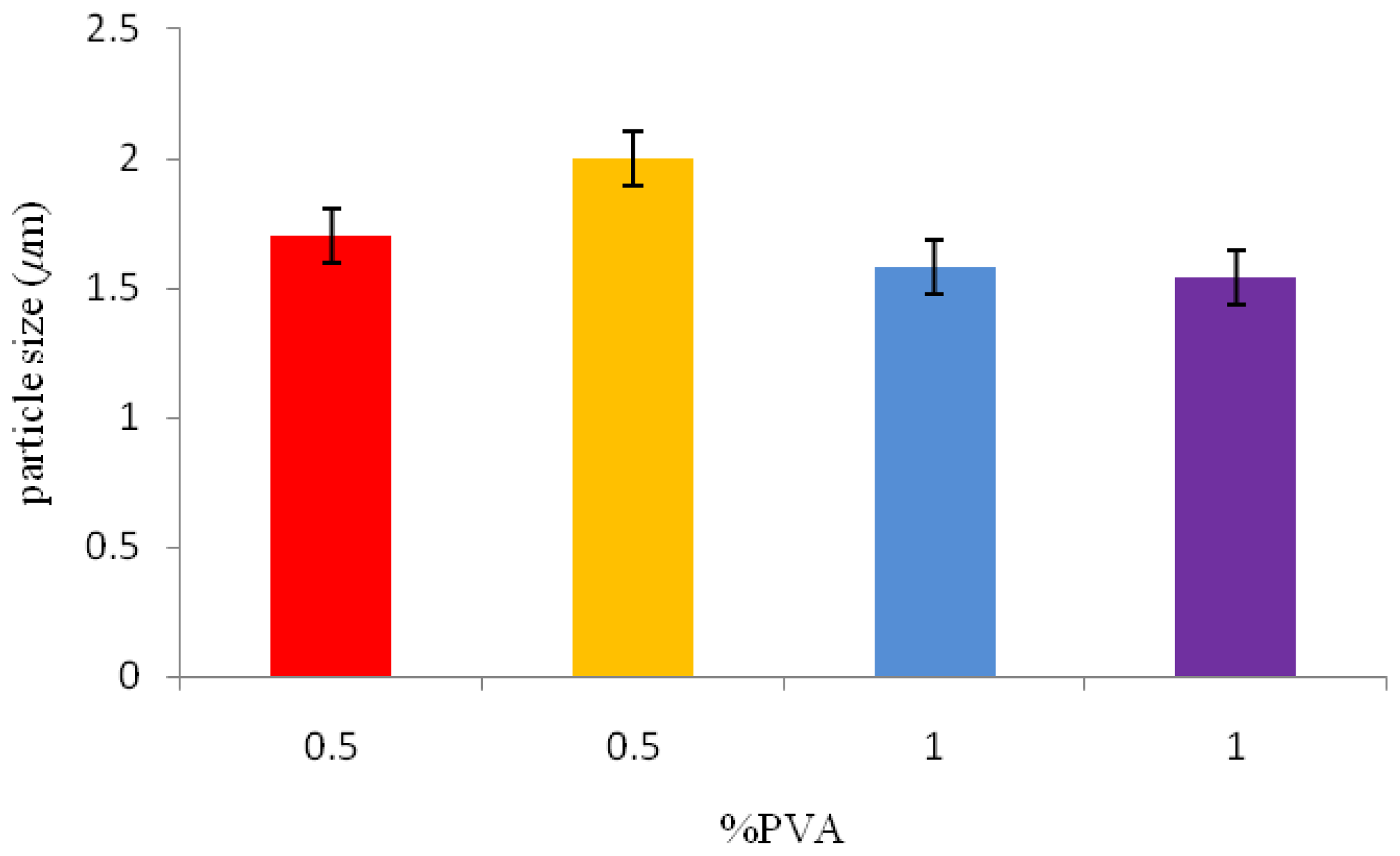
A solid-in-oil-in-water emulsion technique was used for P(3HB) microsphere preparation. Quantities of 1 or 3 g of P(3HB) were dissolved in 8 mL of chloroform and agitated for 3 min. This mixture was then transferred into the first solid-in-oil emulsion (w/o) of 40 mL of 1% or 0.5% w/v aqueous polyvinyl alcohol (PVA) solution, and stirred at 1000 rpm for 3 min. This solution was then added to a second solid-in-oil-in-water emulsion (w/o/w) of 500 mL of 0.5% or 0.05% w/v aqueous PVA solution, forming the second oil-in-water emulsion. This emulsion was stirred either at 300 rpm or 800 rpm for 4 h to eliminate the chloroform used as solvent and then to form P(3HB) microspheres. The resulting microspheres were isolated by centrifugation at 3680 g for 5 min and then washed with distilled water three times, air dried and stored in a desiccator until further use.
Table 1 shows the varying processing conditions (amount of surfactant, polymer concentration and stirring rate) used for the synthesis of P(3HB) microspheres.
2.3. Gentamicin Loaded P(3HB) Microsphere Production
Gentamicin-loaded P(3HB) microspheres were produced using similar methods as described in section 2.2. In this case, 2 mg/g of P(3HB) mixed with gentamicin were dissolved in 8 mL of chloroform and agitated for 3 min. This solution was then added to the aqueous first solid-in-oil emulsion of 40 mL of 1% or 0.5% w/v aqueous polyvinyl alcohol (PVA) solution, and stirred at 1000 rpm for 3 min. This solution was then added to a second solid-in-oil-in-water emulsion of 500 mL of 0.5% or 0.05% w/v aqueous PVA solution, forming the second oil-in-water emulsion. This emulsion was stirred either at 300 rpm or 800 rpm for 4 h to eliminate the chloroform used as solvent and to form P(3HB) microspheres. The resulting microspheres were isolated by centrifugation at 3680 g for 5 min and then freeze-dried (Savant Modulyo D Freeze-drier, Thermo Electron Corp, K) and stored at 4 °C until further use.
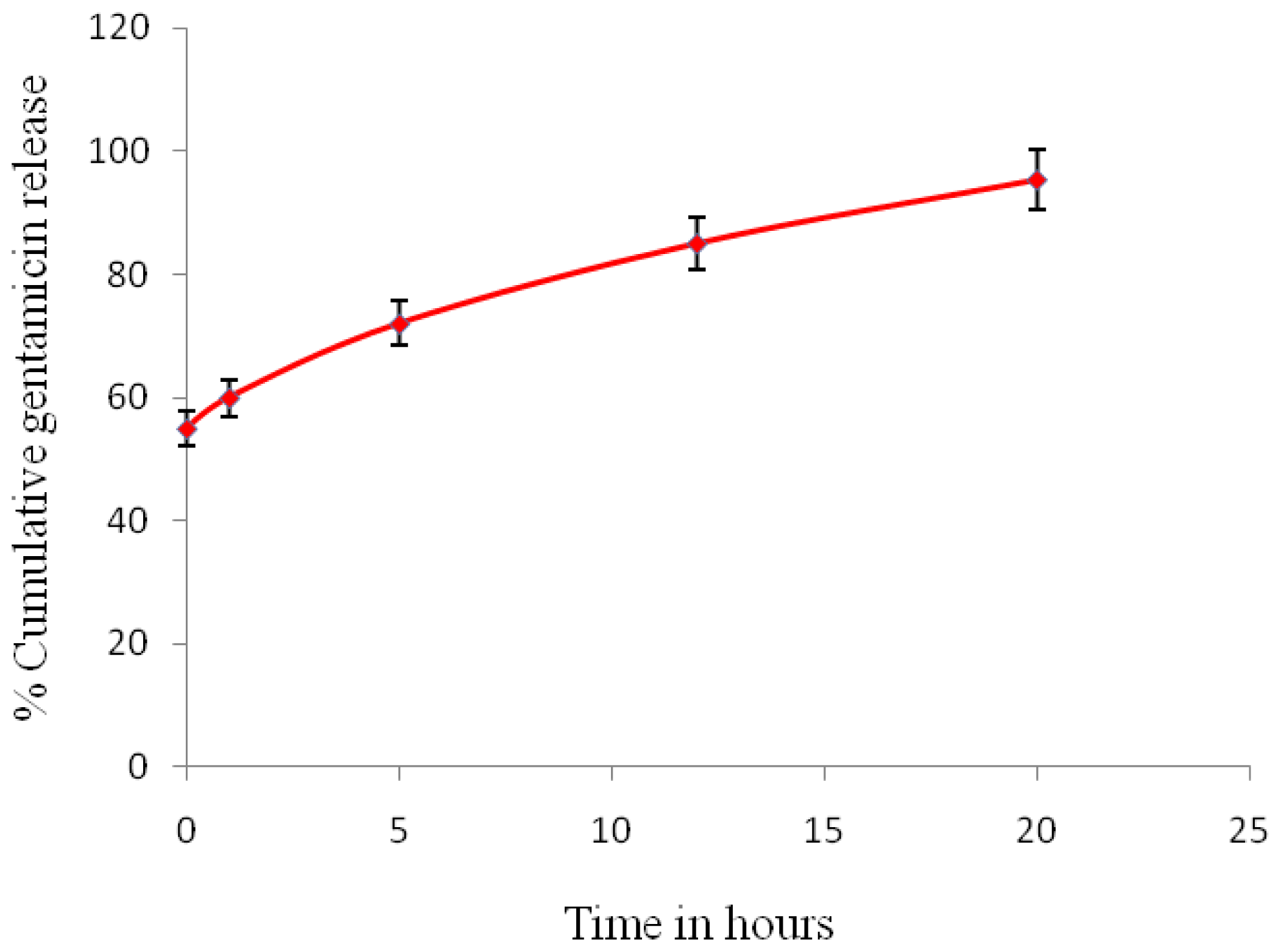
2.4. In Vitro Drug Release Studies
The
in vitro drug release experiments were performed in an incubator at 37 °C for 24 h. 10 mg of the gentamicin-loaded microspheres were immersed in 2 mL of simulated body fluid (SBF) and 1 mL samples were collected at regular intervals, up to a final duration of 24 h. SBF was prepared following the study of Kukobo
et al. [
13]. SBF was used as it has similar ionic concentration as blood plasma. It is therefore important to identify whether or not the gentamicin encapsulated within the microspheres was released in a controlled manner when implanted
in vivo. Each aliquot was replaced with fresh buffer and the tubes returned to the shaker. At each time point the samples were taken out in triplicates and the results averaged. The drug content was determined by injecting 15 μL of the samples onto a high-performance liquid chromatography (HPLC) column and the gentamicin concentration quantified using the standard curve. The experiments were repeated three times.
2.5. Drug Quantification Methods
The encapsulation efficiency (EE%) of the drug-loaded microspheres prepared under different conditions as mentioned in Section 2.2. was determined using
Equation 1.
To determine the EE values, 5 mg of the drug loaded microspheres were dissolved in 1 mL of chloroform, to which 5 mL of water was added after the microspheres were well dissolved. Drugs used in this study, being hydrophilic in nature, separated into the water phase on vortexing. The water phase was then analysed for the drug content using liquid chromatography-mass spectrometry (LC–MS).
2.5.1. Liquid Chromatography Mass Spectrometry
Liquid Chromatography Mass Spectrometry (LC-MS) was used for analysing the drug content from the release buffer. 15 μL samples collected at different time points were injected onto the HPLC column (Dionex HPLC) coupled with an atmospheric pressure (AP)-electrospray ionization (ESI) mass spectrometer (Dionex). Separation was carried out at 50 °C on a reversed-phase C18 60 RP column of dimensions 250 mm × 4 mm [
7].
2.5.2. Mobile Phase used for Gentamicin Quantification
The mobile phase used for gentamicin quantification was an isocratic flow of 60% pentafluoropropionic acid (20 mM in ultra-pure water from Fluka Chemicals, Bucks, Switzerland) and 40% methanol. The standard curve was plotted for gentamicin concentrations ranging between 1.0 and 2000 μg mL
−1 [
7].
2.6. P(3HB) Microsphere Characterization Techniques
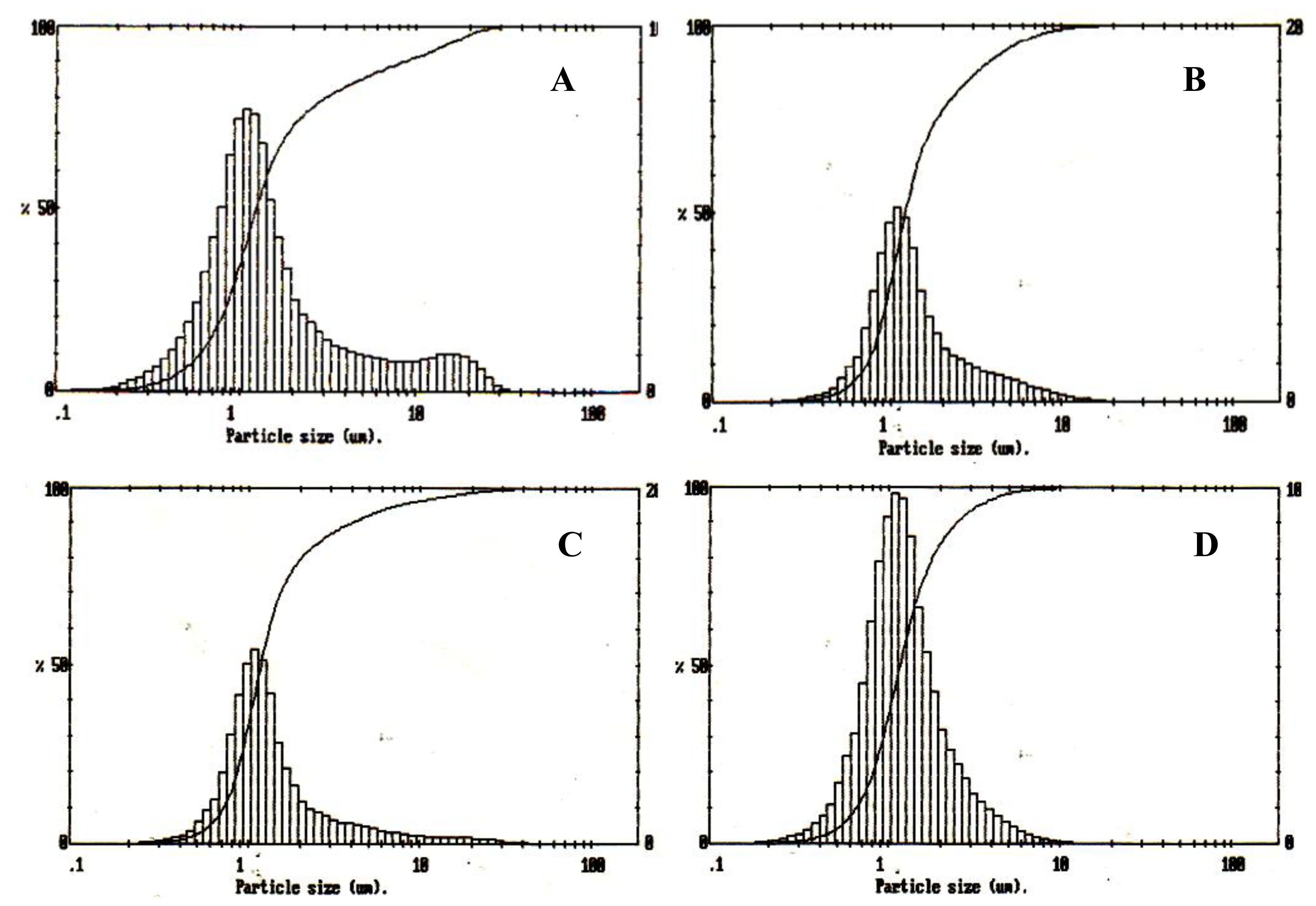
2.6.1. Particle size Analysis
Malvern Mastersizer particle size analyser (Worcestershire, UK) was used to measure the particle size of the microspheres. Microspheres were dispersed well in water and following a background measurement; the suspensions were added drop wise to the analyser until the ideal concentration was reached.
2.6.2. Surface Morphology and Microstructure Characterization
P(3HB) microspheres were observed using a JEOL 5610LV scanning electron microscope (SEM). The samples were placed on 8 mm diameter aluminum stubs using a sticky tag to hold the sample. A gold sputtering device (EMITECH-K550) was used to coat the samples, operating at a pressure of 7 × 10−2 bar and deposition current of 20 mA for 2 min; images were taken at various magnifications to analyse the samples.
2.6.3. Porosity
The porosity (ɛ) of the microspheres was measured using the liquid displacement method. Briefly, 5 mL of ethanol was used as the displacement liquid in a measuring cylinder and weighed. The P(3HB) microspheres immersed in ethanol in the cylinder were sonicated in a water bath to assist penetration of ethanol within the pores.
W1 is the weight of the cylinder filled with ethanol before the immersion of the microsphere sample, W2 is the weight of the cylinder, the ethanol and the sample after removing the excess ethanol above the 5 mL mark, W3 is the weight of the cylinder and ethanol after removing the microsphere sample saturated with ethanol, and Ws is the weight of the microsphere sample used in the measurement.
2.6.4. Determination of Residual PVA Content
The % residual PVA content present on the surface of the microspheres prepared using different conditions were determined by the formation of a coloured complex between two adjacent hydroxyl groups of PVA and an iodine molecule [
14]. Briefly, 2 mg of the lyophilized microsphere samples prepared using the different conditions were treated with 2 mL of 0.5 M sodium hydroxide (NaOH) for 15 min at 60 °C. Each of the samples was then neutralized with 900 μL of 1 N hydrochloric acid (HCl) and the volume was adjusted to 5 mL with distilled water. To each of the samples, 3 mL of 0.65 M solution of boric acid, 0.5 mL of iodine solution (I
2)/Potassium iodide (KI) (0.05 M/0.15 M) and 1.5 mL of distilled water were added. Finally, the absorbance of the samples was measured at 690 nm (Novaspec II Visible spectrophotometer, UK) after 15 min of incubation. A standard graph for quantification of PVA was also prepared under identical conditions [
14].
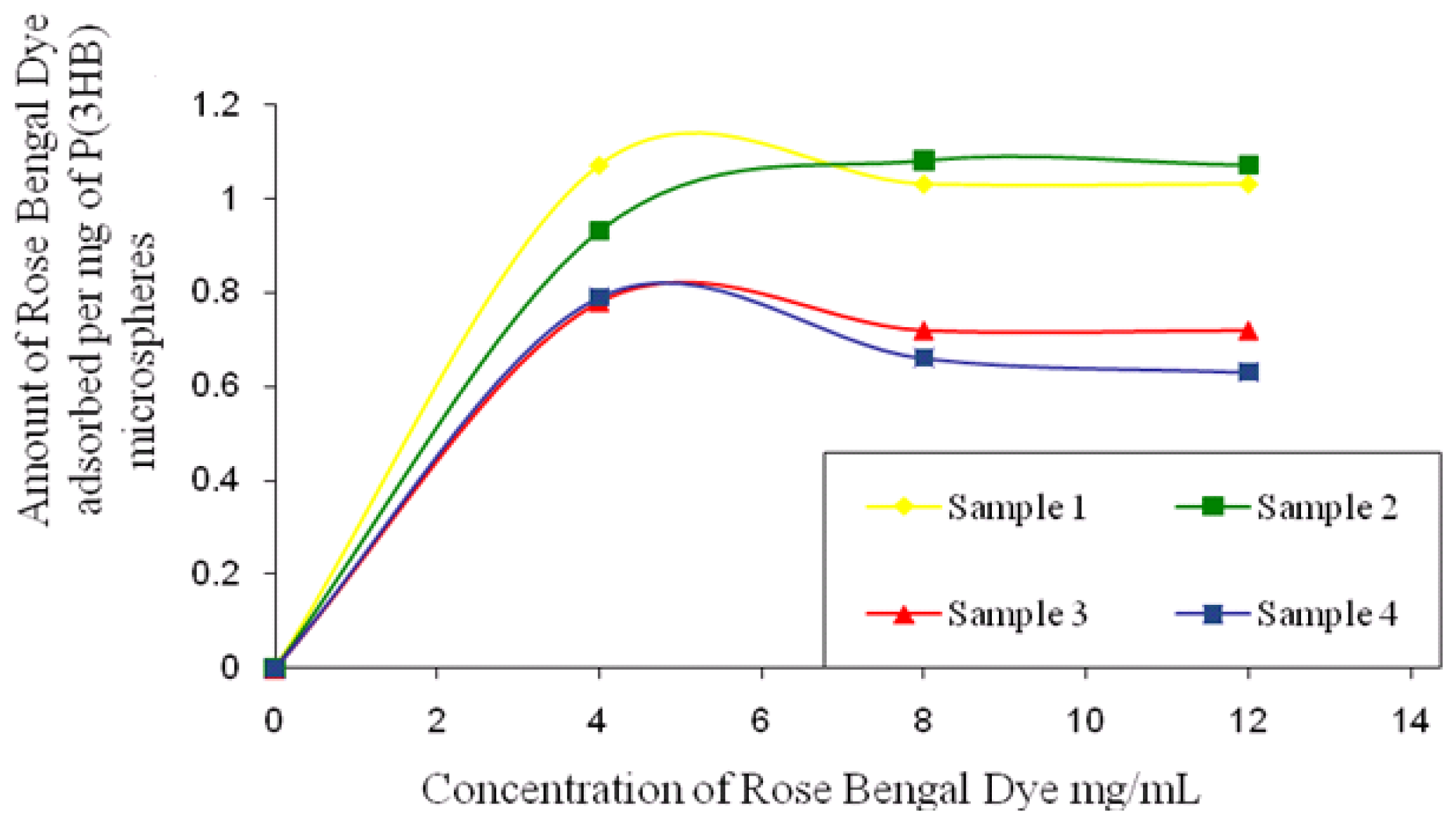
2.6.5. Determination of Surface Hydrophobicity
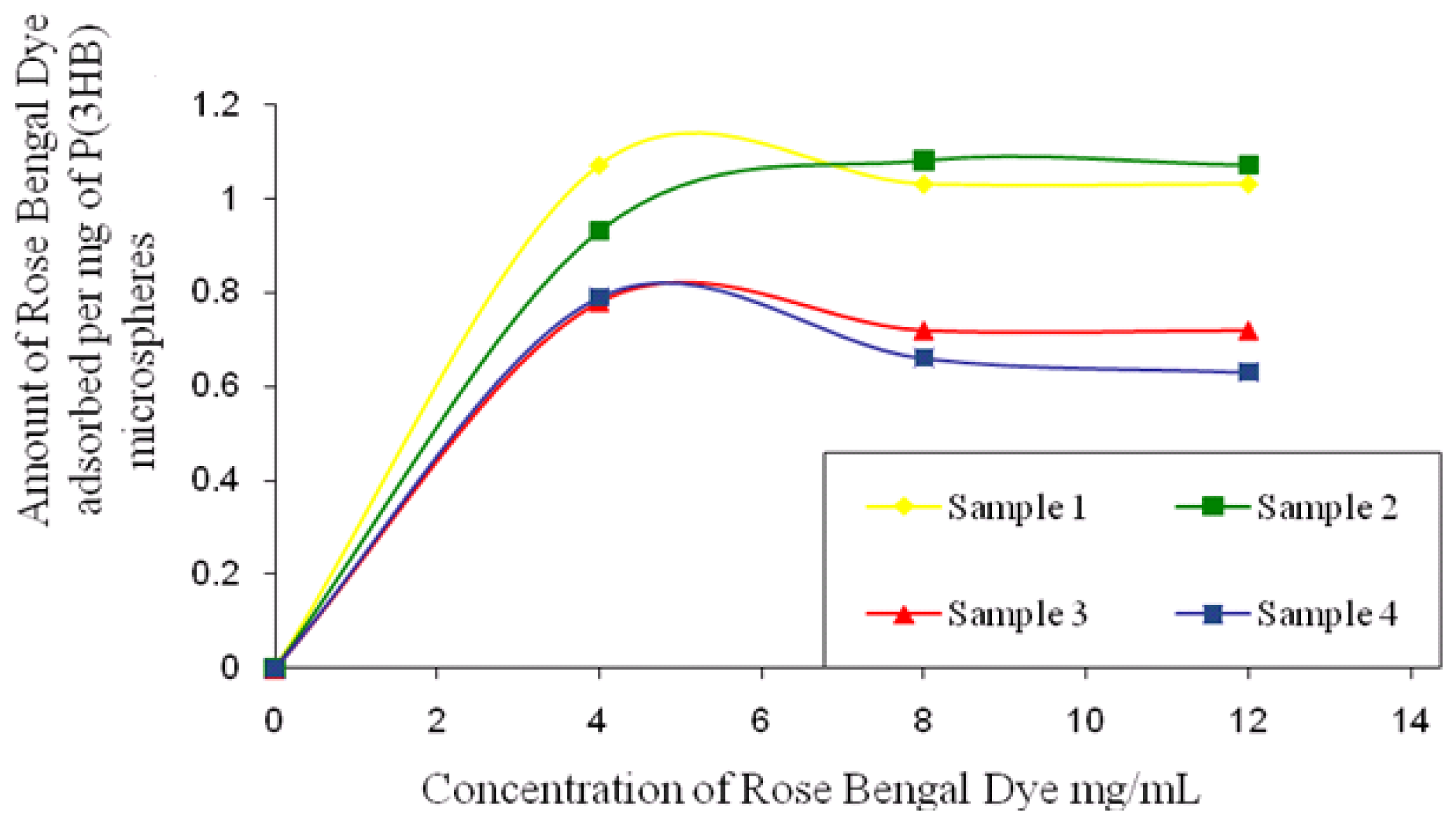
1 mg microsphere samples prepared using different conditions, as mentioned in Section 2.1, were incubated with different concentrations of Rose Bengal dye (4–20 μg/mL) for 3 h at room temperature. The samples were then centrifuged at 110,000 g for 30 min in a microcentrifuge (Sorvall legend RT, UK) to spin down the particles. The supernatant from each of the samples was analysed at 542.7 nm (Novaspec II Visible spectrophotometer, UK) to determine the unbound dye. The dye solution without any microspheres was used as a control and run each time under the same condition to account for the dye bound to the centrifuge tubes [
14].
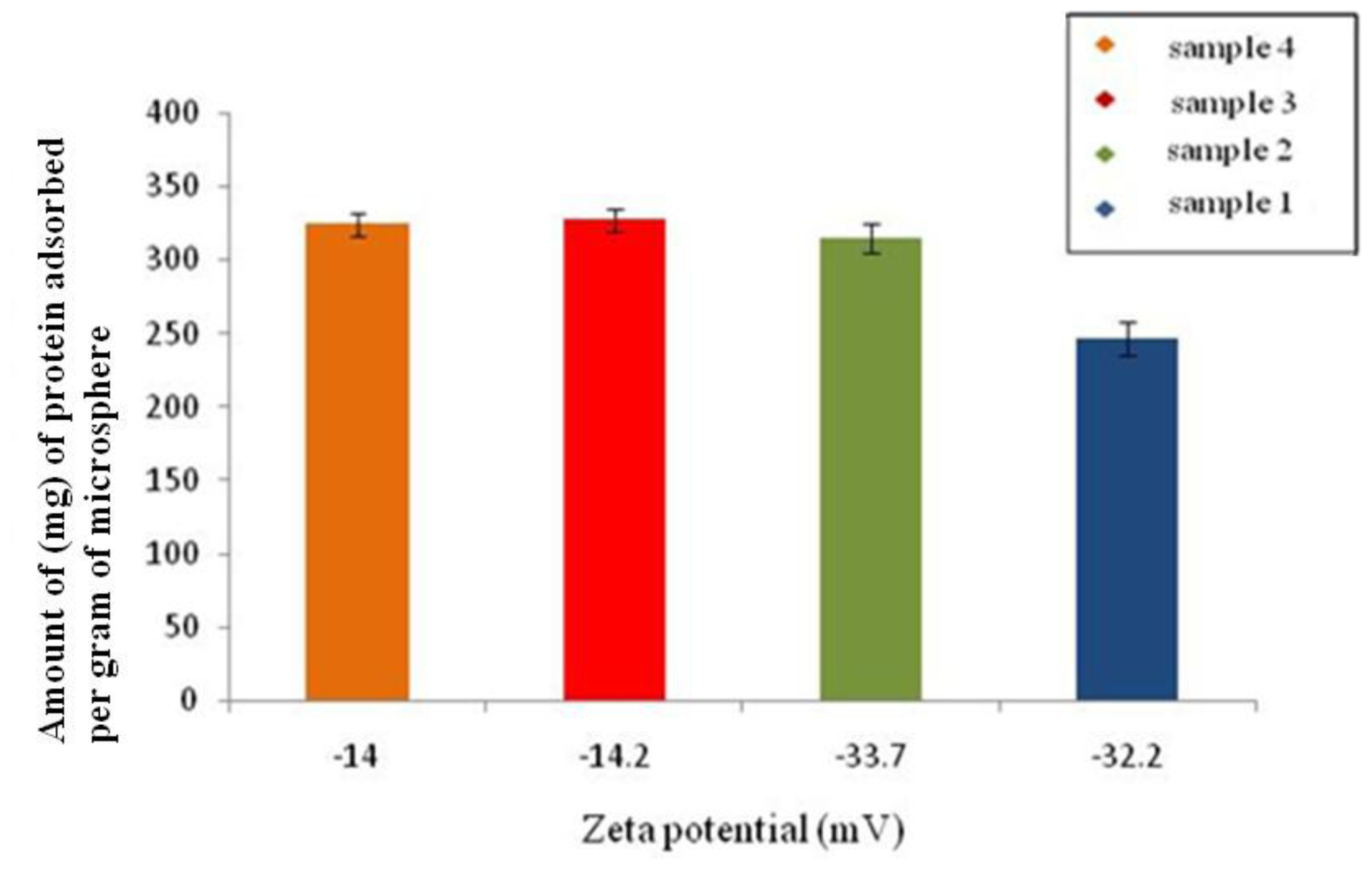
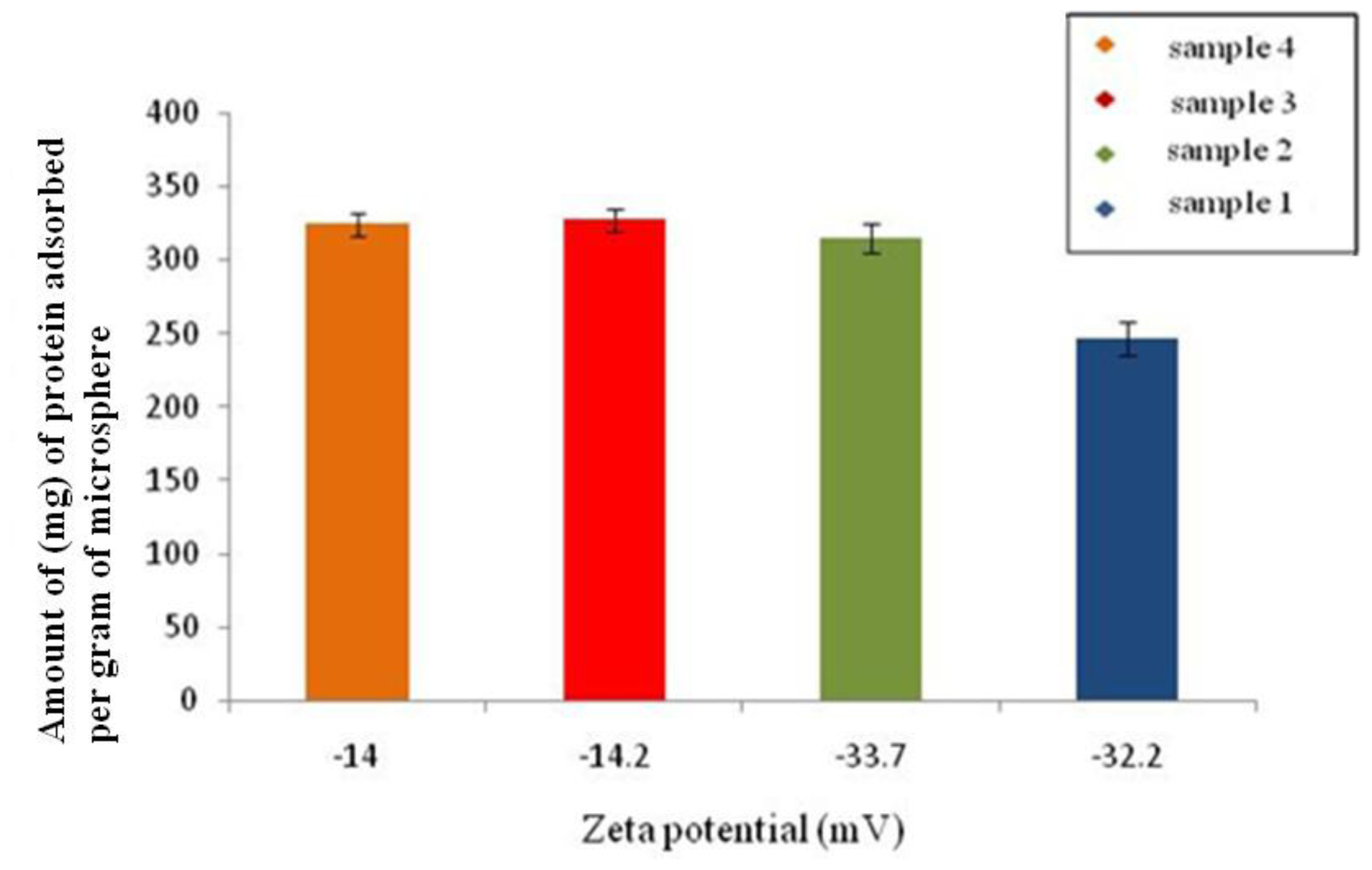
2.6.6. Bovine Serum Albumin (BSA) Adsorption Test
In order to determine the protein adsorption onto the surface of the microspheres 25 mg of the microspheres prepared using different conditions, as mentioned in Section 2.2, were added to 5 mL of distilled water containing 1 mg of BSA. The microspheres were then removed by centrifugation and the concentration of BSA in the supernatant after adsorption on the surface of the microspheres was determined using UV spectroscopy (Eppendorf Biophotometer, UK). The absorbance was measured at 280 nm wavelength. A calibration curve was prepared using known concentrations of BSA. The protein adsorbed (
q) on the surface of the microspheres was calculated using
Equation 3.
where
Ci and
Cf are the initial and final BSA concentrations concentration in the supernatant after adsorption studies, respectively;
V is the total volume of the solution (5 mL); and
m is the weight of the microspheres added into the solution [
15].
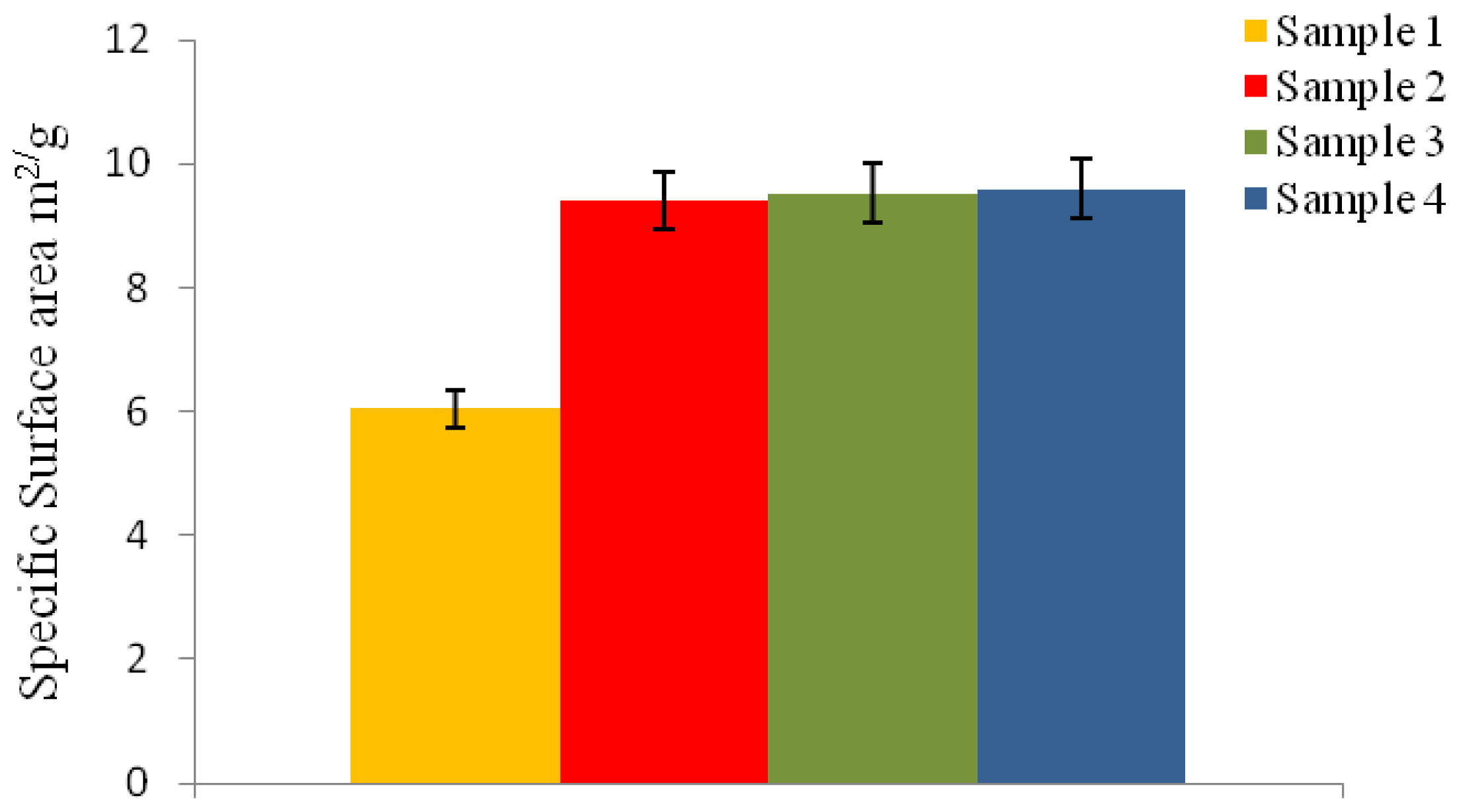
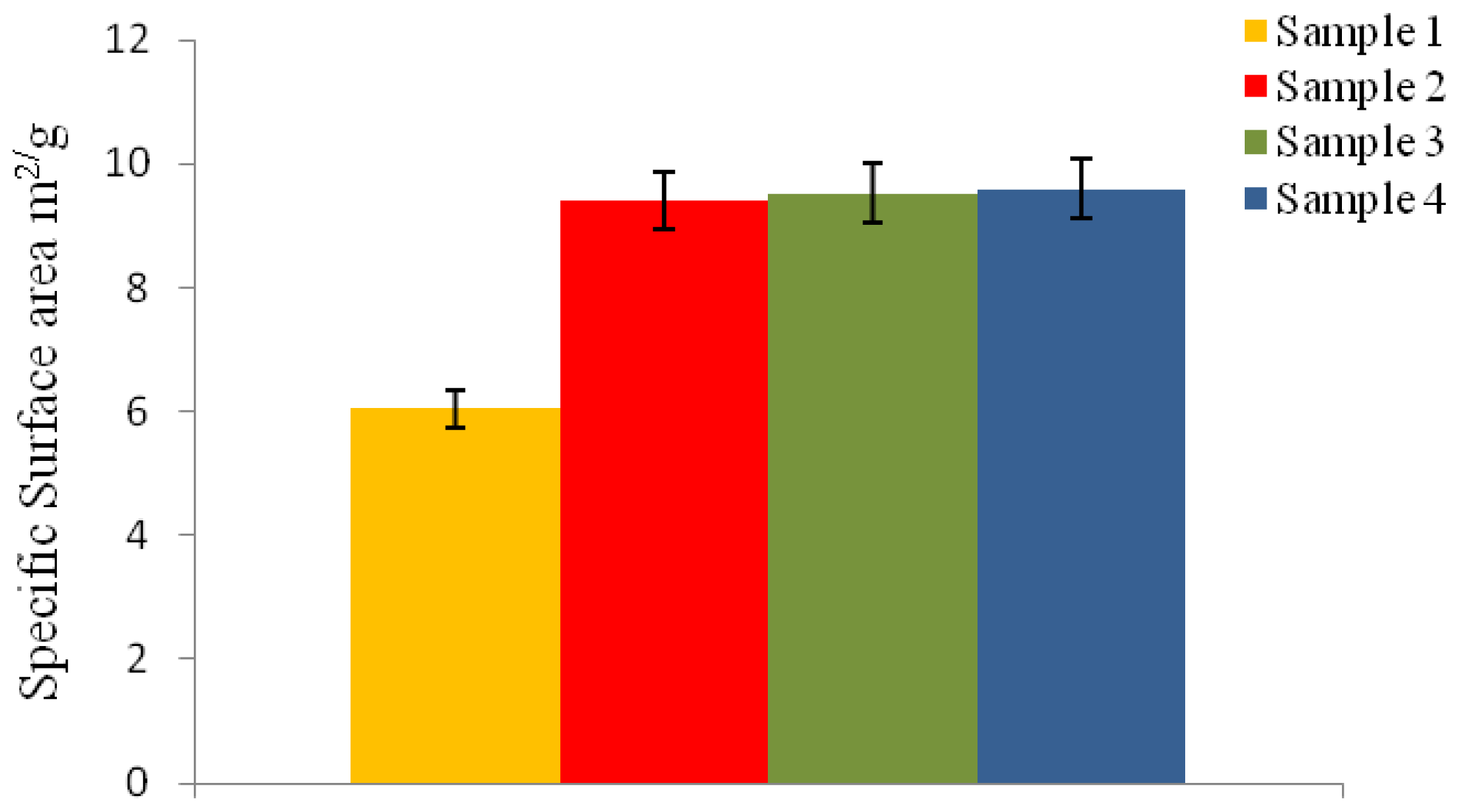
2.6.7. Specific Surface Area Measurement
The specific surface area (SSA) of the microspheres was measured according to Brunauer-Emmett-Teller (BET) method, with nitrogen adsorption at 77 K. The physical adsorption of nitrogen gas molecules on a solid surface was measured using a Micromeritics Tristar instrument after degassing for 1 h at 150 °C. The particle diameter (d
BET) for the P(3HB) microspheres was calculated from the SSA value, using
Equation 6:
where ρ = density of P(3HB), i.e., 1.26 g/cm3 and SSA = specific surface area.
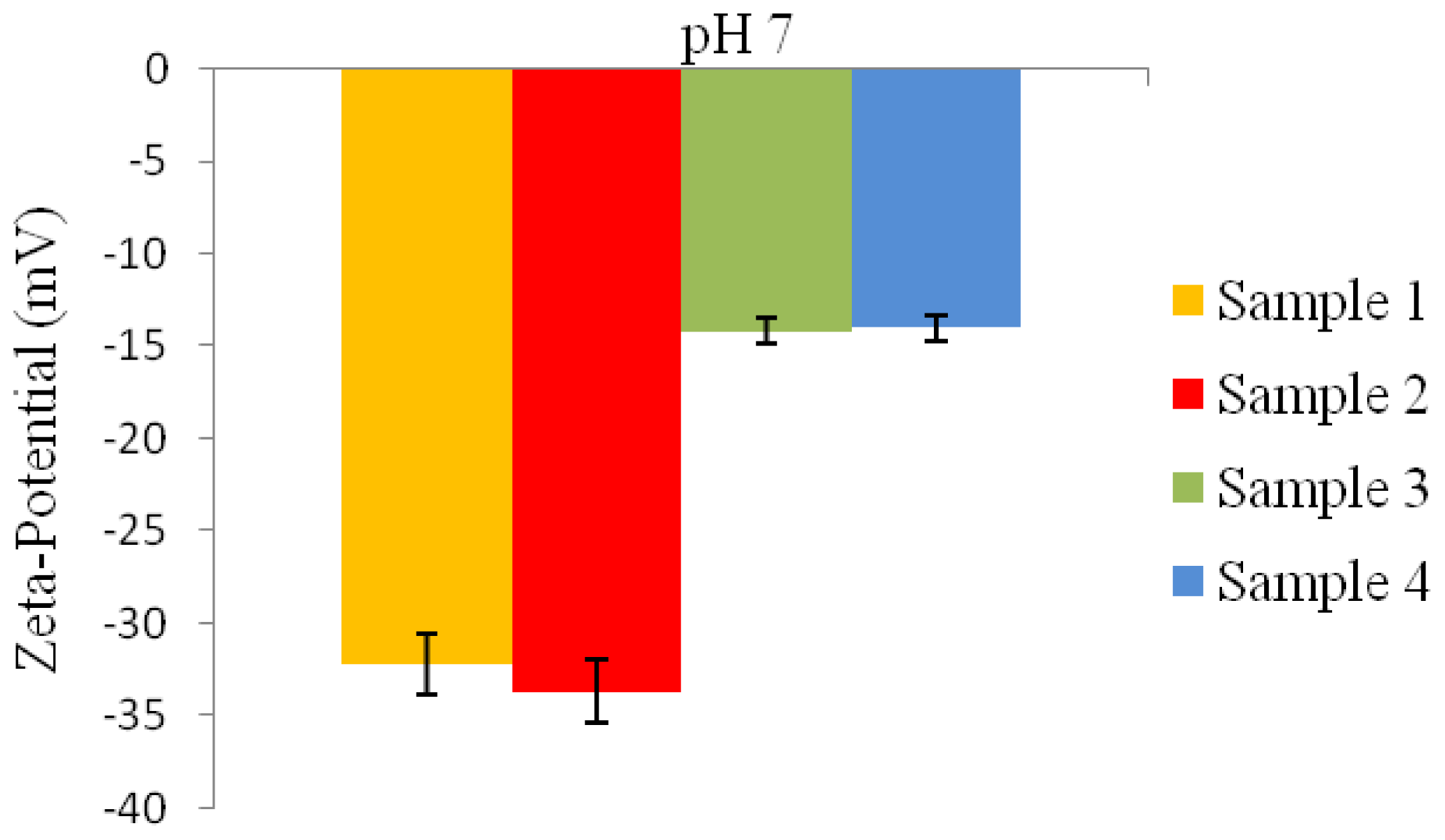
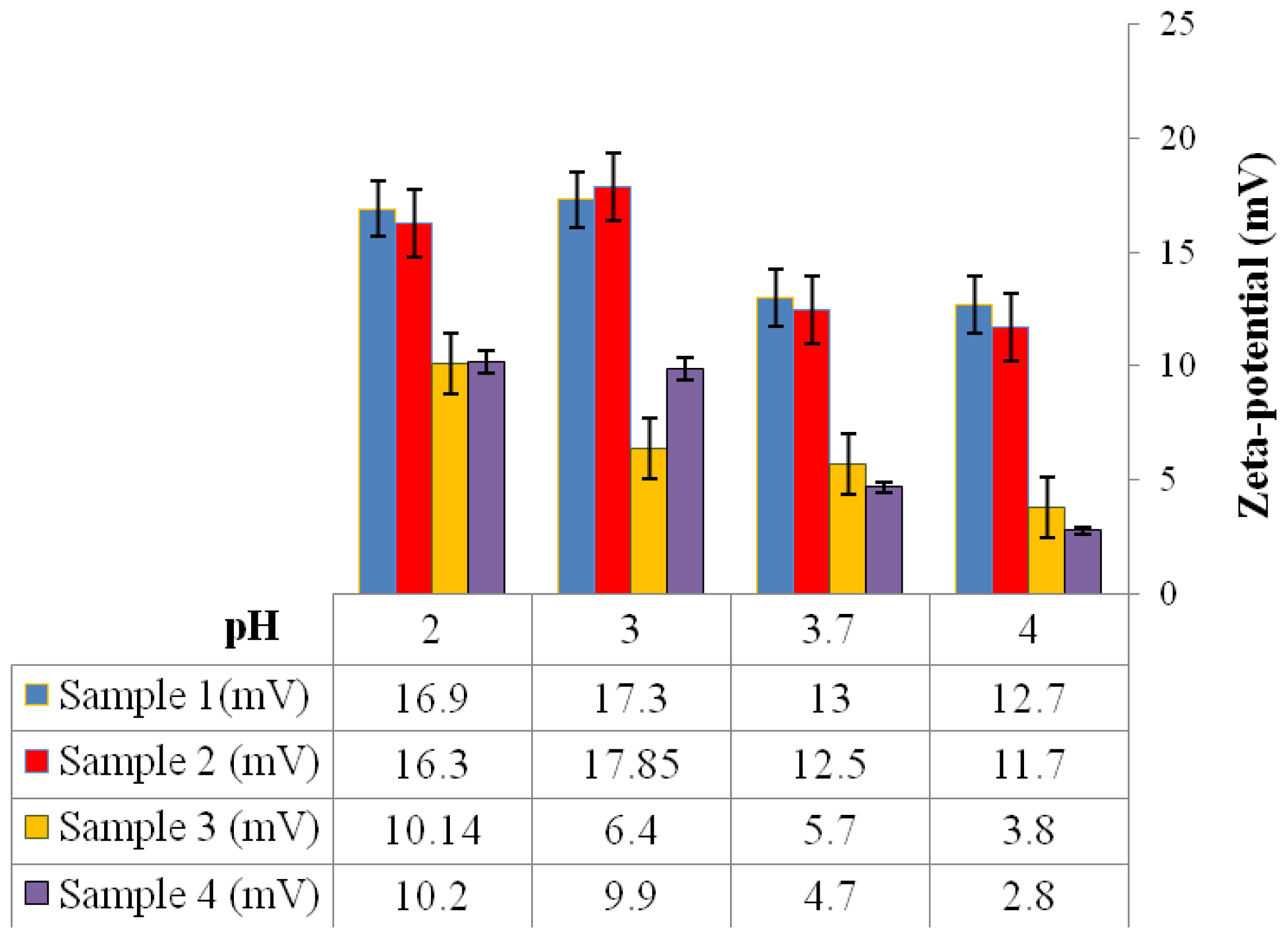
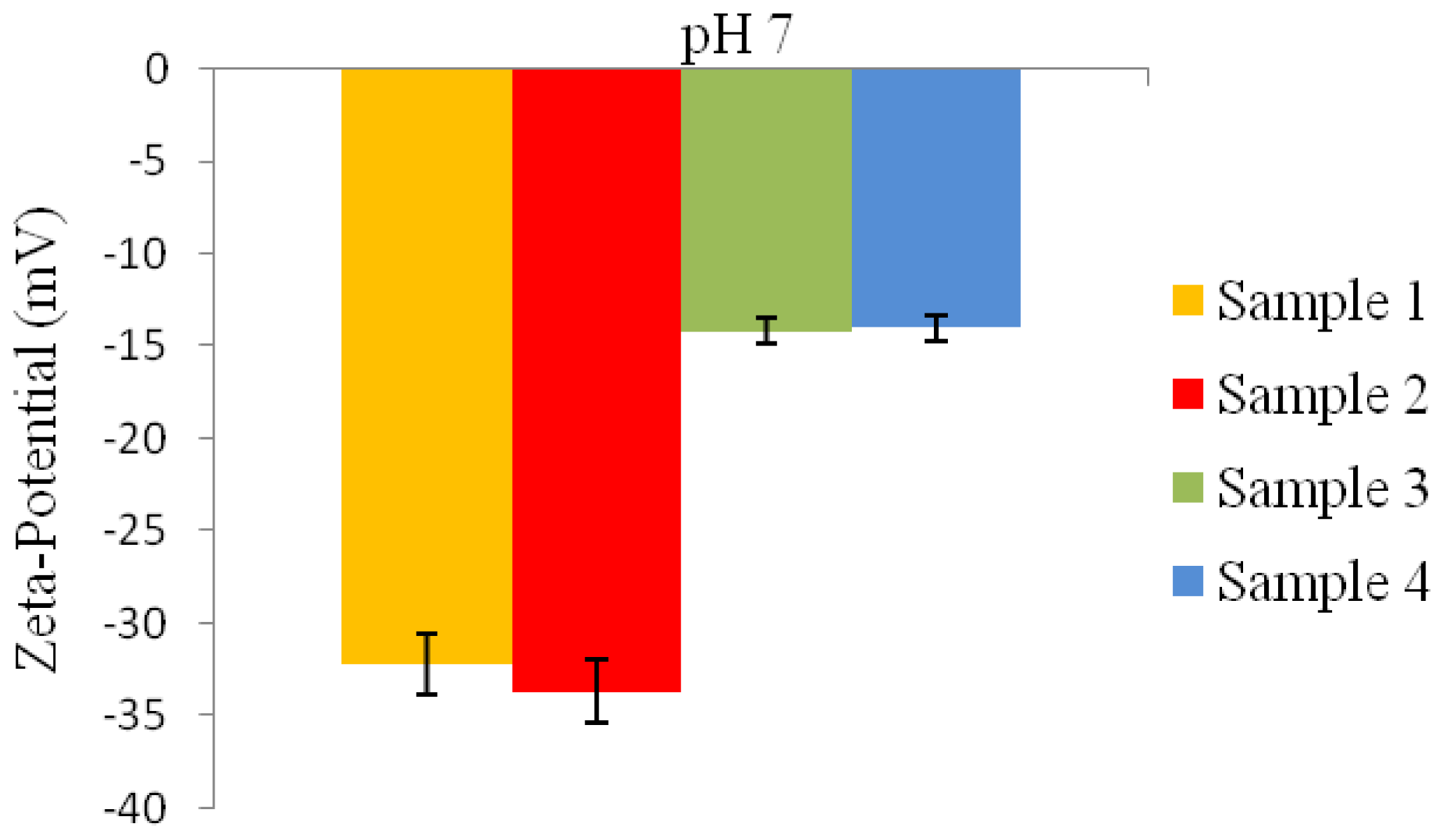
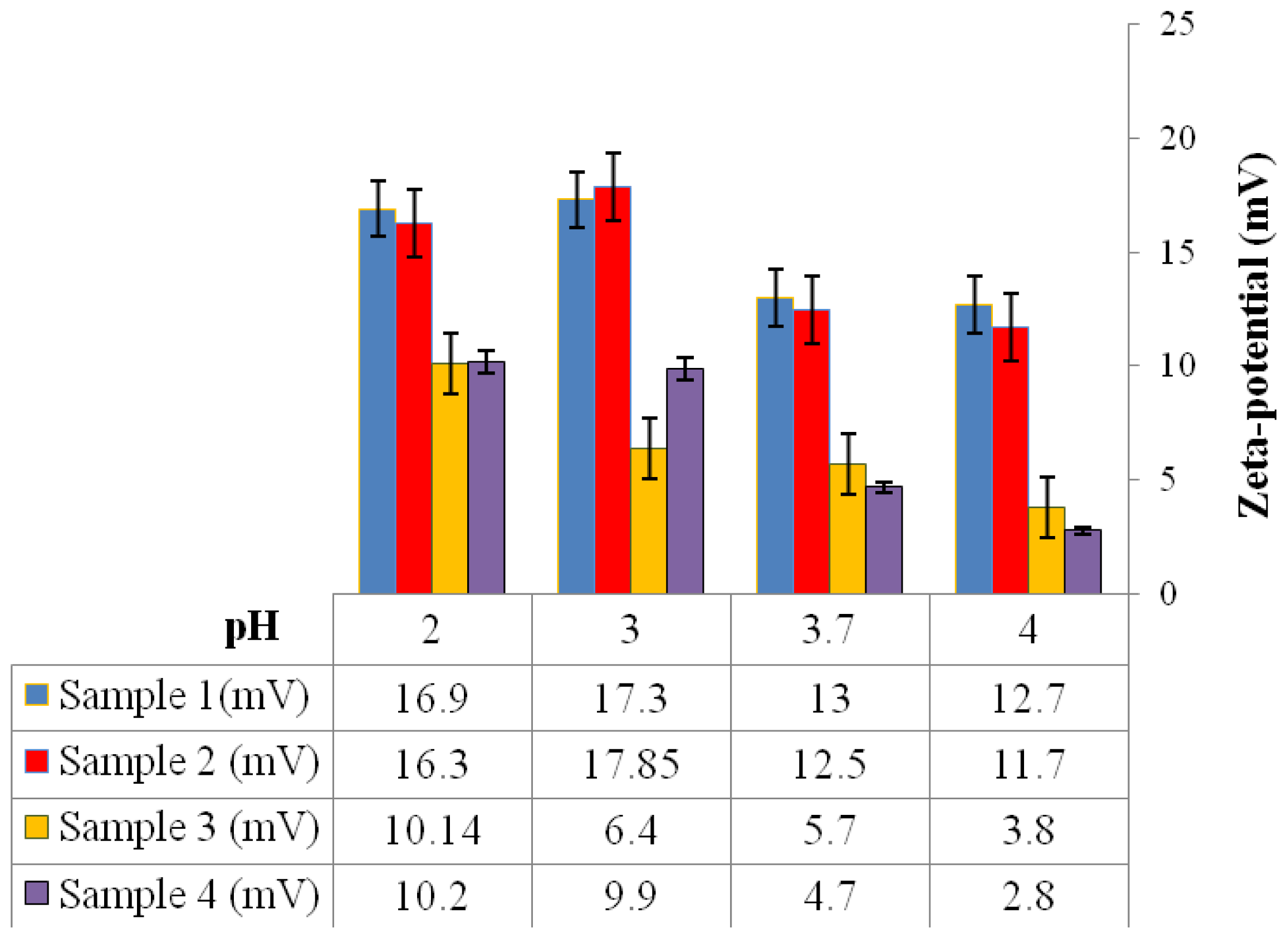
2.6.8. Zeta-Potential Analysis
2 g of the P(3HB) microspheres mixed with water were stirred using a magnetic stirrer for 30 min. Following that, the microspheres were centrifuged and re-dispersed in 5 mL of water and stirred once again for 30 min. Water was used as the dispersant for the microspheres to measure their zeta-potential. The zeta-potential analysis of the microspheres was measured using an Agilent 7020 Zeta Probe (Foster City, USA). 1N NaOH and 1N HCl solutions were added as necessary to adjust the pH [
15].
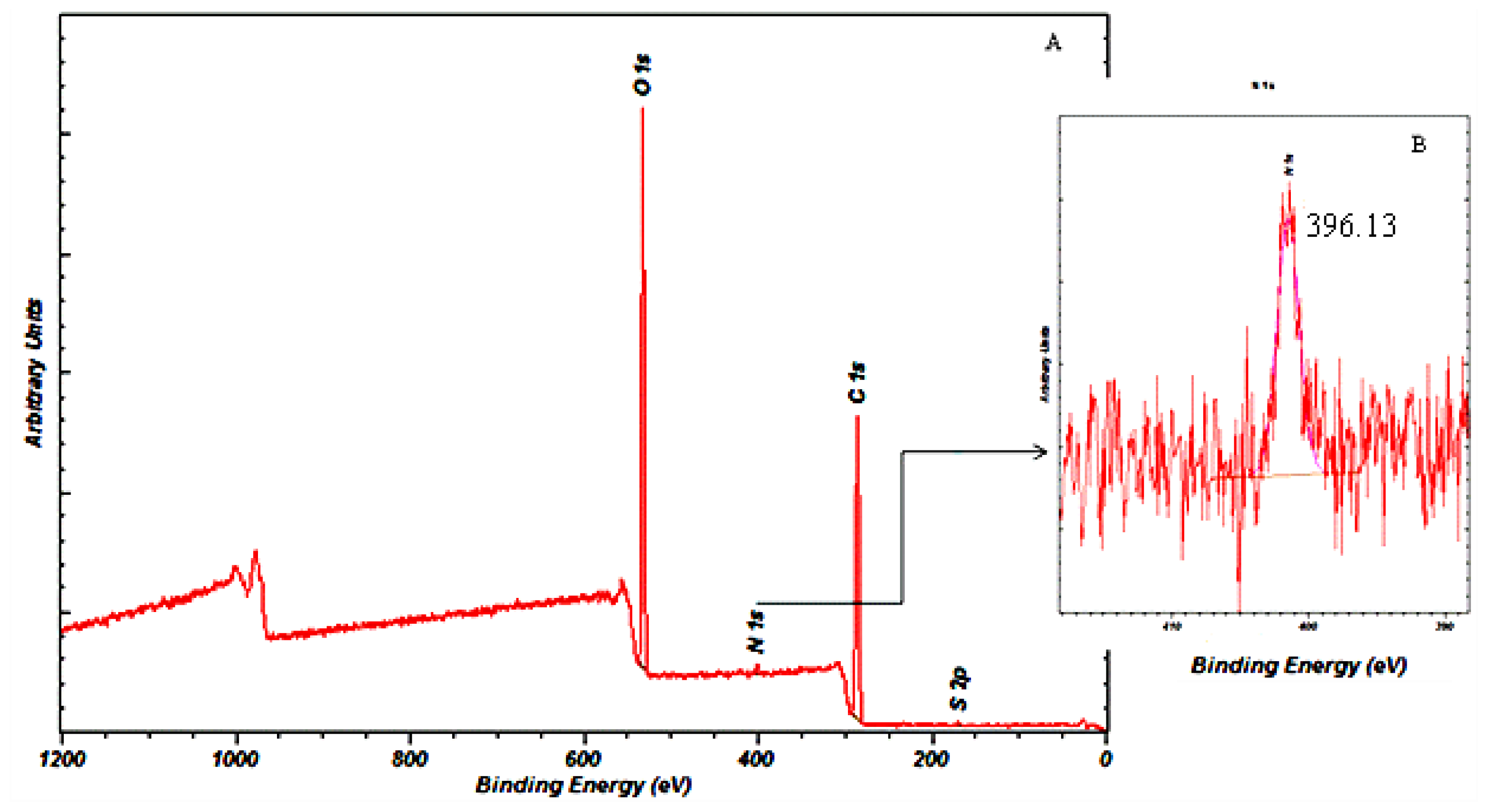
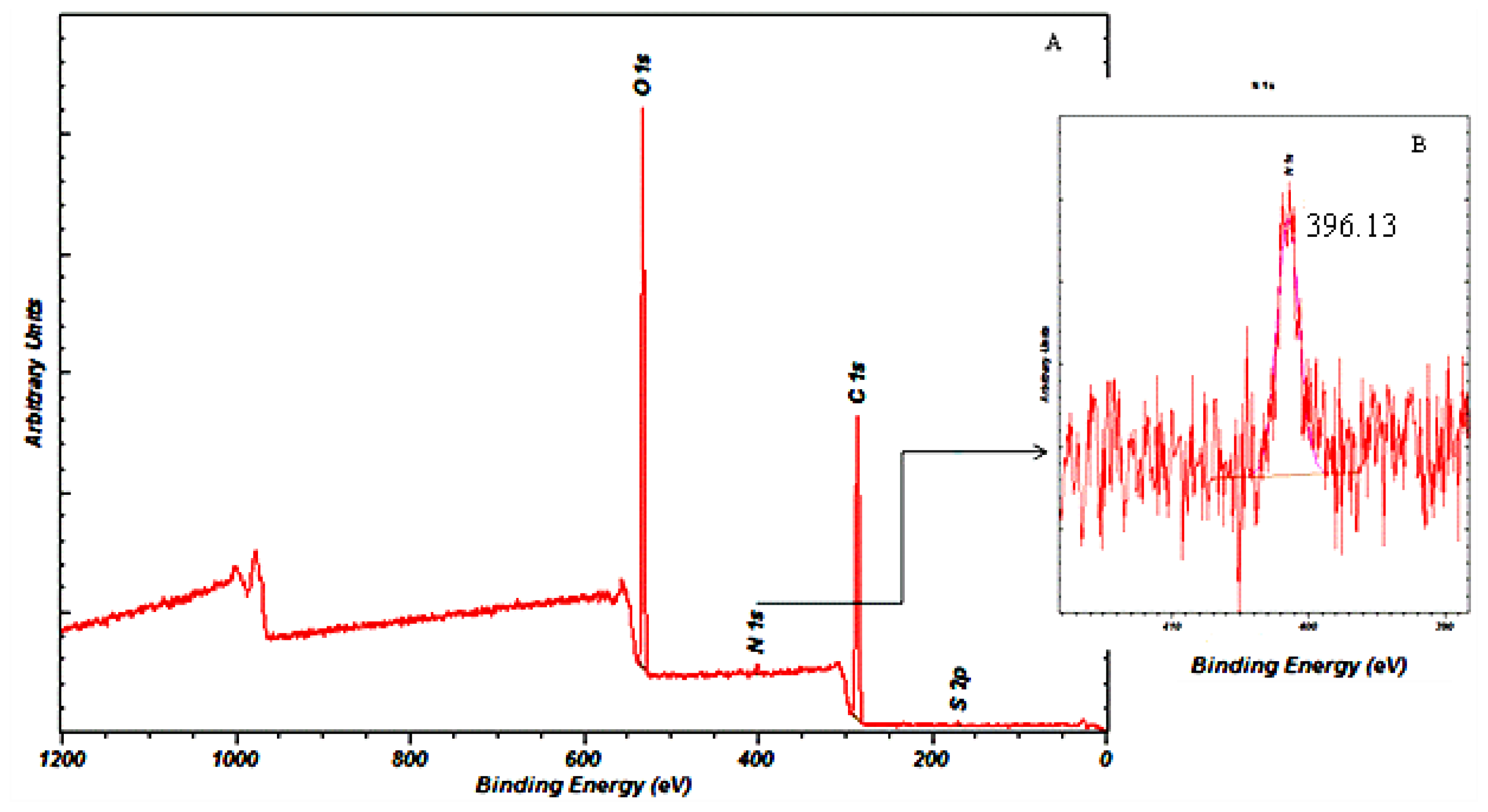
2.6.9. X-ray Photoelectron Spectroscopy (XPS)
The surface chemistry of the gentamicin loaded microspheres was analysed using XPS (Cardiff Catalysis Institute, Cardiff). An Axis Ultra DLD system (Kratos Axis Ultra., Cardiff, UK) with the following acquisition parameters; monochromated 120 W X-ray Power (10 mA emission × 12 kV), 20 eV Pass energy, Analysis Area (700 × 300 μm2) was used to analyse the samples.


 and
and 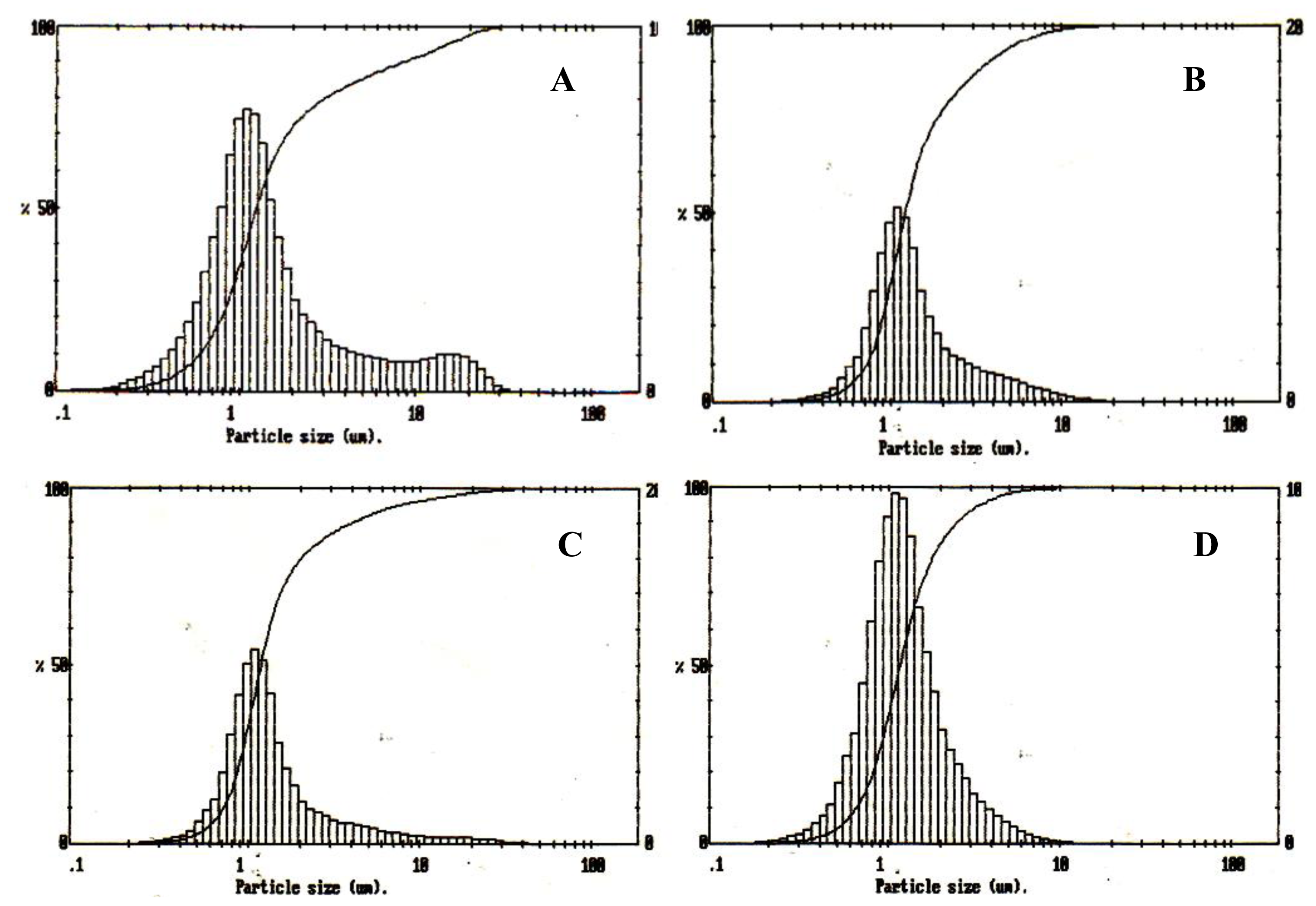


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}