Green Aspects of Techniques for the Determination of Currently Used Pesticides in Environmental Samples

Abstract
:1. Introduction
2. Currently Used Pesticides
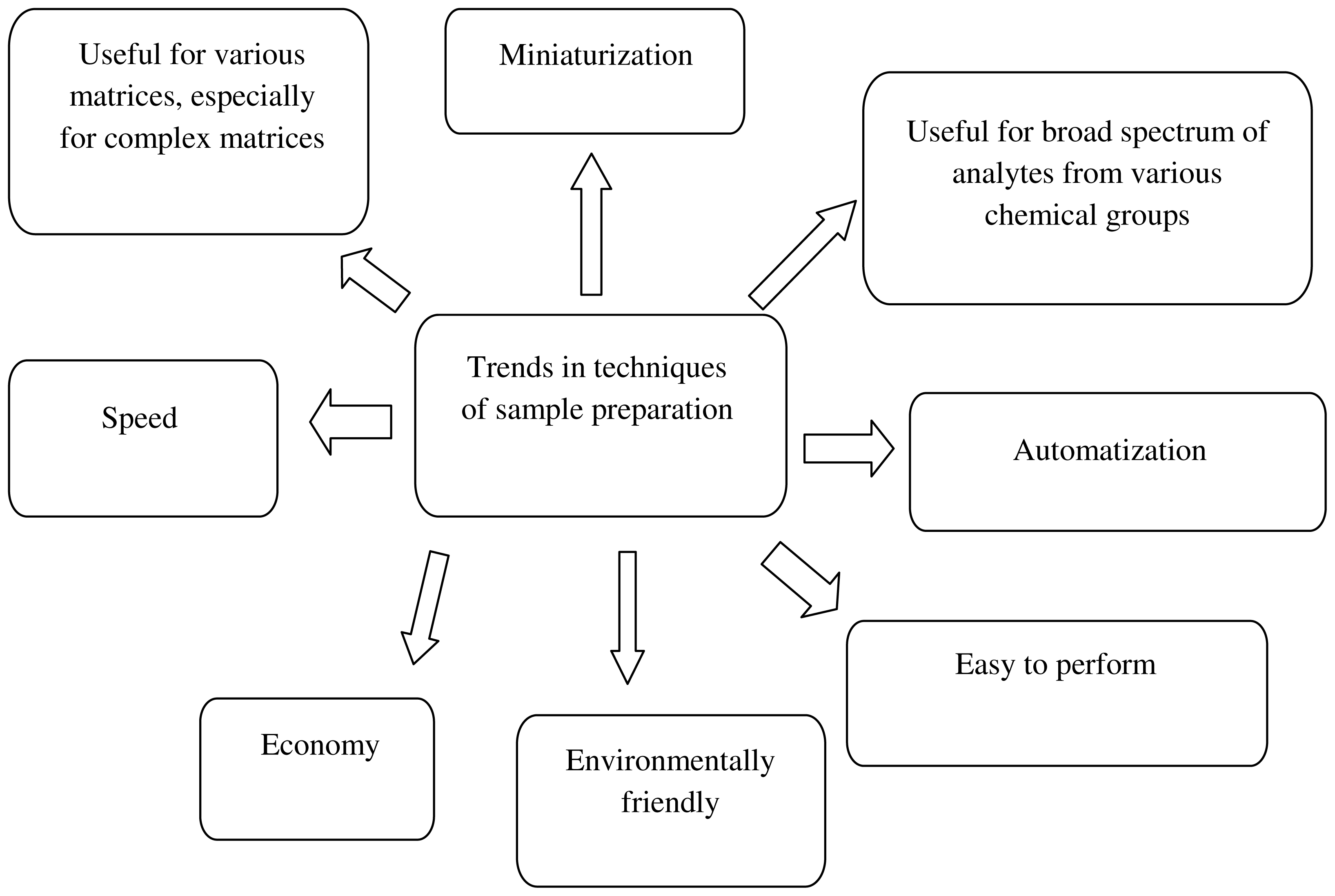
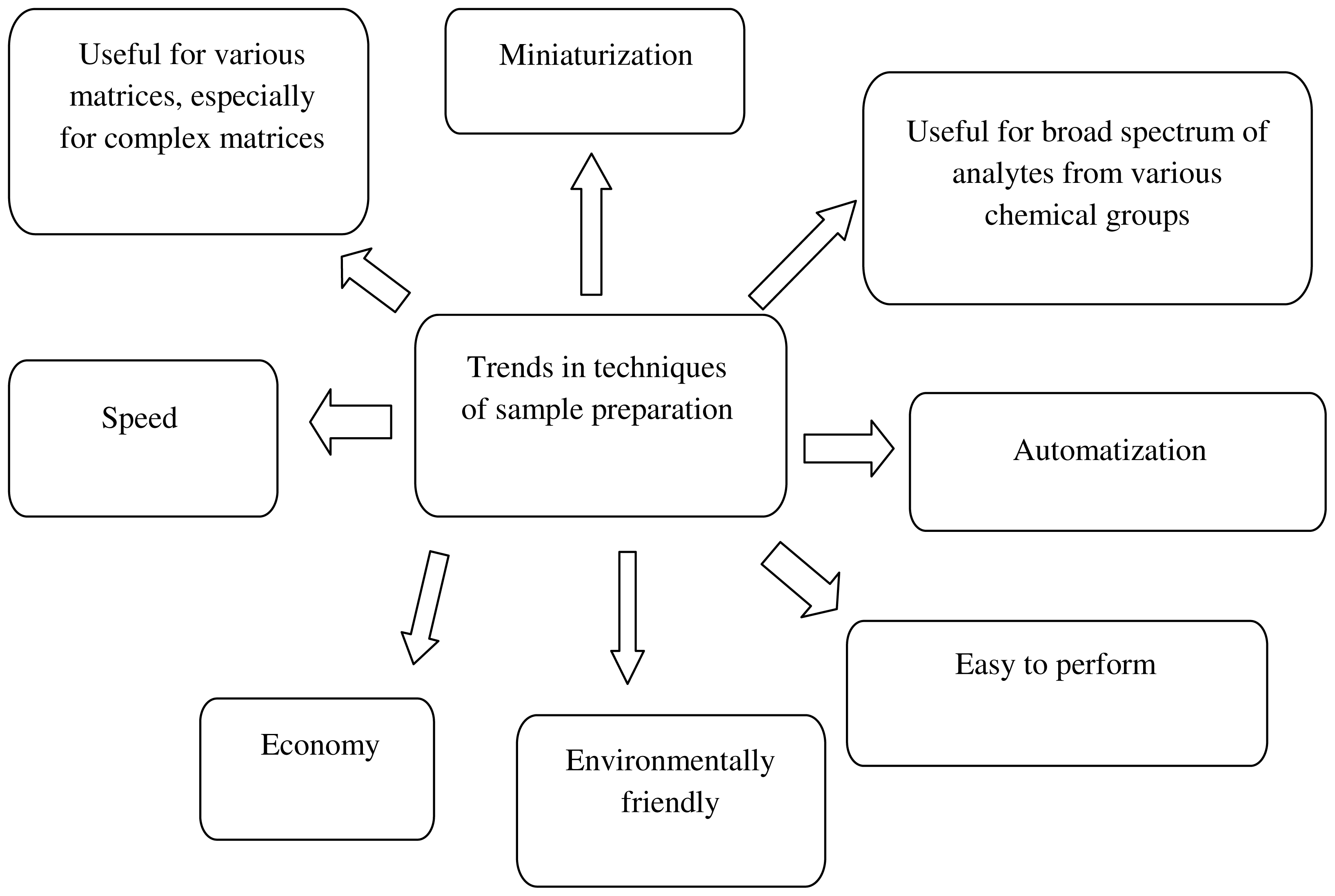
3. Green Analytical Chemistry
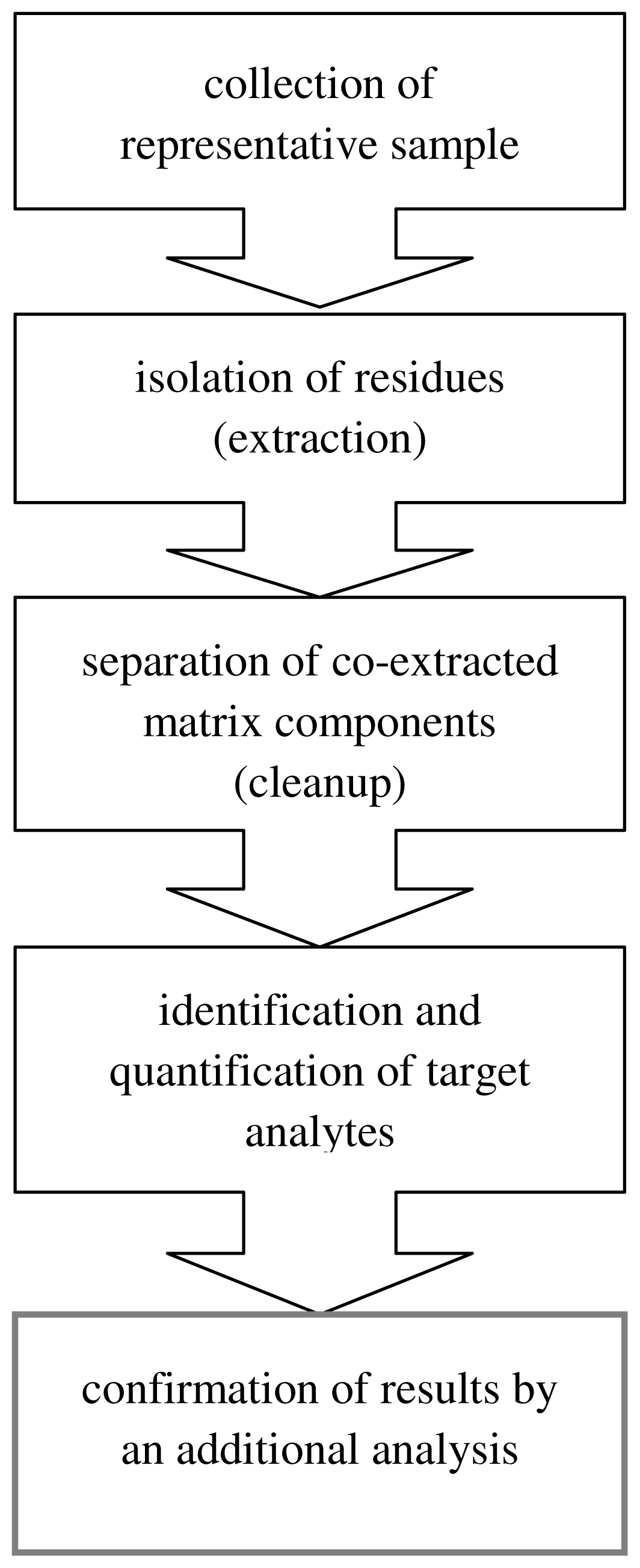
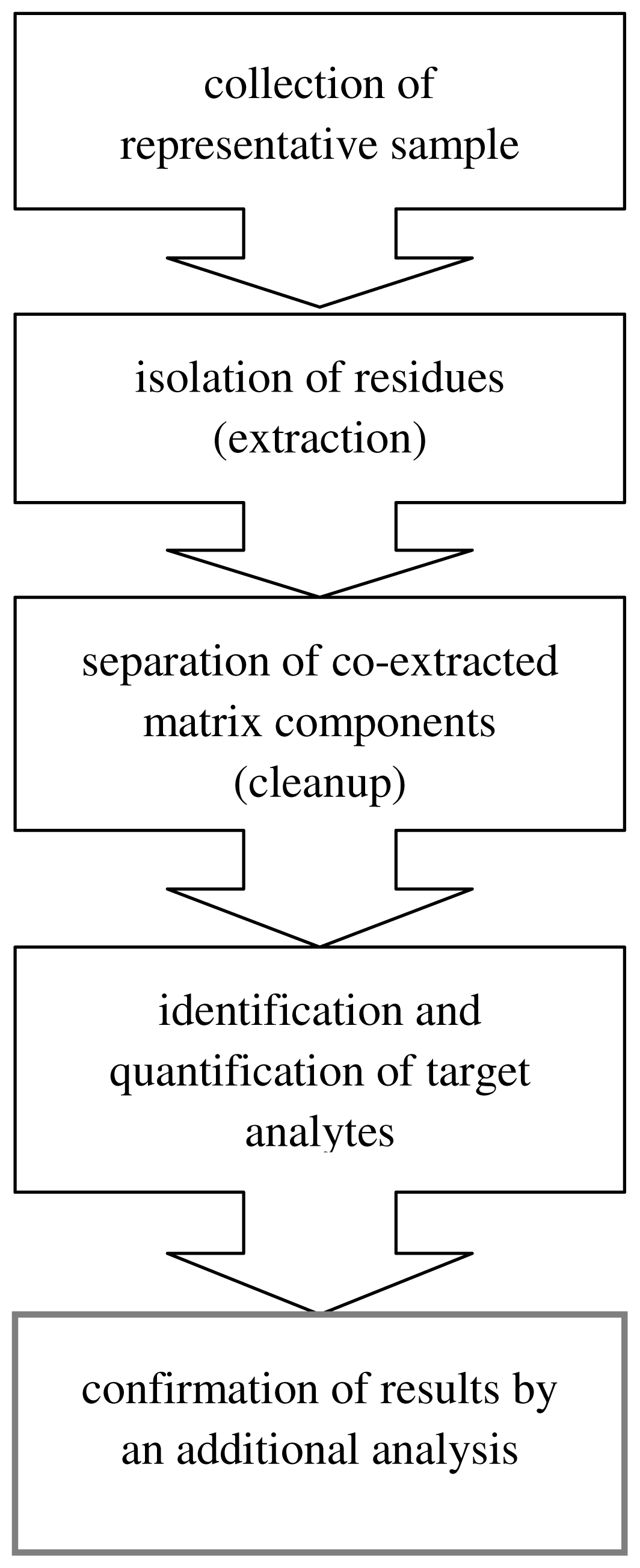
4. Green Aspects in Analytical Methodologies
- ensure maximum removal of interferents from extracts,
- give large recoveries of target compounds, high sensitivity and good precision,
- be environmentally-friendly, i.e., require the smallest possible quantities of samples and chemical reagents, especially organic solvents,
- be cheap, quick and easy to carry out.
- ➢ ECD (Electron Capture Detector)—highly sensitive in relation to compounds containing electronegative atoms,
- ➢ FPD (Flame Photometric Detector)—applied in the determination of organophosphorus compounds,
- ➢ NPD (Nitrogen Phosphorus Detector)—used for the simultaneous determination of organonitrogen and -phosphorus pesticides.
5. Conclusions
Acronyms
| CFME | Continuous-Flow Microextraction |
| CW | Carbowax |
| DAD | Diode Array Detector |
| DI | Direct Immersion |
| DLLME | Dispersive Liquid-Liquid Microextraction |
| DVB | Polydivinylbenzene |
| ECD | Elektron Capture Detector |
| EI | Elektron Impact Ionization |
| ESI | Electrospray Ionization |
| FI | Flow-Injection |
| FPD | Flame Photometric Detector |
| GC | Gas Chromatography |
| GCB | Grapfized Karbon Black |
| GCxGC | Two-Dimensional Gas Chromatography |
| HF(2)ME | Hollow Fiber-protected two-phase Solvent Microextraction |
| HPLC | High Performance Liquid Chromatography |
| HS | Head Space |
| ISTD | Two Different Internal Standards |
| LLE | Liquid-Liquid Extraction |
| LOD | Limit of Detection |
| LPME | Liquid- Phase Microextraction |
| MASE | Membrane-Assisted Solvent Extraction |
| MLLE | Micro Liquid-Liquid Extraction |
| MMLLE | Membrane Micro Liquid-Liquid Extraction |
| MRM | Multiresidue Methods |
| MS | Mass Spectrometry |
| MS/MS | Tandem Mass Spectometry |
| NPD | Nitrogen-Phosphorus Detector |
| PDMS | Polydimethylsiloxane |
| PSA | Primary Secondary Amine |
| SBSE | Stir Bar Sorptive Extraction |
| SDME | Single-Drop Microextraction |
| SPE | Solid-Phase Extraction |
| SPME | Solid Phase Microextraction |
| TSD | Thermionic Specific Detector |
| UV | Ultra-Violet |
References
- About Pesticides; EPA-United States Environmental Protection Agency: Honolulu, HI, USA, 2011. Available online: http://www.epa.gov/pesticides/about/index.htm accessed on 7 August 2011.
- Tankiewicz, M.; Fenik, J.; Biziuk, M. Determination of organophosphorus and organonitrogen pesticides in water samples. Trends Anal. Chem 2010, 29, 1050–1063. [Google Scholar]
- Biuletyn Informacji Publicznej Ministerstwa Rolnictwa i Rozwoju Wsi, Tabela 1-Agregacja według rodzajów środków ochrony roślin. Available online: http://www.bip.minrol.gov.pl accessed on 7 August 2011.
- Ware, G.W.; Whitacre, D.M. The Pesticide Book, 6th ed; MeisterPro Information Resources: Willoughby, OH, USA, 2004. [Google Scholar]
- Beyer, A.; Biziuk, M. Przegląd metod oznaczania pozostałości pestycydów i polichlorowanych bifenyli w próbkach żywności. Ecol. Chem. Eng 2007, 14, 35–58. [Google Scholar]
- Namieśnik, J.; Górecki, T. Preparation of environmental samples for the determination of trace constituents. Pol. J. Environ. Stud 2001, 10, 77–84. [Google Scholar]
- EPA-United States Environmental Protection Agency, Standardized Analytical Methods for Environmental Restoration Following Homeland Security Events—Revision 5.0; EPA-United States Environmental Protection Agency: Cincinnati, OH, USA, 2009.
- Jakość wody. Oznaczanie wybranych chloroorganicznych insektycydów, polichlorowanych bifenyli i chlorobenzenów. Metoda chromatografii gazowej po ekstrakcji ciecz-ciecz. PN-EN ISO 6468:2002. Available online: http://enormy.pl/?m=doc&nid=PN-13.060.50-00246 access on 9 November 2011.
- Jakość wody. Oznaczanie wybranych związków azoto- i fosforoorganicznych. Metody chromatografii gazowej. PN-EN ISO 10695:2004. Available online: http://enormy.pl/?m=doc&v=met&nid=PN-13.060.50-00228&key= access on 9 November 2011.
- Jakość wody. Oznaczanie parationu, parationu metylowego i kilku innych związków fosforoorganicznych w wodzie metodą chromatografii gazowej po ekstrakcji dichlorometanem. PN-EN 12918:2004. Available online: http://enormy.pl/?m=doc&v=met&nid=PN-13.060.50-00181&key= access on 9 November 2011.
- Jakość wody. Oznaczanie wybranych środków ochrony roślin. Metoda z zastosowaniem wysokosprawnej chromatografii cieczowej z detekcją UV po ekstrakcji ciało stałe-ciecz. PN-EN ISO 11369:2002.Available online: http://enormy.pl/?m=doc&nid=PN-13.060.50-00248 access on 9 November 2011.
- Greulich, K.; Alder, L. Fast multiresidue screening of 300 pesticides in water for human consumption by LC-MS/MS. Anal. Bioanal. Chem 2008, 391, 183–197. [Google Scholar]
- Tobiszewski, M.; Mechlinska, A.; Zygmunt, B.; Namiesnik, J. Green analytical chemistry in sample preparation for determination of trace organic pollutants. Trends Anal. Chem 2009, 28, 943–951. [Google Scholar]
- Armenta, S.; Garrigues, S.; Guardia, M. Green analytical chemistry. Trends Anal. Chem 2008, 27, 497–511. [Google Scholar]
- Molina-Diaz, A.; Garcia-Reyes, J.F.; Lopez, B.G. Solid-phase spectroscopy from the point of view of green analytical chemistry. Trends Anal. Chem 2010, 29, 654–666. [Google Scholar]
- Escuderos-Morenas, M.L.; Santos-Delgado, M.J.; Rubio-Barroso, S.; Polo-Diez, L.M. Direct determination of monolinuron, linuron and chlorbromuron residues in potato samples by gas chromatography with q nitrogen–phosphorus detection. J. Chromatogr. A 2003, 1011, 143–153. [Google Scholar]
- Sankararamakrishnan, N.; Sharma, A.K.; Sanghi, R. Organochlorine and organophosphorous pesticide residues in ground water and surface waters of Kanpur, Uttar Pradesh, India. Environ. Int 2005, 31, 113–120. [Google Scholar]
- Pirard, C.; Widart, J.; Nguyen, B.K.; Deleuze, C.; Heudt, L.; Haubruge, E.; De Pauw, E.; Focant, J.F. Development and validation of a multi-residue method for pesticide determination in honey using on-column liquid-liquid extraction and liquid chromatography-tandem mass spectrometry. J. Chromatogr. A 2007, 1152, 116–123. [Google Scholar]
- Tse, H.; Comba, M.; Alaee, M. Method for the determination of organophosphate insecticides in water, sediment and biota. Chemosphere 2004, 54, 41–47. [Google Scholar]
- Sabik, H.; Jeannot, R. Determination of organonitrogen pesticides in large volumes of surface water by liquid-liquid and solid-phase extraction using gas chromatography with nitrogen-phosphorus detection and liquid chromatography with atmospheric pressure chemical ionization mass spectrometry. J. Chromatogr. A 1998, 818, 197–207. [Google Scholar]
- Tahboub, Y.R.; Zaater, M.F.; Al-Talla, Z.A. Determination of the limits of identification and quantitation of selected organochlorine and organophosphorous pesticide residues in surface water by full-scan gas chromatography/mass spectrometry. J. Chromatogr. A 2005, 1098, 150–155. [Google Scholar]
- Fatoki, O.S.; Awofolu, R.O. Methods for selective determination of persistent organochlorine pesticide residues in water and sediments by capillary gas chromatography and electron-capture detection. J. Chromatogr. A 2003, 983, 225–236. [Google Scholar]
- Mahara, B.M.; Borossay, J.; Torkos, K. Liquid-liquid extraction for sample preparation prior to gas chromatography and gas chromatography-mass spectrometry determination of herbicide and pesticide compounds. Microchem. J 1998, 8, 31–38. [Google Scholar]
- Ridgway, K.; Lalljie, S.P.D.; Smith, R.M. Sample preparation techniques for the determination of trace residues and contaminants in foods. J. Chromatogr. A 2007, 1153, 36–53. [Google Scholar]
- David, F.; Sandra, P. Stir bar sorptive extraction for trace analysis. J. Chromatogr. A 2007, 1152, 54–69. [Google Scholar]
- Giordano, A.; Fernández-Franzón, M.; Ruiz, M.J.; Font, G.; Picó, Y. Pesticide residue determination in surface waters by stir bar sorptive extraction and liquid chromatography/tandem mass spectrometry. Anal. Bioanal. Chem 2009, 393, 1733–1743. [Google Scholar]
- David, F.; Hoeck, E.; Sandra, P. Stir bar sorptive extraction for the determination of pyrethroids in water samples. A comparison between thermal desorption in a dedicated thermal desorber, in a split/splitless inlet and by liquid desorption. J. Chromatogr. A 2007, 1157, 1–9. [Google Scholar]
- Quintana, J.B.; Rodriguez, I. Strategies for the microextraction of polar organic contaminants in water samples. Anal. Bioanal. Chem 2006, 384, 1447–1461. [Google Scholar]
- Hyötyläinen, T. Principles, developments and applications of on-line coupling of extraction with chromatography. J. Chromatogr. A 2007, 1153, 14–28. [Google Scholar]
- Lambropoulou, D.A.; Albanis, T.A. Liquid-phase micro-extraction techniques in pesticide residue analysis. J. Biochem. Biophys. Methods 2007, 70, 195–228. [Google Scholar]
- Pinto, M.I.; Sontag, G.; Bernardino, R.J.; Noronha, J.P. Pesticides in water and the performance of the liquid-phase microextraction based techniques. A review. Microchem. J 2010, 96, 225–237. [Google Scholar]
- Zhou, Q.; Liu, J.; Cai, Y.; Liu, G.; Jiang, G. Micro-porous membrane liquid-liquid extraction as an enrichment step prior to nonaqueous capillary electrophoresis determination of sulfonylurea herbicides. Microchem. J 2003, 74, 157–163. [Google Scholar]
- Zapf, A.; Heyer, R.; Stan, H.J. Rapid micro liquid-liquid extraction method for trace analysis of organic contaminants in drinking water. J. Chromatogr. A 1995, 694, 453–461. [Google Scholar]
- Xiao, Q.; Hu, B.; Yu, Ch.; Xia, L.; Jiang, Z. Optimization of a single-drop microextraction procedure for the determination of organophosphorus pesticides in water and fruit juice with gas chromatography-flame photometric detection. Talanta 2006, 69, 848–855. [Google Scholar]
- Ahmadi, F.; Assadi, Y.; Milani Hosseini, S.M.R.; Rezaee, M. Determination of organophosphorus pesticides in water samples by single drop microextraction and gas chromatography-flame photometric detector. J. Chromatogr. A 2006, 110, 307–312. [Google Scholar]
- Lambropoulou, D.A.; Psillakis, E.; Albanis, T.A.; Kalogerakis, N. Single-drop microextraction for the analysis of organophosphorous insecticides in water. Anal. Chim. Acta 2004, 516, 205–211. [Google Scholar]
- Bagheri, H.; Khalilian, F. Immersed solvent microextraction and gas chromatography-mass spectrometric detection of s-triazine herbicides in aquatic media. Anal. Chim. Acta 2005, 537, 81–87. [Google Scholar]
- Solvent Microextraction, Theory and Practice; Kokosa, J.M.; Przyjazny, A.; Jeannot, M.A. (Eds.) Wiley: Horboken, NJ, USA, 2009.
- Nagaraju, D.; Huang, S.D. Determination of triazine herbicides in aqueous samples by dispersive liquid-liquid microextraction with gas chromatography-ion trap mass spectrometry. J. Chromatogr. A 2007, 1161, 89–97. [Google Scholar]
- Rezaee, M.; Yamini, Y.; Faraji, M. Evolution of dispersive liquid-liquid microextraction method. J. Chromatogr. A 2009, 1217, 2342–2357. [Google Scholar]
- He, L.; Luo, X.; Xie, H.; Wang, C.; Lu, X.J.K. Ionic liquid-based dispersive liquid-liquid microextraction followed high-performance liquid chromatography for the determination of organophosphorus pesticides in water sample. Anal. Chim. Acta 2009, 655, 52–59. [Google Scholar]
- Huddleston, J.G.; Visser, A.E.; Reichert, W.M.; Willauer, H.D.; Broker, G.A.; Rogers, R.D. Characterization and comparison of hydrophilic and hydrophobic room temperature ionic liquids incorporating the imidazolium cation. Green Chem 2001, 3, 156–164. [Google Scholar]
- Visser, A.E.; Swatloski, R.P.; Reichert, W.M.; Griffin, S.T.; Rogers, R.D. Traditional extractants in nontraditional solvents: Groups 1 and 2 extraction by crown ethers in room-temperature ionic liquids. Ind. Eng. Chem. Res 2000, 39, 3596–3604. [Google Scholar]
- He, L.J.; Zhang, W.Z.; Zhao, L.; Jiang, X.S. Effect of 1-alkyl-3-methylimidazolium-based ionic liquids as the eluent on the separation of ephedrines by liquid chromatography. J. Chromatogr. A 2003, 1007, 39–45. [Google Scholar]
- Xiao, X.H.; Zhao, L.; Liu, X.; Jiang, S.X. Ionic liquids as additives in high performance liquid chromatography. Analysis of amines and the interaction mechanism of ionic liquids. Anal. Chim. Acta 2004, 519, 207–211. [Google Scholar]
- Basheer, C.; Alnedhary, A.A.; Rao, B.S.M.; Lee, H.K. Determination of organophosphorous pesticides in wastewater samples using binary-solvent liquid-phase microextraction and solid-phase microextraction: A comparative study. Anal. Chim. Acta 2007, 605, 147–152. [Google Scholar]
- Khalili-Zanjani, M.R.; Yaminia, Y.; Yazdanfar, N.; Shariati, S. Extraction and determination of organophosphorus pesticides in water samples by a new liquid phase microextraction-gas chromatography-flame photometric detection. Anal. Chim. Acta 2008, 606, 202–208. [Google Scholar]
- Khalili-Zanjani, M.R.; Yaminia, Y.; Shariati, S.; Jőnsson, J.Å. A new liquid-phase microextraction method based on solidification of floating organic drop. Anal. Chim. Acta 2007, 585, 286–293. [Google Scholar]
- Lüthje, K.; Hyötyläinen, T.; Riekkola, M.L. Comparison of different trapping methods for pressurised hot water extraction. J. Chromatogr. A 2004, 1025, 41–49. [Google Scholar]
- Hyötyläinen, T.; Lüthje, K.; Rautiainen-Rämä, M.; Riekkola, M.L. Determination of pesticides in red wines with on-line coupled microporous membrane liquid-liquid extraction-gas chromatography. J. Chromatogr. A 2004, 1056, 267–271. [Google Scholar]
- Hu, Y.; Wang, Y.; Hu, Y.; Li, G. Liquid-liquid-solid microextraction based on membrane-protected molecularly imprinted polymer fiber for trace analysis of triazines in complex aqueous. J. Chromatogr. A 2009, 1216, 8304–8311. [Google Scholar]
- Hu, X.; Ye, T.; Yu, Y.; Cao, Y.; Guo, C. Novel liquid-liquid-solid microextraction method with molecularly imprinted polymer-coated stainless steel fiber for aqueous sample pretreatment. J. Chromatogr. A 2011, 1218, 3935–3939. [Google Scholar]
- Chen, J.; Duan, C.; Guan, Y. Sorptive extraction techniques in sample preparation for organophosphorus pesticides in complex matrices. J. Chromatogr. B 2010, 878, 1216–1225. [Google Scholar]
- Fontanals, N.; Marce, R.M.; Borrull, F. New hydrophilic materials for solid-phase extraction. Trends Anal. Chem 2005, 24, 394–406. [Google Scholar]
- Fontanals, N.; Marce, R.M.; Borrull, F. New materials in sorptive extraction techniques for polar compounds. J. Chromatogr. A 2007, 1152, 14–31. [Google Scholar]
- Yoshioka, N.; Ichihashi, K. Determination of 40 synthetic food colors in drinks and candies by high-performance liquid chromatography using a short column with photodiode array detection. Talanta 2008, 74, 1408–1413. [Google Scholar]
- Lv, Y.Q.; Lin, Z.X.; Feng, W.; Zhou, X.; Tan, T.W. Selective recognition and large enrichment of dimethoate from tea leaves by molecularly imprinted polymers. Biochem. Eng 2007, 36, 221–229. [Google Scholar]
- Yang, R.Z.; Wei, X.L.; Gao, F.F.; Wang, L.S.; Zhang, H.J.; Xu, Y.J.; Li, C.H.; Ge, Y.X.; Zhang, J.J.; Zhang, J. Simultaneous analysis of anthocyanins and flavonols in petals of lotus (Nelumbo) cultivars by high-performance liquid chromatography-photodiode array detection/electrospray ionization mass spectrometry. J. Chromatogr. A 2009, 1216, 106–112. [Google Scholar]
- Le Moullec, S.; Begos, A.; Pichon, V.; Bellier, B. Selective extraction of organophosphorus nerve agent degradation products by molecularly imprinted solid-phase extraction. J. Chromatogr. A 2006, 1108, 7–13. [Google Scholar]
- Le Moullec, S.; Truong, L.; Montauban, C.; Begos, A.; Pichon, V.; Bellier, B. Extraction of alkyl methylphosphonic acids from aqueous samples using a conventional polymeric solid-phase extraction sorbent and a molecularly imprinted polymer. J. Chromatogr. A 2007, 1139, 171–177. [Google Scholar]
- Kugimiya, A.; Takei, H. Selectivity and recovery performance of phosphate-selective molecularly imprinted polymer. Anal. Chim. Acta 2008, 606, 252–256. [Google Scholar]
- Gonçalves, C.; Alpendurada, M.F. M ultiresidue method for the simultaneous determination of four groups of pesticides in ground and drinking waters, using solid-phase microextraction-gas chromatography with electron-capture and thermionic specific detection. J. Chromatogr. A 2002, 968, 177–190. [Google Scholar]
- Yao, Z.; Jiang, G.; Liu, J.; Cheng, W. Application of solid-phase microextraction for the determination of organophosphorous pesticides in aqueous samples by gas chromatography with flame photometric detector. Talanta 2001, 55, 807–814. [Google Scholar]
- Frías, S.; Rodríguez, M.A.; Conde, J.E.; Pérez-Trujillo, J.P. Optimisation of a solid-phase microextraction procedure for the determination of triazines in water with gas chromatography-mass spectrometry detection. J. Chromatogr. A 2003, 1007, 127–135. [Google Scholar]
- Su, P.; Huang, S.D. Determination of organophosphorus pesticides in water by solid-phase microextraction. Talanta 1999, 49, 393–402. [Google Scholar]
- Rocha, C.; Pappas, E.A.; Huang, C. Determination of trace triazine and chloroacetamide herbicides in tile-fed drainage ditch water using solid-phase microextraction coupled with GC-MS. Environ. Pollut 2008, 152, 239–244. [Google Scholar]
- Magdic, S.; Boyd-Boland, A.; Jinno, K.; Pawliszyn, J. Analysis of organophosphorus insecticides from environmental samples using solid-phase microextraction. J. Chromatogr. A 1996, 736, 219–228. [Google Scholar]
- Basheer, Ch.; Jegadesan, S.; Valiyaveettil, S.; Kee Lee, H. Sol-gel-coated oligomers as novel stationary phases for solid-phase microextraction. J. Chromatogr. A 2005, 1087, 252–258. [Google Scholar]
- Bicchi, C.; Cordero, Ch.; Liberto, E.; Rubiolo, P.; Sgorbini, B.; David, F.; Sandra, P. Dual-phase twisters: A new approach to headspace sorptive extraction and stir bar sorptive extraction. J. Chromatogr. A 2005, 1094, 9–16. [Google Scholar]
- Chen, J.; Duan, Ch.; Guan, Y. Sorptive extraction techniques in sample preparation for organophosphorus pesticides in complex matrices. J. Chromatogr. B 2010, 878, 1216–1225. [Google Scholar]
- Kawaguchi, M.; Ito, R.; Sakui, N.; Okanouchi, N.; Saito, K.; Nakazawa, H. Dual derivatization-stir bar sorptive extraction-thermal desorption-gas chromatography-mass spectrometry for determination of 17β-estradiol in water sample. J. Chromatogr. A 2006, 1105, 140–147. [Google Scholar]
- Anastassiades, M.; Lehotay, S.J.; Štajnbaher, D.; Schenck, F.J. Fast and easy multiresidue method employing acetonitrile extraction/partitioning and dispersive solid-phase extraction for the determination of pesticide residues in produce. J. AOAC Int 2003, 86, 412–431. [Google Scholar]
- Albero, B.; Sanchez-Brunete, C.; Tadeo, J.L. Multiresidue determination of pesticides in juice by solid-phase extraction and gas chromatography-mass spectrometry. Talanta 2005, 66, 917–924. [Google Scholar]
- Gonzalez-Curbelo, M.A.; Hernandez-Borges, J.; Ravelo-Perez, L.M.; Rodriguez-Delgado, M.A. Insecticides extraction from banana leaves using a modified QuEChERS method. Food Chem 2011, 125, 1083–1090. [Google Scholar]
- Nguyen, T.D.; Yu, J.E.; Lee, D.M.; Lee, G.-H. A multiresidue method for the determination of 107 pesticides in cabbage and radish using QuEChERS sample preparation method and gas chromatography mass spectrometry. Food Chem 2008, 110, 207–213. [Google Scholar]
- Koesukwiwat, U.; Lehotay, S.J.; Miaoc, S.; Leepipatpiboon, N. High throughput analysis of 150 pesticides in fruits and vegetables using QuEChERS and low-pressure gas chromatography-time-of-flight mass spectrometry. J. Chromatogr. A 2010, 1217, 6692–6703. [Google Scholar]
- Lehotay, S.J.; Sonb, K.A.; Kwon, H.; Koesukwiwat, U.; Fud, W.; Mastovska, K.; Hoha, E.; Leepipatpiboon, N. Comparison of QuEChERS sample preparation methods for the analysis of pesticide residues in fruits and vegetables. J. Chromatogr. A 2010, 1217, 2548–2560. [Google Scholar]
- Park, J.-Y.; Choi, J.-H.; Abd El-Aty, A.M.; Kim, B.M.; Oh, J.-H.; Do, J.-A.; Kwon, K.S.; Shim, K.-H.; Choi, O.-J.; Shin, S.C.; Shim, J.-H. Simultaneous multiresidue analysis of 41 pesticide residues in cooked foodstuff using QuEChERS: Comparison with classical method. Food Chem 2011, 128, 241–253. [Google Scholar]
- Lehotay, S.J.; Mastovska, K.; Lightfield, A.R.; Gates, R.A. Multi-Analyst, multi-matrix performance of the QuEChERS approach for pesticide residues in foods and feeds using HPLC/MS/MS analysis with different calibration techniques. J. AOAC Int 2010, 93, 355–367. [Google Scholar]
- Nguyen, T.D.; Yu, J.E.; Lee, D.M.; Lee, G.H. A multiresidue method for the determination of 107 pesticides in cabbage and radish using QuEChERS sample preparation method and gas chromatography mass spectrometry. Food Chem 2008, 110, 207–213. [Google Scholar]
- Godula, M.; Hajslová, J.; Alterová, K. Pulsed splitless injection and the extent of matrix effects in the analysis of pesticides. J. High Resolut. Chromatogr 1999, 23, 395–402. [Google Scholar]
- Hernando, M.D.; Agüera, A.; Fernández-Alba, A.R.; Piedra, L.; Contreras, M. Gas chromatographic determination of pesticides in vegetable samples by sequential positive and negative chemical ionization and tandem mass spectrometric fragmentation using an ion trap analyser. Analyst 2001, 126, 46–51. [Google Scholar]
- Martínez Vidal, J.L.; Arrebola, F.J.; Mateu-Sánchez, M. Validation of a gas chromatographic-tandem mass spectrometric method for analysis of pesticide residues in six food commodities selection of a reference matrix for calibration. Chromatographia 2004, 59, 321–327. [Google Scholar]
- Stepan, R.; Ticha, J.; Hajslova, J.; Kovalczuk, T.; Kocourek, V. Baby food production chain: Pesticide residues in fresh apples and products. Food Addit. Contam 2005, 22, 1231–1242. [Google Scholar]
- Mastovska, K.; Hajslova, J.; Lehotay, S.J. Ruggedness and other performance characteristics of low-pressure gas chromatography-mass spectrometry for the fast analysis of multiple pesticide residues in food crops. J. Chromatogr. A 2004, 1054, 335–349. [Google Scholar]
- Garcia-Reyes, J.F.; Ferrer, C.; Gomez-Ramos, M.J.; Fernandez-Alba, A.R.; Molina-Diaz, A. Determination of pesticide residues in olive oil and olives. Trends Anal. Chem 2007, 26, 239–251. [Google Scholar]
- Leandro, C.C.; Hancock, P.; Fussell, R.J.; Keely, B.J. Comparison of ultra-performance liquid chromatography and high-performance liquid chromatography for the determination of priority pesticides in baby foods by tandem quadrupole mass spectrometry. J. Chromatogr. A 2006, 1103, 94–101. [Google Scholar]
- Sadowska-Rociek, A.; Cieślik, E. Stosowane techniki i najnowsze trendy w oznaczaniu pozostałości pestycydów w żywności metodś chromatografii gazowej. Metrologia 2008, 13, 33–38. [Google Scholar]
- Walorczyk, S. Różne możliwości wykorzystania chromatografii gazowej połączonej ze spektrometrią mas w analizie pozostałości środków ochrony roślin. Progr. Plant Protect 2007, 47, 111–114. [Google Scholar]
- Korytar, P.; Janssen, H.G.; Matisova, E.; Brinkman, U.A.T. Practical fast gas chromatography: Methods, instrumentation and applications. Trends Anal. Chem 2002, 21, 558–572. [Google Scholar]
- Mastovska, K.; Lehotay, S.J. Practical approaches to fast gas chromatography-mass spectrometry. J. Chromatogr. A 2003, 1000, 153–180. [Google Scholar]
- Namieśnik, J. Modern trends in monitoring and analysis of environmental pollutants. Pol. J. Environ. Stud 2001, 10, 127–140. [Google Scholar]
- Beyer, A.; Biziuk, M. Methods for determining pesticides and polychlorinated biphenyls in food samples-problems and challenges. Crit. Rev. Food Sci 2008, 48, 888–904. [Google Scholar]
- Alder, L.; Greulich, K.; Kempe, G.; Vieth, B. Residue analysis of 500 high priority pesticides: Better by GC-MS or LC-MS/MS? Mass Spectrom. Rev 2006, 25, 838–865. [Google Scholar]


{kind=link}
{kind=link}
{kind=link}
| Type of pesticide | World market | US market | US percentage of world market [%] |
|---|---|---|---|
| 2006 | |||
| herbicides | 2018 | 498 | 25 |
| insecticides | 955 | 99 | 10 |
| fungicides | 519 | 73 | 14 |
| other | 1705 | 457 | 27 |
| total | 5197 | 1127 | 22 |
| 2007 | |||
| herbicides | 2096 | 531 | 25 |
| insecticides | 892 | 93 | 10 |
| fungicides | 518 | 70 | 14 |
| other | 1705 | 439 | 26 |
| total | 5211 | 1133 | 22 |
| Technique of Sample Preparation | Volume of Organic Solvent | Description | Literature |
|---|---|---|---|
| MLLE (micro liquid-liquid extraction) | about 1 mL per 1 L of sample | It is possible to decrease the consumption of organic solvents by miniaturization and proper design of extraction vessel. The most commonly used solvents for microextraction are dichloromethane, toluene and methyl-tert-butyl ether. | [32,33] |
| SDME (single drop microextraction) | 0.9–1.5 μL | The extraction phase is a drop of organic solvent (e.g., n-hexane, toluene, butyl acetate) suspended at the tip of microsyringe, so it is practically a solvent-free method. It can be carried out in two different ways by direct immersion (DI) or from the headspace (HS). Analyte isolation and preconcentration take place in a single step. The extraction process is assisted by mixing. When the extraction is complete, the microdroplet is directly injected into a gas chromatograph (GC) or high-performance liquid chromatograph (HPLC) for further analysis. The universality of SDME makes it widely applicable to the analysis of pesticides in samples with a complex composition containing target analytes in trace amounts. | [34–37] |
| CFME (continuousflow microextraction) | 1–5 μL | This technique is similar to SDME. The drop of extraction solvent is injected by microsyringe into a glass chamber (0.5 mL) and held at the outlet tip of a polyetheretherketone (PEEK) connecting tube. The sample solution flows past the tube and through the glass extraction unit to waste. Extraction takes place continuously between the organic drop and the flowing sample solution. Because the drop of solvent makes full contact with the sample solution, the technique achieves higher concentration factor than static SDME. | [30] |
| DLLME (dispersive liquid-liquid microextraction) | disperser solvent 0.5–2 mL; extraction solvent 10–50 μL | The mixture of extraction solvent (e.g., chlorobenzene, carbon tetrachloride, tetrachloroethylene, carbon disulfide) and disperser solvent (e.g., acetone or methanol) is rapidly injected into an aqueous sample, resulting in the formation of a cloudy solution. The DLLME procedure is very convenient to operate and extraction could be completed in a few seconds. DLLME has advantages of simplicity of operation, rapidity and low cost. DLLME can be coupled with GC and HPLC. The non-selective characteristic of the extraction solvents can be sometimes a disadvantage. Recently He et al. used as extraction solvent ionic liquid 1-octyl-3- methylimidazolium hexafluorophosphate ([C8MIM][PF6]) for the determination of organophosphorus pesticides in water sample. Ionic liquids belong to non-molecular solvents with unique properties such as negligible vapor pressure associated to a high thermal stability. Hydrophobic ionic liquids incorporating the imidazolium cation and hexafluorophosphate anion have higher density than water. Compared with commonly used solvents they are more compatible with reversed-phase HPLC due to the non-harmfulness to column. | [38–45] |
| HF(2)ME (hollow fiber-protected two-phase solvent microextraction) | 2–3 μL | The method is straightforward, quick, inexpensive and eliminates necessity of extract cleanup prior to final determination. Toluene, hexane or 1-octanol are usually used for the extraction of pesticides. It is based on the partition of analytes between the aqueous solution and the small quantity of organic solvent in a microporous tube (the rod configuration). The hollow fiber can be also in the U-shape configuration. The process is assisted by stirring. About 1–1.5 μL of extract is taken for further analysis using appropriate chromatographic techniques. For more complex matrices and moderately polar pesticides. Basheer et al. developed binary solvent based on HF(2)ME with GC-MS. The mixture (1:1) toluene: hexane was used as solvent. The limits of detection (LODs) were in the range of 0.3–11.4 ng L−1 and relative standard deviations (RSD) were 9–13%. This technique gave higher analytes enrichment, especially when applied to complex matrices (wastewater). | [40,46] |
| LPME-SFO (liquid-phase microextraction based on the solidification of a floating organic drop) | 10 μL | The small volume of an extraction solvent (usually 1-undecanol) is floated on the surface of aqueous solution. The process is assisted by stirring. After the extraction, the floated extractant droplet can be collected easily by solidifying it at low temperature. The solidified organic solvent can be melted quickly at room temperature, which is then determined by either chromatographic or spectrometric methods. The technique is cheap, quick and sensitive, but the rate of extraction is slightly slow. | [47,48] |
| MMLLE (microporous membrane liquid-liquid extraction) | 0.2 mL | Advantages of this technique compared to LLE are small sample volumes, the lack of emulsion formation, the clean extracts obtained and it can be coupled online to gas chromatography. The flat-sheet membrane extraction unit consisted of two blocks, one made of poly(tetrafluoroethylene) (PTFE) and the other of poly(etheretherketone) (PEEK). The membrane constitutes a barrier between two phases: acceptor (usually toluene) and the aqueous donor solution (sample). The donor solution is pumped to the donor channel of the membrane block, while the acceptor is stagnant during the extraction period. | [49,50] |
| LLSME (liquid-liquidsolid microextraction) | 6–100 μL | This technique combines the advantages of solid-phase microextraction and liquid-phase microextraction. The molecularly imprinted polymer (MIP)—coated silica fiber is protected with a length of porous polypropylene hollow fiber membrane which is filled with water-immiscible organic phase (usually toluene). This technique is a three-phase microextraction approach. It is fast, selective and sensitive method for trace analysis of pesticides in complex aqueous samples. | [51,52] |
| Technique of Sample Preparation | Volume of Organic Solvent | Description | Literature |
|---|---|---|---|
| SPE (solid-phase extraction) | <15 mL | The advantages of this method are: requires a lower volume of solvent than traditional LLE, involves simple manipulations which are not time consuming, the SPE cartridges can be used for short-term storage of the species and provides high enhancement factors proportional to the volume of water passed through the SPE cartridge. Conventional sorbents such as C18 silica, graphitized carbon black and macroporous polystyrene divinylbenzene (PS-DVB), show low retention for polar compounds. In order to improve the extraction efficiency for polar compounds, the development of new adsorbents and modification of the adsorbents by introducing the polar groups become a major research direction. Nanomaterials are one kind of novel adsorbents. Carbon nanotubes (CNTs), including single-walled carbon nanotubes (SWCNTs) and multi-walled carbon nanotubes (MWCNTs), are a kind of carbonaceous nanomaterial and have received significant attention in many fields. In recent years, molecular imprinting polymer (MIP) technology with high selectivity evolves rapidly. MIP technology is now well established for the preparation of tailor-made polymers with cavities capable to extract or clean-up of OPPs. | [53–61] |
| SPME (solid phase microextraction) | solvent-free extraction | This technique uses polymer-coated fibers to extract analytes from aqueous or gaseous samples. After extraction, the analytes are either desorbed thermally by exposing the fiber in the injection port of a GC or chemically desorbed and analyzed by LC. SPME does not require the use of organic solvents. It is quick, universal, sensitive and convenient for use in the field and is simply applied in sample preparation. However the fiber is comparatively expensive, fragile and has limited lifetime. The materials used for coating fibers include: polydimethylsiloxane (PDMS), polyacrylate (PA), and also mixtures of: polydimethylsiloxane and polydivinylbenzene (PDMS-DVB), carbowax and polydivinylbenzene (CW-DVB), carbowax and molecularly imprinted resin (CW-TP). Depending on where the fiber is situated in relation to the sample, SPME can be carried out in two different ways by direct immersion (DI) or from the headspace (HS). The advantage of this method is that the limited capacity of the adsorbent precludes column overloading. | [62–68] |
| SBSE (stir bar sorptive extraction) | solvent-free extraction | This techniques uses a 1.5 cm long glass magnetic stirrer coated with a thick layer of polydimethylsiloxane (PDMS) where sorption usually takes place. Its sorption capacity is a hundred times greater in comparison with sorption capacity of SPME fibers. Its main advantage is high sensitivity and a wide application range that includes volatile aromatics, halogenated solvents, polycyclic aromatic hydrocarbons, polychlorinated biphenyls, pesticides or organotion compounds. Because of the non-polar character of PDMS, the SBSE cannot be used to extract strong polar compounds unless derivatization was utilized. | [69–71] |
| Chemical Class | Number of Pesticides in That Class | Not Detected by GC-MS | Not Detected by LC-MS/MS |
|---|---|---|---|
| organophosphorus | 81 | 0 | 1 |
| carbamate | 43 | 17 | 1 |
| organochlorine | 40 | 0 | 33 |
| sulfonylurea | 26 | 26 | 0 |
| triazole | 24 | 1 | 0 |
| triazine | 23 | 6 | 0 |
| urea | 22 | 16 | 0 |
| pyrethroid | 19 | 0 | 2 |
| aryloxyphenoxy-propionate | 12 | 4 | 0 |
| aryloxyalkanoic acid | 10 | 9 | 0 |
| other | 200 | 56 | 12 |
| Total number | 500 | 135 | 49 |
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Stocka, J.; Tankiewicz, M.; Biziuk, M.; Namieśnik, J. Green Aspects of Techniques for the Determination of Currently Used Pesticides in Environmental Samples. Int. J. Mol. Sci. 2011, 12, 7785-7805. https://doi.org/10.3390/ijms12117785
Stocka J, Tankiewicz M, Biziuk M, Namieśnik J. Green Aspects of Techniques for the Determination of Currently Used Pesticides in Environmental Samples. International Journal of Molecular Sciences. 2011; 12(11):7785-7805. https://doi.org/10.3390/ijms12117785
Chicago/Turabian StyleStocka, Jolanta, Maciej Tankiewicz, Marek Biziuk, and Jacek Namieśnik. 2011. "Green Aspects of Techniques for the Determination of Currently Used Pesticides in Environmental Samples" International Journal of Molecular Sciences 12, no. 11: 7785-7805. https://doi.org/10.3390/ijms12117785
APA StyleStocka, J., Tankiewicz, M., Biziuk, M., & Namieśnik, J. (2011). Green Aspects of Techniques for the Determination of Currently Used Pesticides in Environmental Samples. International Journal of Molecular Sciences, 12(11), 7785-7805. https://doi.org/10.3390/ijms12117785