A Brief Review of Molecular Techniques to Assess Plant Diversity

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. DNA Sequencing
2.1. Conventional Sequencing Technique
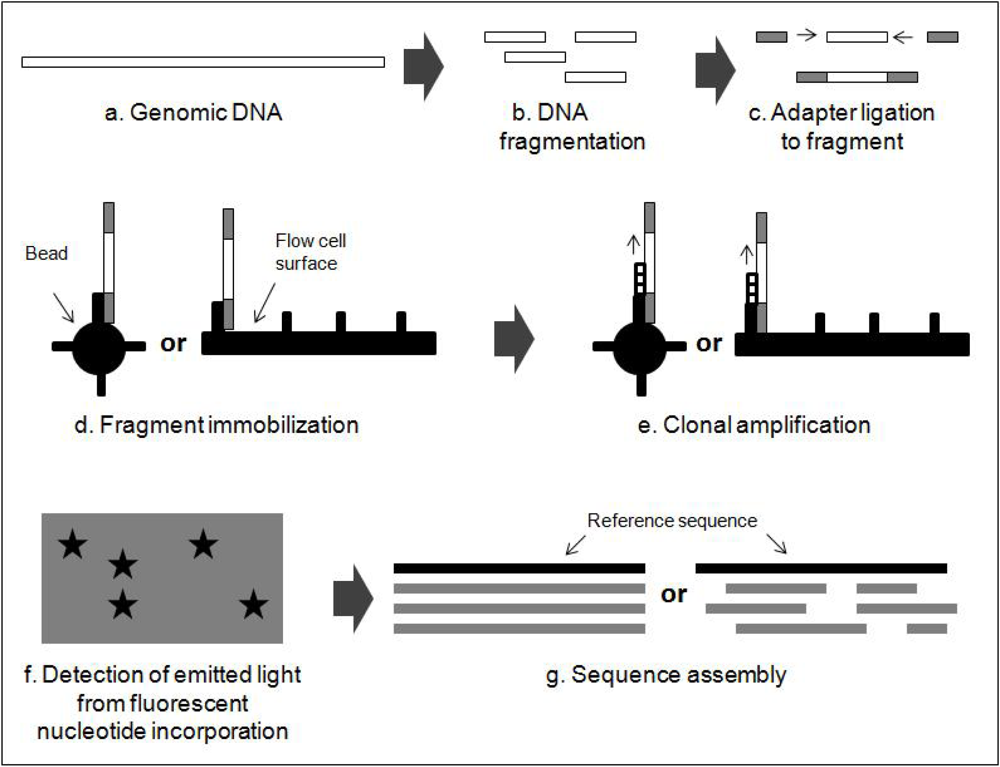
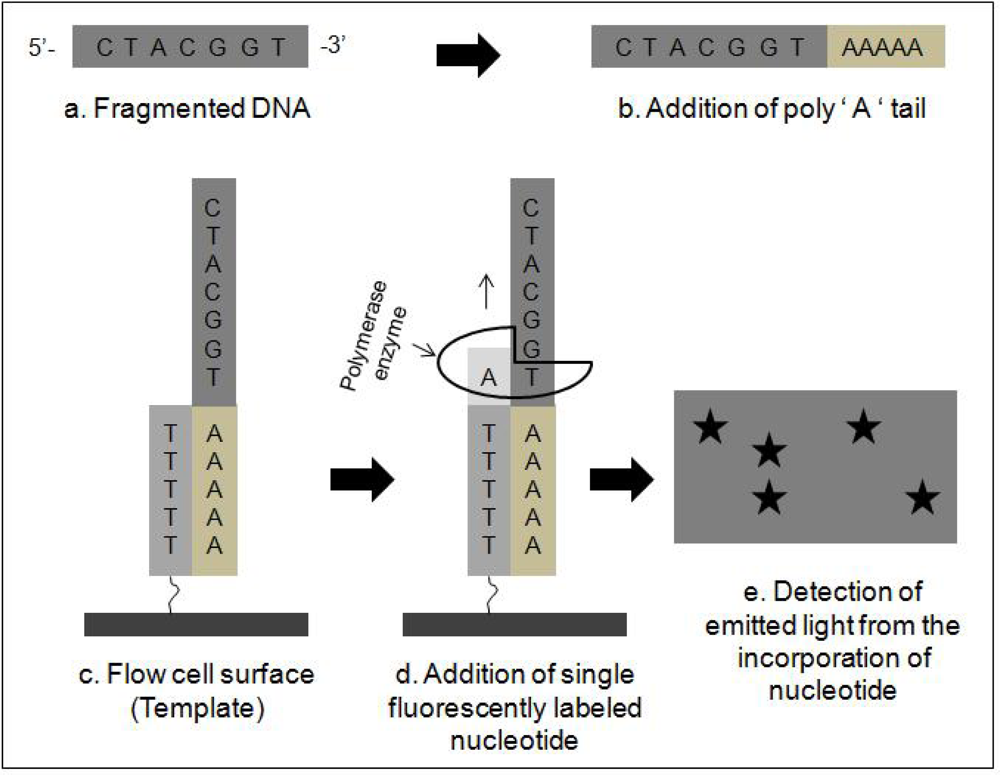
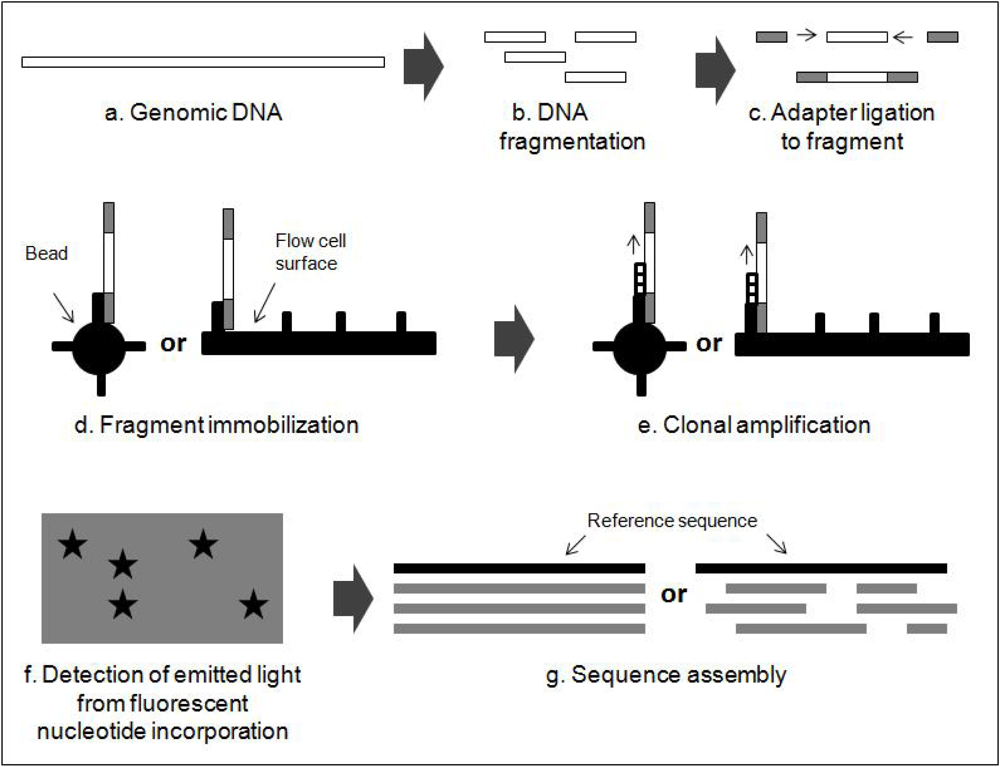
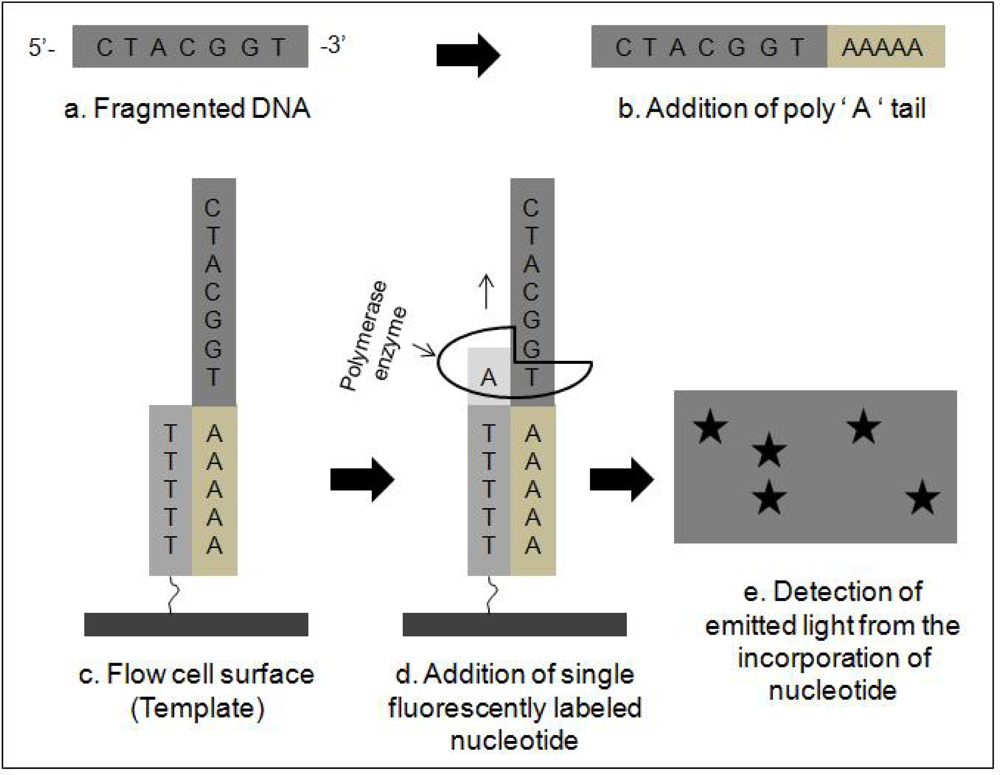
2.2. Next Generation Sequencing Techniques
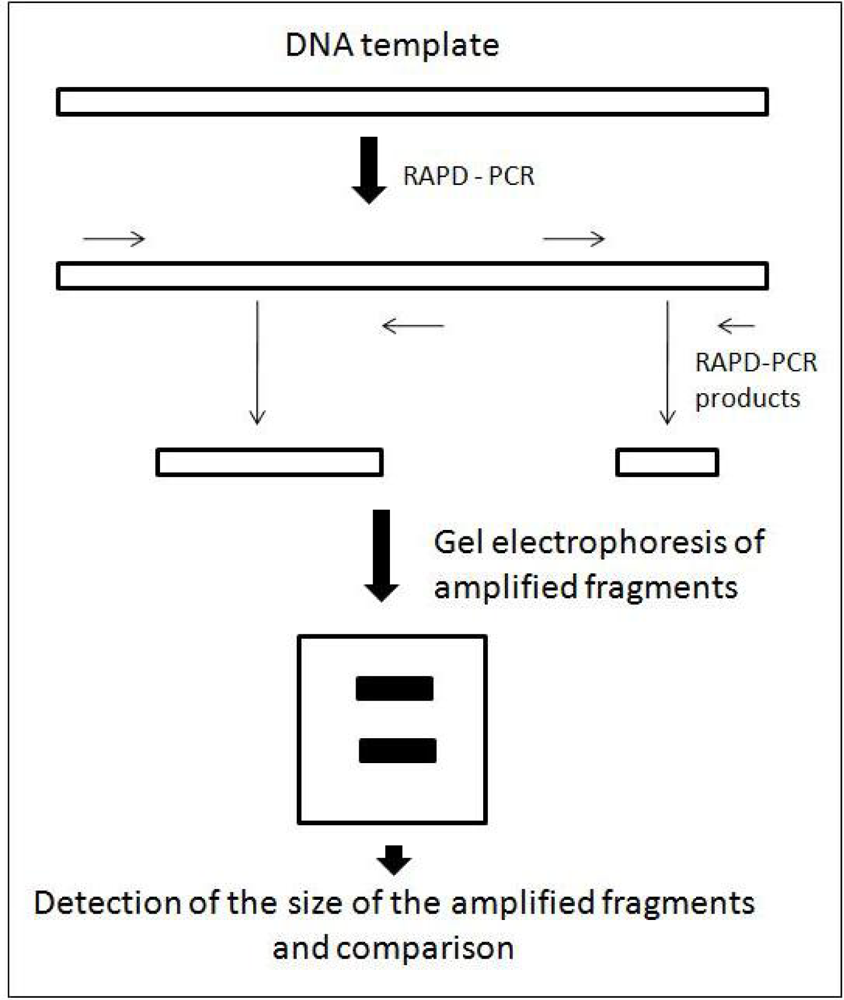
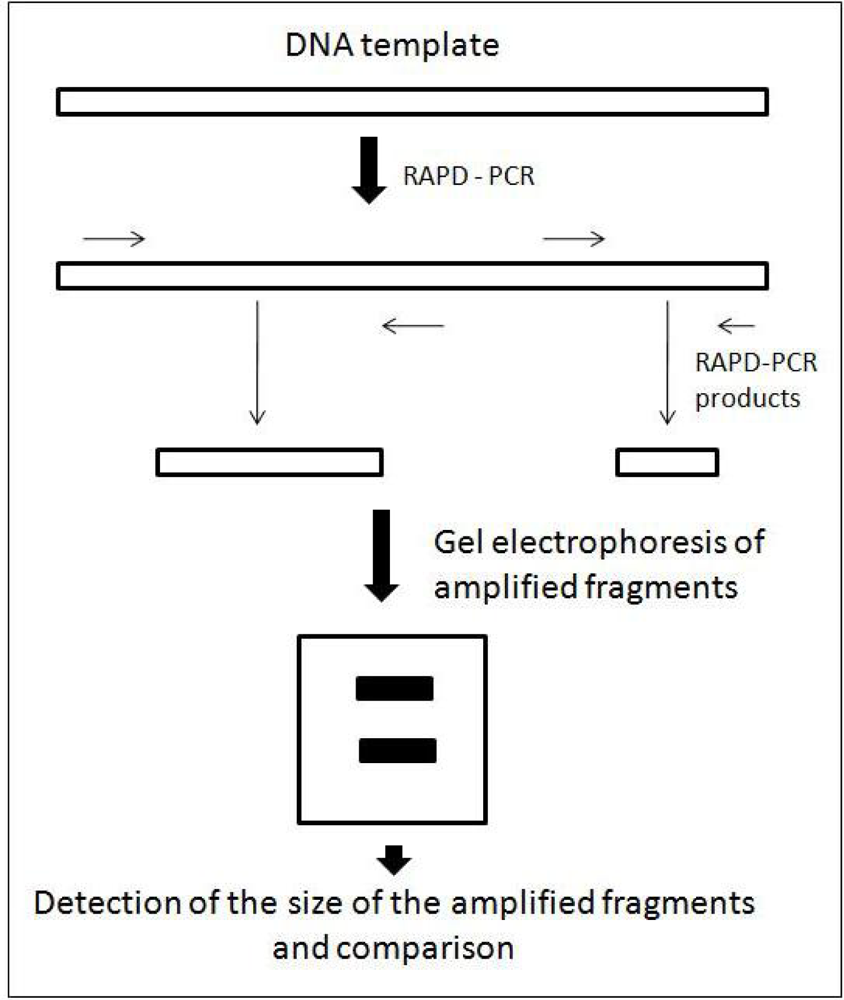
3. Random Amplified Polymorphic DNA (RAPD)
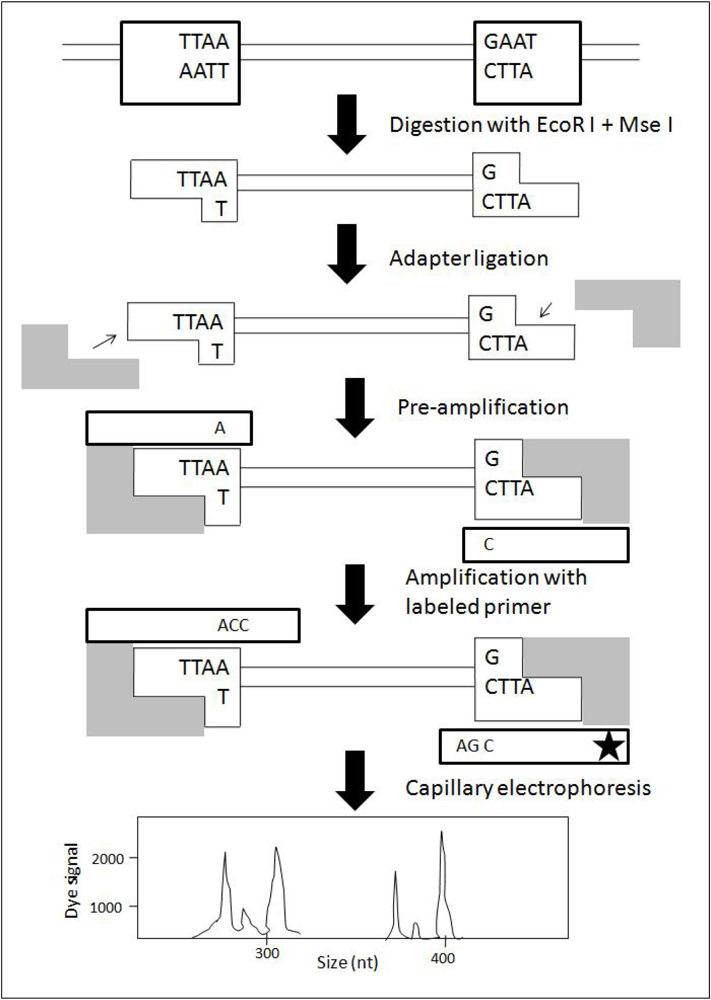
4. Amplified Fragment Length Polymorphism (AFLP)
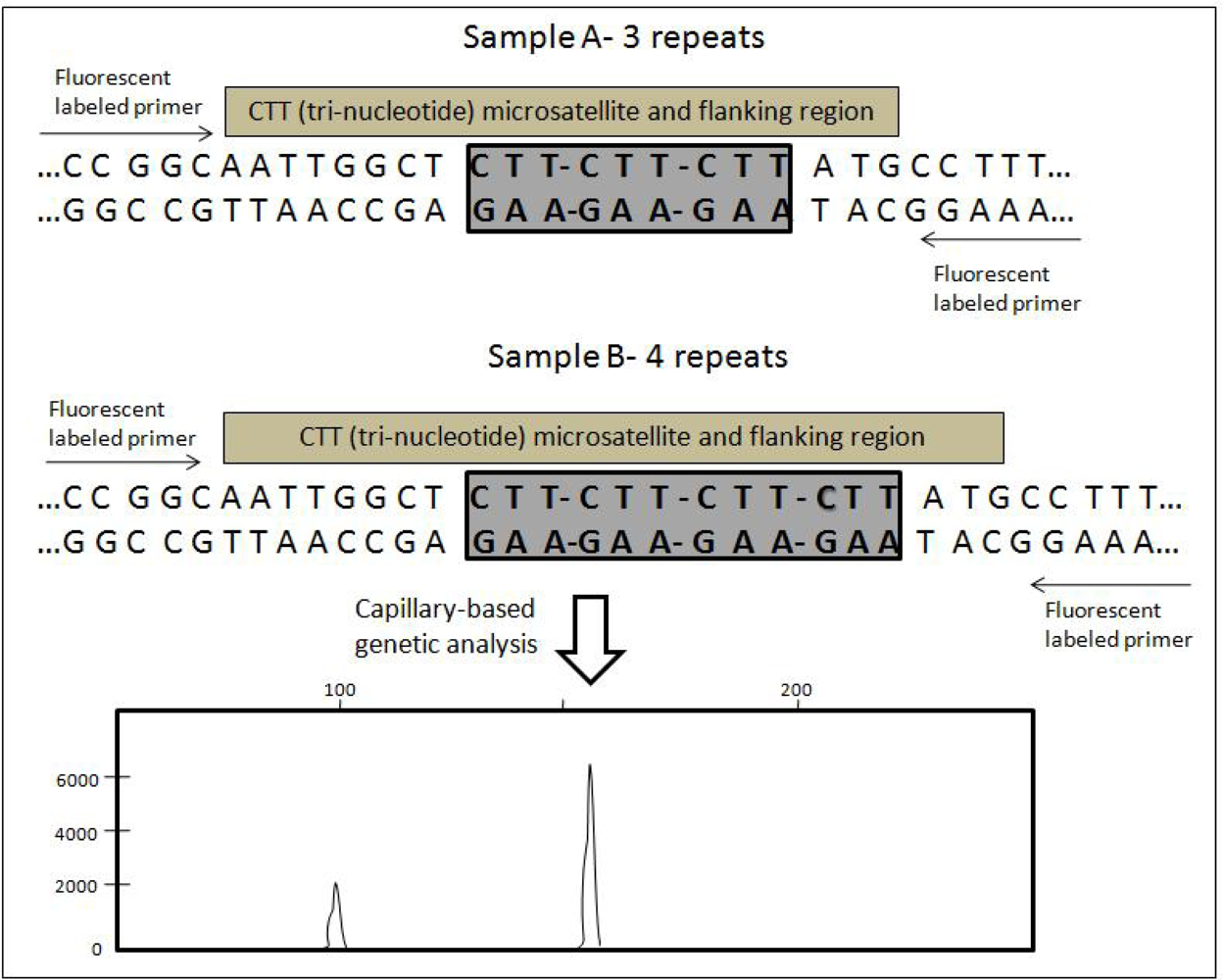
5. Microsatellites
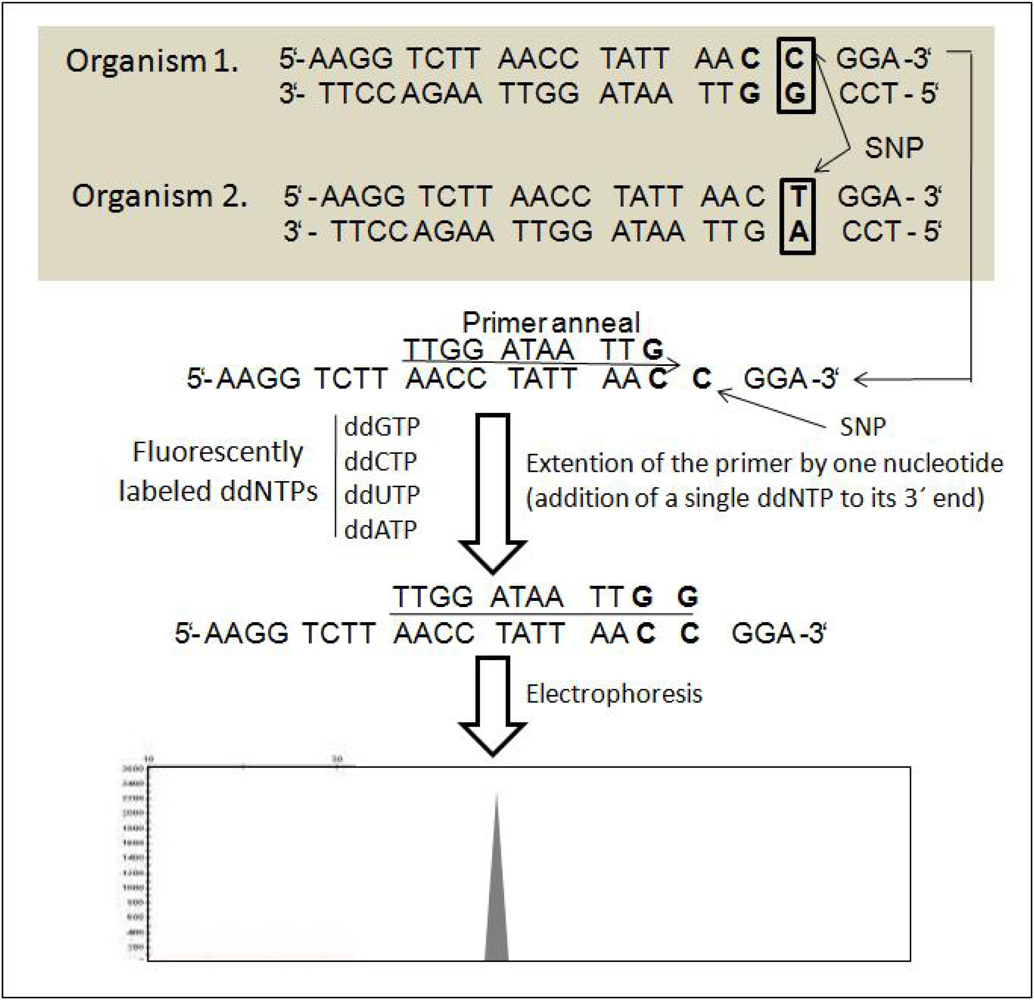
6. Single Nucleotide Polymorphism (SNP)
7. Concluding Remarks
References
- Botstein, D; White, RL; Skolnick, MH; Davis, RW. Construction of a genetic map in man using restriction fragment length polymorphisms. Am. J. Hum. Genet 1980, 32, 314–331. [Google Scholar]
- Karp, A; Seberg, O; Buiatti, M. Molecular Techniques in the Assessment of Botanical Diversity. Ann. Bot 1996, 78, 143–149. [Google Scholar]
- Karp, A; Kresovich, S; Bhat, KV; Ayad, WG; Hodgkin, T. Molecular Tools in Plant Genetic Resources Conservation: A Guide to the Technologies. IPGRI Technical Bulletin No. 2; International Plant Genetic Resources Institute: Rome, Italy, 1997. [Google Scholar]
- Joshi, SP; Ranjanekar, PK; Gupta, VS. Molecular markers in plant genome analysis. Curr. Sci 1999, 77, 230–240. [Google Scholar]
- Robinson, JP; Harris, SA. Which DNA Marker for Which Purpose; Gillet, EM, Ed.; Institut für Forstgenetik und Forstpflanzenzüchtung, Universität Göttingen: Göttingen, Germany, 1999; pp. 1–27. [Google Scholar]
- Struwig, M; Mienie, CMS; van den Berg, J; Mucina, L; Buys, MH. AFLPs are incompatible with RAPD and morphological data in Pennisetum purpureum (Napier grass). Biochem. Sys. Ecol 2009, 37, 645–652. [Google Scholar]
- Hultman, T; Stahl, S; Hornes, E; Uhlen, M. Direct solid phase sequencing of genomic and plasmid DNA using magnetic beads as solid support. Nucl. Acids Res 1989, 17, 4937–4946. [Google Scholar]
- Brytting, M; Wahlberg, J; Lundeberg, J; Wahren, B; Uhlen, M; Sundqvist, VA. Variations in the cytomegalovirus major immediate-early gene found by direct genomic sequencing. J. Clin. Microbiol 1992, 30, 955–960. [Google Scholar]
- Olsvik, O; Wahlberg, J; Petterson, B; Uhlén, M; Popovic, T; Wachsmuth, IK; Fields, PI. Use of automated sequencing of polymerase chain reaction-generated amplicons to identify three types of cholera toxin subunit B in Vibrio cholerae O1 strains. J. Clin. Microbiol 1993, 31, 22–25. [Google Scholar]
- Prober, JM; Trainor, GL; Dam, RJ; Hobbs, FW; Robertson, CW; Zagursky, RJ; Cocuzza, AJ; Jensen, MA; Baumeister, K. A system for rapid DNA sequencing with fluorescent chain-terminating dideoxynucleotides. Science 1987, 238, 336–341. [Google Scholar]
- Kress, WJ; Wurdack, KJ; Zimmer, EA; Weigt, LA; Janzen, DH. Use of DNA barcodes to identify flowering plants. Proc. Natl. Acad. Sci 2005, 102, 8369–8374. [Google Scholar]
- Altschul, SF; Gish, W; Miller, W; Myers, EW; Lipman, DJ. Basic local alignment search tool. J. Mol. Biol 1990, 215, 403–410. [Google Scholar]
- Kress, WJ; Erickson, DL; Jones, FA; Swenson, NG; Perez, R; Sanjur, O; Bermingham, E. Plant DNA barcodes and a community phylogeny of a tropical forest dynamics plot in Panama. Proc. Natl. Acad. Sci. USA 2009, 106, 18621–18626. [Google Scholar]
- CBOL Plant Working Group. A DNA barcode for land plants. Proc. Natl. Acad. Sci. USA 2009, 106, 12794–12797. [Google Scholar]
- Despres, L; Gielly, L; Redoutet, B; Taberlet, P. Using AFLP to resolve phylogenetic relationships in a morphologically diversified plant species complex when nuclear and chloroplast sequences fail to reveal variability. Mol. Phylogenet. Evol 2003, 27, 185–196. [Google Scholar]
- Chase, MW; Soltis, DE; Olmstead, RG; Morgan, D; Les, DH; Mishler, BD; Duvall, MR; Price, RA; Hills, HG; Qiu, YL; et al. Phylogenetics of seed plants: An analysis of nucleotide sequences from the plastid gene rbcL. Ann. Missouri Bot. Gard 1993, 80, 528–580. [Google Scholar]
- Soltis, DE; Morgan, DR; Grable, A; Soltis, PS; Kuzoff, R. Molecular systematics of Saxifragaceae sensu stricto. Am. J. Bot 1993, 80, 1056–1081. [Google Scholar]
- Gielly, L; Taberlet, P. A phylogeny of the European gentians inferred from chloroplast trnL UAA intron sequences. Bot. J. Linn. Soc 1996, 120, 57–75. [Google Scholar]
- Baldwin, BG. Phylogenetic utility of the internal transcribed spacers of nuclear ribosomal DNA in plants: An example from the Compositae. Mol. Phylogenet. Evol 1992, 1, 3–16. [Google Scholar]
- Yuan, YM; Küpfer, P; Doyle, J. Infrageneric phylogeny of the genus Gentiana (Gentianaceae) inferred from nucleotide sequences of the internal transcribed spacers ITSs of nuclear ribosomal DNA. Am. J. Bot 1996, 83, 641–652. [Google Scholar]
- Koopman, WJM; Zevenbergen, MJ; Van-den-Berg, RG. Species relationships in Lactuca S.L. (Lactuceae, Asteraceae) inferred from AFLP fingerprints. Am. J. Bot 2001, 88, 1881–1887. [Google Scholar]
- Sun, M. Comparative analysis of phylogenetic relationships of grain amaranths and their wild relatives (Amaranthus; amaranthaceae) using internal transcribed spacer, amplified length polymorphism, and double-primer fluorescent intersimple sequence repeat markers. Mol. Phylogenet. Evol 2001, 21, 372–387. [Google Scholar]
- Zhang, LB; Comes, HP; Kadereit, JW. Phylogeny and quaternary history of the European montane/alpine endemic Soldanella (Primulaceae) based on ITS and AFLP variation. Am. J. Bot 2001, 88, 2331–2345. [Google Scholar]
- Sanger, F; Nicklen, S; Coulson, AR. DNA sequencing with chain-terminating inhibitors. Proc. Natl. Acad. Sci. USA 1977, 74, 5463–5467. [Google Scholar]
- Schuster, SC. Next-generation sequencing transforms today’s biology. Nat. Methods 2008, 5, 16–18. [Google Scholar]
- Margulies, M; Egholm, M; Altman, WE; Attiya, S; Bader, JS; Bemben, LA; Berka, J; Braverman, MS; Chen, YJ; Chen, Z; et al. Genome Sequencing in Open Microfabricated High Density Picoliter Reactors. Nature 2005, 437, 376–380. [Google Scholar]
- Ronaghi, M; Karamohamed, S; Pettersson, B; Uhlén, M; Nyrén, P. Real-time DNA sequencing using detection of pyrophosphate release. Anal. Biochem 2006, 242, 84–89. [Google Scholar]
- Nyrén, P. The history of pyrosequencing. Methods Mol. Biol 2007, 373, 1–14. [Google Scholar]
- Lerner, HRL; Fleischer, RC. Prospects for the use of next-generation sequencing methods in Ornithology. The Auk 2010, 127, 4–15. [Google Scholar]
- Blow, N. DNA sequencing: Generation next-next. Nat. Methods 2008, 5, 267–274. [Google Scholar]
- Braslavsky, I; Hebert, B; Kartalov, E; Quake, SR. Sequence information can be obtained from single DNA molecules. Proc. Natl. Acad. Sci. USA 2003, 100, 3960–3964. [Google Scholar]
- Clarke, J; Wu, HC; Jayasinghe, L; Patel, A; Reid, S; Bayley, H. Continuous base identification for single-molecule nanopore DNA sequencing. Nat. Nanotechnol 2009, 4, 265–270. [Google Scholar]
- Ozsolak, F; Platt, AR; Jones, DR; Reifenberger, JG; Sass, LE; McInerney, P; Thompson, JF; Bowers, J; Jarosz, M; Milos, PM. Direct RNA sequencing. Nature 2009, 461, 814–818. [Google Scholar]
- Miller, JR; Koren, S; Sutton, G. Assembly algorithms for next gereration sequencing data. Genomics 2010. [Google Scholar]
- Yan, Y; Wang, Z; Tian, W; Dong, Z; Spencer, D. Generation and analysis of expressed sequence tags from the medicinal plant Salvia miltiorrhiza. Sci. Chin. Life Sci 2010, 53, 273–285. [Google Scholar]
- Buggs, RJA; Chamala, S; Wu, W; Gao, L; May, GD; Schnable, PS; Soltis, DE; Soltis, PS; Barbazuk, WB. Characterization of duplicate gene evolution in the recent natural allopolyploid Tragopogon miscellus by next-generation sequencing and Sequenom iPLEX MassARRAY genotyping. Mol. Ecol 2010, 19, 132–146. [Google Scholar]
- Zhao, CZ; Xia, H; Frazier, TP; Yao, YY; Bi, YP; Li, AQ; Li, MJ; Li, CS; Zhang, BH; Wang, XJ. Deep sequencing identifies novel and conserved microRNAs in peanuts (Arachis hypogaea L.). BMC Plant Biol 2010, 10, 1–12. [Google Scholar]
- Acharjee, S; Sarmah, BK; Kumar, PA; Olsen, K; Mahon, R; Moar, WJ; Moore, A; Higgins, TJV. Transgenic chickpeas (Cicer arietinum L.) expressing a sequence-modified cry2Aa gene. Plant Sci 2010, 178, 333–339. [Google Scholar]
- Williams, JGK; Kubelik, AR; Livak, KJ; Rafalski, JA; Tingey, SV. DNA polymorphisms amplified by arbitrary primers are useful as genetic markers. Nucl. Acids Res 1990, 18, 6531–6535. [Google Scholar]
- Charney, J; Vershon, D; Bill Sofer, B. RAPD PCR, Genes, Genomes and Human Genetics.
- Lin, KH; Lai, YC; Li, HC; Lo, SF; Chen, LFO; Lo, HF. Genetic variation and its relationship to root weight in the sweet potato as revealed by RAPD analysis. Sci. Hort 2009, 120, 2–7. [Google Scholar]
- da-Mata, TL; Segeren, MI; Fonseca, AS; Colombo, CA. Genetic divergence among gerbera accessions evaluated by RAPD. Sci. Hort 2009, 121, 92–96. [Google Scholar]
- Singh, N; Lal, RK; Shasany, AK. Phenotypic and RAPD diversity among 80 germplasm accessions of the medicinal plant isabgol (Plantago ovata), Plantaginaceae. Genet. Mol. Res 2009, 8, 1273–1284. [Google Scholar]
- Zheng, W; Wang, L; Meng, L; Liu, J. Genetic variation in the endangered Anisodus tanguticus (Solanaceae), an alpine perennial endemic to the Qinghai-Tibetan Plateau. Genetica 2008, 132, 123–129. [Google Scholar]
- Liu, P; Yang, YS; Hao, CY; Guo, WD. Ecological risk assessment using RAPD and distribution pattern of a rare and endangered species. Chemosphere 2007, 68, 1497–1505. [Google Scholar]
- Wang, ZS; An, SQ; Liu, H; Leng, X; Zheng, JW; Liu, YH. Genetic structure of the endangered plant Neolitsea sericea (Lauraceae) from the Zhoushan archipelago using RAPD markers. Ann. Bot 2005, 95, 305–313. [Google Scholar]
- Lu, HP; Cai, YW; Chen, XY; Zhang, X; Gu, YJ; Zhang, GF. High RAPD but no cpDNA sequence variation in the endemic and endangered plant, Heptacodium miconioides Rehd. (Caprifoliaceae). Genetica 2006, 128, 409–417. [Google Scholar]
- Vos, P; Hogers, R; Bleeker, M; Reijans, M; van de Lee, T; Hornes, M; Frijters, A; Pot, J; Peleman, J; Kuiper, M; Zabeau, M. AFLP: A new technique for DNA fingerprinting. Nucl. Acids Res 1995, 23, 4407–4414. [Google Scholar]
- Romero, G; Adeva, C; Battad, Z. Genetic fingerprinting: Advancing the frontiers of crop biology research. Philipp. Sci. Lett 2009, 2, 8–13. [Google Scholar]
- Amplified Fragment Length Polymorphism (AFLP) Analysis on Applied Biosystems Capillary Electrophoresis Systems, Application Note AFLP on the 3130/3730, ; Applied Biosystems: Foster City, CA, USA, 2005.
- Mba, C; Tohme, J. Use of AFLP markers in surveys of plant diversity. Methods Enzymol 2005, 395, 177–201. [Google Scholar]
- Tatikonda, L; Wani, SP; Kannan, S; Beerelli, N; Sreedevi, TK; Hoisington, DA; Devi, P; Varshney, RK. AFLP-based molecular characterization of an elite germplasm collection of Jatropha curcas L.: A biofuel plant. Plant Sci 2009, 176, 505–513. [Google Scholar]
- Elameen, A; Klemsdal, SS; Dragland, S; Fjellheim, S; Rognli, OA. Genetic diversity in a germplasm collection of roseroot (Rhodiola rosea) in Norway studied by AFLP. Biochem. Syst. Ecol 2008, 36, 706–715. [Google Scholar]
- Teyer, FS; Salazar, MS; Esqueda, M; Barraza, A; Robert, ML. Genetic variability of wild Agave angustifolia populations based on AFLP: A basic study for conservation. J. Arid Environ 2009, 73, 611–616. [Google Scholar]
- van Ee, BW; Jelinski, N; Berry, PE; Hipp, AL. Phylogeny and biogeography of Croton alabamensis (Euphorbiaceae), a rare shrub from Texas and Alabama, using DNA sequence and AFLP data. Mol. Ecol 2006, 15, 2735–2751. [Google Scholar]
- Ronikier, M. The use of AFLP markers in conservation genetics-a case study on Pulsatilla vernalis in the Polish lowlands. Cell. Mol. Biol. Lett 2002, 7, 677–684. [Google Scholar]
- Li, X; Ding, X; Chu, B; Zhou, Q; Ding, G; Gu, S. Genetic diversity analysis and conservation of the endangered Chinese endemic herb Dendrobium officinale Kimura et Migo (Orchidaceae) based on AFLP. Genetica 2008, 133, 159–166. [Google Scholar]
- Zawko, G; Krauss, SL; Dixon, KW; Sivasithamparam, K. Conservation genetics of the rare and endangered Leucopogon obtectus (Ericaceae). Mol. Ecol 2001, 10, 2389–2396. [Google Scholar]
- Bidichandani, S; Ashizawa, T; Patel, PI. The GAA triplet-repeat expansion in Friedreich ataxia interferes with transcription and may be associated with an unusual DNA structure. Am. J. Hum. Genet 1998, 62, 111–121. [Google Scholar]
- Queller, DC; Strassman, JE; Hughes, CR. Microsatellites and Kinship. Trends Ecol. Evolut 1993, 8, 285–288. [Google Scholar]
- Allender, C. Microsatellite Information Exchange for Brassica; HRI: Wellesbourne, UK, 2004.
- Jarne, P; Lagoda, PJL. Microsatellites, from molecules to populations and back. Trends Ecol. Evol 1996, 11, 424–429. [Google Scholar]
- Noda, A; Nomura, N; Mitsui, Y; Setoguchi, H. Isolation and characterisation of microsatellite loci in Calystegia soldanella (Convolvulaceae), an endangered coastal plant isolated in Lake Biwa, Japan. Conserv. Genet 2009, 10, 1077–1079. [Google Scholar]
- Setoguchi, H; Mitsui, Y; Ikeda, H; Nomura, N; Tamura, A. Development and characterization of microsatellite loci in the endangered Tricyrtis ishiiana (Convallariaceae), a local endemic plant in Japan. Conserv. Genet 2009, 10, 705–707. [Google Scholar]
- Mcglaughlin, ME; Riley, L; Helenurm, K. Isolation of microsatellite loci from the endangered plant Galium catalinense subspecies acrispum (Rubiaceae). Mol. Ecol. Resources 2009, 9, 984–986. [Google Scholar]
- Foster, JT; Allan, GJ; Chan, AP; Rabinowicz, PD; Ravel, J; Jackson, PJ; Keim, P. Single nucleotide polymorphisms for assessing genetic diversity in castor bean (Ricinus communis). BMC Plant Biol 2010, 10, 13–23. [Google Scholar]
- Brumfield, RT; Beerli, P; Nickerson, DA; Edwards, SV. The utility of single nucleotide polymorphisms in inferences of population history. Trends Ecol. Evol. (Amst.) 2003, 18, 249–256. [Google Scholar]
- Tsuchihashi, Z; Dracopoli, NC. Progress in high-throughput SNP genotyping methods. Pharmacogenomics J 2002, 2, 103–110. [Google Scholar]
- Coles, ND; Coleman, CE; Christensen, SA; Jellen, EN; Stevens, MR; Bonifacio, RA; Rojas-Beltran, JA; Fairbanks, DJ; Maughan, PJ. Development and use of an expressed sequenced tag library in quinoa (Chenopodium quinoa Willd.) for the discovery of single nucleotide polymorphisms. Plant Soc 2005, 168, 439–447. [Google Scholar]
- Gómez, JMJ; Maloo, JN. Sequence diversity in three tomato species: SNPs, markers and molecular evolution. BMC Plant Biol 2009, 9, 85. [Google Scholar]
- Miller, PT; Henry, R. Single-nucleotide polymorphism detection in plants using a single-stranded pyrosequencing protocol with a universal biotinylated primer. Anal. Biochem 2003, 317, 166–170. [Google Scholar]
- Jaccoud, D; Peng, K; Feinstein, D; Kilian, A. Diversity arrays: A solid state technology for sequence information independent genotyping. Nucl. Acids Res 2001, 29, e25. [Google Scholar]
- van-Eijk, MJT; Broekhof, JLN; van-der-Poel, HJA; Hogers, RCJ; Schneiders, H; Verstege, KJE; van-Aart, JW; Geerlings, H; Buntjer, JB; van-Oeveren, AJ; Vos, P. SNPWave: A flexible multiplexed SNP genotyping technology. Nucl. Acids Res 2004, 32, e47. [Google Scholar]
- Rapley, R; Harbron, S. Molecular Analysis and Genome Discovery; John Wiley & Sons Ltd: Chichester, UK, 2001. [Google Scholar]
- Ingelsson, M; Shin, Y; Irizarry, MC; Hyman, BT; Lilius, L; Forsell, C; Graff, C. Genotyping of apolipoprotein E: Comparative evaluation of different protocols. Curr Protoc Hum Genet 2003, 9, 914:1–914:13. [Google Scholar]
- Coenye, T; Vandamme, P; Govan, JRW; LiPuma, JL. Taxonomy and Identification of the Burkholderia cepacia Complex. J. Clin. Microbiol 2001, 39, 3427–3436. [Google Scholar]
- Ferri, L; Perrin, E; Campana, S; Tabacchioni, S; Taccetti, G; Cocchi, P; Ravenni, N; Dalmastri, C; Chiarini, L; Bevivino, A; Manno, G; Mentasti, M; Fani, R. Application of multiplex single nucleotide primer extension (mSNuPE) to the identification of bacteria: The Burkholderia cepacia complex case. J. Microbiol. Methods 2010, 80, 251–256. [Google Scholar]
- Rafalski, JA; Tingey, SV. Genetic diagnostics in plant breeding: RAPDs, microsatellites and machines. Trends Genet 1993, 9, 275–280. [Google Scholar]
- Rafalski, JA. Novel genetic mapping tools in plants: SNPs and LD-based approaches. Plant Soc 2002, 162, 329–333. [Google Scholar]
- Wilson, AC. The molecular basis of evolution. Sci. Am 1985, 253, 164–173. [Google Scholar]
- Morozova, O; Marra, MA. Applications of next-generation sequencing technologies in functional genomics. Genomics 2008, 92, 255–264. [Google Scholar]
- Nei, M. Molecular Evolutionary Genetics; Columbia University Press: New York, NY, USA, 1987. [Google Scholar]
- Nadler, SA. Advantages and disadvantages of molecular phylogenetics: A case study of ascaridoid nematodes. J. Nematol 1995, 27, 423–432. [Google Scholar]
- Munthali, M; Ford-Lloyd, BV; Newbury, HJ. The random amplification of polymorphic DNA for fingerprinting plants. PCR Methods Appl 1992, 1, 274–276. [Google Scholar]
- Lowe, AJ; Hanotte, O; Guarino, L. Standardization of molecular genetic techniques for the characterization of germplasm collections: The case of random amplified polymorphic DNA (RAPD). Plant Genet. Resour. Newsl 1996, 107, 50–54. [Google Scholar]
- Krauss, SL. Complete exclusion of nonsires in an analysis of paternity in a natural plant population using amplified fragment length polymorphism (AFLP). Mol. Ecol 1999, 8, 217–226. [Google Scholar]
- Law, JR; Donini, P; Koebner, RMD; Jones, CR; Cooke, RJ. DNA profiling and plant variety registration III: The statistical assessment of distinctness in wheat using amplified fragment length polymorphisms. Euphytica 1998, 102, 335–342. [Google Scholar]
- Hokanson, SC; Szewc-McFadden, AK; Lamboy, WF; McFerson, JR. Microsatellite (SSR) markers reveal genetic identities, genetic diversity and relationships in a Malus x domestica Borkh. core subset collection. Theor. Appl. Genet 1998, 97, 671–683. [Google Scholar]
- Tautz, D; Ellegren, H; Weigel, D. Next generation molecular ecology. Mol. Ecol 2010, 19, 1–3. [Google Scholar]
- Sobrino, B; Brión, M; Carracedo, A. SNPs in forensic genetics: A review on SNP typing methodologies. Forensic Sci Int 2005. [Google Scholar]
- Engle, LJ; Simpson, CL; Landers, JE. Using high-throughput SNP technologies to study cancer. Oncogene 2006, 25, 1594–1601. [Google Scholar]
- Varshney, RK; Chabane, K; Hendre, PS; Aggarwal, RK; Graner, A. Comparative assessment of EST-SSR, EST-SNP and AFLP markers for evaluation of genetic diversity and conservation of genetic resources using wild, cultivated and elite barleys. Plant Soc 2007, 173, 638–649. [Google Scholar]
© 2010 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Arif, I.A.; Bakir, M.A.; Khan, H.A.; Al Farhan, A.H.; Al Homaidan, A.A.; Bahkali, A.H.; Al Sadoon, M.; Shobrak, M. A Brief Review of Molecular Techniques to Assess Plant Diversity. Int. J. Mol. Sci. 2010, 11, 2079-2096. https://doi.org/10.3390/ijms11052079
Arif IA, Bakir MA, Khan HA, Al Farhan AH, Al Homaidan AA, Bahkali AH, Al Sadoon M, Shobrak M. A Brief Review of Molecular Techniques to Assess Plant Diversity. International Journal of Molecular Sciences. 2010; 11(5):2079-2096. https://doi.org/10.3390/ijms11052079
Chicago/Turabian StyleArif, Ibrahim A., Mohammad A. Bakir, Haseeb A. Khan, Ahmad H. Al Farhan, Ali A. Al Homaidan, Ali H. Bahkali, Mohammad Al Sadoon, and Mohammad Shobrak. 2010. "A Brief Review of Molecular Techniques to Assess Plant Diversity" International Journal of Molecular Sciences 11, no. 5: 2079-2096. https://doi.org/10.3390/ijms11052079
APA StyleArif, I. A., Bakir, M. A., Khan, H. A., Al Farhan, A. H., Al Homaidan, A. A., Bahkali, A. H., Al Sadoon, M., & Shobrak, M. (2010). A Brief Review of Molecular Techniques to Assess Plant Diversity. International Journal of Molecular Sciences, 11(5), 2079-2096. https://doi.org/10.3390/ijms11052079