Conformationally Constrained Histidines in the Design of Peptidomimetics: Strategies for the χ-Space Control



Abstract
:1. Introduction
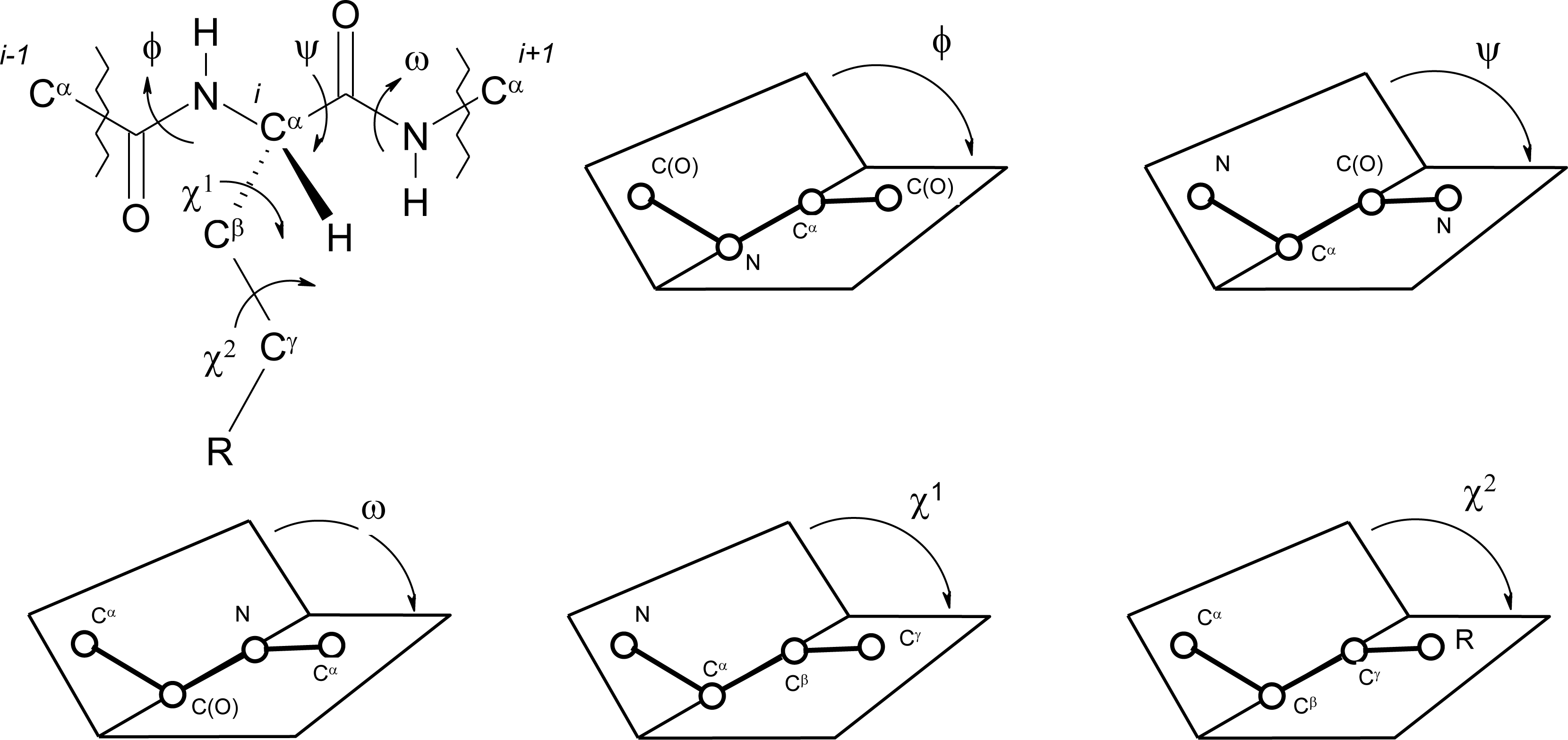
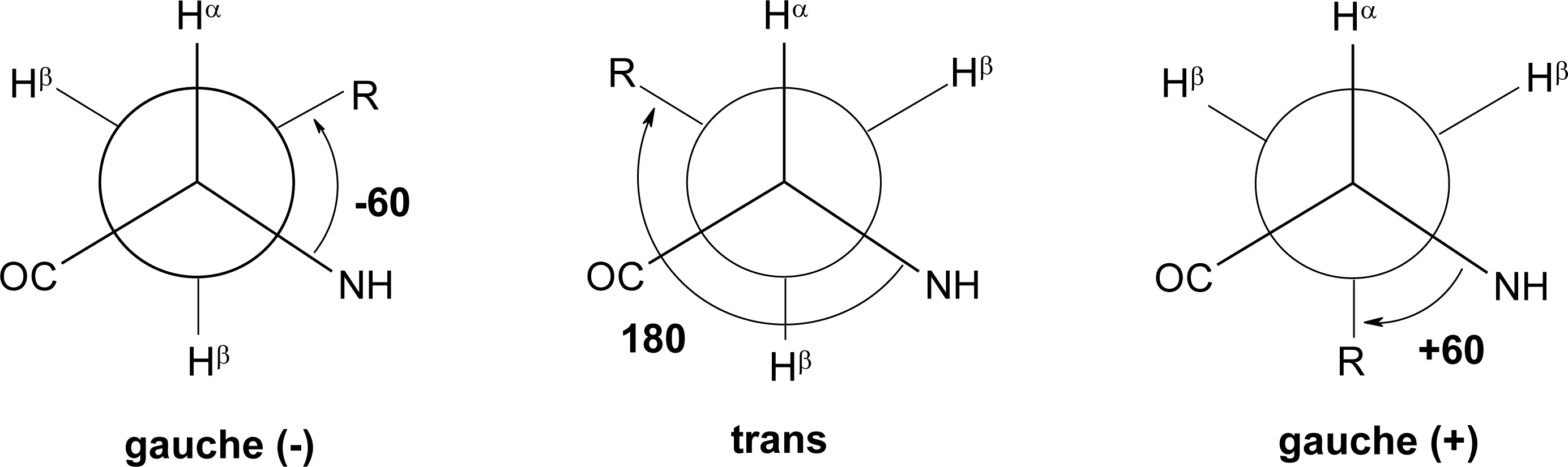
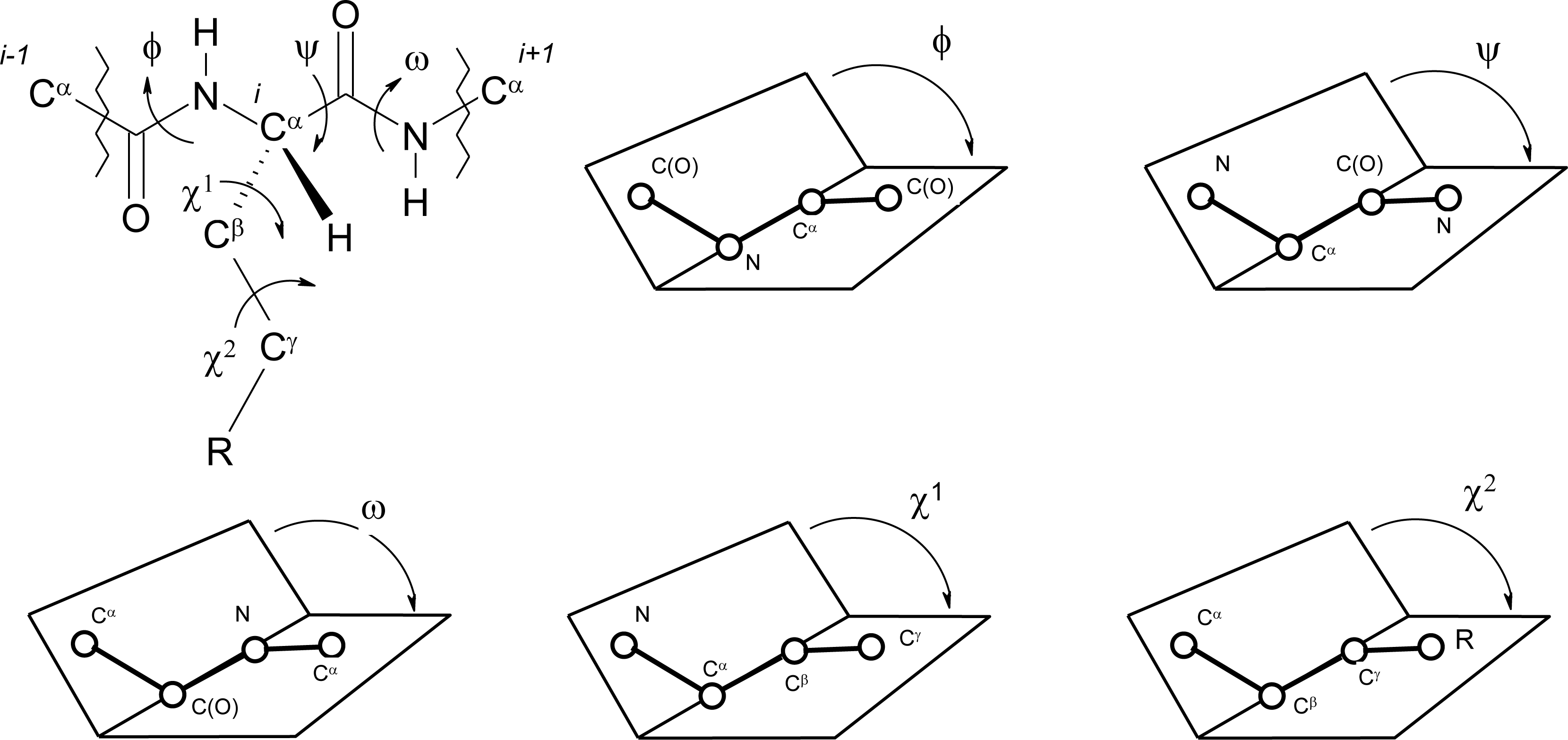
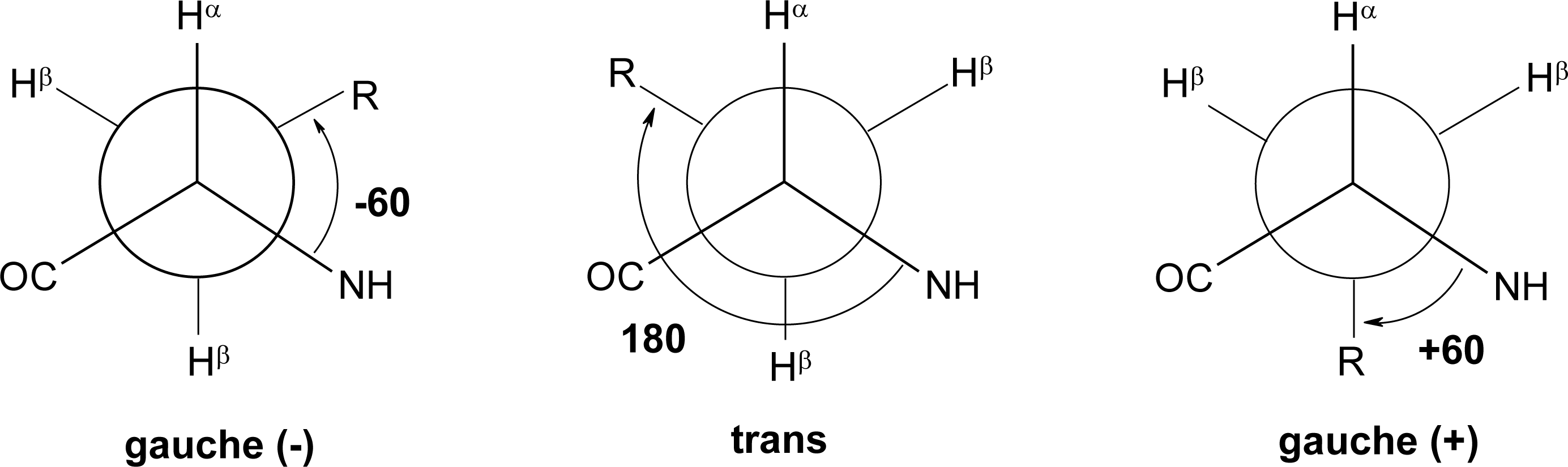
Peptide Dihedral Angles
2. Background
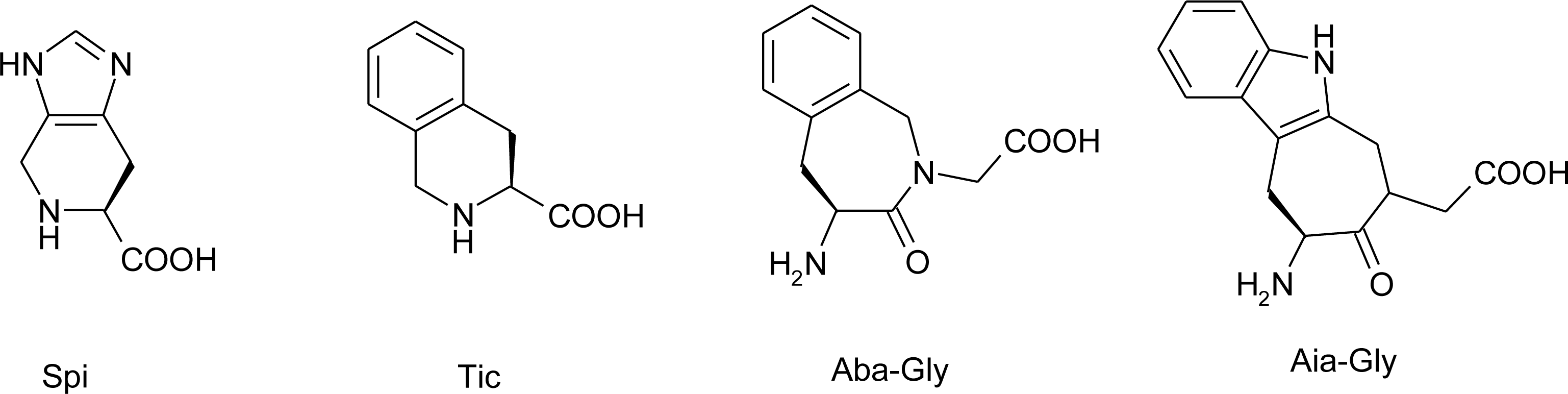
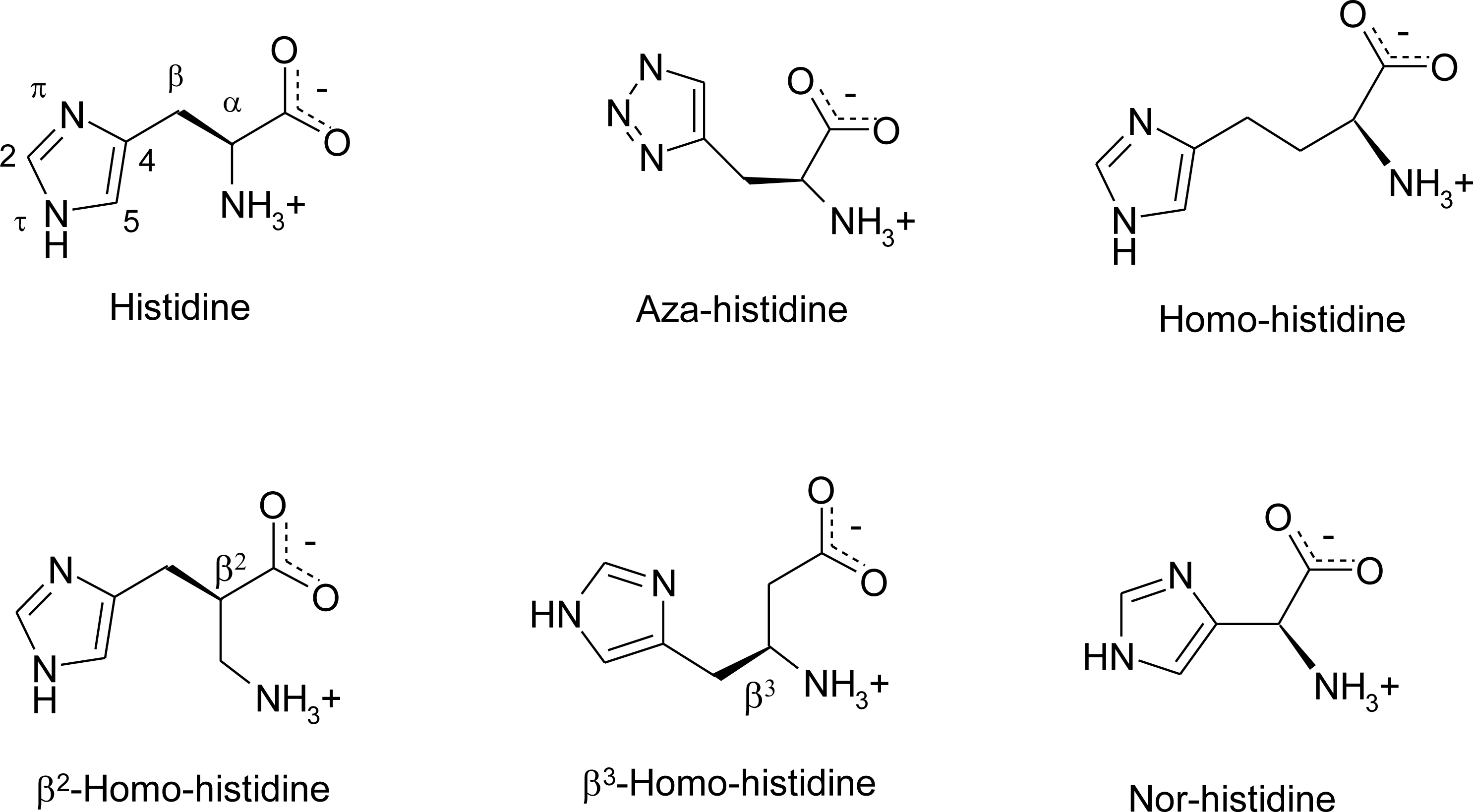
3. Nomenclature
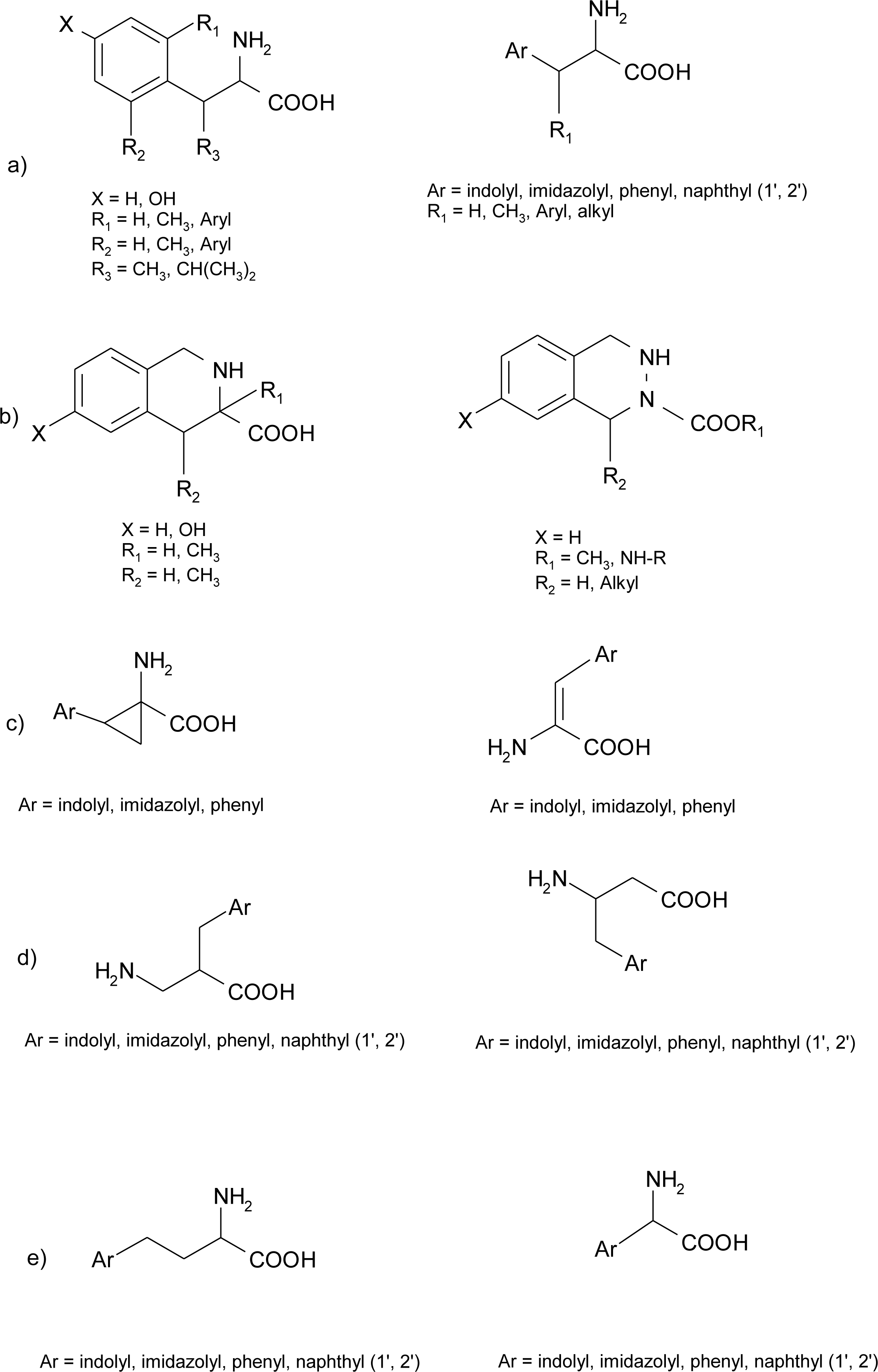
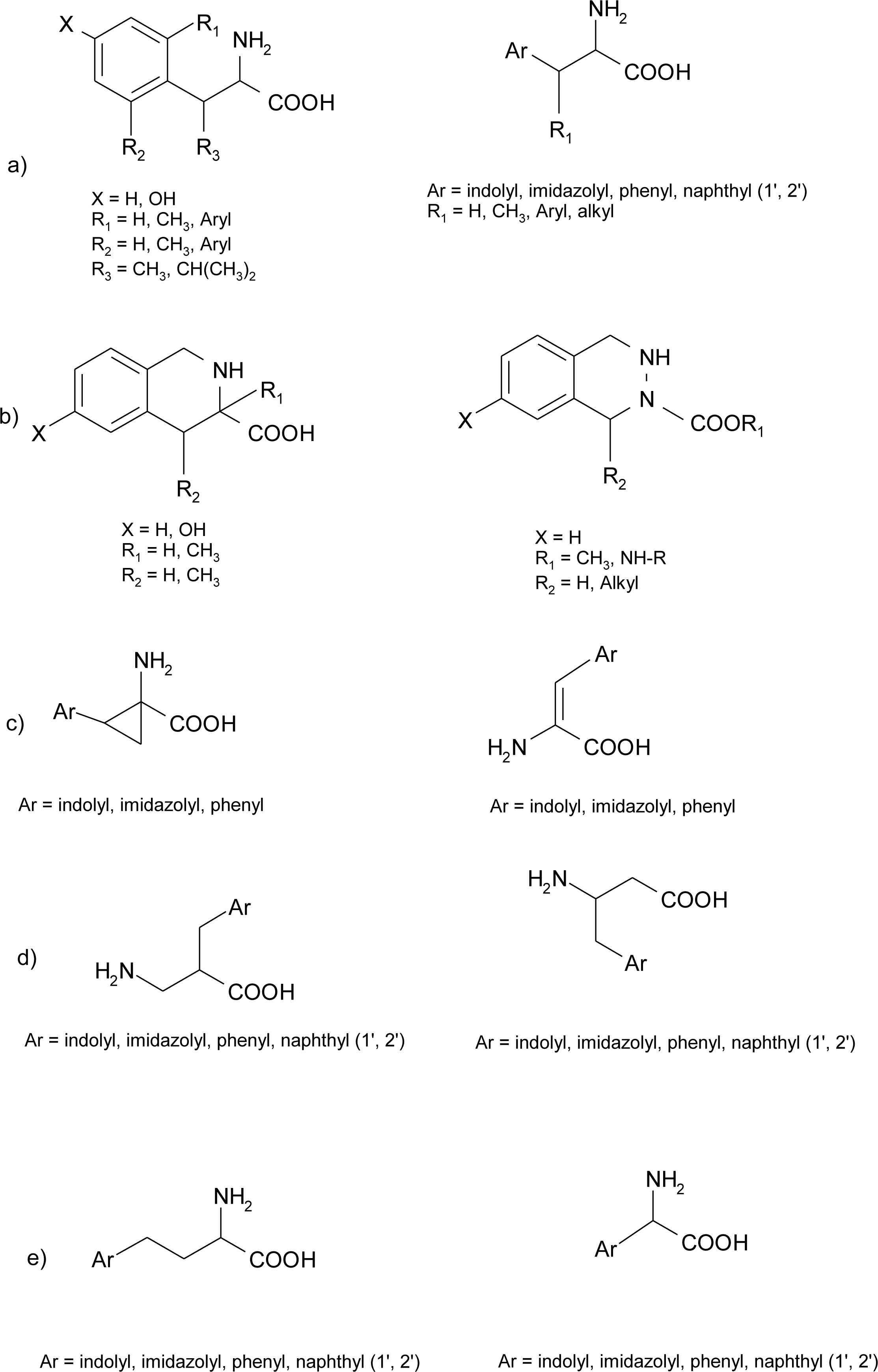
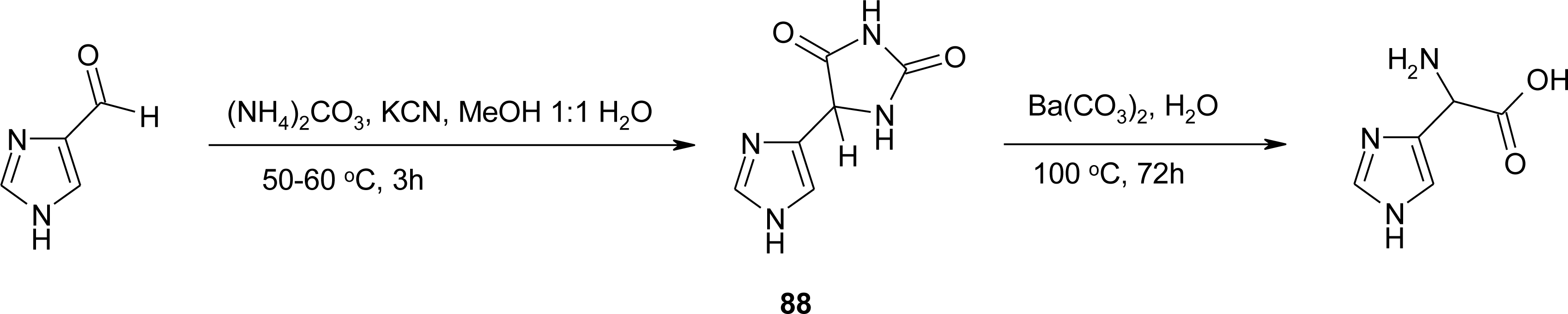
Histidine Derivatives
4. Chemistry
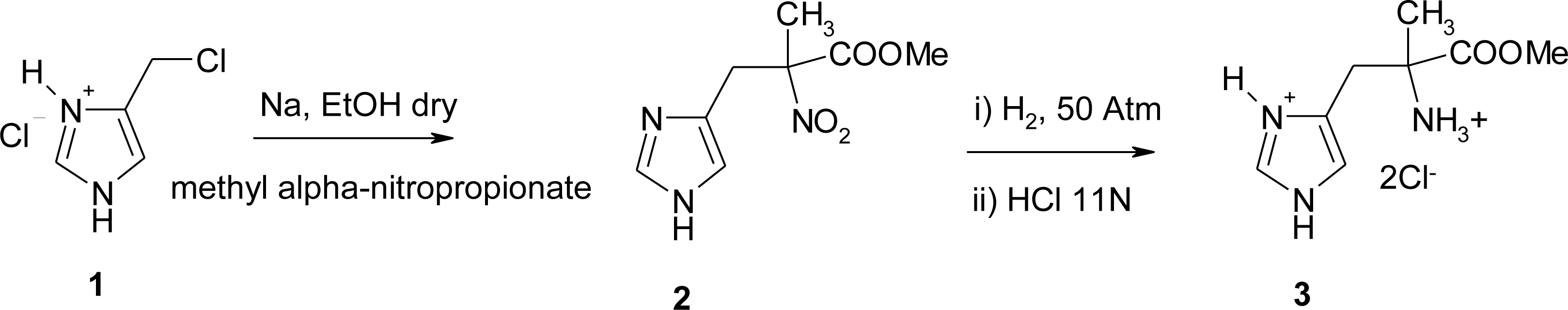
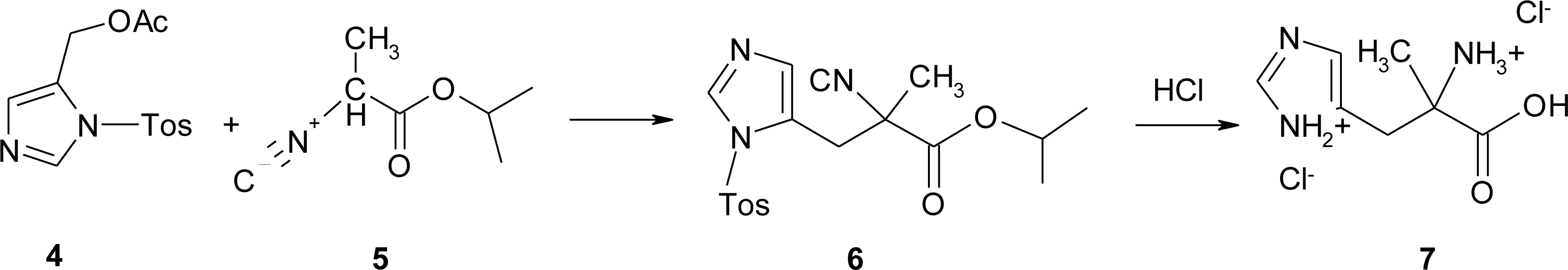
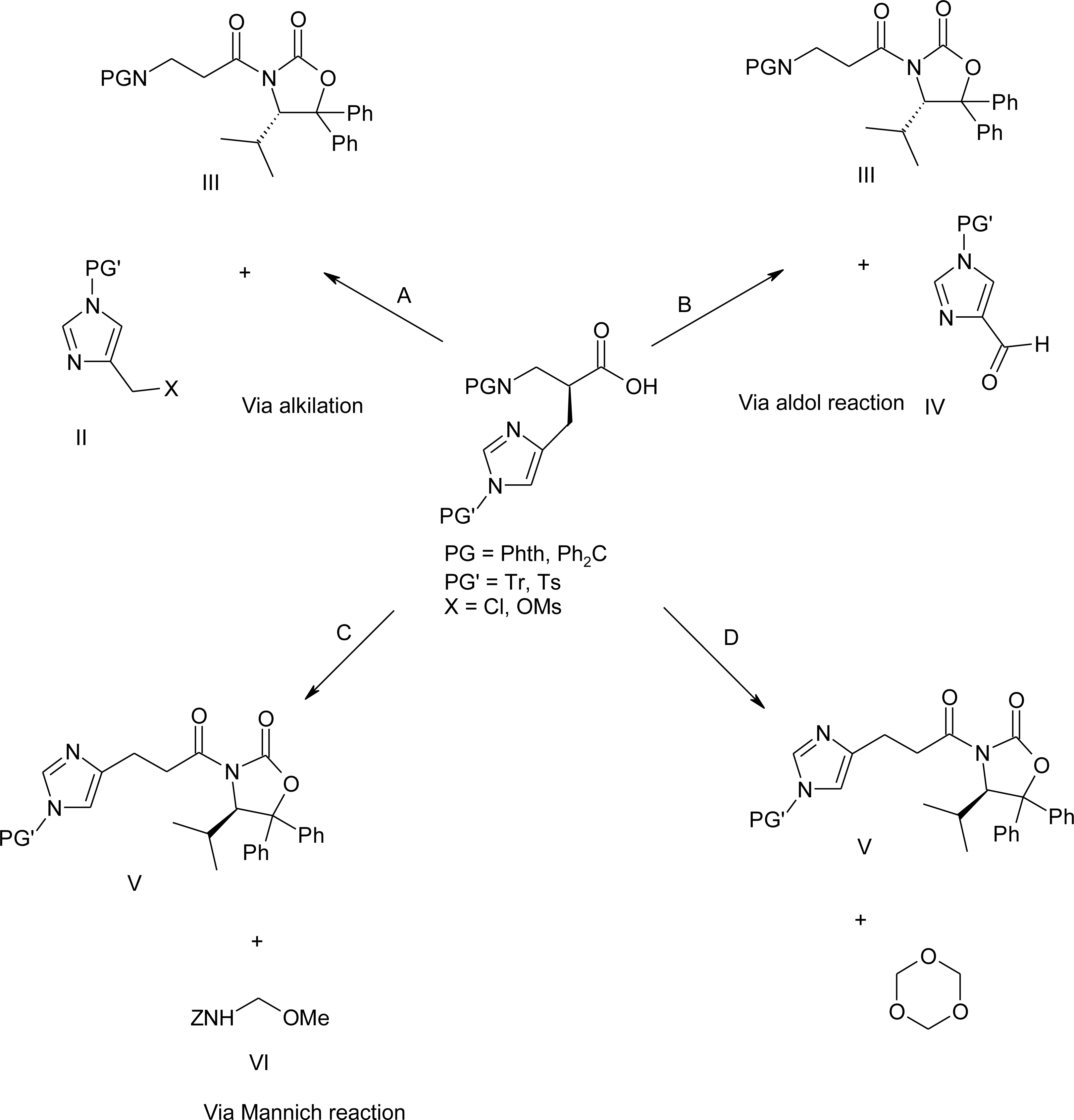
4.1. α-Alkyl Substitution
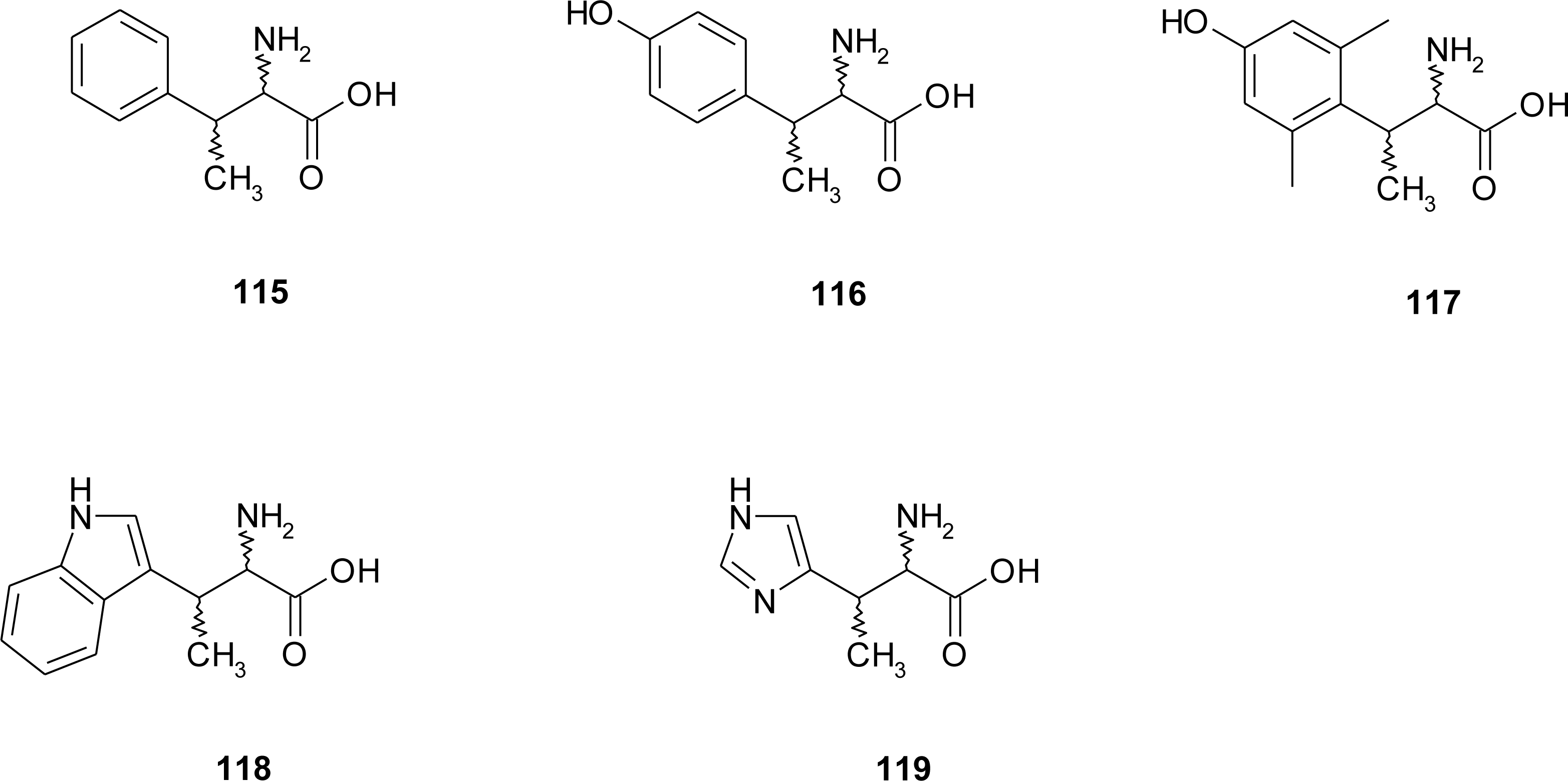
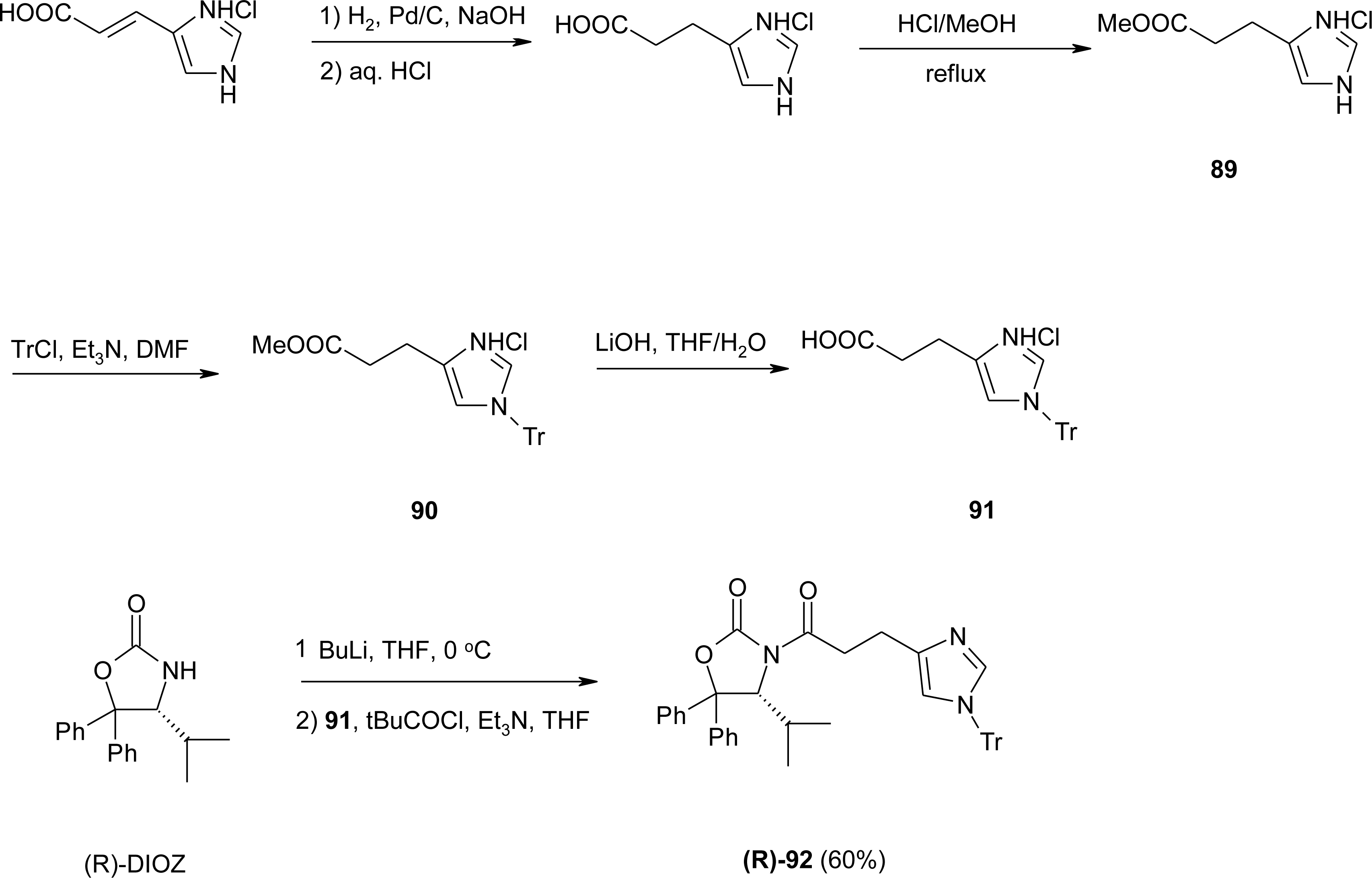
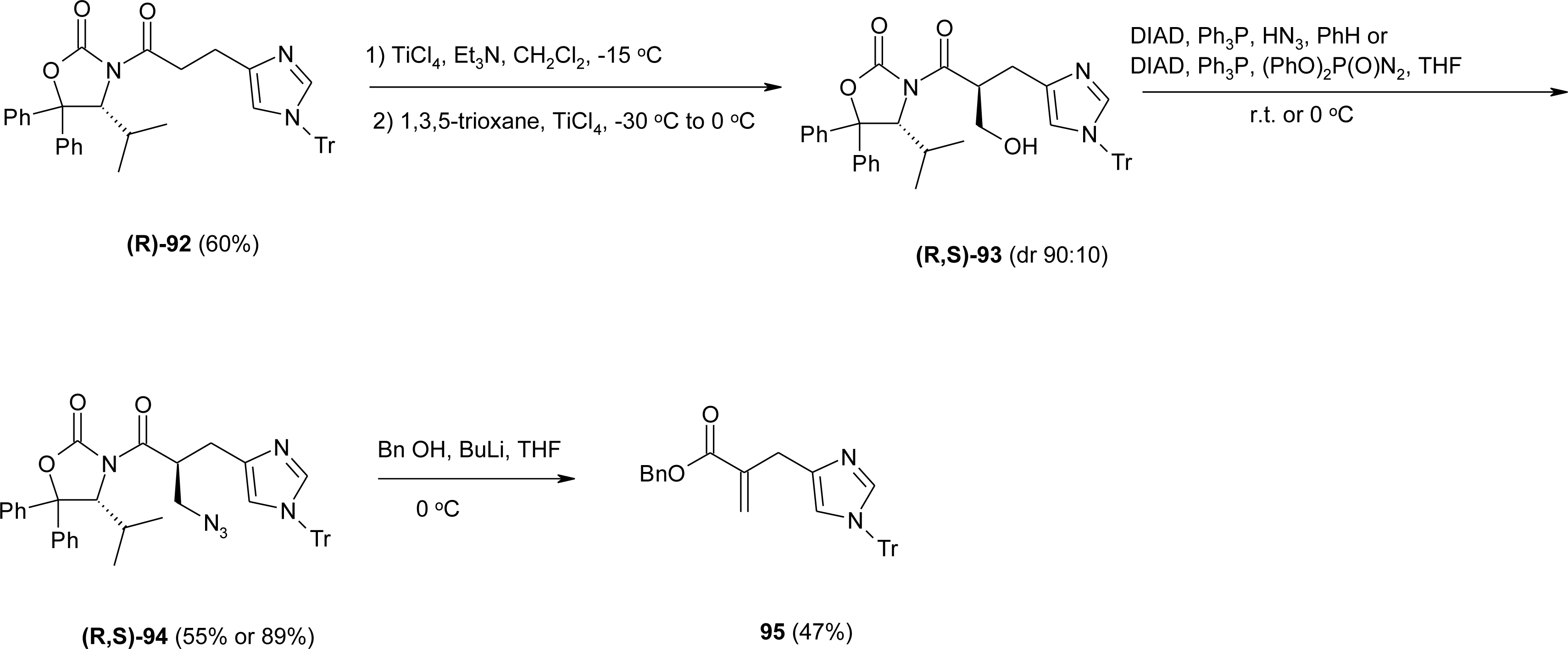
4.2. β-Alkyl Substitutions
4.3. β,β–Dimethyl Substitution
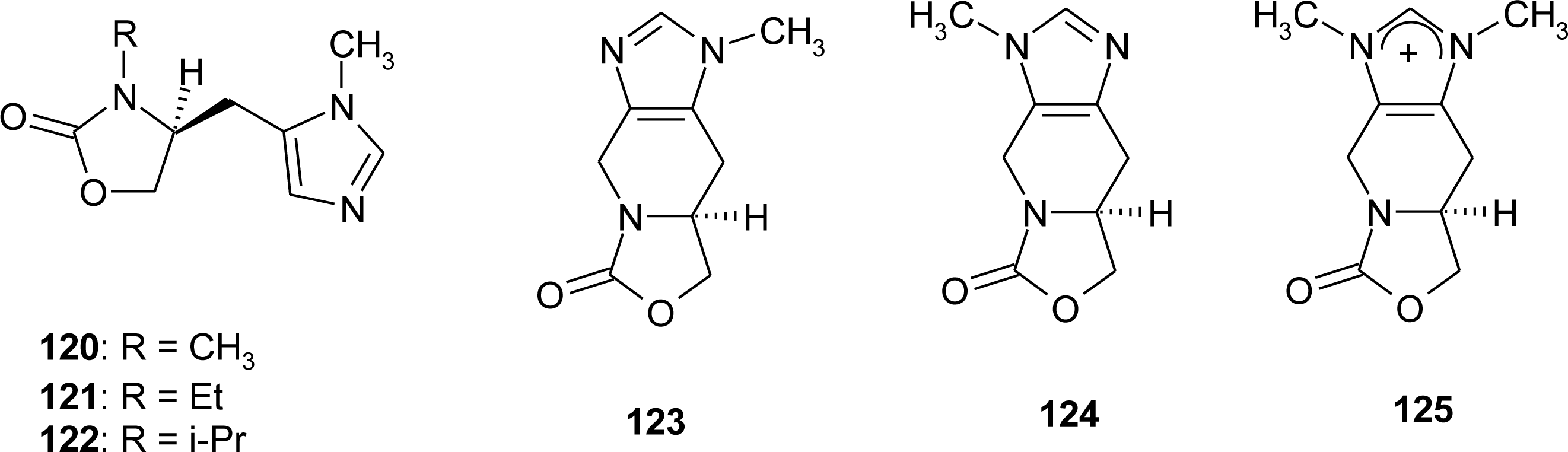
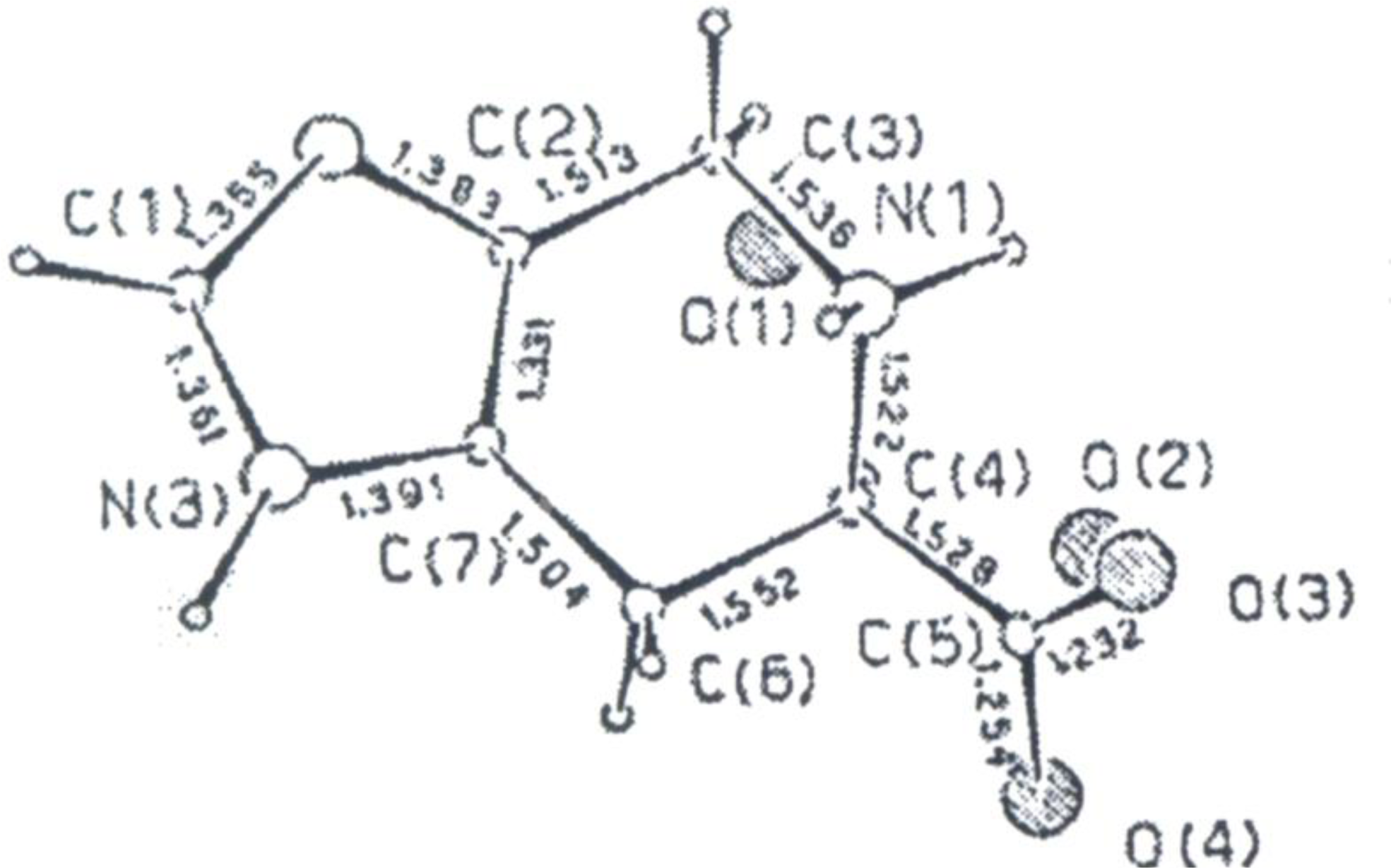
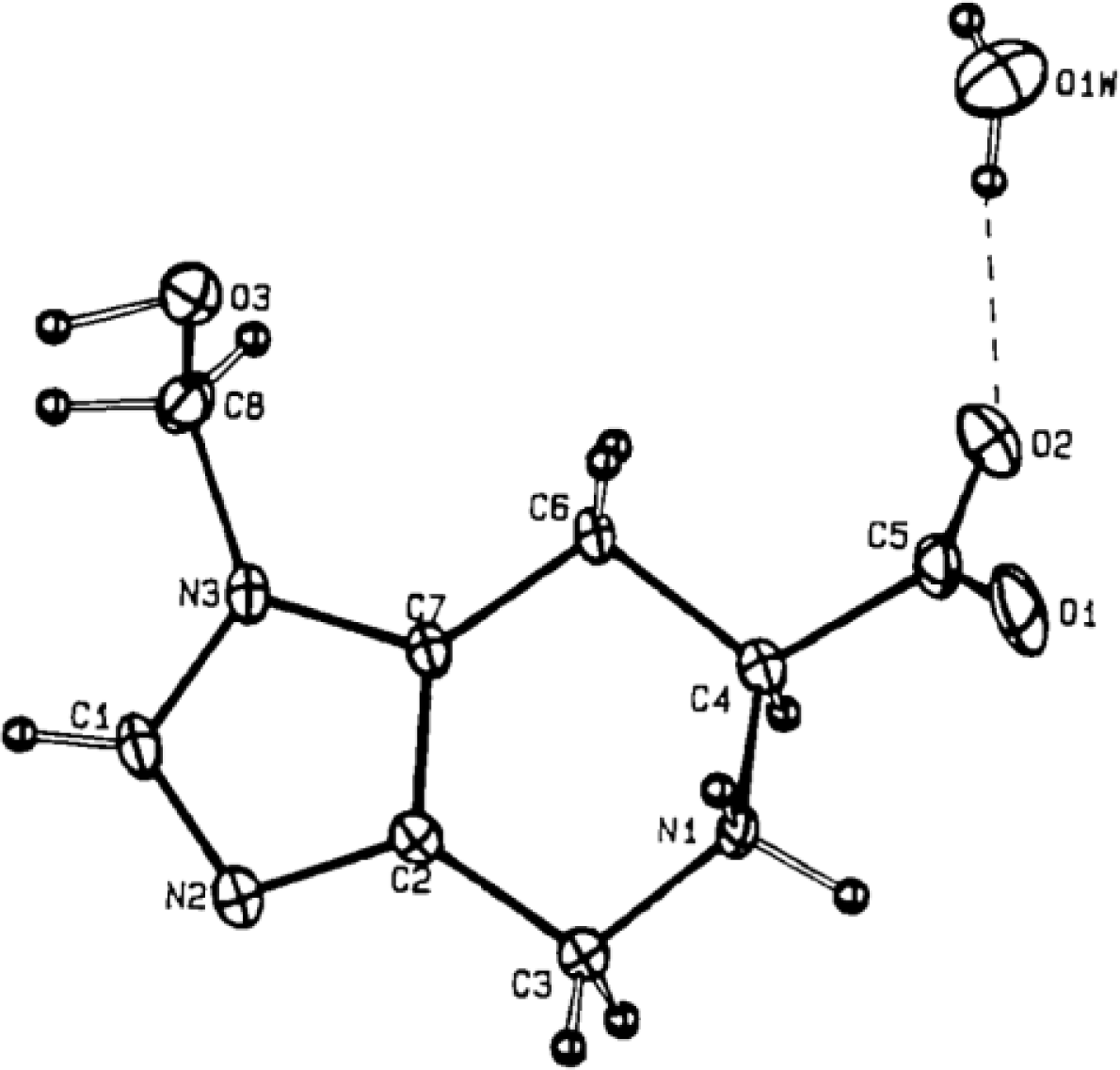
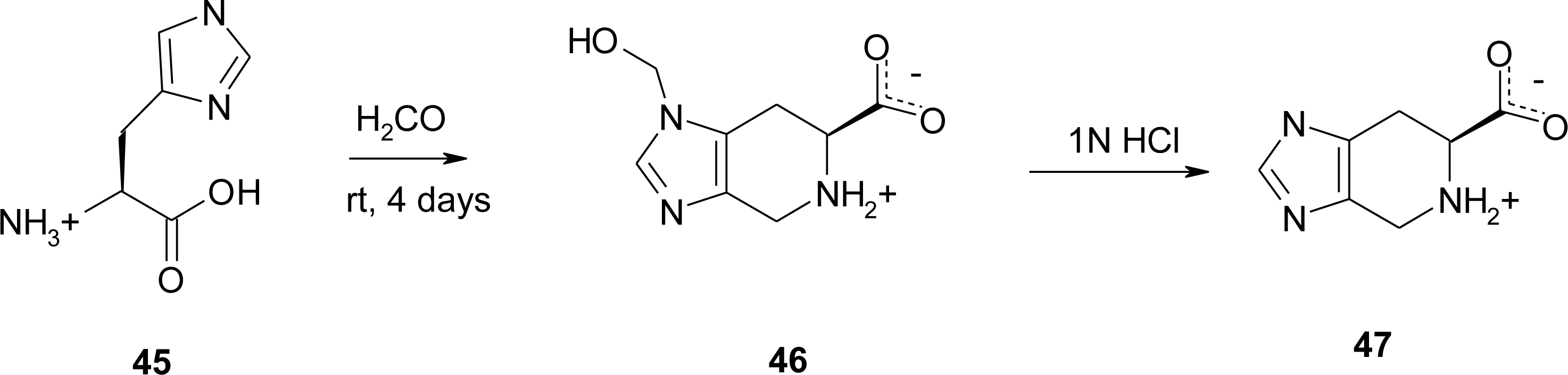
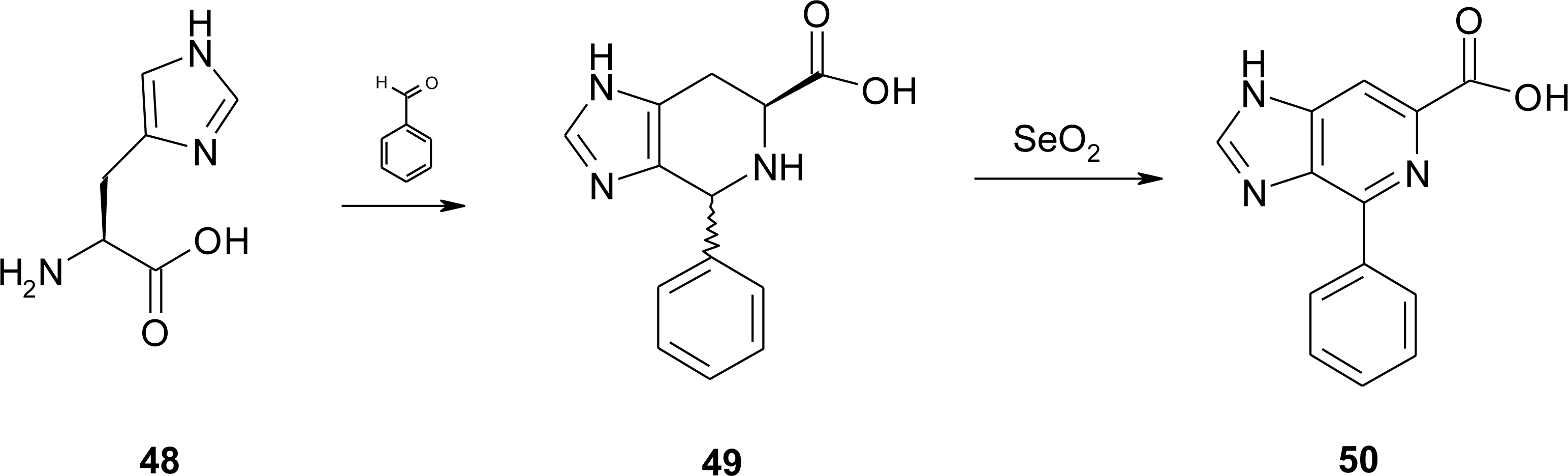
4.4. Constrained “Imino Acids” 1,2,3,4-Tetrahydroquinoline Derivatives (Spinacines)
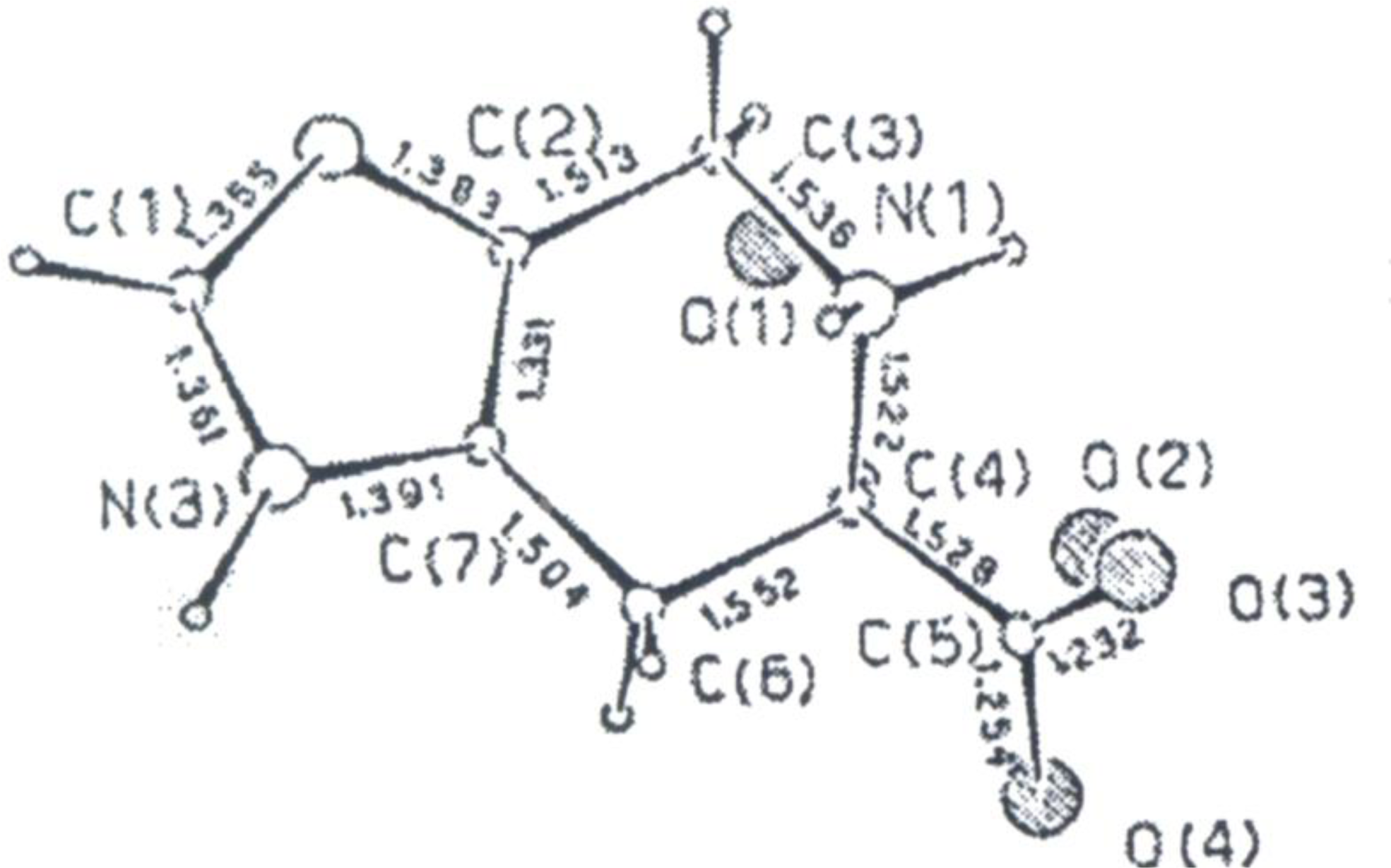
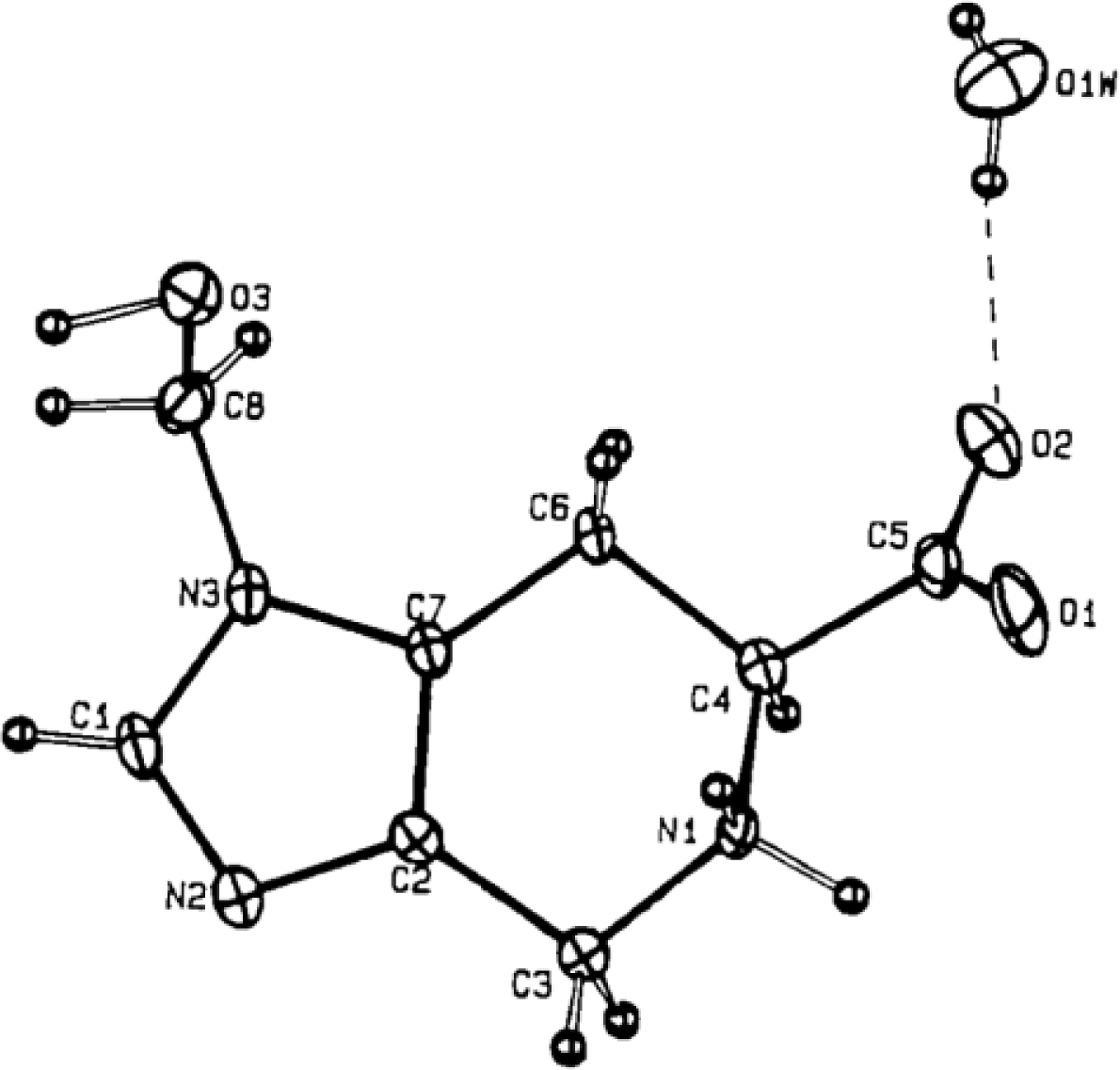
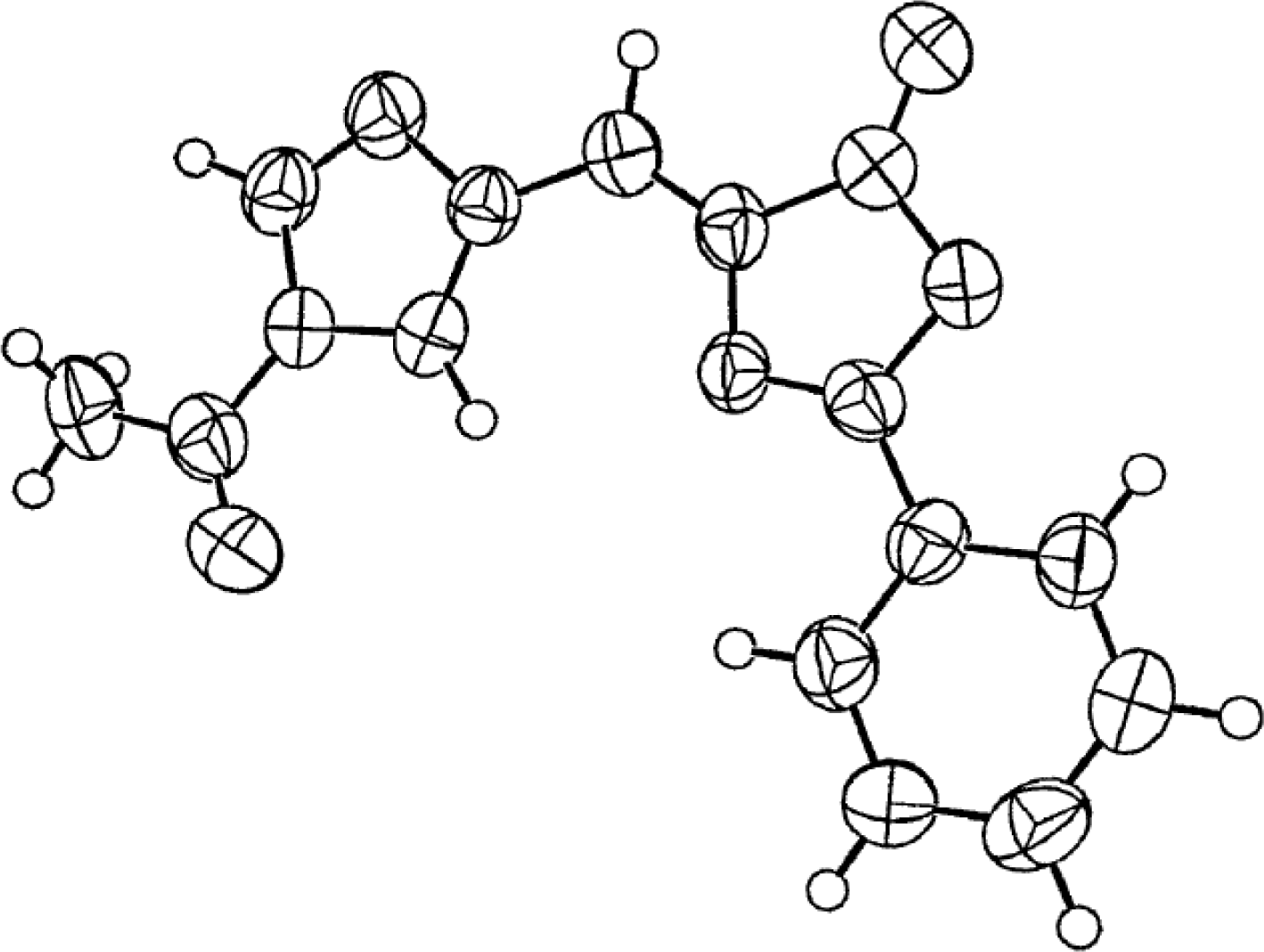
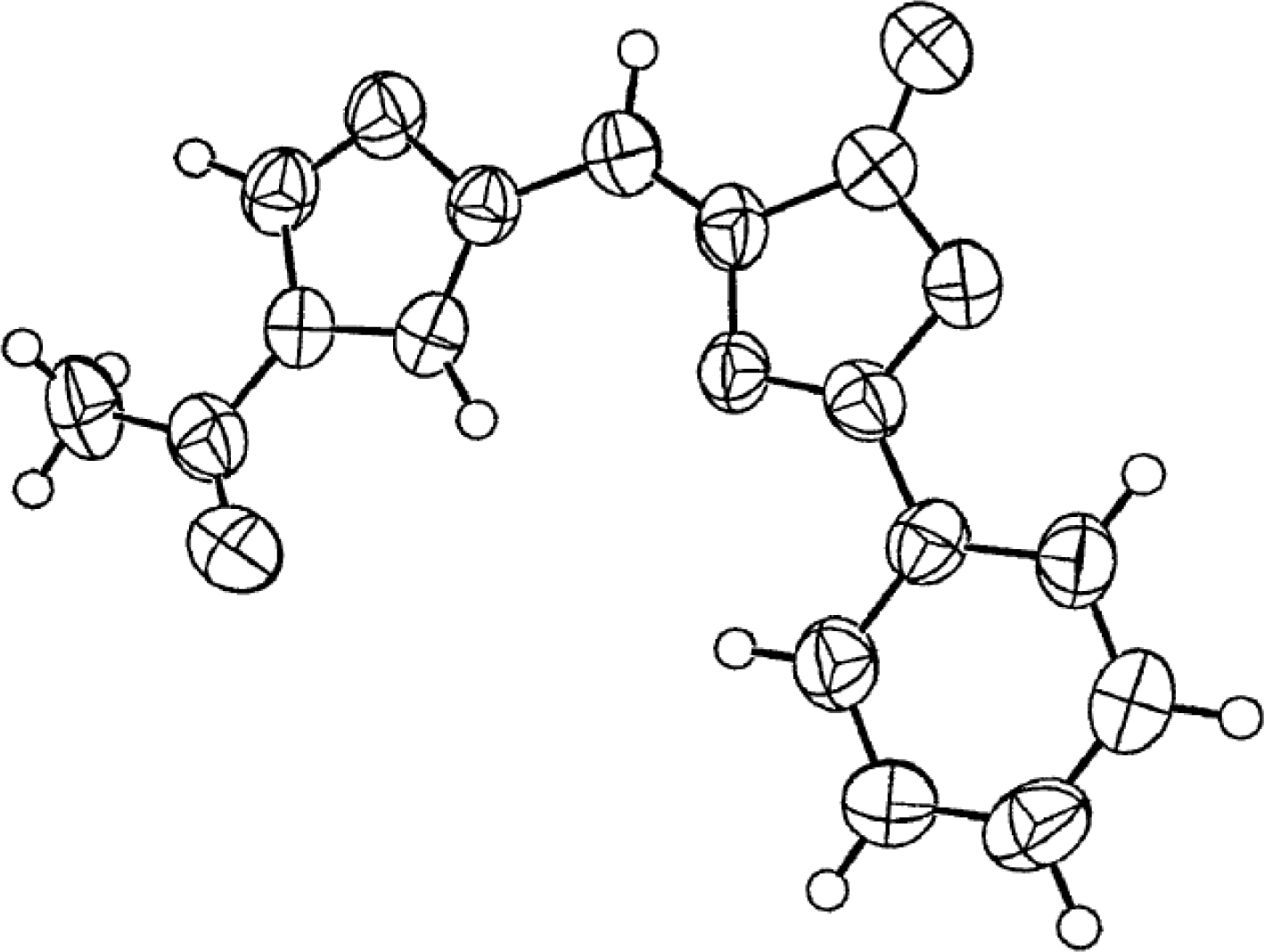
X-ray Studies on Spinacine Derivatives
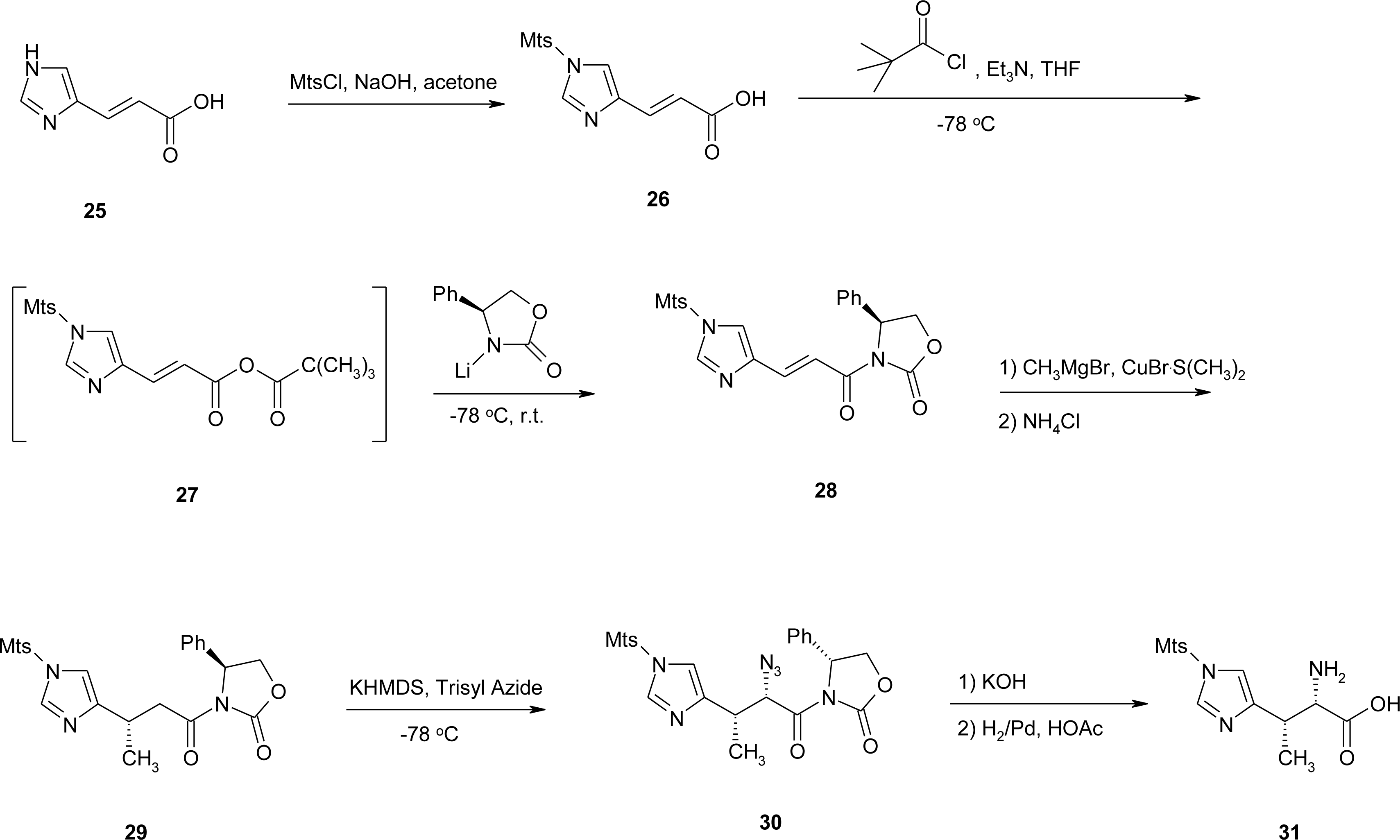
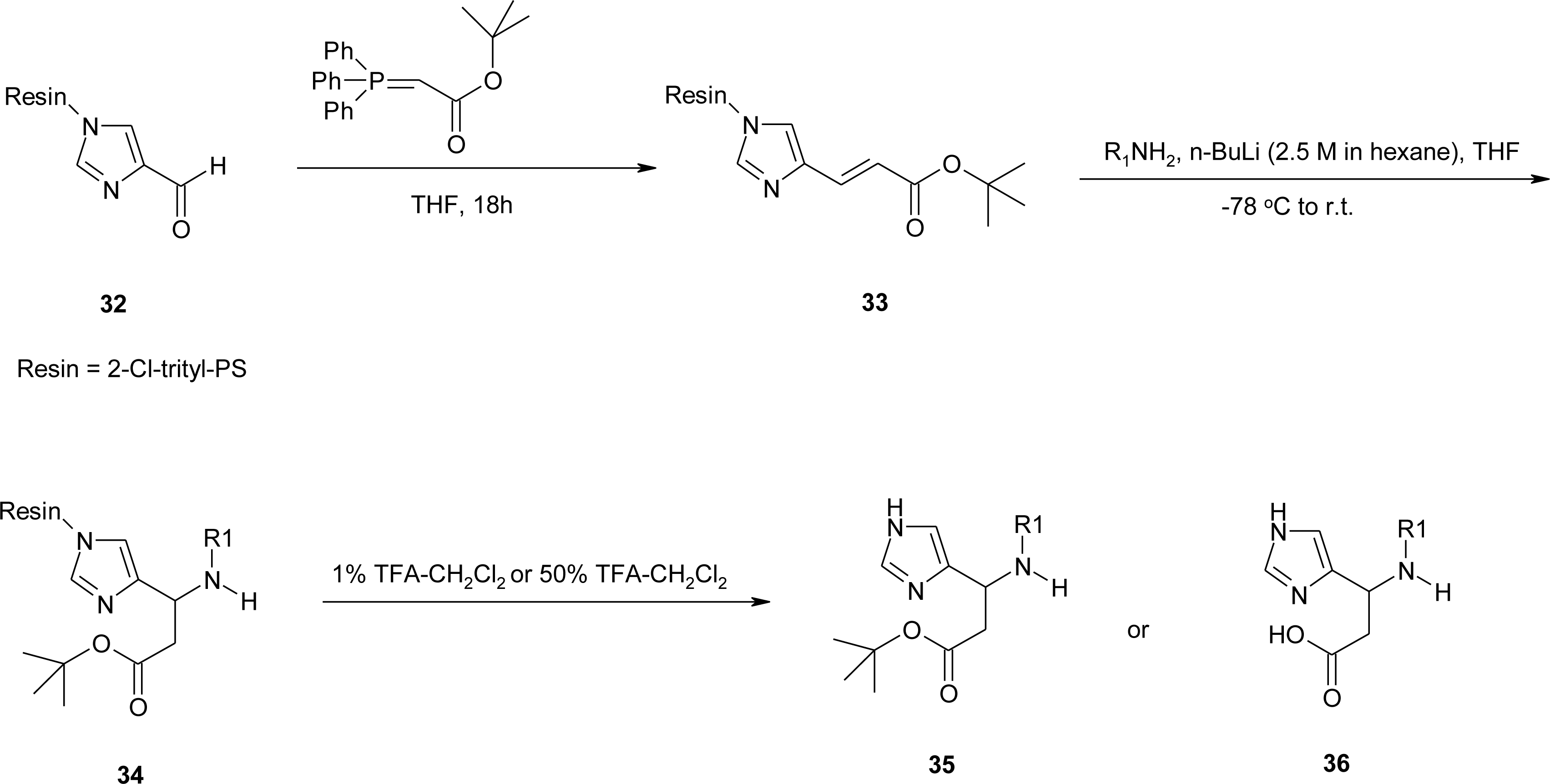
4.5. 1-Amino-2-(4-imidazolyl)cyclopropanecarboxylic Acid Derivatives (ACC)
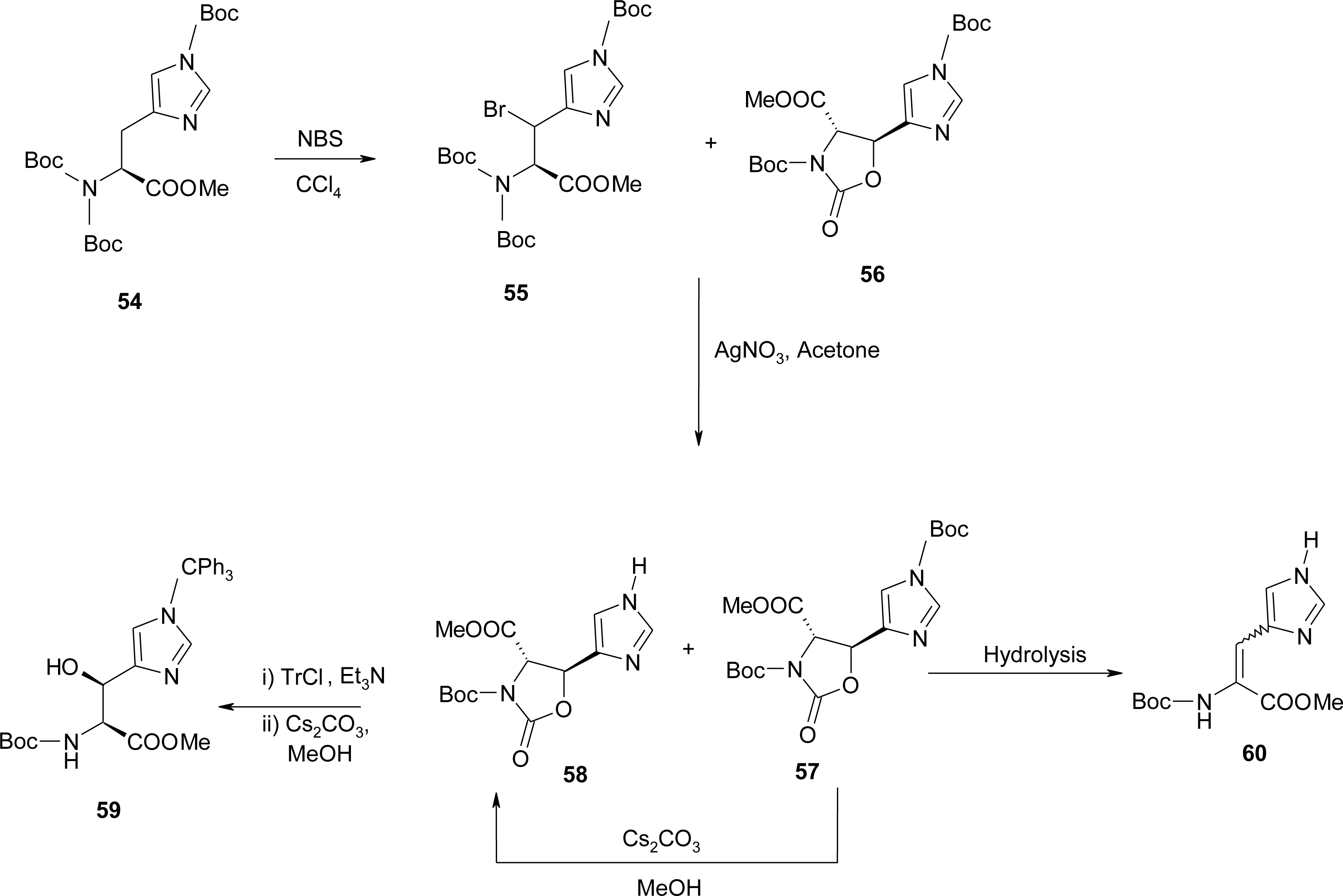
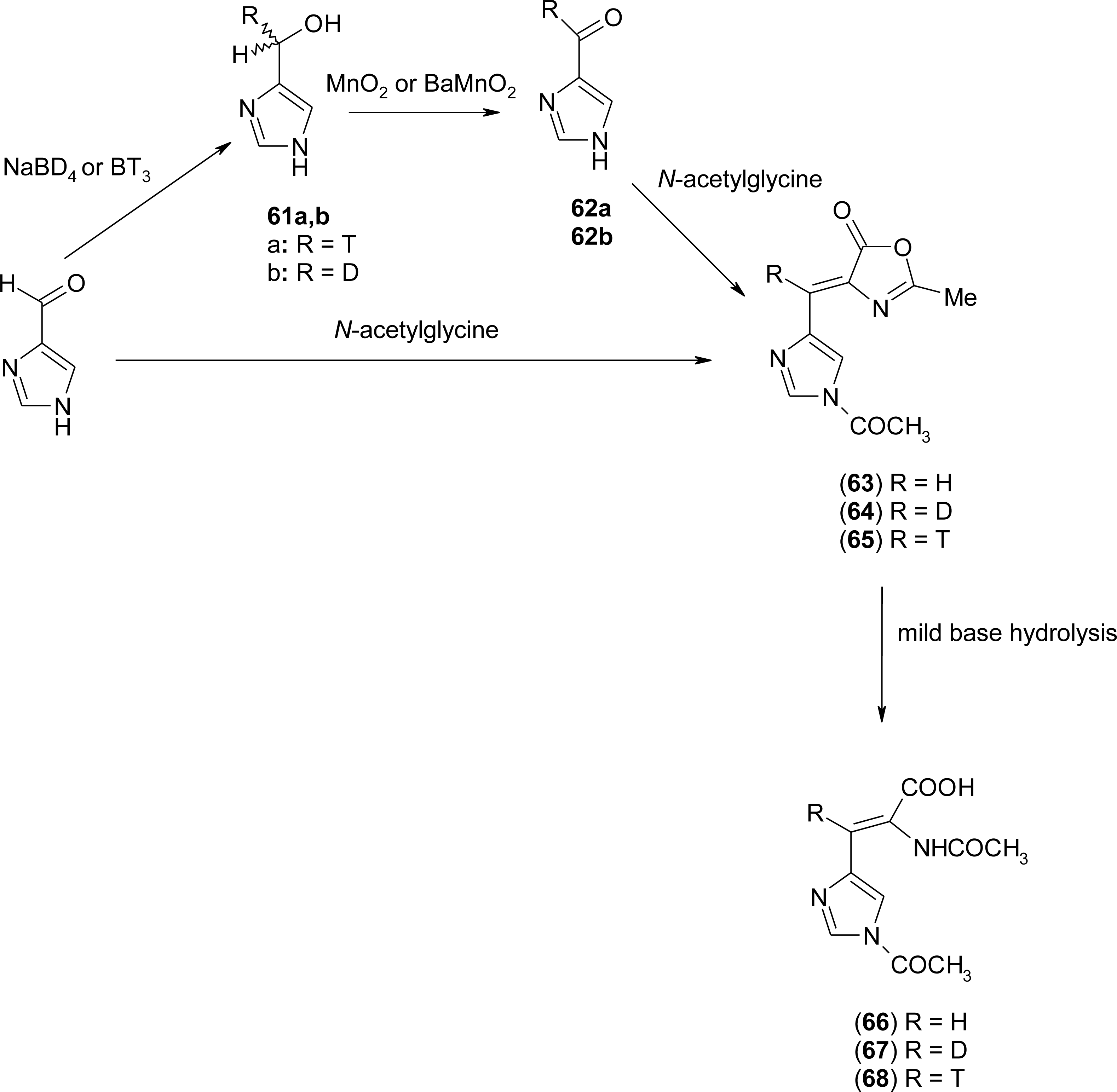
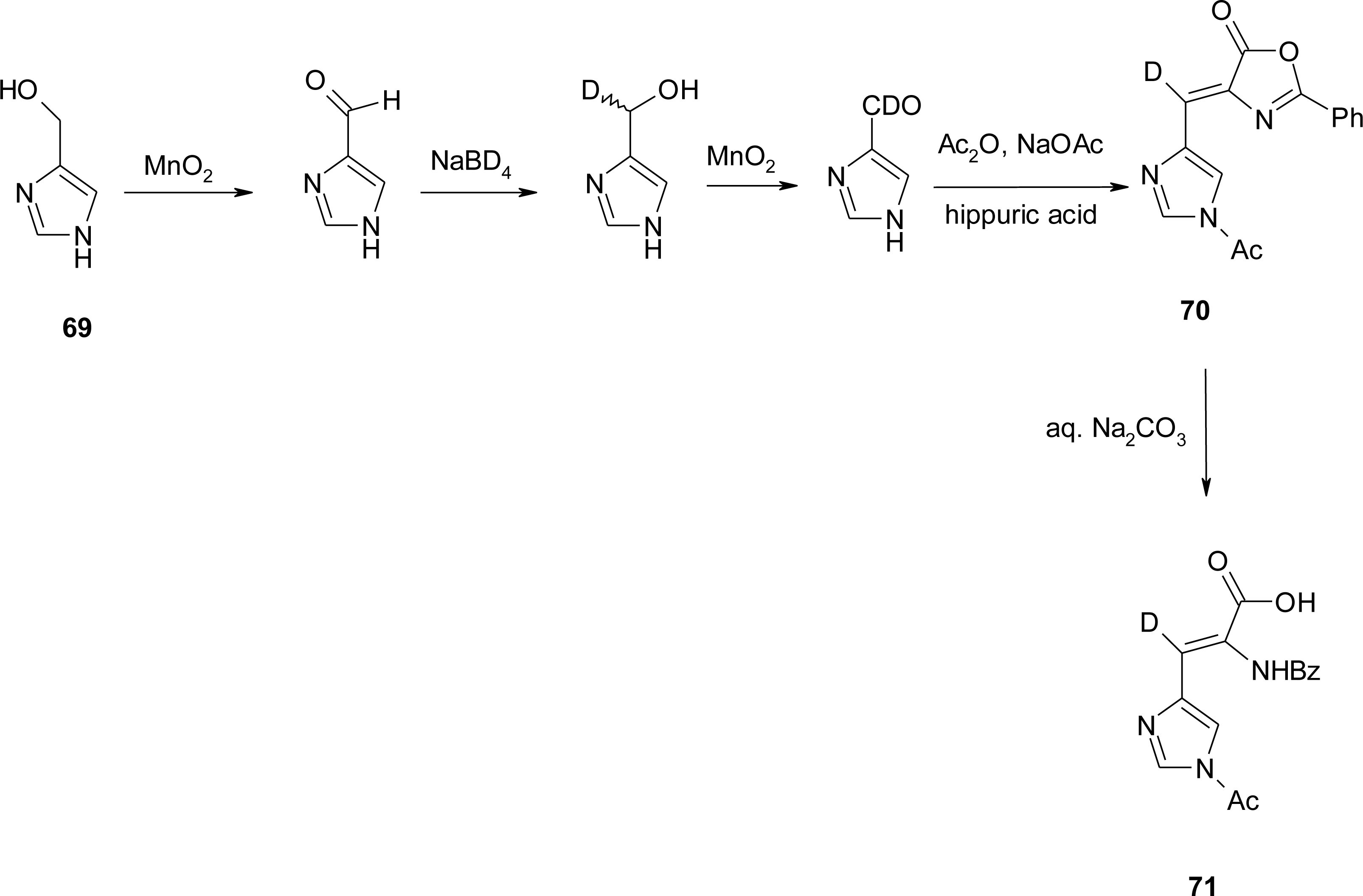
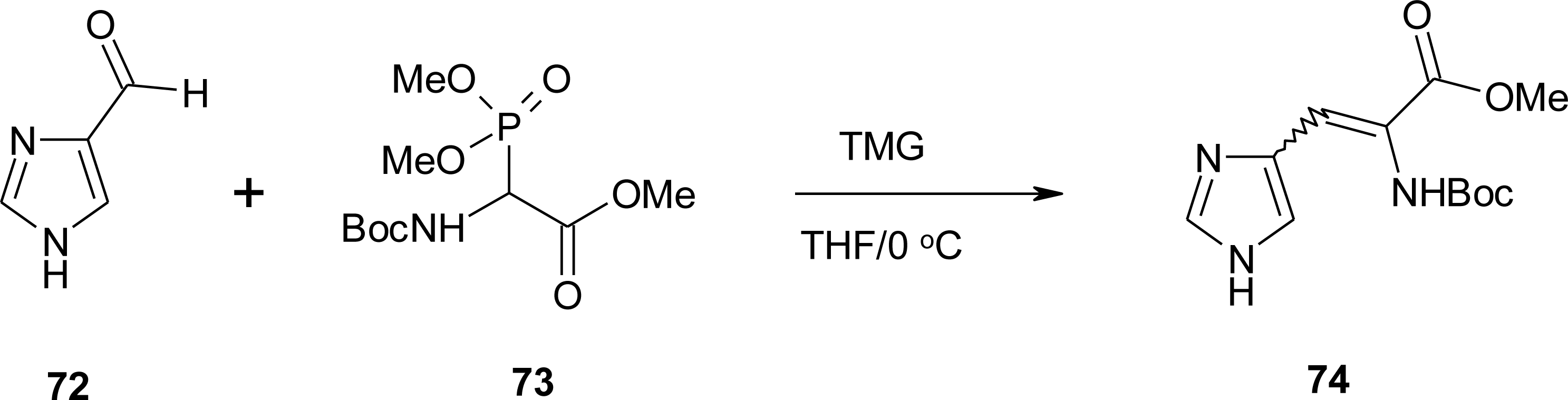
4.6. α,β-Dehydro Amino Acids
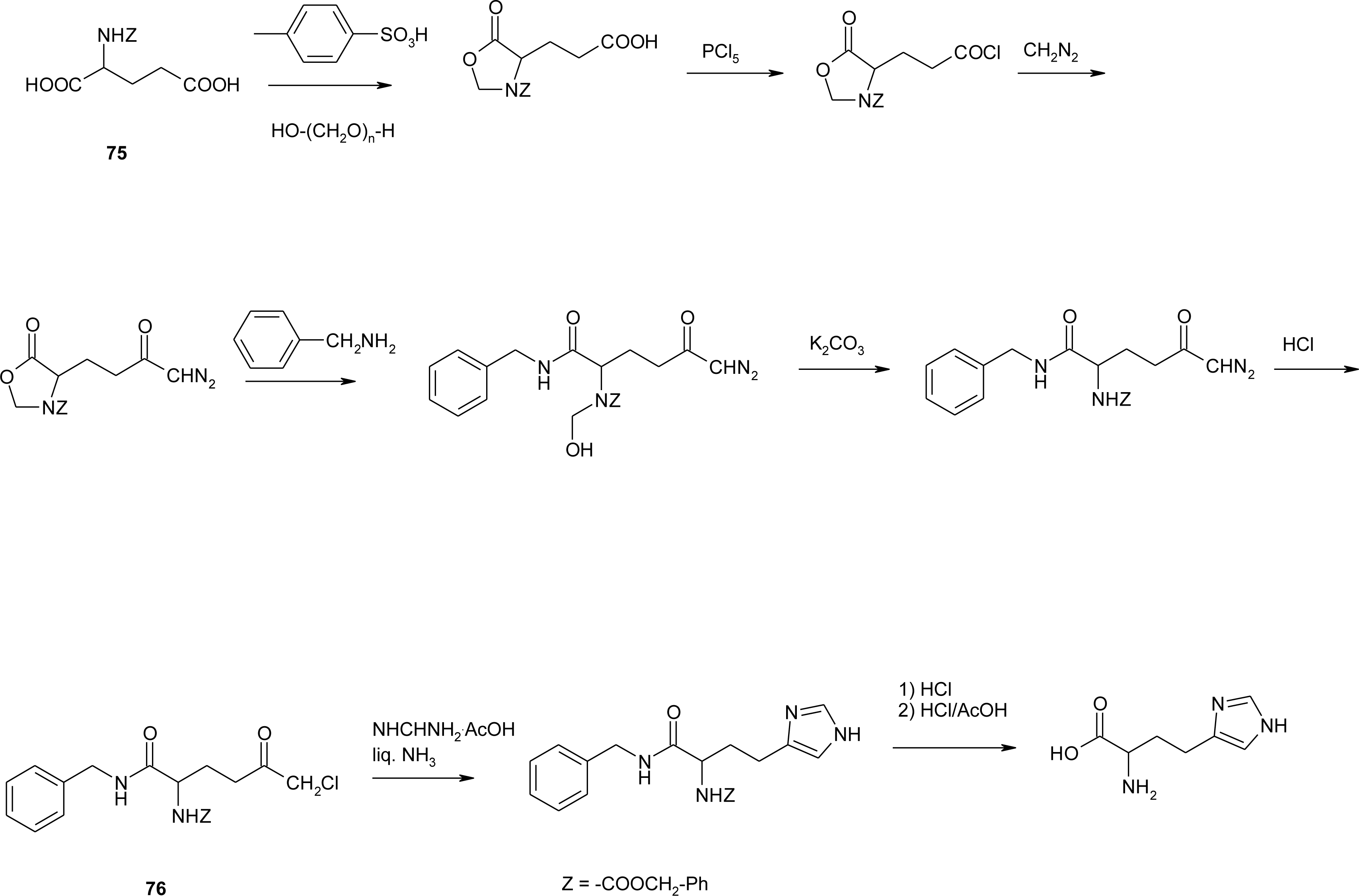
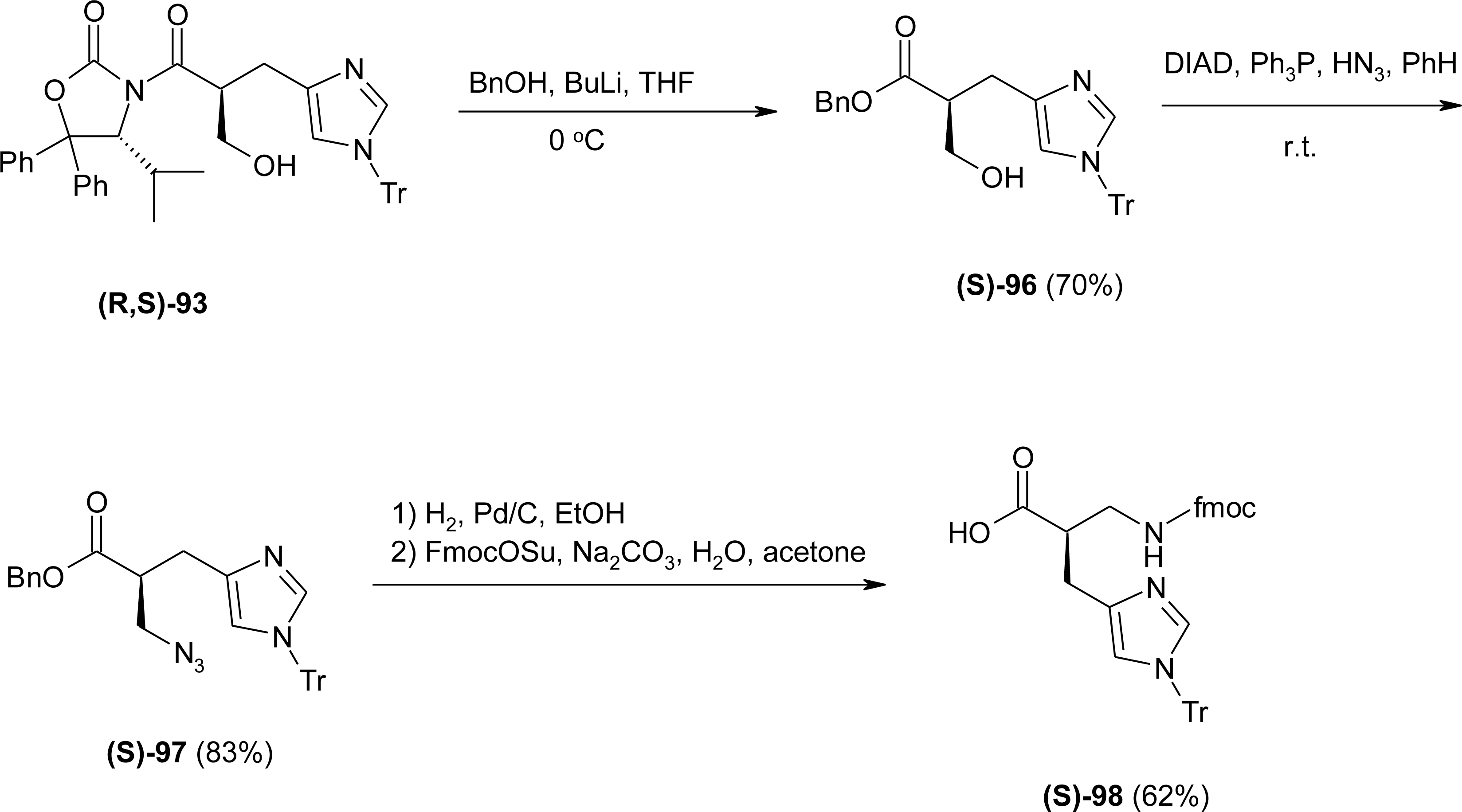
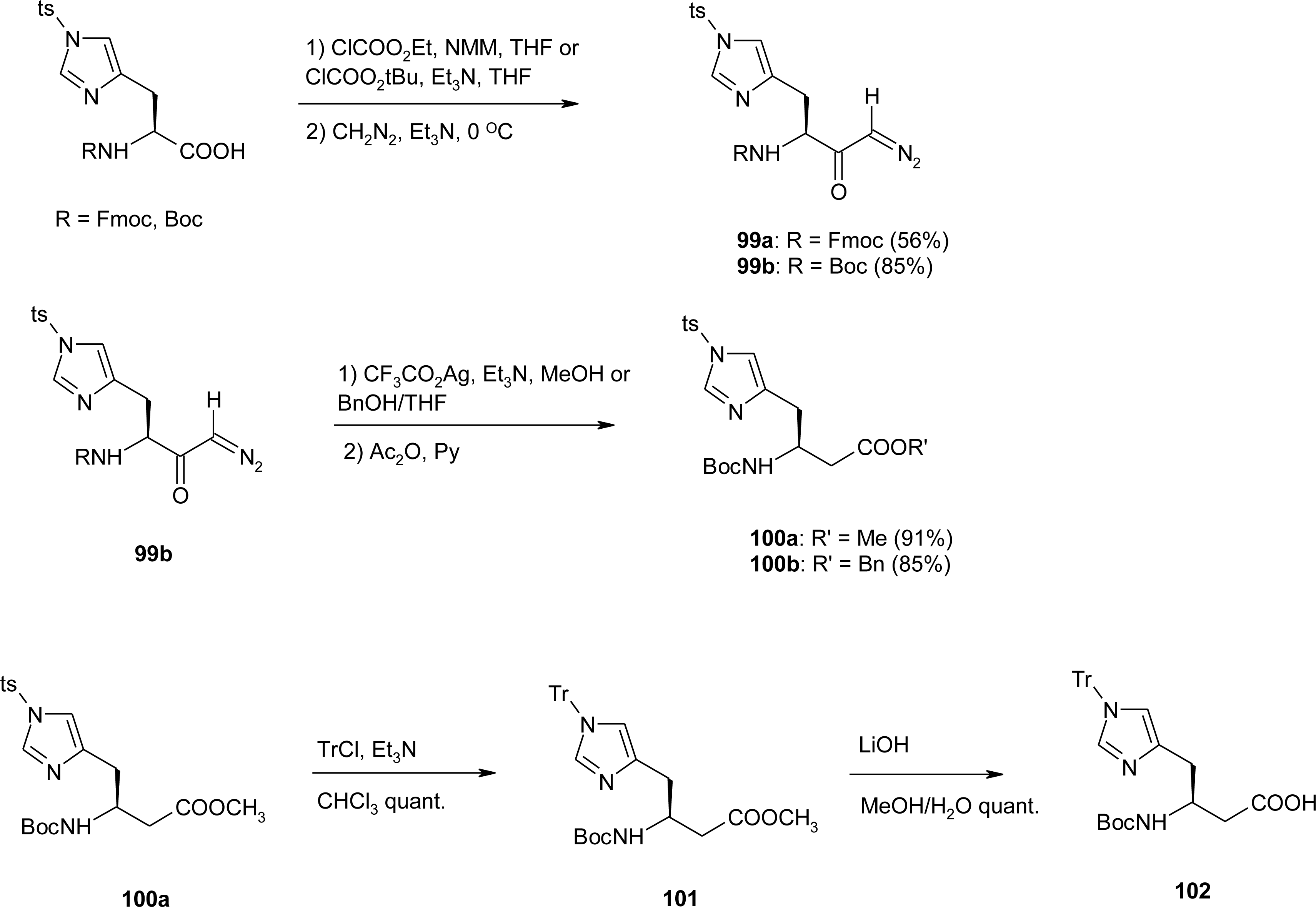
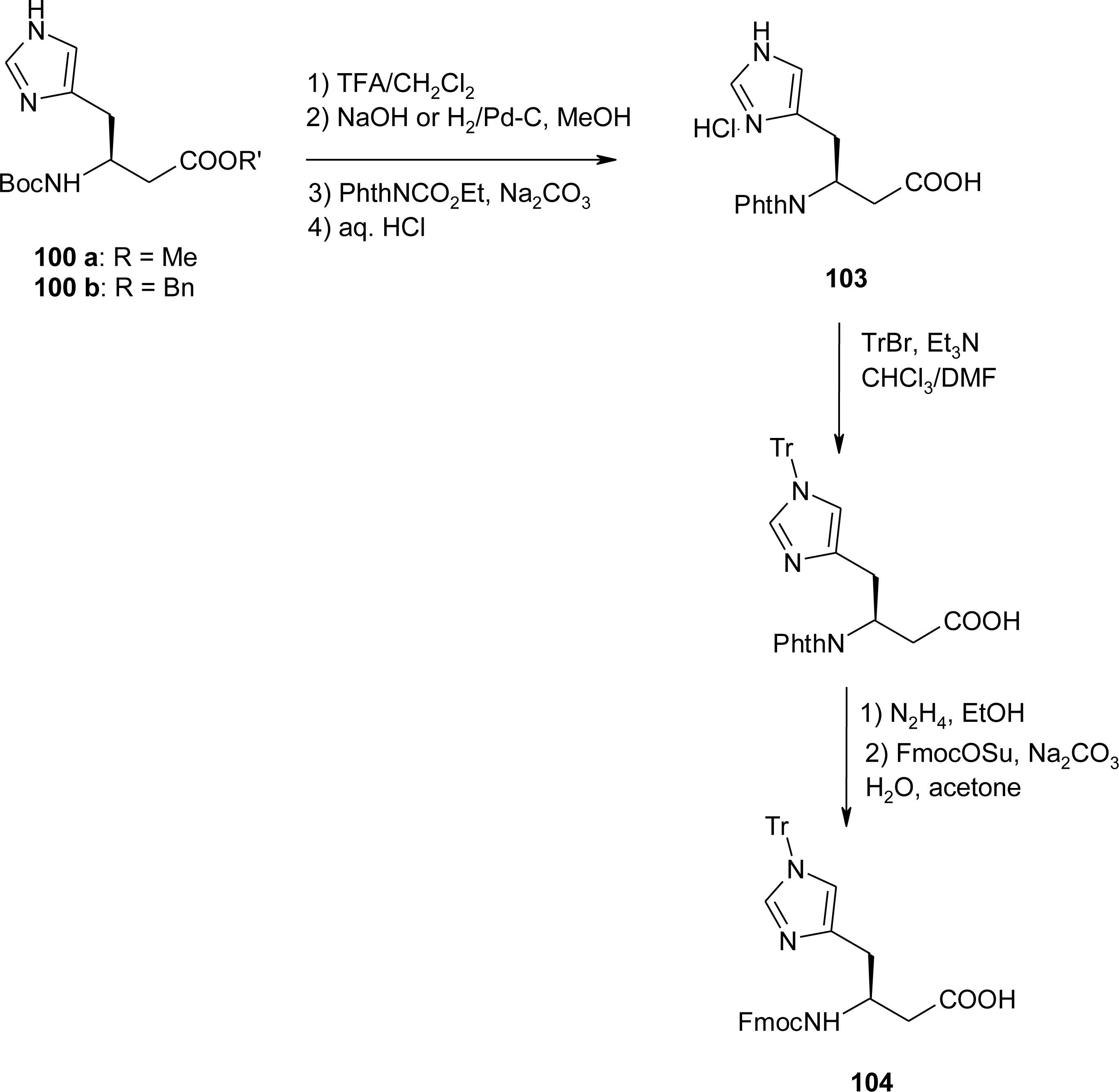
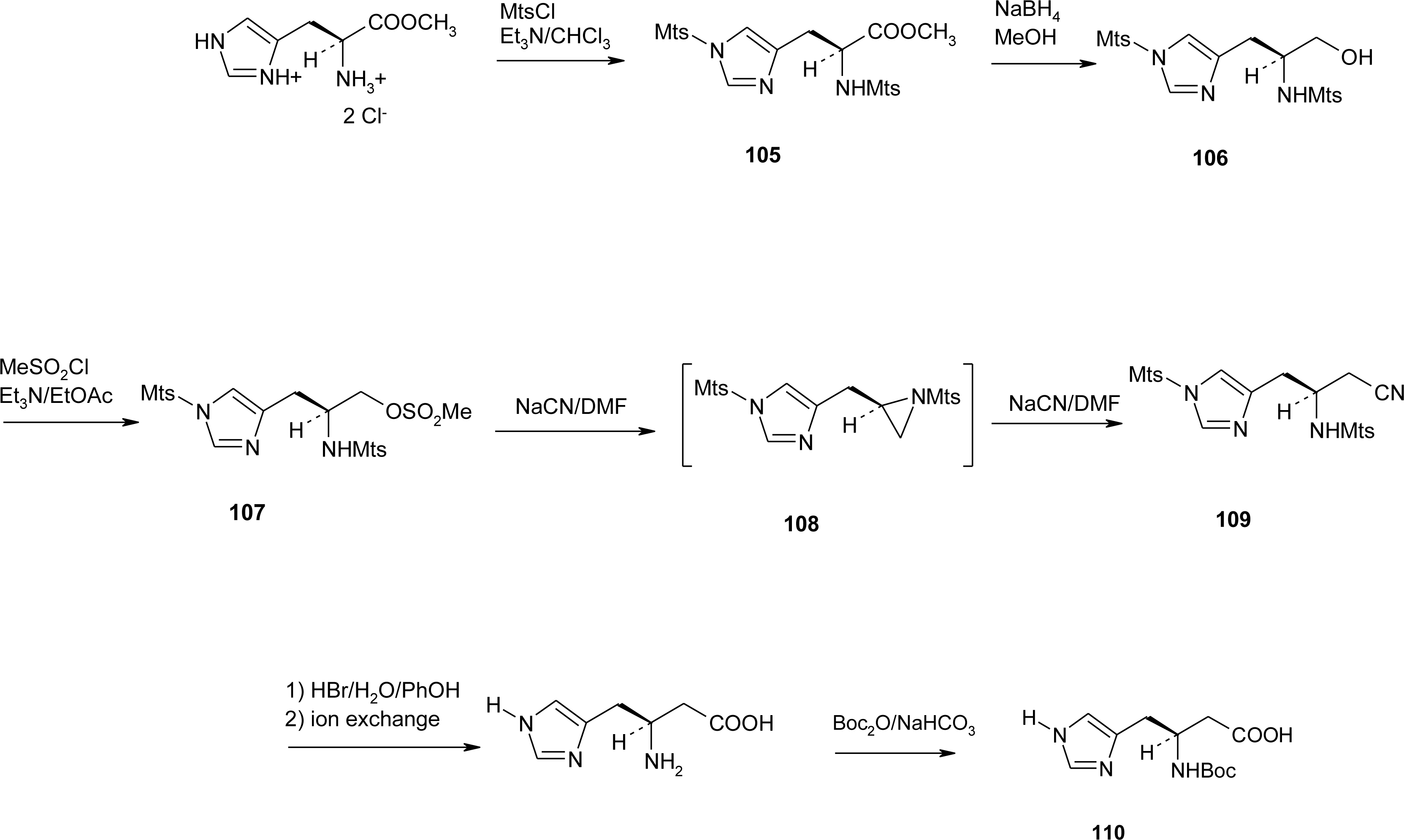
4.7. Homo-Histidine
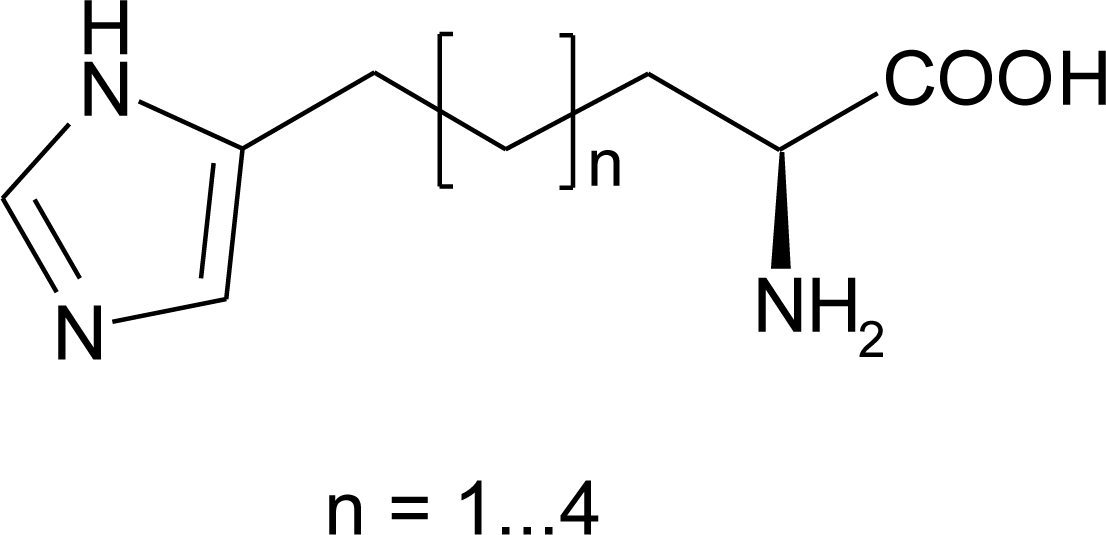
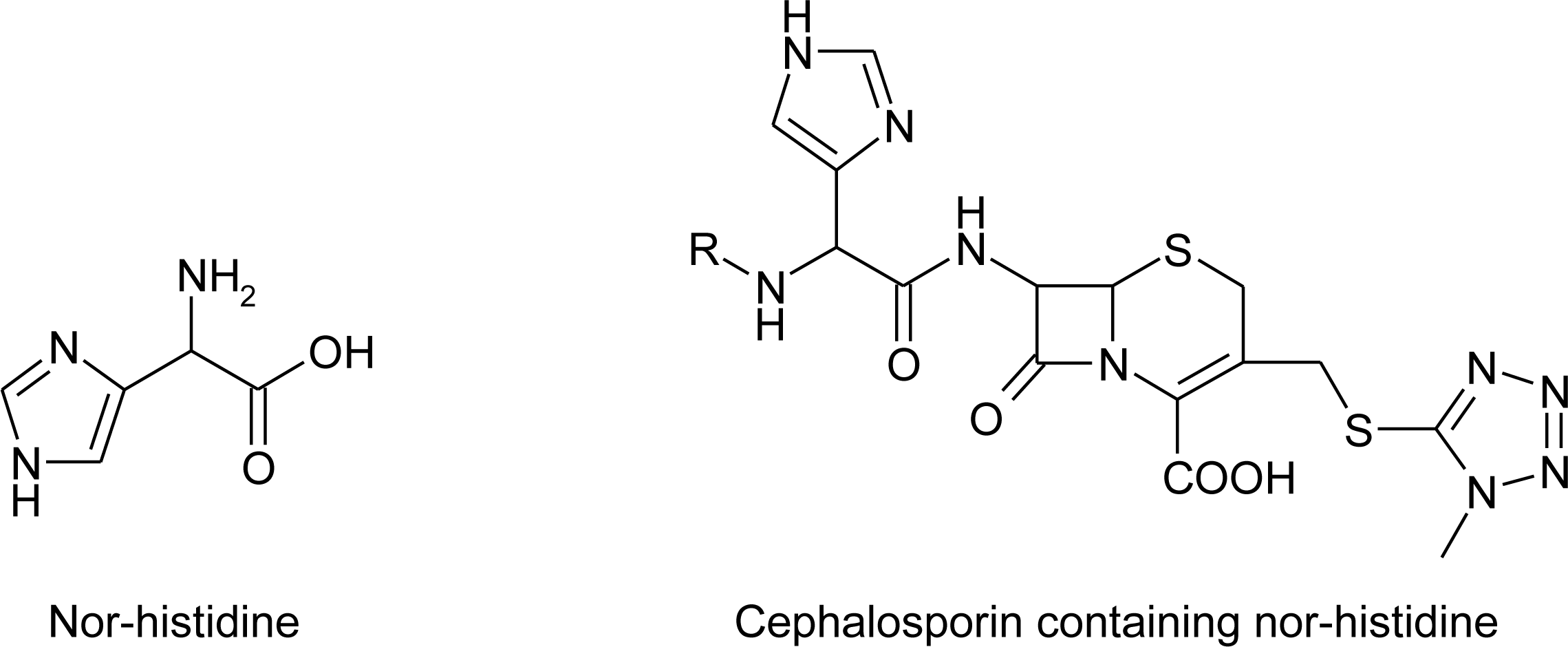
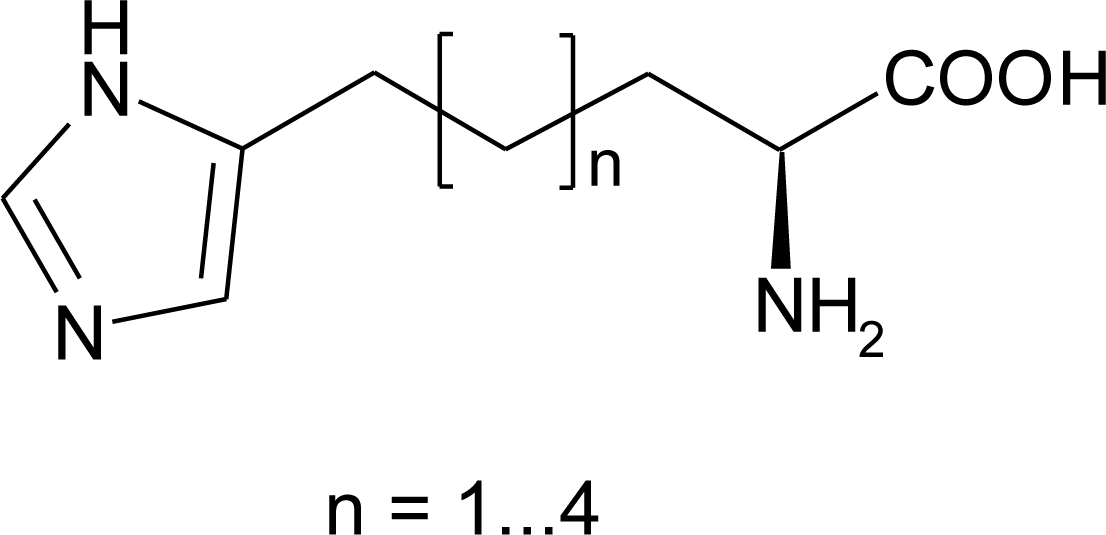
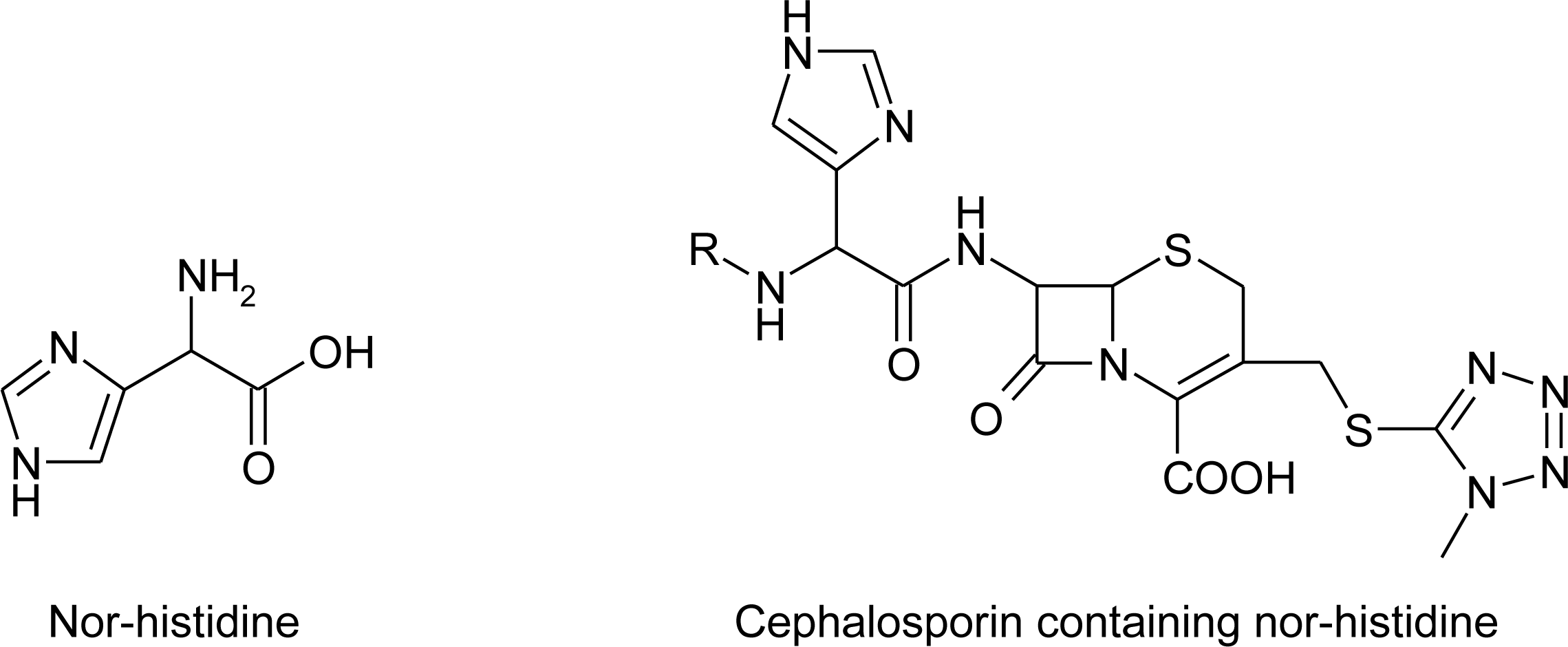
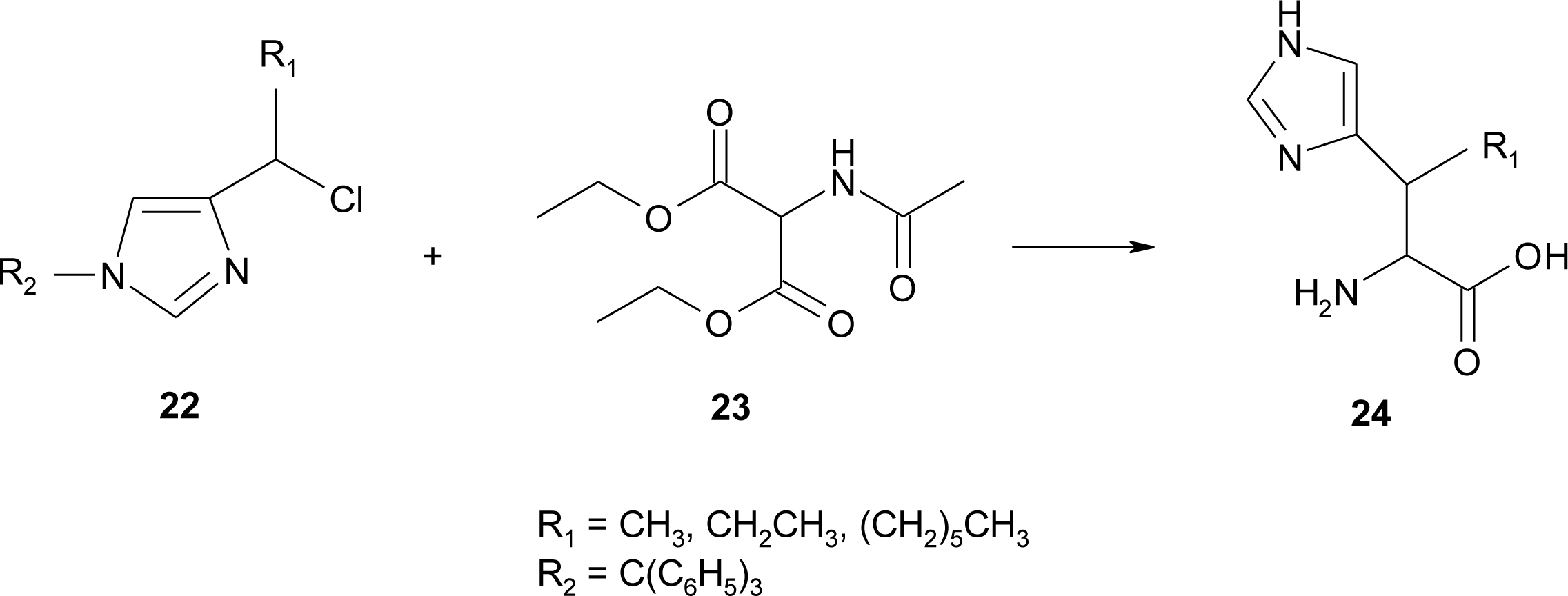
4.8. Nor-Histidine
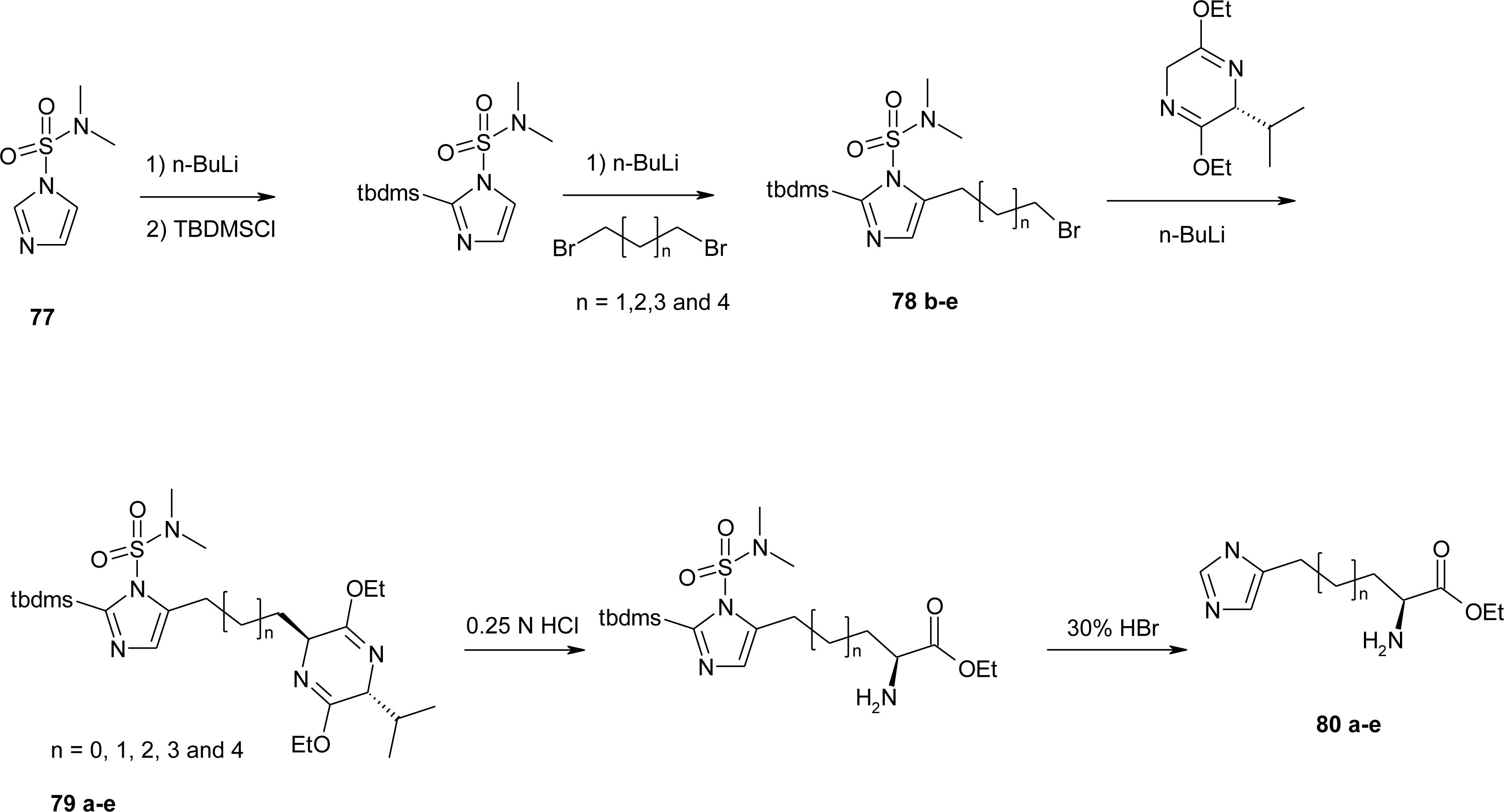
4.9. β2-Homo-Histidine
4.10. β3-Homo-Histidine
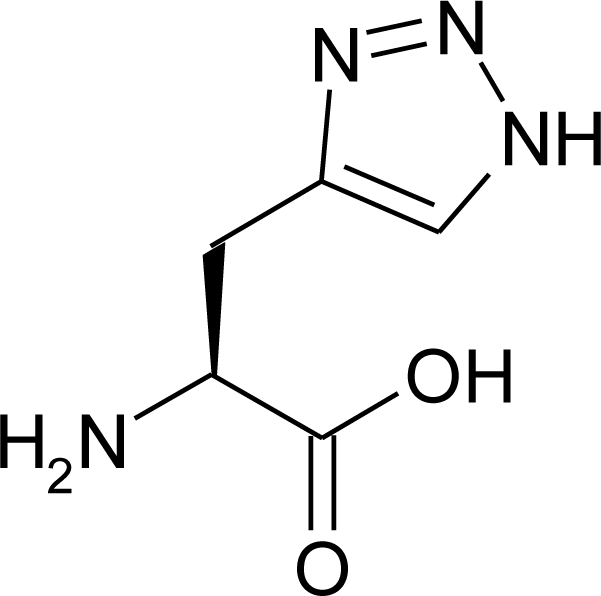
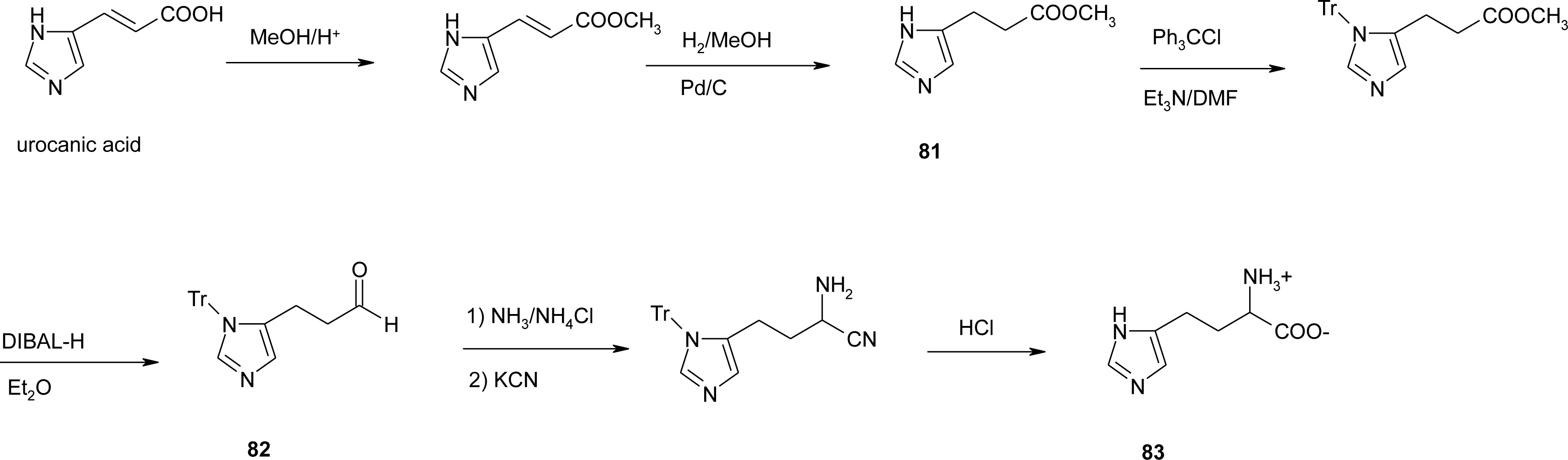
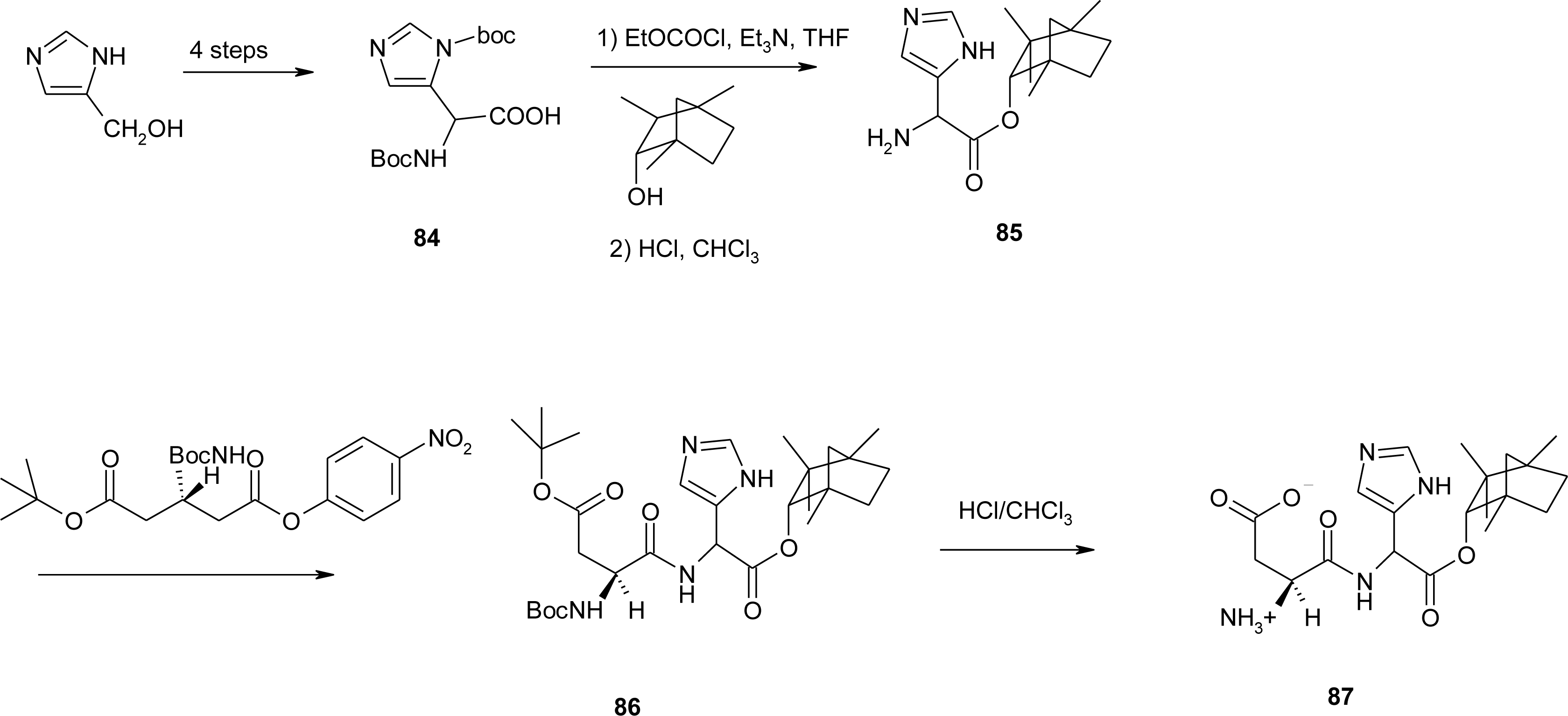
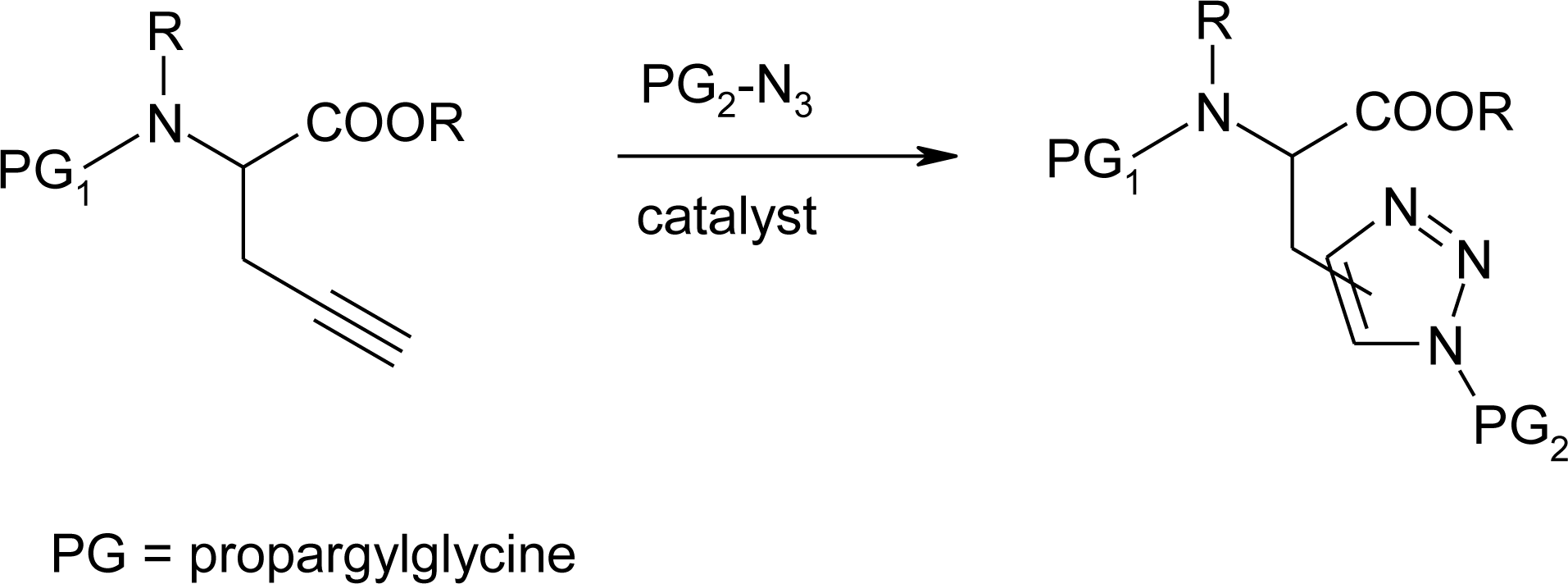
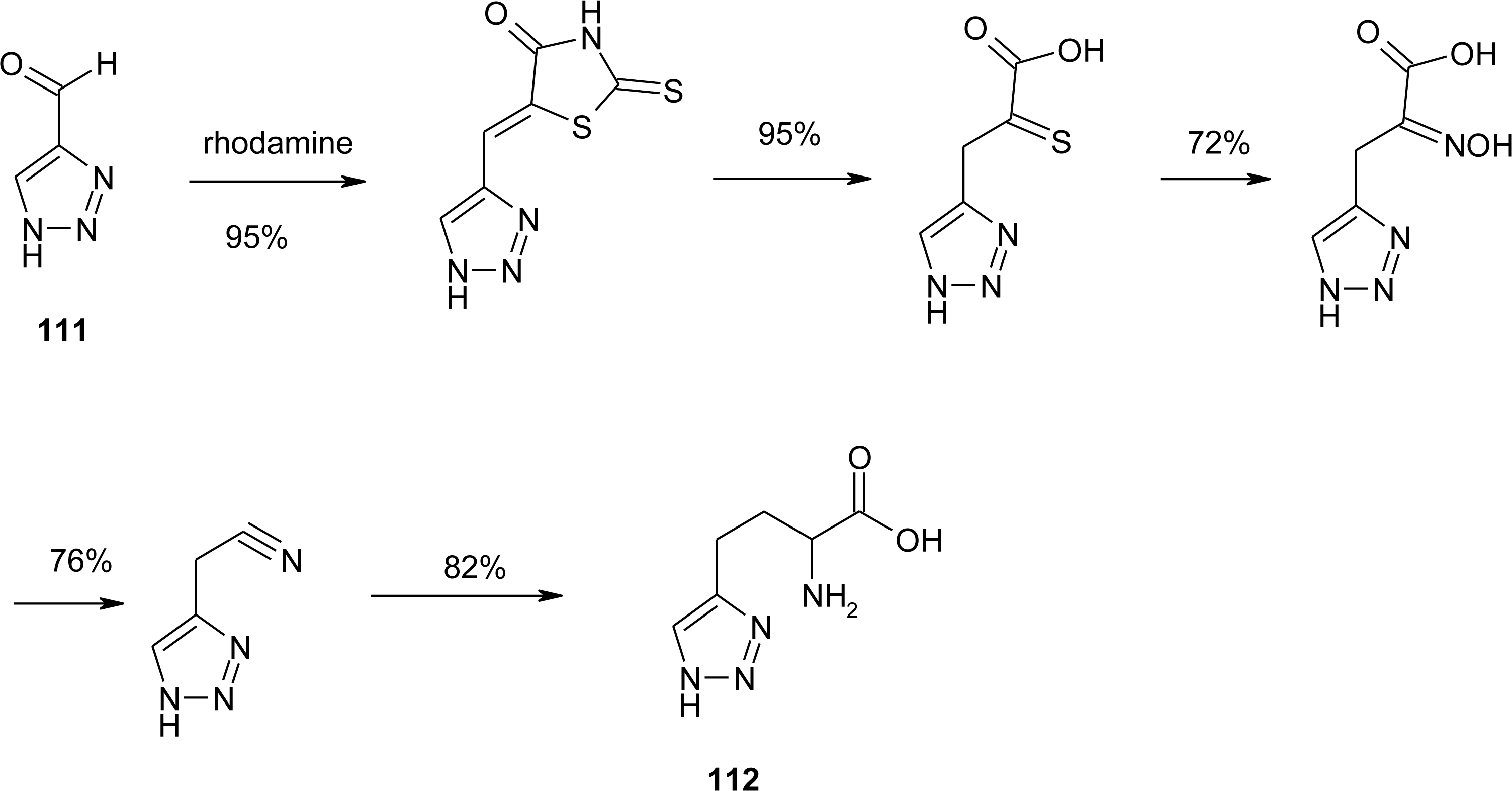
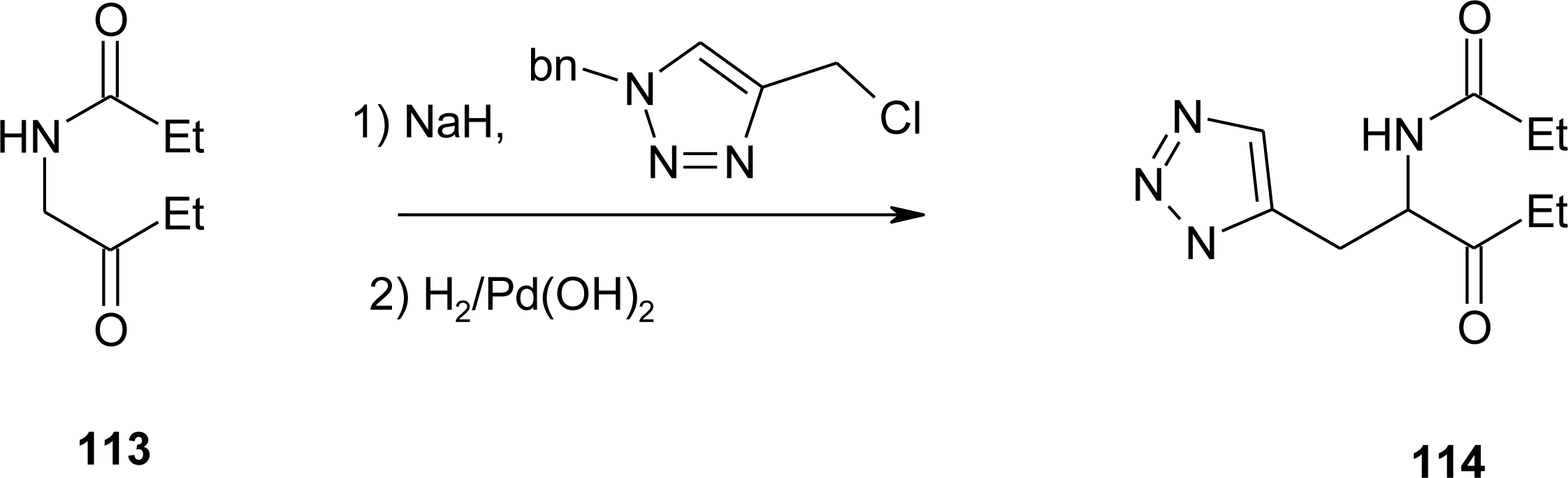
4.11. Aza-Histidine
5. Conclusions
The Importance of Histidine Amino Acid as Residue in Bioactive Molecules and in the Synthesis of Biologically Active Compounds
References
- Deschamps, JR; Flippen-Anderson, JL; George, C. X-ray studies on ligands. Biopolymers 2003, 66, 287–293. [Google Scholar]
- Hruby, VJ; Li, G; Haskell-Luevano, C; Shenderovich, MD. Design of peptides, proteins, and peptidomimetics in χ space. Biopolymers 1997, 43, 219–266. [Google Scholar]
- Pinnen, F; Zanotti, G; Lucente, G; Cerrini, S; Fedeli, W; Gavuzzo, E. Cyclodepsitripeptides. Synthesis, Crystal Structure and Molecular Conformations of cyclo(-l-2-Hydroxyisovaleryl-l-prolyl-l-prolyl-) and cyclo(-d-2-Hydroxyisovaleryl-l-prolyl-l-prolyl-). J. Chem. Soc. Perkin Trans. II 1985, 12, 1931–1937. [Google Scholar]
- Cerrini, S; Gavuzzo, E; Lucente, G; Luisi, G; Pinnen, F; Radics, L. Ten-membered cyclotripeptides. Int. J. Peptide Prot. Res 1991, 38, 289–297. [Google Scholar]
- Ramachandran, GN; Sasisekharan, V. Conformation of polypeptides and proteins. Adv. Protein Chem 1968, 23, 283–438. [Google Scholar]
- Hruby, VJ. Conformational and topographical considerations in the design of biologically active peptides. Biopolymers 1993, 33, 1073–1082. [Google Scholar]
- Hruby, VJ. Peptides: Chemistry, Structure and Biology; Hodges, RS, Wieland, T, Karger, S, Eds.; Karger: Basel, Switzerland, 1995; pp. 207–220. [Google Scholar]
- Wang, S; Tang, X; Hruby, VJ. First stereoselective synthesis of an optically pure β-substituted histidine: (2S,3S)-β-methylhistidine. Tetrahedron Lett 2000, 41, 1307–1310. [Google Scholar]
- Schiavone, MT; Santos, RA; Brosnihan, KB; Khosla, MC; Ferrario, CM. Release of vasopressin from the rat hypothalamo-neurohypophysial system by angiotensin-(1–7) heptapeptide. Proc. Natl. Acad. Sci. USA 1988, 85, 4095–4098. [Google Scholar]
- Seeburg, PH; Adelman, JP. Characterization of cDNA for precursore of human luteinizing hormone releasing hormone. Nature 1984, 311, 666–668. [Google Scholar]
- Pickart, L; Freedman, JH; Loker, WJ; Peisach, J; Perkins, CM; Stenkamp, RE; Weinstein, B. Growth-modulating plasma trypeptide may function by facilitating cooper uptake into cells. Nature 1980, 288, 715–717. [Google Scholar]
- Sundberg, RJ; Martin, RB. Interactions of histidine and other imidazole derivatives with transition metal ions in chemical and biological system. Chem. Rev 1974, 74, 471–517. [Google Scholar]
- McNaught, AD; Wilkinson, A. Compendium of Chemical Terminology: IUPAC Reccomandations, 2nd ed; Blackwell Scientific Publications: Oxford, UK, 1997; p. 43. [Google Scholar]
- Morera, E; Nalli, M; Mollica, A; Paradisi, MP; Aschi, M; Gavuzzo, E; Mazza, F; Lucente, G. Peptides containing 4-amino-1,2-dithiolane-4-carboxylic acid (Adt): Conformation of Boc-Adt-Adt-NHMe and NH...S interactions. J. Pept. Sci 2005, 11, 104–112. [Google Scholar]
- Robinson, B; Shepherd, DM. The preparation of dl-α-methylhistidine dihydrochloride. J Chem Soc 1961, 503–508. [Google Scholar]
- Snyder, HR; Smith, CW; Curtius, W; Stewart, JM. C-Alkylation with quaternary ammonium salts. A new approach to the synthesis of compounds containing the β-indolemethylene group. J. Am. Chem. Soc 1944, 66, 200–204. [Google Scholar]
- Weidenhagen, R; Harrmann, R. A new synthesis of imidazole derivatives. Chem. Ber 1935, 68, 1953–1961. [Google Scholar]
- Turner, RA; Huebner, CF; Scholz, CR. Studies on imidazole compounds. I. 4-Methylimidazole and related compounds. J. Am. Chem. Soc 1949, 71, 2801–2803. [Google Scholar]
- Schmidt, KF. The imine residue. Chem. Ber 1924, 57, 704–706. [Google Scholar]
- Sletzinger, M; Pfister, K. α-alkyl histidines. US Patent 3,169,971. 1965. [Google Scholar]
- Kornblum, N; Blackwood, RK. Ethyl-α-nitrobutyrate. Org. Synth 1957, 37, 44–46. [Google Scholar]
- O’Donnell, MJ; Rusterholz, DB. Synthesis of α-methylhistidine by catalytic phase-transfer alkylations. Synt. Commun 1989, 19, 1157–1165. [Google Scholar]
- Matsumoto, K; Miyahara, T; Suzuki, M; Miyoshi, M. Synthesis of amino acids and related compounds. 8. Improved synthesis of dl-histidine. Agric. Biol. Chem 1974, 38, 1097–1115. [Google Scholar]
- Kelley, JL; Miller, CA; McLean, EW. Attempted inhibition of histidine decarboxylase with β-alkyl analogs of histidine. J. Med. Chem 1977, 20, 721–723. [Google Scholar]
- Albertson, NF; Archer, S. Use of Et acetamidomalonate in the synthesis of amino acids. Preparation of dl-histidine, dl-phenilalanine and dl-leucine. J. Am. Chem. Soc 1945, 67, 308–310. [Google Scholar]
- Evans, DA; Weber, AE. Asymmetric glycine enolate aldol reactions: Synthesis of cyclosporin’s unusual amino acid, MeBmt. J. Am. Chem. Soc 1986, 108, 6757–6761. [Google Scholar]
- Pyman, FL. Derivatives of glyoxaline-4(or 5)-formaldehyde and glyoxaline-4(or 5)-carboxylic acid. A new synthesis of histidine. J. Chem. Soc 1916, 109, 186–202. [Google Scholar]
- Saha, AK; End, DW. Novel β-(imidazol-4-yl)-β-amino acids: Solid-phase synthesis and study of their inhibitory activity against geranylgeranyl protein transferase type I. Biorg. Med. Chem. Lett 2005, 15, 1713–1719. [Google Scholar]
- Davies, SG; Ichihara, O. Asymmetric synthesis of R-β-amino butanoic acid and S-β-tyrosine: Homochiral lithium amide equivalents for Michael additions to α,β-unsaturated esters. Tetrahedron: Asimmetry 1991, 2, 183–186. [Google Scholar]
- De Graw, JI; Engstrom, J; Ellis, M; Johnson, HL. Potential histidine decarboxylase inhibitors. I. α- and β-substituted histidine analogues. J. Med. Chem 1977, 20, 1671–1674. [Google Scholar]
- Hirsch, A; Richardson, K. Reactions of Histidine. J. Appl. Chem 1969, 19, 83–85. [Google Scholar]
- Goodson, LH; Honigberg, IL; Lehman, JJ; Burton, WH. Potential growth antagonists. I. Hydantoins and di-substituted glycine. J. Org. Chem 1960, 25, 1920–1924. [Google Scholar]
- Pictet, A; Spengler, T. Formation of isoquinoline derivatives by the action of methylal on phenylethylamine, phenylalanine and tyrosine. Ber. Dtsch. Chem. Ges 1911, 44, 2030–2036. [Google Scholar]
- Whaley, WM; Govindachari, TR. The Pictet-Spengler of tetrahydroisoquinolines and related compounds. Org. React 1951, 6, 151–190. [Google Scholar]
- Verschueren, K; Toth, G; Tourwe, D; Lelb, M; van Binst, G; Hruby, VJ. A facile synthesis of 1,2,3,4-tetrahydro-7-hydroxyisoquinoline-3-carboxyilic acid, a conformationally constrained tyrosine analog. Synthesis 1992, 458–460. [Google Scholar]
- Shinkai, H; Toi, K; Kumashiro, I; Seto, Y; Fukuma, M; Dan, K; Toyoshima, S. N-acylphenylalanines and related compounds. A new class of oral hypoglycemic agents. J. Med. Chem 1988, 31, 2092–2097. [Google Scholar]
- Hayashi, K; Ozaki, Y; Nunami, K; Uchida, T; Kato, J; Kinashi, K; Yoneda, N. Studies on angiotensin-converting enzyme inhibitors. I. Syntheses and angiotensin-converting enzyme inhibitory activity of 2-(3-mercaptopropionyl)-1,2,3,4-tetrahydroisoquinoline-3-carboxylic acid derivatives. Chem. Pharm. Bull 1983, 31, 570–576. [Google Scholar]
- Remelli, M; Munerato, C; Pulidori, F. Binary and ternary copper (II) complexes of Nτ- and Nπ- methyl-l-histidine in aqueous solution. J. Chem. Soc. Dalton Trans 1994, 14, 2049–2056. [Google Scholar]
- Remelli, M; Rossi, S; Guerrini, R; Pulidori, F. Binary and ternary copper (II) complexes of l-spinacine in aqueous solution. Ann. Chim 1995, 85, 503–518. [Google Scholar]
- Salvadori, S; Guerrini, R; Forlani, V; Bryant, SD; Attila, M; Lazarus, LH. Prerequisite for His4 in deltorphin A for high δ-opioid receptor selectivity. Amino Acids 1994, 7, 291–304. [Google Scholar]
- Wellisch, J. Synthetic alkaloids from tyrosine, tryptophane and histidine. Biochem. Z 1913, 49, 173–194. [Google Scholar]
- Klutchko, S; Hodges, JC; Blankley, CJ; Colbry, NL. 4,5,6,7-Tetrahydro-1H-imidazo[4,5-c]pyridine-6-carboxylic acids (Spinacines). J. Heterocycl. Chem 1991, 28, 97–108. [Google Scholar]
- Remelli, M; Pulidori, F; Guerrini, R; Bertolasi, V. Synthesis of spinacine and spinacine derivatives: Crystal and molecular structures of Nπ-hydroxy-methyl spinacine and Nα-methyl spinaceamine. J. Chem. Crystallogr 1997, 27, 507–513. [Google Scholar]
- Guzman, F; Cain, M; Larscheid, P; Hagen, T; Cook, JM; Schweri, M; Skolnick, P; Paul, SM. Biomimetic approach to potential benzodiazepine. Receptor agonists and antagonists. J. Med. Chem 1984, 27, 564–570. [Google Scholar]
- Wille, MA. The base-catalyzed Pictet-Spengler reaction Synthesis of nitrogen heterocyclic compounds from the reaction of histamine and tryptamine analogs with aromatic aldehydes in alkaline medium; PhD Thesis. University of Pennsylvania, Philadelphia, PA, USA; 1969. [Google Scholar]
- Andreetti, GD; Cavalca, L; Sgarabotto, P. Crystal and molecular structure of the amino acid spinacine dihydrate [4,5,6,7-tetrahydro-1H-imidazo[4,5-c]pyridine-6-carboxylic acid dihydrate]. Gazz. Chim. Ital 1971, 101, 625–634. [Google Scholar]
- McMullan, RK; Epstein, J; Ruble, JR; Craven, BM. The crystal structure of imidazole at 103 K by neutron diffraction. Acta Crystallogr 1979, B35, 688–691. [Google Scholar]
- Madden, JL; McGandy, EL; Seeman, NC. The crystal structure of the monoclinic form of l-Histidine. Acta Crystallogr 1972, B28, 2377–2382. [Google Scholar]
- Cremer, D; Pople, JA. General definition of ring puckering coordinates. J. Am. Chem. Soc 1975, 97, 1354–1358. [Google Scholar]
- Nardelli, M. Ring asymmetry parameters from out-of plane atomic displacements. Acta Crystallogr 1983, C39, 1141–1142. [Google Scholar]
- Gibson, SE; Guillo, N; Tozer, MJ. Towards control of χ-space: Conformationally constrained analogues of Phe, Tyr, Trp and His. Tetrahedron 1999, 55, 585–615. [Google Scholar]
- Pages, RA; Burger, A. 1-Amino-2-(4-imidazolyl)cyclopropanecarboxylic acid. J. Med. Chem 1966, 9, 766–768. [Google Scholar]
- Awad, WI; Fateen, AK; Zayed, MA. 2-Phenyl-4-arylidene-5-oxazolones. Tetrahedron 1964, 20, 891–896. [Google Scholar]
- Mustafa, A; Asker, W; Harhash, AH; Fleifel, AM. Reactivity of unsaturated centers in heterocycles and chalcones towards diazoalkanes. Tetrahedron 1965, 21, 2215–2229. [Google Scholar]
- Easton, CJ; Hutton, CA; Roslet, PD; Tiekink, ERT. A stereocontrolled synthesis of β-hydroxyphenylalanine and β-hydroxytyrosine derivatives. Tetrahedron 1994, 50, 7327–7340. [Google Scholar]
- Easton, CJ; Hutton, CA; Tan, EW; Tiekink, ERT. Synthesis of homochiral hydroxyl-α-amino acid derivatives. Tetrahedron Lett 1990, 31, 7059–7062. [Google Scholar]
- Hutton, CA. Substituent effects in the stereoconvergent synthesis of β-hydroxyphenylalanine derivatives. Tetrahedron Lett 1997, 38, 5899–5902. [Google Scholar]
- Croft, AK; Easton, CJ; Kociuba, K; Radom, L. Strategic use of amino acid N-substituents to limit α-carbon-centered radical formation and consequent loss of stereochemical integrity. Tetrahedron: Asymmetry 2003, 14, 2919–2126. [Google Scholar]
- Crich, D; Banerjee, A. Expedient synthesis of threo-β-hydroxy-α-amino acid derivatives: Phenylalanine, Tyrosine, Histidine and Tryptophan. J. Org. Chem 2006, 71, 7106–7109. [Google Scholar]
- Xu, J; Yadan, JC. Synthesis of l-(+)-Ergothioneine. J. Org. Chem 1995, 60, 6296–6301. [Google Scholar]
- Battersby, AR; Nicoletti, M; Staunton, J; Vleggaar, R. Studies of enzyme-mediated reactions. Part 13. Stereochemical course of the formation of histamine by decarboxylation of (2S)-histidine with enzymes from Clostridium welchii and Lactobacillus 30a. J Chem Soc Perkin I 1980, 43–51. [Google Scholar]
- Cativiela, C; Diaz de Villegas, MD; Garcia, JI; Mayoral, JA; Melendez, E. Stereoselective synthesis of (Z)-2-(acylamino)-3-hetaryl-2-propenoic acids. An. Quim 1985, 81, 56–61. [Google Scholar]
- Parker, AR; Moore, JA; Schwab, JM; Jo Davisson, V. Escherichia Coli imidazoleglycerol phosphate dehydratase: Spectroscopic characterization of the enzymic product and the steric course of the reaction. J. Am. Chem. Soc 1995, 117, 10605–10613. [Google Scholar]
- Toshiyuki, SJ. New imidazole derivative, method for producing the same, and method for producing histidine amide derivative using the imidazole derivative. JP Patent 2010031004 2010. [Google Scholar]
- Bloemhoff, W; Kerling, KET. Polypeptides, X.V. Synthesis of l- and d-homo-histidine. Rec. Trav. Chim. Pays-Bas 1975, 94, 182–185. [Google Scholar]
- Altman, J; Wilchek, M; Lipp, R; Schunack, W. An improved synthesis of l-homo-histidine. Synth. Commun 1989, 19, 2069–2076. [Google Scholar]
- Lee, Y; Martasek, P; Roman, LJ; Masters, BSS; Silverman, RB. Imidazole-containing amino acids as selective inhibitors of nitric Oxide synthase. Biorg. Med. Chem 1999, 7, 1941–1951. [Google Scholar]
- Pirrung, MC; Pei, TJ. Synthesis of (+)-Homo-histidine. J. Org. Chem 2000, 65, 2229–2230. [Google Scholar]
- Browne, LJ; Gude, C; Rodriguez, H; Steele, RE; Bhatnager, A. Fadrozole hydrochloride: A potent, selective, nonsteroidal inhibitor of aromatase for the treatment of estrogen-dependent desease. J. Med. Chem 1991, 34, 725–736. [Google Scholar]
- Court, TT; Chester, PW; Ila, S. Amino-substituted heterocycles as rennin inhibitors. US Patent 5,643,879 1997. [Google Scholar]
- Mazur, RH; Schlatter, JM; Goldkamp, AH. Structure-taste relationships of some dipeptides. J. Am. Chem. Soc 1969, 91, 2684–2691. [Google Scholar]
- Janusz, JM; Young, PA; Blum, RB; Riley, CM. High-Potency Dipeptide Sweeteners. 2. l-Aspartylfuryl-, Thienyl-, and Imidazolylglycine Esters. J. Med. Chem 1990, 33, 1676–1682. [Google Scholar]
- van Batenburg, OD; Kerling, KET; Havinga, E. Studies on polypeptides. Part XIX. Synthesis and enzymic activity of an RNase S’ analog in which the 4-imidazolylglycyl residue takes the position and the role of histidine-12. FEBS Lett 1976, 68, 228–230. [Google Scholar]
- van Batenburg, OD; Kerling, KET. Improved synthesis of tert-butyloxycarbonyl-l-histidine. Int. J. Pept. Prot. Res 1976, 8, 1–2. [Google Scholar]
- Furukawa, M; Arimoto, M; Nakamura, S; Ejima, A; Higashi, O; Tagawa, H. Semisynthetic β-lactam antibiotics. I. Synthesis and and antibacterial activity of 7β-[2-aryl-2-(aminoacetamido)acetamido]cephalosporins. J. Antibiot 1986, 9, 1225–1235. [Google Scholar]
- Etoga, JLG; Ahmed, SK; Patel, S; Bridges, RJ; Thompson, CM. Conformationally-restricted amino acid analogues bearing a distal sulfonic acid show selective inhibition of system x_c over the vesicular glutamate transporter. Bioorg. Med. Chem. Lett 2010, 20, 2680–2683. [Google Scholar]
- Chruma, JJ; Liu, L; Zhou, W; Breslow, R. Hydrophobic and electronic factors in the design of dialkylglycine decarboxylase mimics. Bioorg. Med. Chem 2005, 13, 5873–5883. [Google Scholar]
- Nenajdenko, VG; Zakurdaev, EP; Prusov, EV; Balenkova, ES. Convenient synthesis of melatonin analogues: 2- and 3-substituted-N-acetylindolylalkylamines. Tetrahedron 2004, 60, 11719–11724. [Google Scholar]
- Stalker, RA; Munsch, TE; Tran, JD; Nie, X; Warmuth, R; Beatty, A; Aakeröy, CB. Asymmetric synthesis of two new conformationally constrained lysine derivatives. Tetrahedron 2002, 58, 4837–4849. [Google Scholar]
- Lelais, G; Micuch, P; Lefebvre, DJ; Rossi, F; Seeback, D. Preparation of Protected β2- and β3-Homo-cysteine, β2- and β3-Homo-histidine, and β2-Homo-serine for solid-phase Syntheses. D. Helv. Chim. Acta 2004, 87, 3131–3159. [Google Scholar]
- Kumar, A; Ghilagaber, S; Knight, J; Wyatt, PB. The homologation of Histidine. Tetrahedron Lett 2002, 43, 6991–6994. [Google Scholar]
- Roux, S; Ligeti, M; Buisson, DA; Rousseaux, B; Cintrat, JC. Synthesis of orthogonally protected aza-histidine: Application to the synthesis of a GHK analogue. Amino Acids 2010, 38, 279–286. [Google Scholar]
- Ikeda, Y; Kawahara, S; Taki, M; Kuno, A; Hasegawa, T; Taira, K. Synthesis of a novel histidine analog and its efficient incorporation into a protein in vivo. Protein Eng 2003, 16, 699–706. [Google Scholar]
- Sheenan, JC; Robinson, CA. The synthesis of triazole analogues of histamine and related compounds. J. Am. Chem. Soc 1949, 71, 1436–1440. [Google Scholar]
- Boyd, RE; Press, JB; Rasmussen, CR; Raffa, RB. α2 Adrenoceptor Agonists as Potential Analgesic Agents. 1. (Imidazolylmethyl)oxazoles and thiazoles. J. Med. Chem 1999, 42, 5064–5071. [Google Scholar]
- Hruby, VJ; Boteju, LW. Molecular Biology and Biotechnology; Meyers, RA, Ed.; VCH: New York, NY, USA, 1995; pp. 658–664. [Google Scholar]
- Qian, X; Köver, KE; Shenderovich, MD; Lou, BS; Misicka, A; Zalewska, T; Horvath, R; Davis, P; Bilsky, EJ; Porreca, F; et al. Newly discovered stereochemical requirements in the side-chain conformation of δ-opioid agonists for recognizing opioid δ-receptors. J. Med. Chem 1994, 37, 1746–1757. [Google Scholar]
- Nikiforovich, GV; Prakash, OM; Gehrig, CA; Hruby, VJ. Solution conformations of the peptide backbone for DPDPE and its β-MePhe4-substituted analogues. Int. J. Peptide Prot. Res 1993, 41, 347–361. [Google Scholar]
- Huang, Z; He, YB; Raynor, K; Tallent, M; Reisine, T; Goodman, M. Main chain and side chain chiral methylated somatostatin analogs: Syntheses and conformational analyses. J. Am. Chem. Soc 1992, 114, 9390–9401. [Google Scholar]
- Tóth, G; Russell, KC; Landis, G; Kramer, TH; Fang, L; Knapp, R; Davis, P; Burks, TF; Yamamura, HI; Hruby, VJ. Ring substituted and other conformationally constrained tyrosine analogues of [D-Pen2, D-Pen5] enkephalin with δ opioid receptor selectivity. J. Med. Chem 1992, 35, 238491. [Google Scholar]
- Hruby, VJ; Krystenansky, JL; McKee, R; Pelton, JT. Signal Transduction. In Hormonal Control of Gluconeogenesis; CRC Press: Boca Raton, FL, USA, 1986; pp. 2–20. [Google Scholar]
- Sànchez, MS; Alberdi, LMT; Rioseras, MJ; Ferriera, MR; Gonzales, FB. The Pictet-Spengler reaction on l-Histidine. Preparation of Conformationally restricted (+)-Pilocarpine analogs. Bull. Chem. Soc. Jpn 1998, 66, 191–195. [Google Scholar]
- Lakaszuk, A; Demaegdt, H; Feytens, D; Vanderheyden, P; Vauquelin, G; Tourwè, DJ. The replacement of His(4) in Angiotensin IV by Conformationally residues provides highly potent and selective analogues. J. Med. Chem 2009, 52, 5612–5618. [Google Scholar]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}



© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Stefanucci, A.; Pinnen, F.; Feliciani, F.; Cacciatore, I.; Lucente, G.; Mollica, A. Conformationally Constrained Histidines in the Design of Peptidomimetics: Strategies for the χ-Space Control. Int. J. Mol. Sci. 2011, 12, 2853-2890. https://doi.org/10.3390/ijms12052853
Stefanucci A, Pinnen F, Feliciani F, Cacciatore I, Lucente G, Mollica A. Conformationally Constrained Histidines in the Design of Peptidomimetics: Strategies for the χ-Space Control. International Journal of Molecular Sciences. 2011; 12(5):2853-2890. https://doi.org/10.3390/ijms12052853
Chicago/Turabian StyleStefanucci, Azzurra, Francesco Pinnen, Federica Feliciani, Ivana Cacciatore, Gino Lucente, and Adriano Mollica. 2011. "Conformationally Constrained Histidines in the Design of Peptidomimetics: Strategies for the χ-Space Control" International Journal of Molecular Sciences 12, no. 5: 2853-2890. https://doi.org/10.3390/ijms12052853
APA StyleStefanucci, A., Pinnen, F., Feliciani, F., Cacciatore, I., Lucente, G., & Mollica, A. (2011). Conformationally Constrained Histidines in the Design of Peptidomimetics: Strategies for the χ-Space Control. International Journal of Molecular Sciences, 12(5), 2853-2890. https://doi.org/10.3390/ijms12052853