MALDI Imaging Mass Spectrometry (MALDI-IMS)―Application of Spatial Proteomics for Ovarian Cancer Classification and Diagnosis


Abstract
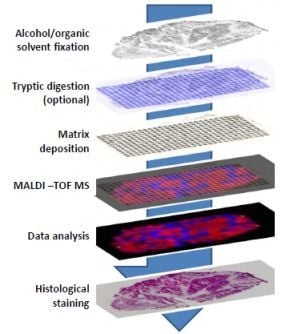
:
1. Epidemiology of Ovarian Cancer
2. Early Detection of Ovarian Cancer
3. Molecular Classification of Ovarian Carcinomas
4. Application of Proteomics to Ovarian Cancer
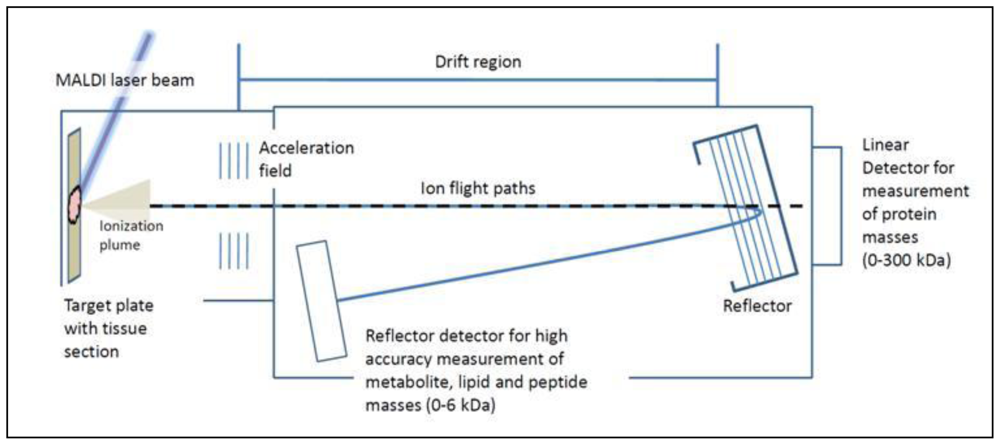
5. Tissue Analysis by Mass Spectrometry
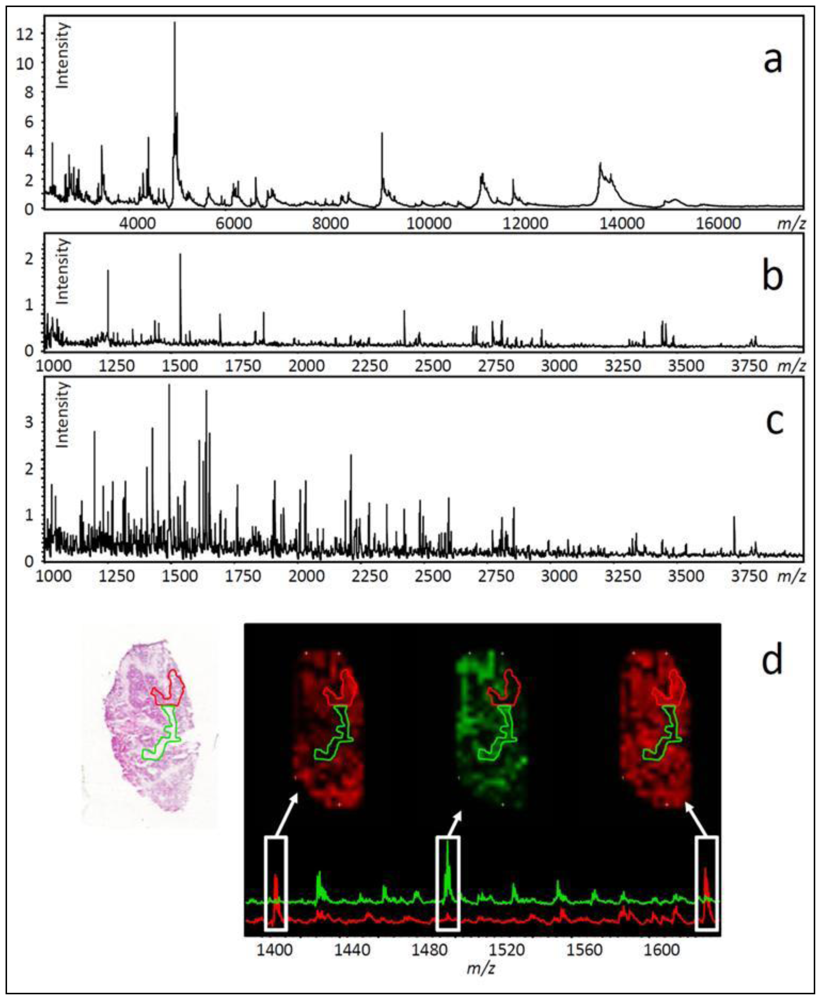
- Several hundred molecular features can be measured in a single experiment (see Figure 3a–c).
- No preliminary knowledge about tissue composition is required.
- No antibodies are required.
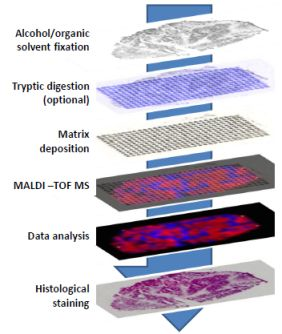
6. Methods for in Situ MALDI-TOF Analysis of Ovarian Cancer Tissue
7. Profiling Cancer Tissues Using MALDI-TOF MS
8. Profiling vs. Imaging
9. Software for Data Analysis
10. Automated Sample Preparation for Imaging Cancer Tissues
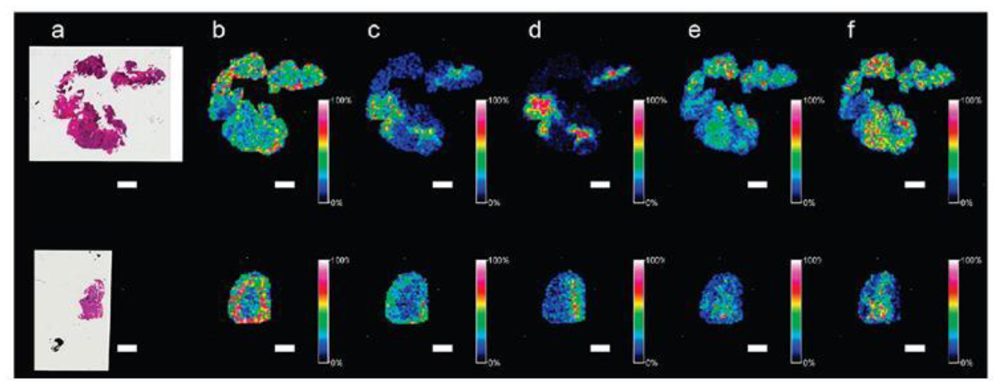
11. Peptide Imaging Provides Data Complementary to Protein Imaging
12. Using Histology to Guide Imaging Mass Spectrometry
13. Ovarian Cancer Biomarker Discovery Using Imaging Mass Spectrometry
14. Application of Tryptic Digestion to Formalin-Fixed Paraffin Embedded Ovarian Tissues
15. Conclusions and Future Prospects
References
- Hennessy, BT; Coleman, RL; Markman, M. Ovarian cancer. Lancet 2009, 374, 1371–1382. [Google Scholar]
- Menon, U; Gentry-Maharaj, A; Hallett, R; Ryan, A; Burnell, M; Sharma, A; Lewis, S; Davies, S; Philpott, S; Lopes, A; Godfrey, K; Oram, D; Herod, J; Williamson, K; Seif, MW; Scott, I; Mould, T; Woolas, R; Murdoch, J; Dobbs, S; Amso, NN; Leeson, S; Cruickshank, D; McGuire, A; Campbell, S; Fallowfield, L; Singh, N; Dawnay, A; Skates, SJ; Parmar, M; Jacobs, I. Sensitivity and specificity of multimodal and ultrasound screening for ovarian cancer, and stage distribution of detected cancers: Results of the prevalence screen of the UK Collaborative Trial of Ovarian Cancer Screening (UKCTOCS). Lancet Oncol 2009, 10, 327–340. [Google Scholar]
- Moore, RG; MacLaughlan, S; Bast, RC, Jr. Current state of biomarker development for clinical application in epithelial ovarian cancer. Gynecol. Oncol 2010, 116, 240–245. [Google Scholar]
- Mor, G; Visintin, I; Lai, Y; Zhao, H; Schwartz, P; Rutherford, T; Yue, L; Bray-Ward, P; Ward, DC. Serum protein markers for early detection of ovarian cancer. Proc Natl Acad Sci USA 2005, 102, 7677–7682. [Google Scholar]
- Muller, CY. Doctor, should I get this new ovarian cancer test-OVA1? Obstet. Gynecol 2010, 116, 246–247. [Google Scholar]
- Anderson, NL; Anderson, NG. The human plasma proteome: History, character, and diagnostic prospects. Mol Cell Proteomics 2002, 1, 845–867. [Google Scholar]
- Poschmann, G; Sitek, B; Sipos, B; Hamacher, M; Vonend, O; Meyer, HE; Stuhler, K. Cell-based proteome analysis: The first stage in the pipeline for biomarker discovery. Biochim. Biophys Acta 2009, 1794, 1309–1316. [Google Scholar]
- Eriksson, J; Fenyo, D. Improving the success rate of proteome analysis by modeling protein-abundance distributions and experimental designs. Nat. Biotechnol 2007, 25, 651–655. [Google Scholar]
- Cancer of the ovary—SEER stat fact sheets. Surveillance Epidemiology and End Results. Available online: http://seer.cancer.gov/statfacts/html/ovary.html accessed on 17 January 2010.
- Benedet, JL; Bender, H; Jones, H, III; Ngan, HY; Pecorelli, S. FIGO staging classifications and clinical practice guidelines in the management of gynecologic cancers. FIGO Committee on Gynecologic Oncology. Int. J. Gynaecol. Obstet 2000, 70, 209–262. [Google Scholar]
- Kosary, CL. FIGO stage, histology, histologic grade, age and race as prognostic factors in determining survival for cancers of the female gynecological system: An analysis of 1973–1987 SEER cases of cancers of the endometrium, cervix, ovary, vulva, and vagina. Semin. Surg. Oncol 1994, 10, 31–46. [Google Scholar]
- Shimizu, Y; Kamoi, S; Amada, S; Akiyama, F; Silverberg, SG. Toward the development of a universal grading system for ovarian epithelial carcinoma: Testing of a proposed system in a series of 461 patients with uniform treatment and follow-up. Cancer 1998, 82, 893–901. [Google Scholar]
- Sagae, S; Saito, T; Satoh, M; Ikeda, T; Kimura, S; Mori, M; Sato, N; Kudo, R. The reproducibility of a binary tumor grading system for uterine endometrial endometrioid carcinoma, compared with FIGO system and nuclear grading. Oncology 2004, 67, 344–350. [Google Scholar]
- Vang, R; Shih Ie, M; Kurman, RJ. Ovarian low-grade and high-grade serous carcinoma: Pathogenesis, clinicopathologic and molecular biologic features, and diagnostic problems. Adv. Anat. Pathol 2009, 16, 267–282. [Google Scholar]
- Ali-Fehmi, R; Semaan, A; Sethi, S; Arabi, H; Bandyopadhyay, S; Hussein, YR; Diamond, MP; Saed, G; Morris, RT; Munkarah, AR. Molecular typing of epithelial ovarian carcinomas using inflammatory markers. Cancer 2010. [Google Scholar] [CrossRef]
- Cho, KR. Ovarian cancer update: Lessons from morphology, molecules, and mice. Arch. Pathol. Lab. Med 2009, 133, 1775–1781. [Google Scholar]
- Lu, P; Vogel, C; Wang, R; Yao, X; Marcotte, EM. Absolute protein expression profiling estimates the relative contributions of transcriptional and translational regulation. Nat. Biotechnol 2007, 25, 117–124. [Google Scholar]
- Gygi, SP; Rochon, Y; Franza, BR; Aebersold, R. Correlation between protein and mRNA abundance in yeast. Mol. Cell Biol 1999, 19, 1720–1730. [Google Scholar]
- Kim, H; Wu, R; Cho, KR; Thomas, DG; Gossner, G; Liu, JR; Giordano, TJ; Shedden, KA; Misek, DE; Lubman, DM. Comparative proteomic analysis of low stage and high stage endometrioid ovarian adenocarcinomas. Proteomics Clin. Appl 2008, 2, 571–584. [Google Scholar]
- Washburn, MP; Wolters, D; Yates, JR, III. Large-scale analysis of the yeast proteome by multidimensional protein identification technology. Nat. Biotechnol 2001, 19, 242–247. [Google Scholar]
- Wilkins, MR; Pasquali, C; Appel, RD; Ou, K; Golaz, O; Sanchez, JC; Yan, JX; Gooley, AA; Hughes, G; Humphery-Smith, I; Williams, KL; Hochstrasser, DF. From proteins to proteomes: Large scale protein identification by two-dimensional electrophoresis and amino acid analysis. Biotechnology 1996, 14, 61–65. [Google Scholar]
- Mitulovic, G; Mechtler, K. HPLC techniques for proteomics analysis—a short overview of latest developments. Brief. Funct Genomic Proteomic 2006, 5, 249–260. [Google Scholar]
- Condina, MR; Gustafsson, JO; Klingler-Hoffmann, M; Bagley, CJ; McColl, SR; Hoffmann, P. EZYprep. LC-coupled MALDI-TOF/TOF MS: An improved matrix spray application for phosphopeptide characterisation. Proteomics 2010, 10, 2516–2530. [Google Scholar]
- Zhu, Y; Wu, R; Sangha, N; Yoo, C; Cho, KR; Shedden, KA; Katabuchi, H; Lubman, DM. Classifications of ovarian cancer tissues by proteomic patterns. Proteomics 2006, 6, 5846–5856. [Google Scholar]
- Caprioli, RM; Farmer, TB; Gile, J. Molecular imaging of biological samples: Localization of peptides and proteins using MALDI-TOF MS. Anal. Chem 1997, 69, 4751–4760. [Google Scholar]
- Amstalden van Hove, ER; Smith, DF; Heeren, RM. A concise review of mass spectrometry imaging. J. Chromatogr A 2010, 1217, 3946–3954. [Google Scholar]
- Meriaux, C; Franck, J; Wisztorski, M; Salzet, M; Fournier, I. Liquid ionic matrixes for MALDI mass spectrometry imaging of lipids. J proteomics 2010, 73, 1204–1218. [Google Scholar]
- Lemaire, R; Menguellet, SA; Stauber, J; Marchaudon, V; Lucot, JP; Collinet, P; Farine, MO; Vinatier, D; Day, R; Ducoroy, P; Salzet, M; Fournier, I. Specific MALDI imaging and profiling for biomarker hunting and validation: Fragment of the 11S proteasome activator complex, Reg alpha fragment, is a new potential ovary cancer biomarker. J. Proteome Res 2007, 6, 4127–4134. [Google Scholar]
- Kang, S; Shim, HS; Lee, JS; Kim, DS; Kim, HY; Hong, SH; Kim, PS; Yoon, JH; Cho, NH. Molecular proteomics imaging of tumor interfaces by mass spectrometry. J. Proteome Res 2010, 9, 1157–1164. [Google Scholar]
- El Ayed, M; Bonnel, D; Longuespee, R; Castelier, C; Franck, J; Vergara, D; Desmons, A; Tasiemski, A; Kenani, A; Vinatier, D; Day, R; Fournier, I; Salzet, M. MALDI imaging mass spectrometry in ovarian cancer for tracking, identifying, and validating biomarkers. Med Sci Monit 2010, 16, BR233–245. [Google Scholar]
- Franck, J; Longuespee, R; Wisztorski, M; van Remoortere, A; van Zeijl, R; Deelder, A; Salzet, M; McDonnell, L; Fournier, I. MALDI mass spectrometry imaging of proteins exceeding 30,000 daltons. Med Sci Monit 2010, 16, BR293–299. [Google Scholar]
- Karas, M; Gluckmann, M; Schafer, J. Ionization in matrix-assisted laser desorption/ionization: Singly charged molecular ions are the lucky survivors. J. Mass Spectrom 2000, 35, 1–12. [Google Scholar]
- Knochenmuss, R; Zenobi, R. MALDI ionization: The role of in-plume processes. Chem. Rev 2003, 103, 441–452. [Google Scholar]
- Knochenmuss, R. Ion formation mechanisms in UV-MALDI. Analyst 2006, 131, 966–986. [Google Scholar]
- Daltonics, B. MALDI Theory Mass Spectrometry; Bruker Daltonics: Bremen, Germany, 2004; p. 18. [Google Scholar]
- Reyzer, ML; Hsieh, Y; Ng, K; Korfmacher, WA; Caprioli, RM. Direct analysis of drug candidates in tissue by matrix-assisted laser desorption/ionization mass spectrometry. J. Mass. Spectrom 2003, 38, 1081–1092. [Google Scholar]
- Chen, R; Hui, L; Sturm, RM; Li, L. Three dimensional mapping of neuropeptides and lipids in crustacean brain by mass spectral imaging. J. Am. Soc. Mass. Spectrom 2009, 20, 1068–1077. [Google Scholar]
- Taban, IM; Altelaar, AF; Van der Burgt, YE; McDonnell, LA; Heeren, RM; Fuchser, J; Baykut, G. Imaging of peptides in the rat brain using MALDI-FTICR mass spectrometry. J. Am. Soc. Mass. Spectrom 2007, 18, 145–151. [Google Scholar]
- Rauser, S; Marquardt, C; Balluff, B; Deininger, SO; Albers, C; Belau, E; Hartmer, R; Suckau, D; Specht, K; Ebert, MP; Schmitt, M; Aubele, M; Hofler, H; Walch, A. Classification of HER2 receptor status in breast cancer tissues by MALDI imaging mass spectrometry. J. Proteome Res 2010, 9, 1854–1863. [Google Scholar]
- Gustafsson, JOR; McColl, SR; Hoffmann, P. Imaging mass spectrometry and its methodological application to Murine tissue. J Proteomics Bioinformatics 2008, 1, 458–463. [Google Scholar]
- Lemaire, R; Wisztorski, M; Desmons, A; Tabet, JC; Day, R; Salzet, M; Fournier, I. MALDI-MS direct tissue analysis of proteins: Improving signal sensitivity using organic treatments. Anal. Chem 2006, 78, 7145–7153. [Google Scholar]
- Seeley, EH; Oppenheimer, SR; Mi, D; Chaurand, P; Caprioli, RM. Enhancement of protein sensitivity for MALDI imaging mass spectrometry after chemical treatment of tissue sections. J. Am. Soc. Mass. Spectrom 2008, 19, 1069–1077. [Google Scholar]
- Schwartz, SA; Reyzer, ML; Caprioli, RM. Direct tissue analysis using matrix-assisted laser desorption/ionization mass spectrometry: Practical aspects of sample preparation. J. Mass. Spectrom 2003, 38, 699–708. [Google Scholar]
- Chaurand, P; Norris, JL; Cornett, DS; Mobley, JA; Caprioli, RM. New developments in profiling and imaging of proteins from tissue sections by MALDI mass spectrometry. J. Proteome Res 2006, 5, 2889–2900. [Google Scholar]
- Ilina, EN; Borovskaya, AD; Malakhova, MM; Vereshchagin, VA; Kubanova, AA; Kruglov, AN; Svistunova, TS; Gazarian, AO; Maier, T; Kostrzewa, M; Govorun, VM. Direct bacterial profiling by matrix-assisted laser desorption-ionization time-of-flight mass spectrometry for identification of pathogenic Neisseria. J. Mol. Diagn 2009, 11, 75–86. [Google Scholar]
- Yanagisawa, K; Shyr, Y; Xu, BJ; Massion, PP; Larsen, PH; White, BC; Roberts, JR; Edgerton, M; Gonzalez, A; Nadaf, S; Moore, JH; Caprioli, RM; Carbone, DP. Proteomic patterns of tumour subsets in non-small-cell lung cancer. Lancet 2003, 362, 433–439. [Google Scholar]
- Caldwell, RL; Holt, GE; Caprioli, RM. Tissue profiling by MALDI mass spectrometry distinguishes clinical grades of soft tissue sarcomas. Cancer Genomics Proteomics 2005, 2, 333–346. [Google Scholar]
- Chaurand, P; Schwartz, SA; Billheimer, D; Xu, BJ; Crecelius, A; Caprioli, RM. Integrating histology and imaging mass spectrometry. Anal. Chem 2004, 76, 1145–1155. [Google Scholar]
- Samsi, SS; Krishnamurthy, AK; Groseclose, M; Caprioli, RM; Lozanski, G; Gurcan, MN. Imaging mass spectrometry analysis for follicular lymphoma grading. Conf. Proc. IEEE Eng. Med. Biol. Soc 2009, 2009, 6969–6972. [Google Scholar]
- Rahman, SM; Shyr, Y; Yildiz, PB; Gonzalez, AL; Li, H; Zhang, X; Chaurand, P; Yanagisawa, K; Slovis, BS; Miller, RF; Ninan, M; Miller, YE; Franklin, WA; Caprioli, RM; Carbone, DP; Massion, PP. Proteomic patterns of preinvasive bronchial lesions. Am. J. Respir. Crit. Care Med 2005, 172, 1556–1562. [Google Scholar]
- Schwamborn, K; Krieg, RC; Reska, M; Jakse, G; Knuechel, R; Wellmann, A. Identifying prostate carcinoma by MALDI-Imaging. Int. J. Mol. Med 2007, 20, 155–159. [Google Scholar]
- Cazares, LH; Troyer, D; Mendrinos, S; Lance, RA; Nyalwidhe, JO; Beydoun, HA; Clements, MA; Drake, RR; Semmes, OJ. Imaging mass spectrometry of a specific fragment of mitogen-activated protein kinase/extracellular signal-regulated kinase kinase kinase 2 discriminates cancer from uninvolved prostate tissue. Clin. Cancer Res 2009, 15, 5541–5551. [Google Scholar]
- Agar, NY; Malcolm, JG; Mohan, V; Yang, HW; Johnson, MD; Tannenbaum, A; Agar, JN; Black, PM. Imaging of meningioma progression by matrix-assisted laser desorption ionization time-of-flight mass spectrometry. Anal. Chem 2010, 82, 2621–2625. [Google Scholar]
- Schwartz, SA; Weil, RJ; Thompson, RC; Shyr, Y; Moore, JH; Toms, SA; Johnson, MD; Caprioli, RM. Proteomic-based prognosis of brain tumor patients using direct-tissue matrixassisted laser desorption ionization mass spectrometry. Cancer Res 2005, 65, 7674–7681. [Google Scholar]
- Rompp, A; Guenther, S; Schober, Y; Schulz, O; Takats, Z; Kummer, W; Spengler, B. Histology by mass spectrometry: Label-free tissue characterization obtained from high-accuracy bioanalytical imaging. Angew. Chem. Int. Ed. Engl 2010, 49, 3834–3838. [Google Scholar]
- Jardin-Mathe, O; Bonnel, D; Franck, J; Wisztorski, M; Macagno, E; Fournier, I; Salzet, M. MITICS (MALDI Imaging Team Imaging Computing System): A new open source mass spectrometry imaging software. J Proteomics 2008, 71, 332–345. [Google Scholar]
- Martens, L; Chambers, M; Sturm, M; Kessner, D; Levander, F; Shofstahl, J; Tang, WH; Rompp, A; Neumann, S; Pizarro, AD; Montecchi-Palazzi, L; Tasman, N; Coleman, M; Reisinger, F; Souda, P; Hermjakob, H; Binz, PA; Deutsch, EW. mzML—a community standard for mass spectrometry data. Mol Cell Proteomics 2010. [Google Scholar] [CrossRef]
- Rompp, A; Schramm, T; Hester, A; Klinkert, I; Both, JP; Heeren, RM; Stockli, M; Spengler, B. imzML: Imaging mass spectrometry markup language: A common data format for mass spectrometry imaging. Methods Mol. Biol 2011, 696, 205–224. [Google Scholar]
- Aerni, HR; Cornett, DS; Caprioli, RM. Automated acoustic matrix deposition for MALDI sample preparation. Anal. Chem 2006, 78, 827–834. [Google Scholar]
- Franck, J; Arafah, K; Barnes, A; Wisztorski, M; Salzet, M; Fournier, I. Improving tissue preparation for matrix-assisted laser desorption ionization mass spectrometry imaging. Part 1: Using microspotting. Anal. Chem 2009, 81, 8193–8202. [Google Scholar]
- Lemaire, R; Tabet, JC; Ducoroy, P; Hendra, JB; Salzet, M; Fournier, I. Solid ionic matrixes for direct tissue analysis and MALDI imaging. Anal. Chem 2006, 78, 809–819. [Google Scholar]
- Groseclose, MR; Massion, PP; Chaurand, P; Caprioli, RM. High-throughput proteomic analysis of formalin-fixed paraffin-embedded tissue microarrays using MALDI imaging mass spectrometry. Proteomics 2008, 8, 3715–3724. [Google Scholar]
- Groseclose, MR; Andersson, M; Hardesty, WM; Caprioli, RM. Identification of proteins directly from tissue: In situ tryptic digestions coupled with imaging mass spectrometry. J. Mass. Spectrom 2007, 42, 254–262. [Google Scholar]
- Gustafsson, JO; Oehler, MK; McColl, SR; Hoffmann, P. Citric acid antigen retrieval (CAAR) for tryptic peptide imaging directly on archived formalin-fixed paraffin-embedded tissue. J. Proteome Res 2010, 9, 4315–4328. [Google Scholar]
- Alexandrov, T; Becker, M; Deininger, SO; Ernst, G; Wehder, L; Grasmair, M; Von Eggeling, F; Thiele, H; Maass, P. Spatial segmentation of imaging mass spectrometry data with edge-preserving image denoising and clustering. J. Proteome Res 2010, 9, 6535–6546. [Google Scholar]
- Oppenheimer, SR; Mi, D; Sanders, ME; Caprioli, RM. Molecular analysis of tumor margins by MALDI mass spectrometry in renal carcinoma. J. Proteome Res 2010, 9, 2182–2190. [Google Scholar]
- Woo, MM; Alkushi, A; Verhage, HG; Magliocco, AM; Leung, PC; Gilks, CB; Auersperg, N. Gain of OGP, an estrogen-regulated oviduct-specific glycoprotein, is associated with the development of endometrial hyperplasia and endometrial cancer. Clin. Cancer Res 2004, 10, 7958–7964. [Google Scholar]
- Elg, SA; Mayer, AR; Carson, LF; Twiggs, LB; Hill, RB; Ramakrishnan, S. Alpha-1 acid glycoprotein is an immunosuppressive factor found in ascites from ovaria carcinoma. Cancer 1997, 80, 1448–1456. [Google Scholar]
- Addis, MF; Tanca, A; Pagnozzi, D; Crobu, S; Fanciulli, G; Cossu-Rocca, P; Uzzau, S. Generation of high-quality protein extracts from formalin-fixed, paraffin-embedded tissues. Proteomics 2009, 9, 3815–3823. [Google Scholar]
- Addis, MF; Tanca, A; Pagnozzi, D; Rocca, S; Uzzau, S. 2-D PAGE and MS analysis of proteins from formalin-fixed, paraffin-embedded tissues. Proteomics 2009, 9, 4329–4339. [Google Scholar]
- Grantzdorffer, I; Yumlu, S; Gioeva, Z; von Wasielewski, R; Ebert, MP; Rocken, C. Comparison of different tissue sampling methods for protein extraction from formalin-fixed and paraffin-embedded tissue specimens. Exp. Mol. Pathol 2010, 88, 190–196. [Google Scholar]
- Hood, BL; Conrads, TP; Veenstra, TD. Mass spectrometric analysis of formalin-fixed paraffin-embedded tissue: Unlocking the proteome within. Proteomics 2006, 6, 4106–4114. [Google Scholar]
- Aoki, Y; Toyama, A; Shimada, T; Sugita, T; Aoki, C; Umino, Y; Suzuki, A; Aoki, D; Daigo, Y; Nakamura, Y; Sato, TA. A novel method for analyzing formalin-fixed paraffin embedded (FFPE) tissue sections by mass spectrometry imaging. Proc. Jpn. Acad. Ser 2007, 83, 205–214. [Google Scholar]
- Lemaire, R; Desmons, A; Tabet, JC; Day, R; Salzet, M; Fournier, I. Direct analysis and MALDI imaging of formalin-fixed, paraffin-embedded tissue sections. J. Proteome Res 2007, 6, 1295–1305. [Google Scholar]
- Stauber, J; Lemaire, R; Franck, J; Bonnel, D; Croix, D; Day, R; Wisztorski, M; Fournier, I; Salzet, M. MALDI imaging of formalin-fixed paraffin-embedded tissues: Application to model animals of parkinson disease for biomarker hunting. J. Proteome Res 2008, 7, 969–978. [Google Scholar]
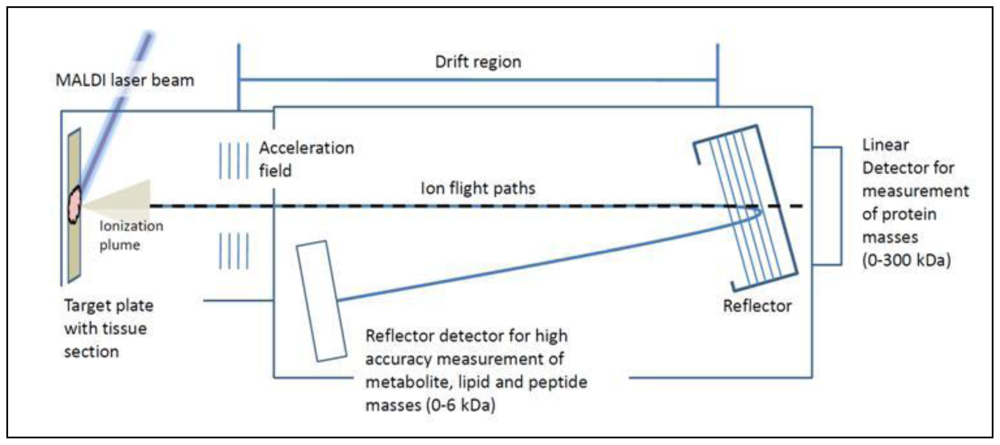

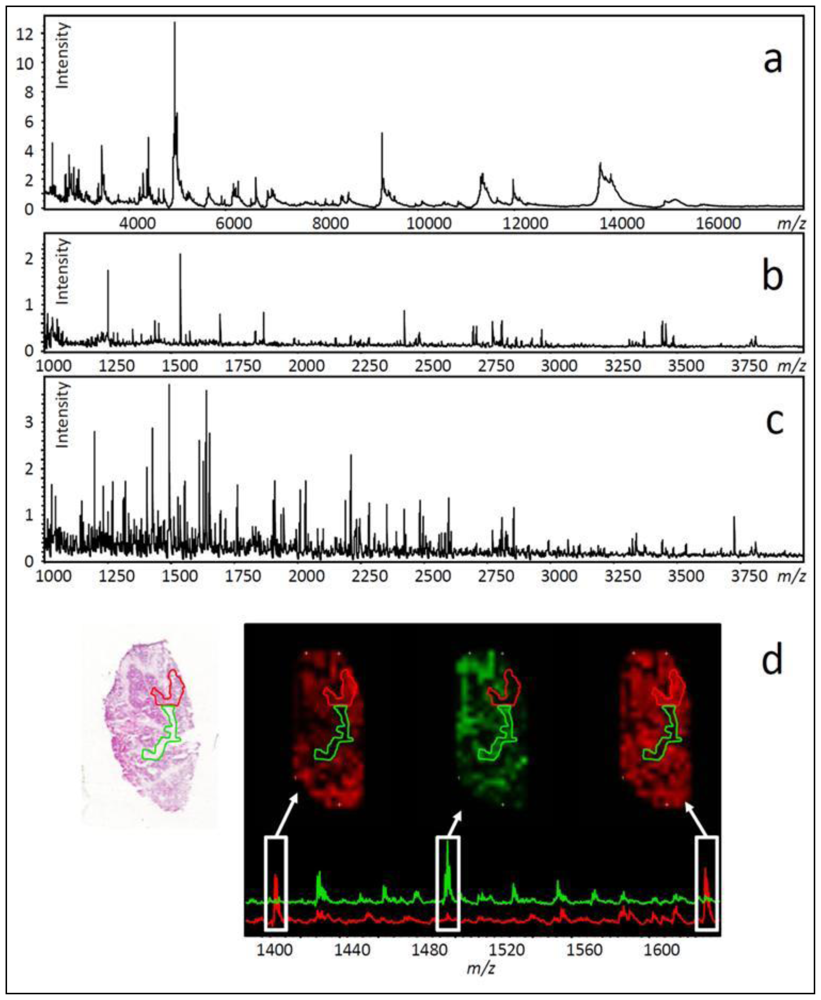
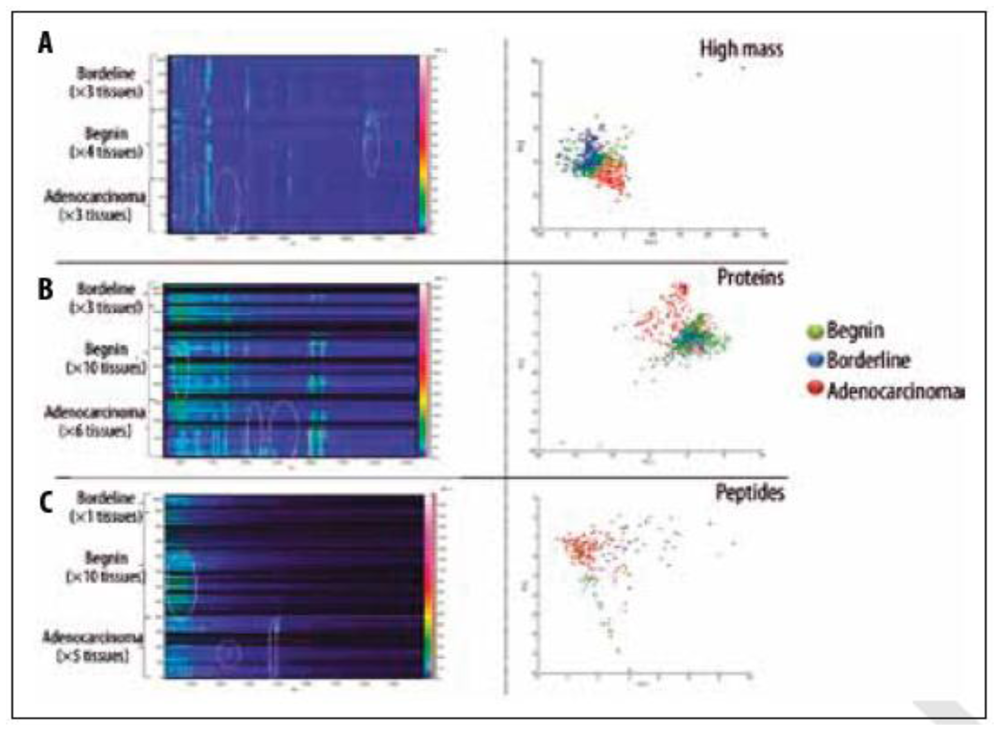



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| FIGO Stage | Prevalence (%) | Anatomical features |
|---|---|---|
| I | 25 | Limited to ovaries |
| II | 11 | Pelvic extension |
| III | 47 | Abdominal extension and/or positive lymph nodes |
| IV | 17 | Distant metastases |
| Grading system | Grade | Key features | Ref. | |||
|---|---|---|---|---|---|---|
| FIGO | 1 | Well differentiated | Grade based on % solid non-squamous growth, grade + 1 if nuclear atypia apparent | <5% solid growth | [10] | |
| 2 | Moderately differentiated | 6–50% solid growth | ||||
| 3 | Poorly differentiated | >50% solid growth | [13] | |||
| 3-tier universal grading | 1 | Grade based on sum of individual feature scores (see right) 1 = 3–5 points 2 = 6–7 points 3 = 8–9 points | Architecture based score | Glandular = 1 point Papillary = 2 points Solid = 3 points | ||
| 2 | Nuclear pleomorphism score | Slight = 1 point Moderate = 2 points Marked = 3 points | [12] | |||
| 3 | Mitotic activity score | 0–9 = 1 point 10–24 = 2 points ≥25 = 3 points | ||||
| 2-tier grading | Serous tumour | Low grade (type I) | Slow development | Low chromosomal instability | Gene mutation–KRAS, BRAF, ERBB2 | [14] |
| High grade (type II) | Rapid development | High chromosomal instability | Gene mutation–P53 | |||
| Endomet roid tumour | Low grade | Well differentiated, no necrosis | Solid glandular architecture | Gene mutation–Wnt, PI3K/Akt | [13] | |
| High grade | Solid growth >50%, necrosis | Diffusely infiltrative or expansive growth, no glandular architecture | Gene mutation–TP53 | [16] | ||
| Histology | IHC | Proteomics | ||
|---|---|---|---|---|
| Fractionation-MS | Direct tissue MS | |||
| Methods | Cellular staining | Antibody directed staining of specific proteins | Liquid phase separation (i.e., liquid chromatography) | Direct measurement of peptides and proteins from tissue section |
| Analysis | Tissue morphology assessment by light microscopy | Protein distribution across tissue sections | MS protein identification | MS profiles of tissue sections |
| Quantitation using protein labelling | Peptide and protein intensity maps showing distribution across tissue sections | |||
| Advantages | Easy staining methods | Highly specific | Highly sensitive | Rapid |
| Cellular microscopy resolution | Cellular microscopy resolution | Thousands of proteins analysed at a time | Spatial proteome information | |
| Well established | Well established | Heavily automated | Measurement of hundreds of molecular features at a time | |
| Clinical personnel already available | Clinical personnel already available | Highly modular workflows | No antibodies required | |
| Disadvantages | Reproducibility issues | Time consuming | Time consuming | Expensive equipment |
| Based on visual assessment of morphology | Labor intensive | Labor intensive | Novel technology | |
| Non-specific | Limited to 3–4 proteins | Removes spatial information | Requires fraction-MS based proteomics to identify peptide and protein features | |
| Analysis is subjective | Dependent on antibody quality | Requires specialist personnel | Analytical resolution limited to a maximum of 20–50 μm | |
| Matrix | Chemical name | Biomolecule specificity |
|---|---|---|
| DHB | 2,5-dihydroxybenzoic acid | Lipids, peptides, <10 kDa proteins |
| DHB/aniline | DHB + aniline | Lipids, peptides, <10 kDa proteins |
| DHB/3-AP | DHB + 3-acetyl pyridine | Lipids, peptides, <10 kDa proteins |
| CHCA | α-cyano-4-hydroxycinnamic acid | Peptides, small proteins (<10 kDa) |
| CHCA/aniline | CHCA + aniline | Peptides, <10 kDa proteins |
| SA | 3,5-dimethoxy-4-hydroxycinnamic acid | Proteins (>10 kDa) |
| SA/aniline | SA + aniline | Proteins (>10 kDa) |
| SA/3-AP | SA + 3-acetyl pyridine | Proteins (>10 kDa) |
| SA/HFIP | SA + 1,1,1,3,3,3-hexafluoro-2-propanol | Proteins (>30 kDa) |
| SA/TFE | SA + 2,2,2-trifluoroethanol | Proteins (>30 kDa) |
| Nebulising instruments | Printers | |||
|---|---|---|---|---|
| Air brush | ImagePrep station | ChIP-1000 | Labcyte Portrait | |
| Reproducibility | Poor | Good | Excellent | Excellent |
| Acquisition resolution | ≥5 μm | ≥20 μm | ≥150 μm | ≥150 μm |
| Advantages | Cheap | Automated | Automated | Automated |
| High resolution MS acquisition | High resolution MS acquisition | Control over reagent volume deposited | Control over reagent volume deposited | |
| Good for start up imaging MS laboratories | Default methods available but methods can be modified by user | High MS sensitivity | High MS sensitivity | |
| Disadvantages | Lower peptide/protein incorporation into matrix | Lower peptide/protein incorporation into matrix | Expensive | Most expensive |
| Requires experienced user | Requires experienced user | Time consuming preparation | Time consuming preparation | |
| Manual preparation | Expensive | Lower data acquisition resolution than nebulised preparations | Lower data acquisition resolution than nebulised preparations | |
© 2011 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Gustafsson, J.O.R.; Oehler, M.K.; Ruszkiewicz, A.; McColl, S.R.; Hoffmann, P. MALDI Imaging Mass Spectrometry (MALDI-IMS)―Application of Spatial Proteomics for Ovarian Cancer Classification and Diagnosis. Int. J. Mol. Sci. 2011, 12, 773-794. https://doi.org/10.3390/ijms12010773
Gustafsson JOR, Oehler MK, Ruszkiewicz A, McColl SR, Hoffmann P. MALDI Imaging Mass Spectrometry (MALDI-IMS)―Application of Spatial Proteomics for Ovarian Cancer Classification and Diagnosis. International Journal of Molecular Sciences. 2011; 12(1):773-794. https://doi.org/10.3390/ijms12010773
Chicago/Turabian StyleGustafsson, Johan O. R., Martin K. Oehler, Andrew Ruszkiewicz, Shaun R. McColl, and Peter Hoffmann. 2011. "MALDI Imaging Mass Spectrometry (MALDI-IMS)―Application of Spatial Proteomics for Ovarian Cancer Classification and Diagnosis" International Journal of Molecular Sciences 12, no. 1: 773-794. https://doi.org/10.3390/ijms12010773
APA StyleGustafsson, J. O. R., Oehler, M. K., Ruszkiewicz, A., McColl, S. R., & Hoffmann, P. (2011). MALDI Imaging Mass Spectrometry (MALDI-IMS)―Application of Spatial Proteomics for Ovarian Cancer Classification and Diagnosis. International Journal of Molecular Sciences, 12(1), 773-794. https://doi.org/10.3390/ijms12010773