Roles of Oxidative Stress, Apoptosis, PGC-1α and Mitochondrial Biogenesis in Cerebral Ischemia

 ,
, 
{kind=link}
{kind=link}
Abstract
:1. Introduction
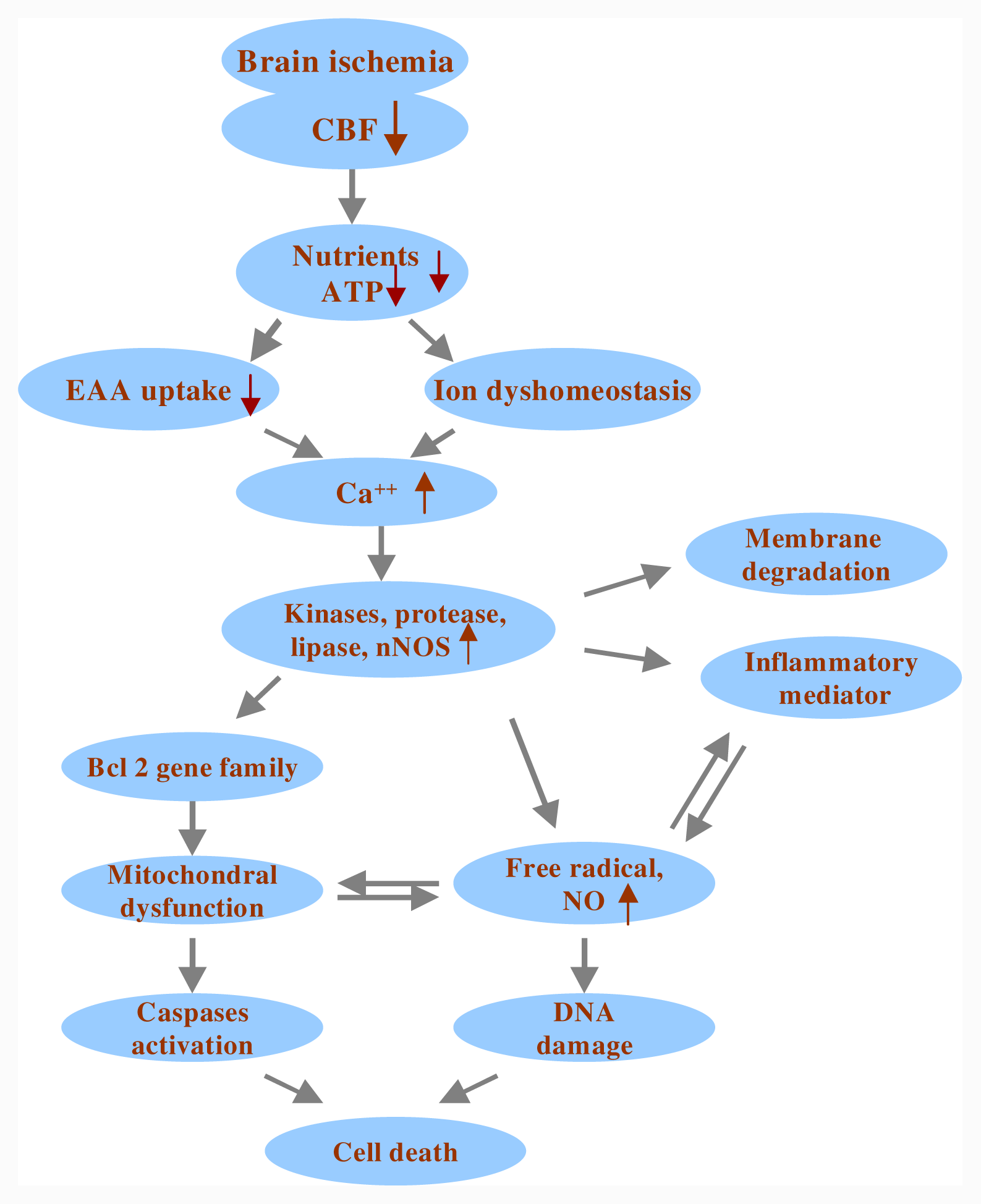
2. Ischemic Cascade Involving Mitochondria and ROS
2.1. Mitochondria and Oxidative Stress in Cerebral Ischemia
2.2. Mitochondria and Nitrosative Stress in Cerebral Ischemia
3. Pro- and Anti-Apoptotic Proteins Associated with Mitochondria-Dependent Apoptosis in Cerebral Ischemia
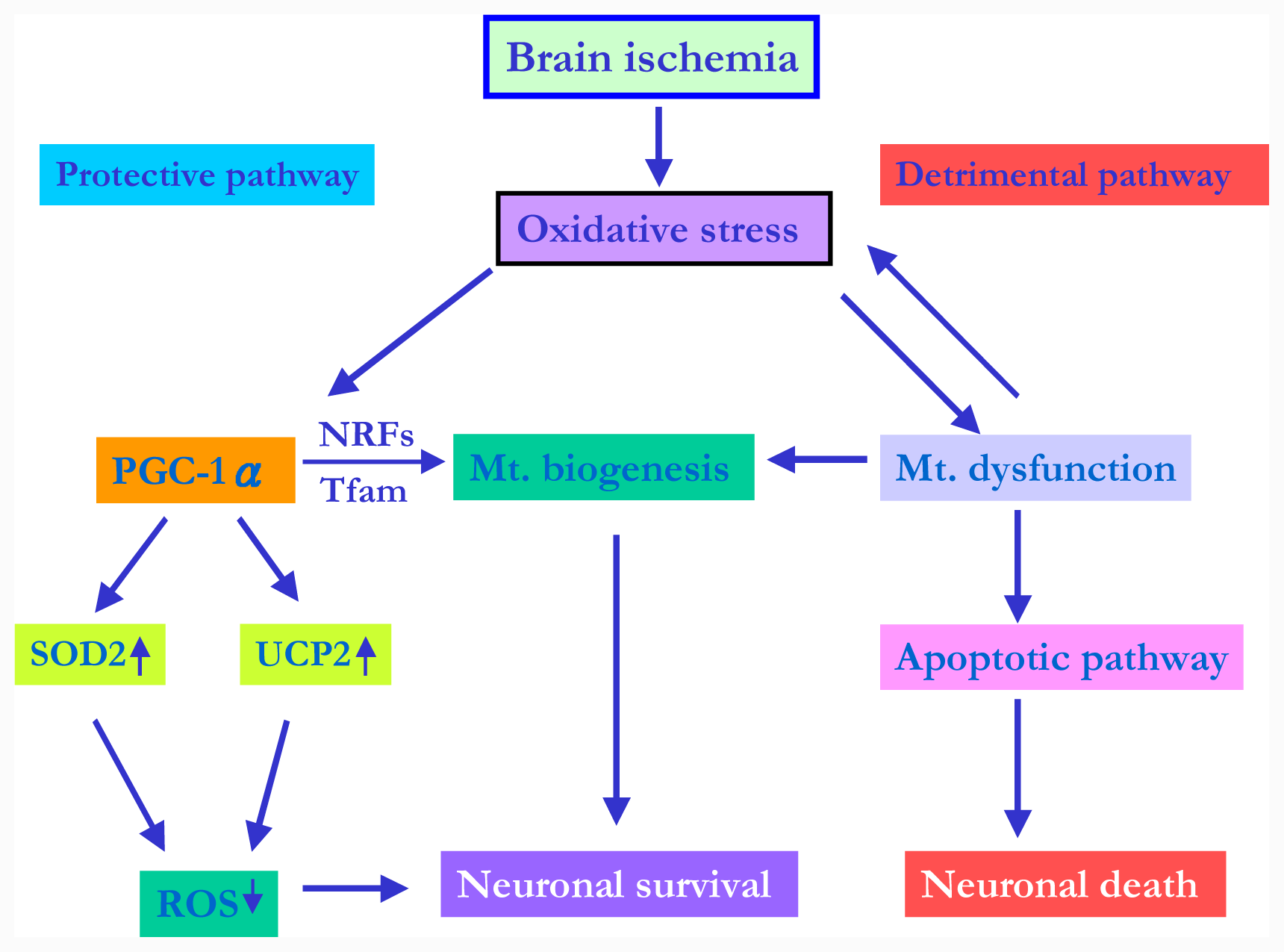
4. PGC-1α–an Endogenous Protective Mechanism Involving ROS and Mitochondria Biogenesis in Cerebral Ischemia
4.1. PGC-1α in Mitochondria-Related ROS Metabolism under Cerebral Ischemia
4.2. PGC-1α Signaling Pathway and Mitochondrial Biogenesis under Cerebral Ischemia
5. Conclusions
Acknowledgement
References
- Hatefi, Y. The mitochondrial electron transport and oxidative phosphorylation system. Annu. Rev. Biochem 1985, 54, 1015–1069. [Google Scholar]
- Ames, A, 3rd. CNS energy metabolism as related to function. Brain Res. Brain Res. Rev 2000, 34, 42–68. [Google Scholar]
- Niizuma, K; Yoshioka, H; Chen, H; Kim, GS; Jung, JE; Katsu, M; Okami, N; Chan, PH. Mitochondrial and apoptotic neuronal death signaling pathways in cerebral ischemia. Biochim. Biophys. Acta 2010, 1802, 92–99. [Google Scholar]
- Franklin, JL. Redox regulation of the intrinsic pathway in neuronal apoptosis. Antioxid. Redox Signal 2011, 14, 1437–1448. [Google Scholar]
- Lin, MT; Beal, MF. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature 2006, 443, 787–795. [Google Scholar]
- Cadenas, E; Davies, KJ. Mitochondrial free radical generation, oxidative stress, and aging. Free Radic. Biol. Med 2000, 29, 222–230. [Google Scholar]
- Valko, M; Leibfritz, D; Moncol, J; Cronin, MT; Mazur, M; Telser, J. Free radicals and antioxidants in normal physiological functions and human disease. Int. J. Biochem. Cell Biol 2007, 39, 44–84. [Google Scholar]
- Guglielmotto, M; Tamagno, E; Danni, O. Oxidative stress and hypoxia contribute to Alzheimer’s disease pathogenesis: Two sides of the same coin. ScientificWorldJournal 2009, 9, 781–791. [Google Scholar]
- Tamagno, E; Guglielmotto, M; Aragno, M; Borghi, R; Autelli, R; Giliberto, L; Muraca, G; Danni, O; Zhu, X; Smith, MA; et al. Oxidative stress activates a positive feedback between the gamma- and beta-secretase cleavages of the beta-amyloid precursor protein. J. Neurochem 2008, 104, 683–695. [Google Scholar]
- Chen, SD; Lee, JM; Yang, DI; Nassief, A; Hsu, CY. Combination therapy for ischemic stroke: Potential of neuroprotectants plus thrombolytics. Am. J. Cardiovasc. Drugs 2002, 2, 303–313. [Google Scholar]
- Niizuma, K; Endo, H; Chan, PH. Oxidative stress and mitochondrial dysfunction as determinants of ischemic neuronal death and survival. J. Neurochem 2009, 109, 133–138. [Google Scholar]
- Chen, SD; Lin, TK; Yang, DI; Lee, SY; Shaw, FZ; Liou, CW; Chuang, YC. Protective effects of peroxisome proliferator-activated receptors gamma coactivator-1alpha against neuronal cell death in the hippocampal CA1 subfield after transient global ischemia. J. Neurosci. Res 2010, 88, 605–613. [Google Scholar]
- St-Pierre, J; Drori, S; Uldry, M; Silvaggi, JM; Rhee, J; Jager, S; Handschin, C; Zheng, K; Lin, J; Yang, W; et al. Suppression of reactive oxygen species and neurodegeneration by the PGC-1 transcriptional coactivators. Cell 2006, 127, 397–408. [Google Scholar]
- Chen, SD; Lin, TK; Lin, JW; Yang, DI; Lee, SY; Shaw, FZ; Liou, CW; Chuang, YC. Activation of calcium/calmodulin-dependent protein kinase IV and peroxisome proliferator-activated receptor gamma coactivator-1alpha signaling pathway protects against neuronal injury and promotes mitochondrial biogenesis in the hippocampal CA1 subfield after transient global ischemia. J. Neurosci. Res 2010, 88, 3144–3154. [Google Scholar]
- Yin, W; Signore, AP; Iwai, M; Cao, G; Gao, Y; Chen, J. Rapidly increased neuronal mitochondrial biogenesis after hypoxic-ischemic brain injury. Stroke 2008, 39, 3057–3063. [Google Scholar]
- Lipton, P. Ischemic cell death in brain neurons. Physiol. Rev 1999, 79, 1431–1568. [Google Scholar]
- Lee, JM; Zipfel, GJ; Choi, DW. The changing landscape of ischaemic brain injury mechanisms. Nature 1999, 399, A7–A14. [Google Scholar]
- Moskowitz, MA; Lo, EH; Iadecola, C. The science of stroke: Mechanisms in search of treatments. Neuron 2010, 67, 181–198. [Google Scholar]
- Lennon, SV; Martin, SJ; Cotter, TG. Dose-dependent induction of apoptosis in human tumour cell lines by widely diverging stimuli. Cell Prolif 1991, 24, 203–214. [Google Scholar]
- Novo, E; Parola, M. Redox mechanisms in hepatic chronic wound healing and fibrogenesis. Fibrog. Tissue Repair 2008, 1, 5. [Google Scholar]
- Chen, SD; Wu, HY; Yang, DI; Lee, SY; Shaw, FZ; Lin, TK; Liou, CW; Chuang, YC. Effects of rosiglitazone on global ischemia-induced hippocampal injury and expression of mitochondrial uncoupling protein 2. Biochem. Biophys. Res. Commun 2006, 351, 198–203. [Google Scholar]
- Bayir, H; Kagan, VE. Bench-to-bedside review: Mitochondrial injury, oxidative stress and apoptosis--there is nothing more practical than a good theory. Crit. Care 2008, 12, 206. [Google Scholar]
- Leonard, JV; Schapira, AH. Mitochondrial respiratory chain disorders II: Neurodegenerative disorders and nuclear gene defects. Lancet 2000, 355, 389–394. [Google Scholar]
- Patel, M. Mitochondrial dysfunction and oxidative stress: Cause and consequence of epileptic seizures. Free Radic. Biol. Med 2004, 37, 1951–1962. [Google Scholar]
- Balaban, RS; Nemoto, S; Finkel, T. Mitochondria, oxidants, and aging. Cell 2005, 120, 483–495. [Google Scholar]
- Iadecola, C. Bright and dark sides of nitric oxide in ischemic brain injury. Trends Neurosci 1997, 20, 132–139. [Google Scholar]
- Lipton, SA. Neuronal protection and destruction by NO. Cell Death Differ 1999, 6, 943–951. [Google Scholar]
- Kunz, A; Park, L; Abe, T; Gallo, EF; Anrather, J; Zhou, P; Iadecola, C. Neurovascular protection by ischemic tolerance: role of nitric oxide and reactive oxygen species. J. Neurosci 2007, 27, 7083–7093. [Google Scholar]
- Giulivi, C; Poderoso, JJ; Boveris, A. Production of nitric oxide by mitochondria. J. Biol. Chem 1998, 273, 11038–11043. [Google Scholar]
- Tatoyan, A; Giulivi, C. Purification and characterization of a nitric-oxide synthase from rat liver mitochondria. J. Biol. Chem 1998, 273, 11044–11048. [Google Scholar]
- Kanai, A; Peterson, J. Function and regulation of mitochondrially produced nitric oxide in cardiomyocytes. Am. J. Physiol. Heart Circ Physiol 2004, 286, H11–H12. [Google Scholar]
- Navarro, A; Boveris, A. Mitochondrial nitric oxide synthase, mitochondrial brain dysfunction in aging, and mitochondria-targeted antioxidants. Adv. Drug Deliv. Rev 2008, 60, 1534–1544. [Google Scholar]
- Finocchietto, PV; Franco, MC; Holod, S; Gonzalez, AS; Converso, DP; Arciuch, VG; Serra, MP; Poderoso, JJ; Carreras, MC. Mitochondrial nitric oxide synthase: A masterpiece of metabolic adaptation, cell growth, transformation, and death. Exp. Biol. Med. (Maywood) 2009, 234, 1020–1028. [Google Scholar]
- Ignarro, LJ. Heart mtNOS, a key mediator of oxidative injury in ischemia/reperfusion. J. Mol. Cell. Cardiol 2007, 43, 409–410. [Google Scholar]
- Valdez, LB; Zaobornyj, T; Bombicino, S; Iglesias, DE; Boveris, A; Donato, M; D’Annunzio, V; Buchholz, B; Gelpi, RJ. Complex I syndrome in myocardial stunning and the effect of adenosine. Free Radic. Biol. Med 2011, 51, 1203–1212. [Google Scholar]
- Gu, Z; Nakamura, T; Lipton, SA. Redox reactions induced by nitrosative stress mediate protein misfolding and mitochondrial dysfunction in neurodegenerative diseases. Mol. Neurobiol 2010, 41, 55–72. [Google Scholar]
- Chen, SD; Chang, AY; Chuang, YC. The potential role of mitochondrial dysfunction in seizure-associated cell death in the hippocampus and epileptogenesis. J. Bioenerg. Biomembr 2010, 42, 461–465. [Google Scholar]
- Lee, BI; Chan, PH; Kim, GW. Metalloporphyrin-based superoxide dismutase mimic attenuates the nuclear translocation of apoptosis-inducing factor and the subsequent DNA fragmentation after permanent focal cerebral ischemia in mice. Stroke 2005, 36, 2712–2717. [Google Scholar]
- Fang, M; Li, J; Tiu, SC; Zhang, L; Wang, M; Yew, DT. N-methyl-D-aspartate receptor and apoptosis in Alzheimer’s disease and multiinfarct dementia. J. Neurosci. Res 2005, 81, 269–274. [Google Scholar]
- Endo, H; Kamada, H; Nito, C; Nishi, T; Chan, PH. Mitochondrial translocation of p53 mediates release of cytochrome c and hippocampal CA1 neuronal death after transient global cerebral ischemia in rats. J. Neurosci 2006, 26, 7974–7983. [Google Scholar]
- Sairanen, T; Szepesi, R; Karjalainen-Lindsberg, ML; Saksi, J; Paetau, A; Lindsberg, PJ. Neuronal caspase-3 and PARP-1 correlate differentially with apoptosis and necrosis in ischemic human stroke. Acta Neuropathol 2009, 118, 541–552. [Google Scholar]
- MacManus, JP; Buchan, AM. Apoptosis after experimental stroke: fact or fashion? J. Neurotrauma 2000, 17, 899–914. [Google Scholar]
- Zhao, H; Steinberg, GK; Sapolsky, RM. General versus specific actions of mild-moderate hypothermia in attenuating cerebral ischemic damage. J. Cereb. Blood Flow Metab 2007, 27, 1879–1894. [Google Scholar]
- Leist, M; Single, B; Castoldi, AF; Kuhnle, S; Nicotera, P. Intracellular adenosine triphosphate (ATP) concentration: A switch in the decision between apoptosis and necrosis. J. Exp. Med 1997, 185, 1481–1486. [Google Scholar]
- Ginsberg, MD. The new language of cerebral ischemia. AJNR Am. J. Neuroradiol 1997, 18, 1435–1445. [Google Scholar]
- Broughton, BR; Reutens, DC; Sobey, CG. Apoptotic mechanisms after cerebral ischemia. Stroke 2009, 40, e331–e339. [Google Scholar]
- Li, L; Peng, L; Zuo, Z. Isoflurane preconditioning increases B-cell lymphoma-2 expression and reduces cytochrome c release from the mitochondria in the ischemic penumbra of rat brain. Eur. J. Pharmacol 2008, 586, 106–113. [Google Scholar]
- Van Hemelrijck, A; Hachimi-Idrissi, S; Sarre, S; Ebinger, G; Michotte, Y. Post-ischaemic mild hypothermia inhibits apoptosis in the penumbral region by reducing neuronal nitric oxide synthase activity and thereby preventing endothelin-1-induced hydroxyl radical formation. Eur. J. Neurosci 2005, 22, 1327–1337. [Google Scholar]
- Green, DR; Kroemer, G. The pathophysiology of mitochondrial cell death. Science 2004, 305, 626–629. [Google Scholar]
- Kroemer, G. Mitochondrial control of apoptosis: An overview. Biochem. Soc. Symp 1999, 66, 1–15. [Google Scholar]
- Kitagawa, K; Matsumoto, M; Tsujimoto, Y; Ohtsuki, T; Kuwabara, K; Matsushita, K; Yang, G; Tanabe, H; Martinou, JC; Hori, M; et al. Amelioration of hippocampal neuronal damage after global ischemia by neuronal overexpression of BCL-2 in transgenic mice. Stroke 1998, 29, 2616–2621. [Google Scholar]
- Liu, D; Lu, C; Wan, R; Auyeung, WW; Mattson, MP. Activation of mitochondrial ATP-dependent potassium channels protects neurons against ischemia-induced death by a mechanism involving suppression of Bax translocation and cytochrome c release. J. Cereb. Blood Flow Metab 2002, 22, 431–443. [Google Scholar]
- Zhao, H; Yenari, MA; Cheng, D; Sapolsky, RM; Steinberg, GK. Bcl-2 overexpression protects against neuron loss within the ischemic margin following experimental stroke and inhibits cytochrome c translocation and caspase-3 activity. J. Neurochem 2003, 85, 1026–1036. [Google Scholar]
- Song, YS; Lee, YS; Narasimhan, P; Chan, PH. Reduced oxidative stress promotes NF-kappaB-mediated neuroprotective gene expression after transient focal cerebral ischemia: Lymphocytotrophic cytokines and antiapoptotic factors. J. Cereb. Blood Flow Metab 2007, 27, 764–775. [Google Scholar]
- Rami, A; Bechmann, I; Stehle, JH. Exploiting endogenous anti-apoptotic proteins for novel therapeutic strategies in cerebral ischemia. Prog. Neurobiol 2008, 85, 273–296. [Google Scholar]
- Saelens, X; Festjens, N; Vande Walle, L; van Gurp, M; van Loo, G; Vandenabeele, P. Toxic proteins released from mitochondria in cell death. Oncogene 2004, 23, 2861–2874. [Google Scholar]
- Webster, KA; Graham, RM; Thompson, JW; Spiga, MG; Frazier, DP; Wilson, A; Bishopric, NH. Redox stress and the contributions of BH3-only proteins to infarction. Antioxid. Redox Signal 2006, 8, 1667–1676. [Google Scholar]
- Inta, I; Paxian, S; Maegele, I; Zhang, W; Pizzi, M; Spano, P; Sarnico, I; Muhammad, S; Herrmann, O; Inta, D; et al. Bim and Noxa are candidates to mediate the deleterious effect of the NF-kappa B subunit RelA in cerebral ischemia. J. Neurosci 2006, 26, 12896–12903. [Google Scholar]
- Niizuma, K; Endo, H; Nito, C; Myer, DJ; Chan, PH. Potential role of PUMA in delayed death of hippocampal CA1 neurons after transient global cerebral ischemia. Stroke 2009, 40, 618–625. [Google Scholar]
- Engel, T; Plesnila, N; Prehn, JH; Henshall, DC. In vivo contributions of BH3-only proteins to neuronal death following seizures, ischemia, and traumatic brain injury. J. Cereb. Blood Flow Metab 2011, 31, 1196–1210. [Google Scholar]
- Crompton, M. Mitochondrial intermembrane junctional complexes and their role in cell death. J. Physiol 2000, 529, 11–21. [Google Scholar]
- Li, P; Nijhawan, D; Budihardjo, I; Srinivasula, SM; Ahmad, M; Alnemri, ES; Wang, X. Cytochrome c and dATP-dependent formation of Apaf-1/caspase-9 complex initiates an apoptotic protease cascade. Cell 1997, 91, 479–489. [Google Scholar]
- Zou, H; Henzel, WJ; Liu, X; Lutschg, A; Wang, X. Apaf-1, a human protein homologous to C. elegans CED-4, participates in cytochrome c-dependent activation of caspase-3. Cell 1997, 90, 405–413. [Google Scholar]
- Yoshida, H; Kong, YY; Yoshida, R; Elia, AJ; Hakem, A; Hakem, R; Penninger, JM; Mak, TW. Apaf1 is required for mitochondrial pathways of apoptosis and brain development. Cell 1998, 94, 739–750. [Google Scholar]
- Ferrer, I; Friguls, B; Dalfo, E; Justicia, C; Planas, AM. Caspase-dependent and caspase-independent signalling of apoptosis in the penumbra following middle cerebral artery occlusion in the adult rat. Neuropathol. Appl. Neurobiol 2003, 29, 472–481. [Google Scholar]
- Saito, A; Hayashi, T; Okuno, S; Ferrand-Drake, M; Chan, PH. Interaction between XIAP and Smac/DIABLO in the mouse brain after transient focal cerebral ischemia. J. Cereb. Blood Flow Metab 2003, 23, 1010–1019. [Google Scholar]
- Culmsee, C; Zhu, C; Landshamer, S; Becattini, B; Wagner, E; Pellecchia, M; Blomgren, K; Plesnila, N. Apoptosis-inducing factor triggered by poly(ADP-ribose) polymerase and Bid mediates neuronal cell death after oxygen-glucose deprivation and focal cerebral ischemia. J. Neurosci 2005, 25, 10262–10272. [Google Scholar]
- Deveraux, QL; Reed, JC. IAP family proteins--suppressors of apoptosis. Genes Dev 1999, 13, 239–252. [Google Scholar]
- Yin, KJ; Lee, JM; Chen, SD; Xu, J; Hsu, CY. Amyloid-beta induces Smac release via AP-1/Bim activation in cerebral endothelial cells. J. Neurosci 2002, 22, 9764–9770. [Google Scholar]
- Chan, PH. Mitochondria and neuronal death/survival signaling pathways in cerebral ischemia. Neurochem. Res 2004, 29, 1943–1949. [Google Scholar]
- Handschin, C; Spiegelman, BM. Peroxisome proliferator-activated receptor gamma coactivator 1 coactivators, energy homeostasis, and metabolism. Endocr. Rev 2006, 27, 728–735. [Google Scholar]
- Schon, EA; Manfredi, G. Neuronal degeneration and mitochondrial dysfunction. J. Clin. Invest 2003, 111, 303–312. [Google Scholar]
- Lin, J; Wu, PH; Tarr, PT; Lindenberg, KS; St-Pierre, J; Zhang, CY; Mootha, VK; Jager, S; Vianna, CR; Reznick, RM; et al. Defects in adaptive energy metabolism with CNS-linked hyperactivity in PGC-1alpha null mice. Cell 2004, 119, 121–135. [Google Scholar]
- Wareski, P; Vaarmann, A; Choubey, V; Safiulina, D; Liiv, J; Kuum, M; Kaasik, A. PGC-1{alpha} and PGC-1{beta} regulate mitochondrial density in neurons. J. Biol. Chem 2009, 284, 21379–21385. [Google Scholar]
- Chinetti, G; Fruchart, JC; Staels, B. Peroxisome proliferator-activated receptors (PPARs): Nuclear receptors at the crossroads between lipid metabolism and inflammation. Inflamm. Res 2000, 49, 497–505. [Google Scholar]
- Delerive, P; Fruchart, JC; Staels, B. Peroxisome proliferator-activated receptors in inflammation control. J. Endocrinol 2001, 169, 453–459. [Google Scholar]
- Ricote, M; Li, AC; Willson, TM; Kelly, CJ; Glass, CK. The peroxisome proliferator-activated receptor-gamma is a negative regulator of macrophage activation. Nature 1998, 391, 79–82. [Google Scholar]
- Fong, WH; Tsai, HD; Chen, YC; Wu, JS; Lin, TN. Anti-apoptotic actions of PPAR-gamma against ischemic stroke. Mol. Neurobiol 2010, 41, 180–186. [Google Scholar]
- Wu, JS; Cheung, WM; Tsai, YS; Chen, YT; Fong, WH; Tsai, HD; Chen, YC; Liou, JY; Shyue, SK; Chen, JJ; et al. Ligand-activated peroxisome proliferator-activated receptor-gamma protects against ischemic cerebral infarction and neuronal apoptosis by 14-3-3 epsilon upregulation. Circulation 2009, 119, 1124–1134. [Google Scholar]
- Yin, KJ; Deng, Z; Hamblin, M; Zhang, J; Chen, YE. Vascular PPARdelta protects against stroke-induced brain injury. Arterioscler. Thromb. Vasc. Biol 2011, 31, 574–581. [Google Scholar]
- Yin, KJ; Deng, Z; Hamblin, M; Xiang, Y; Huang, H; Zhang, J; Jiang, X; Wang, Y; Chen, YE. Peroxisome proliferator-activated receptor delta regulation of miR-15a in ischemia-induced cerebral vascular endothelial injury. J. Neurosci 2010, 30, 6398–408. [Google Scholar]
- Finck, BN; Kelly, DP. PGC-1 coactivators: Inducible regulators of energy metabolism in health and disease. J. Clin. Invest 2006, 116, 615–622. [Google Scholar]
- Knutti, D; Kralli, A. PGC-1, a versatile coactivator. Trends Endocrinol. Metab 2001, 12, 360–365. [Google Scholar]
- Puigserver, P; Spiegelman, BM. Peroxisome proliferator-activated receptor-gamma coactivator 1 alpha (PGC-1 alpha): transcriptional coactivator and metabolic regulator. Endocr. Rev 2003, 24, 78–90. [Google Scholar]
- Arany, Z; Foo, SY; Ma, Y; Ruas, JL; Bommi-Reddy, A; Girnun, G; Cooper, M; Laznik, D; Chinsomboon, J; Rangwala, SM; et al. HIF-independent regulation of VEGF and angiogenesis by the transcriptional coactivator PGC-1alpha. Nature 2008, 451, 1008–1012. [Google Scholar]
- Gutsaeva, DR; Carraway, MS; Suliman, HB; Demchenko, IT; Shitara, H; Yonekawa, H; Piantadosi, CA. Transient hypoxia stimulates mitochondrial biogenesis in brain subcortex by a neuronal nitric oxide synthase-dependent mechanism. J. Neurosci 2008, 28, 2015–2024. [Google Scholar]
- Storey, KB. Mammalian hibernation. Transcriptional and translational controls. Adv. Exp. Med. Biol 2003, 543, 21–38. [Google Scholar]
- Negre-Salvayre, A; Hirtz, C; Carrera, G; Cazenave, R; Troly, M; Salvayre, R; Penicaud, L; Casteilla, L. A role for uncoupling protein-2 as a regulator of mitochondrial hydrogen peroxide generation. FASEB J 1997, 11, 809–815. [Google Scholar]
- Raha, S; McEachern, GE; Myint, AT; Robinson, BH. Superoxides from mitochondrial complex III: the role of manganese superoxide dismutase. Free Radic. Biol. Med 2000, 29, 170–180. [Google Scholar]
- Chan, PH. Reactive oxygen radicals in signaling and damage in the ischemic brain. J. Cereb. Blood Flow Metab 2001, 21, 2–14. [Google Scholar]
- Mattiasson, G; Shamloo, M; Gido, G; Mathi, K; Tomasevic, G; Yi, S; Warden, CH; Castilho, RF; Melcher, T; Gonzalez-Zulueta, M; et al. Uncoupling protein-2 prevents neuronal death and diminishes brain dysfunction after stroke and brain trauma. Nat. Med 2003, 9, 1062–1068. [Google Scholar]
- Deierborg, T; Wieloch, T; Diano, S; Warden, CH; Horvath, TL; Mattiasson, G. Overexpression of UCP2 protects thalamic neurons following global ischemia in the mouse. J. Cereb. Blood Flow Metab 2008, 28, 1186–1195. [Google Scholar]
- Keller, JN; Kindy, MS; Holtsberg, FW; St Clair, DK; Yen, HC; Germeyer, A; Steiner, SM; Bruce-Keller, AJ; Hutchins, JB; Mattson, MP. Mitochondrial manganese superoxide dismutase prevents neural apoptosis and reduces ischemic brain injury: Suppression of peroxynitrite production, lipid peroxidation, and mitochondrial dysfunction. J. Neurosci 1998, 18, 687–697. [Google Scholar]
- Noshita, N; Sugawara, T; Fujimura, M; Morita-Fujimura, Y; Chan, PH. Manganese Superoxide Dismutase Affects Cytochrome c Release and Caspase-9 Activation After Transient Focal Cerebral Ischemia in Mice. J. Cereb. Blood Flow Metab 2001, 21, 557–567. [Google Scholar]
- Valle, I; Alvarez-Barrientos, A; Arza, E; Lamas, S; Monsalve, M. PGC-1alpha regulates the mitochondrial antioxidant defense system in vascular endothelial cells. Cardiovasc. Res 2005, 66, 562–573. [Google Scholar]
- Diaz, F; Moraes, CT. Mitochondrial biogenesis and turnover. Cell Calcium 2008, 44, 24–35. [Google Scholar]
- Lee, HC; Wei, YH. Mitochondrial biogenesis and mitochondrial DNA maintenance of mammalian cells under oxidative stress. Int. J. Biochem. Cell Biol 2005, 37, 822–834. [Google Scholar]
- Scarpulla, RC. Transcriptional paradigms in mammalian mitochondrial biogenesis and function. Physiol. Rev 2008, 88, 611–638. [Google Scholar]
- Ekstrand, MI; Falkenberg, M; Rantanen, A; Park, CB; Gaspari, M; Hultenby, K; Rustin, P; Gustafsson, CM; Larsson, NG. Mitochondrial transcription factor A regulates mtDNA copy number in mammals. Hum. Mol. Genet 2004, 13, 935–944. [Google Scholar]
- Vosler, PS; Graham, SH; Wechsler, LR; Chen, J. Mitochondrial targets for stroke: focusing basic science research toward development of clinically translatable therapeutics. Stroke 2009, 40, 3149–3155. [Google Scholar]

© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Chen, S.-D.; Yang, D.-I.; Lin, T.-K.; Shaw, F.-Z.; Liou, C.-W.; Chuang, Y.-C. Roles of Oxidative Stress, Apoptosis, PGC-1α and Mitochondrial Biogenesis in Cerebral Ischemia. Int. J. Mol. Sci. 2011, 12, 7199-7215. https://doi.org/10.3390/ijms12107199
Chen S-D, Yang D-I, Lin T-K, Shaw F-Z, Liou C-W, Chuang Y-C. Roles of Oxidative Stress, Apoptosis, PGC-1α and Mitochondrial Biogenesis in Cerebral Ischemia. International Journal of Molecular Sciences. 2011; 12(10):7199-7215. https://doi.org/10.3390/ijms12107199
Chicago/Turabian StyleChen, Shang-Der, Ding-I Yang, Tsu-Kung Lin, Fu-Zen Shaw, Chia-Wei Liou, and Yao-Chung Chuang. 2011. "Roles of Oxidative Stress, Apoptosis, PGC-1α and Mitochondrial Biogenesis in Cerebral Ischemia" International Journal of Molecular Sciences 12, no. 10: 7199-7215. https://doi.org/10.3390/ijms12107199
APA StyleChen, S.-D., Yang, D.-I., Lin, T.-K., Shaw, F.-Z., Liou, C.-W., & Chuang, Y.-C. (2011). Roles of Oxidative Stress, Apoptosis, PGC-1α and Mitochondrial Biogenesis in Cerebral Ischemia. International Journal of Molecular Sciences, 12(10), 7199-7215. https://doi.org/10.3390/ijms12107199