Effects of Aflatoxin B1 and Fumonisin B1 on the Viability and Induction of Apoptosis in Rat Primary Hepatocytes

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Experimental Section
2.1. Animals and Cell Isolation
2.2. Cell Culture
2.3. Toxins
2.4. Treatments
2.5. Flow Cytometry Analysis
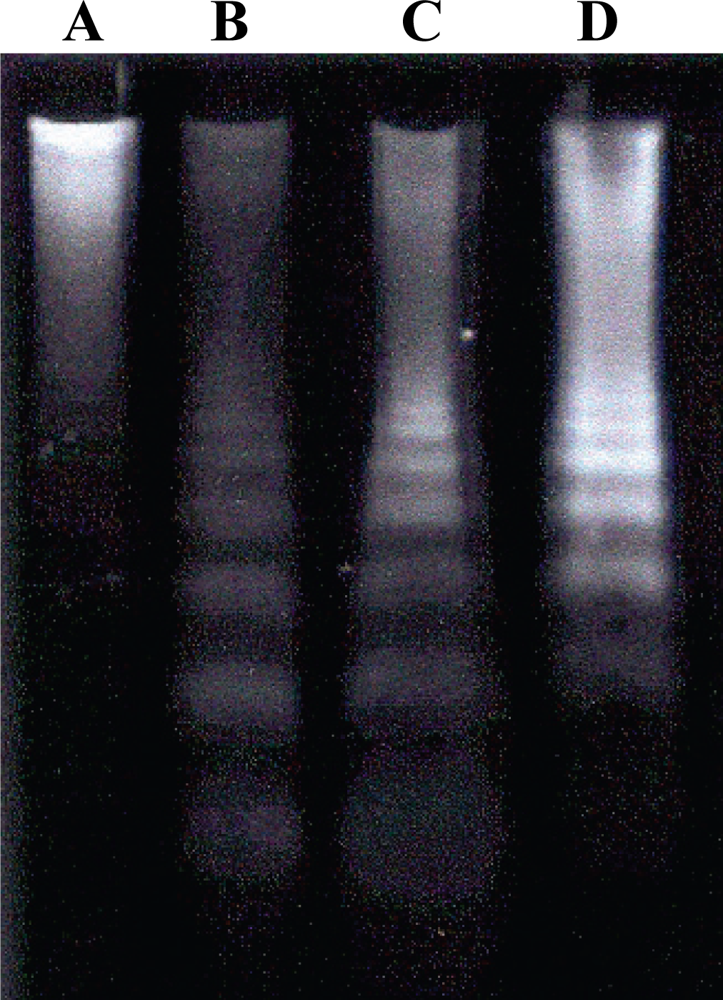
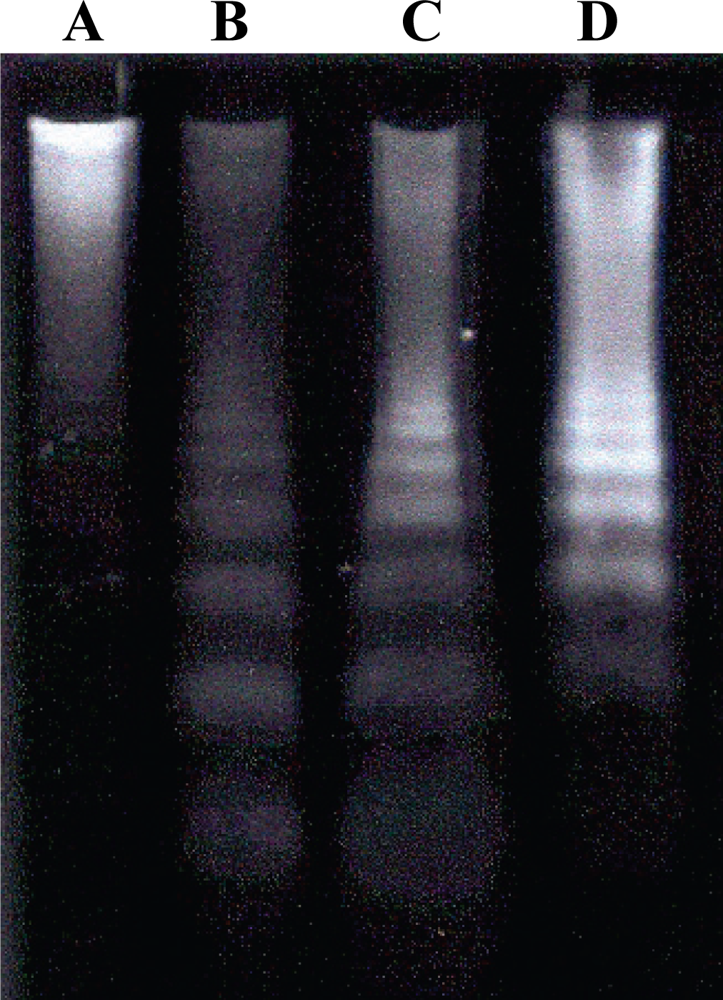
2.6. Detection of DNA Fragmentation by Agarose Gel Electrophoresis
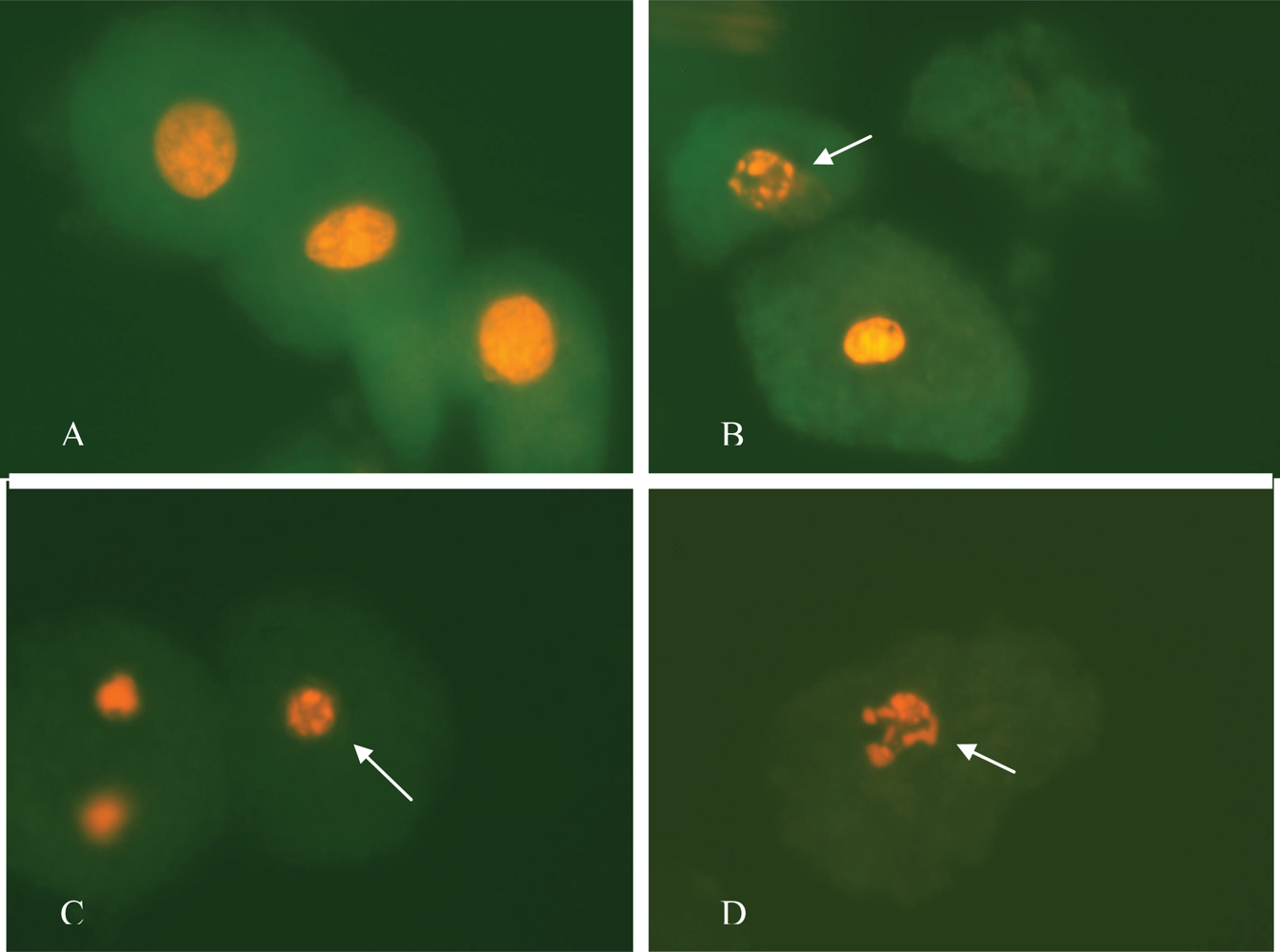
2.7. Observation of Morphological Alterations
2.8. Statistical Analysis
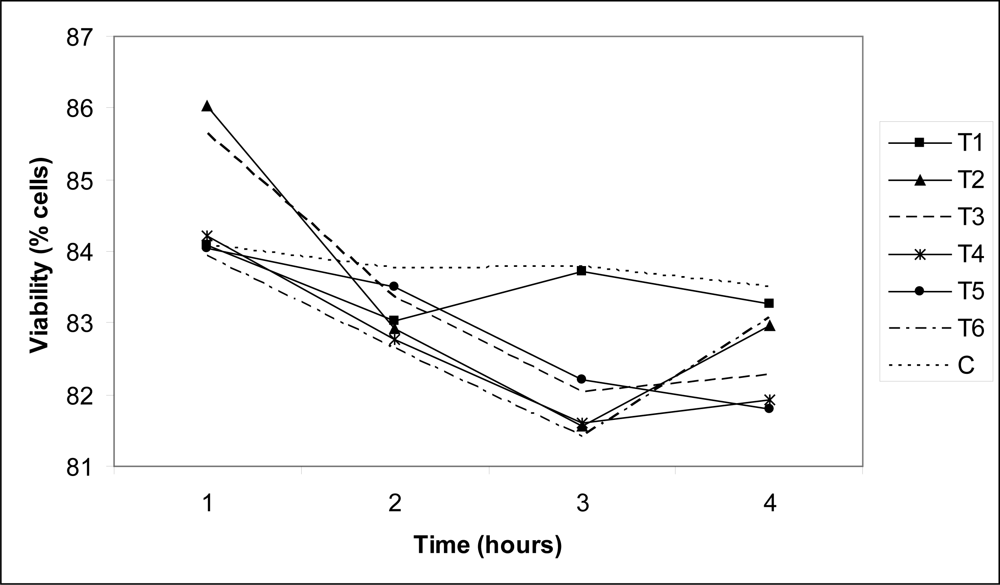
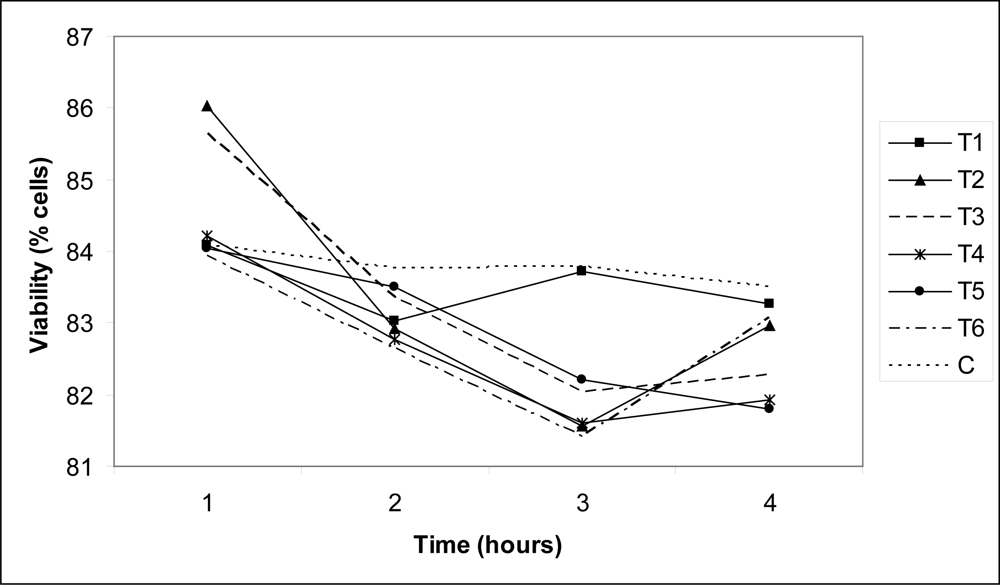
3. Results and Discussion
Acknowledgments
References and Notes
- Mycotoxins: Risks in Plant, Animal, and Human Systems, Task Force Report No.139; Council for Agricultural Science and Technology: Ames, IA, USA, 2003.
- Hayes, JR; Judah, DJ; McLellan, LI; Neal, GE. Contribution of the glutatione-s-transferase to the mechanism of resistence to aflatoxin B1. Pharmac. Ther 1991, 50, 442–472. [Google Scholar]
- Smith, JE; Herderson, SR. Mycotoxin and Animal Food, 3rd ed; CRC Press: Boca Raton, FL, USA, 1991; pp. 1–35. [Google Scholar]
- Bezuidenhout, SC; Gelderblom, WCA; Gorst-Allman, CP; Horak, RM; Marasas, WFO; Spiteller, G; Vleggaar, R. Structure elucidation of the fumonisins, mycotoxins from Fusarium moniliforme. J. Chem. Soc. Chem. Commun 1988, 11, 743–745. [Google Scholar]
- Meredith, FI. Isolation and characterization of fumonisins. Methods Enzymol 1999, 311, 361–373. [Google Scholar]
- Wang, E; Norred, WP; Bacon, CW; Riley, RT; Merril, AH, Jr. Inhibition of sphingolipid biosynthesis by fumonisins. Implications for diseases associated with Fusarium moniliforme. J. Biol. Chem 1991, 266, 1486–1490. [Google Scholar]
- Riley, RT; Voss, KA; Yoo, HS; Gelderblom, WCA; Merril, AH, Jr. Mechanism of Fumonisin Toxicity and Carcinogenesis. J. Food Prot 1994, 57, 638–645. [Google Scholar]
- Dragan, YP; Bidlack, WR; Cohen, SM; Goldsworthy, TL; Hard, GC; Howard, PC; Riley, RT; Voss, KA. Implications of apoptosis for toxicity, carcinogenicity, and risk assessment: fumonisin B1 as an example. Toxicol. Sci 2001, 61, 6–17. [Google Scholar]
- Seefelder, W; Humpf, H-U; Schwerdt, G; Freudinger, R; Gekle, M. Indution os apoptosis in cultures human proximal tubule cells by fumonisins and fumonisin metabolites. Toxicol. Appl. Pharmacol 2003, 192, 146–153. [Google Scholar]
- Smith, ER; Jones, PL; Boss, JM; Merril, AH, Jr. Changing J774A.1 cells to new medium perturbs multiple signaling pathways, including the modulation of protein kinase c by endogenous sphingoid bases. J. Biol. Chem 1997, 272, 5640–5646. [Google Scholar]
- Jones, C; Ciacci-Zanella, JR; Zhang, Y; Henderson, G; Dickman, M. Analysis of fumonisin B1-induced apoptosis. Environ. Health Perspect 2001, 109, 315–320. [Google Scholar]
- Ramasamy, S; Wang, E; Hennig, B; Merril, AH, Jr. Fumonisin B1 alters sphingolipid metabolism and disrupts the barrier function of endothelial cells in culture. Toxicol. Appl. Pharmacol 1995, 133, 343–348. [Google Scholar]
- Cuvillier, O. Sphingosine in apoptosis signaling. Biochim. Biophys. Acta 2002, 1585, 153–162. [Google Scholar]
- Orsi, RB; Correa, B; Pozzi, CR; Schammass, E; Nogueira, JR; Dias, SMC; Mallozzi, M. Mycoflora and occurende of fumonisin in freshly harvested and stored hybrid maize. J. Stored Prod. Res 2001, 36, 75–87. [Google Scholar]
- Gelderblom, WCA; Marasas, WFO; Lebepe-Mazur, S; Swanevelder, S; Vessey, CJ; Hall, P; de la, M. Interaction f fumonisin B1 and Aflatoxin B1 in a short-term carcinogenesis model in rat liver. Toxicology 2002, 171, 161–173. [Google Scholar]
- Freshney, RI. Culture of Animal Cells: A Manual of Basic Techniques, 4th ed; Wiley-Liss Hardcover: New York, NY, USA, 2000; pp. 169–171. [Google Scholar]
- Lin, MT; Dianese, JC. A coconut agar medium for rapid detection of aflatoxin production by Aspergillus spp. Phytopathology 1976, 66, 1466–1469. [Google Scholar]
- Oliveira, CAF; Rosmaninho, JF; Butkersitis, P; Corrêa, B; Reis, TA; Guerra, JL; Albuquerque, R; Moro, MEG. Effect of low levels of dietary aflatoxin B1 on Laying Japanese quail. Poult. Sci 2002, 81, 976–980. [Google Scholar]
- AOAC, Association Official Analytical Chemists Official Methods of Official Analytical Chemists, 16th ed; AOAC: Washington, DC, USA, 1995.
- Parks, DR; Bryan, VM; Oi, VM; Herzenberg, LA. Antigen-specific identification and cloning of hybridomas with a Fluorescence Activated Cell Sorter (FACS). Proc. Natl. Acad. Sci. USA 1979, 76, 1976–1979. [Google Scholar]
- McCullag, PE; Nelder, JA. Generalized Linear Models, 2nd ed; Chapman and Hall: London, UK, 1989. [Google Scholar]
- Shen, HM; Shi, CY; Shen, Y; Ong, CN. Detection of elevated reactive oxygen species level in cultures rat hepatocytes treated with aflatoxin B1. Free Rad. Biol. Med 1996, 21, 139–146. [Google Scholar]
- Kuilman, MEM; Maas, RFM; Fink-Gremels, J. Cytochrome P450-mediated metabolism and cytotoxicity of aflatoxin B1 in bovine hepatocytes. Toxicol. in Vitro 2000, 14, 321–327. [Google Scholar]
- Metcalfe, S; Neal, GE. Some studies on the relationship between the citotoxicity of aflatoxin B1 to the rat hepatocite and the metabolism of the toxin. Carcinogenesis 1983, 4, 1013–1019. [Google Scholar]
- Galvano, F; Russo, A; Cardile, V; Galvano, G; Vanella, A; Renis, M. DNA damage in human fibroblasts exposed to fumonisin B1. Food. Chem. Toxicol 2002, 40, 25–31. [Google Scholar]
- Minervini, F; Fornelli, F; Flynn, KM. Toxicity and apoptosis induced by the mycotoxins nivalenol, deoxinivalenol and fumonisin B1 in a human erythroleukemia cell line. Toxicol. in Vitro 2004, 18, 21–28. [Google Scholar]
- Wu, W; Liu, T; Vesonder, RF. Comparative cytotoxicity of Fumonisin B1 and moniliformin in chicken primary cell cultures. Mycopathology 1995, 132, 111–116. [Google Scholar]
- Rumora, L; Kovacic, S; Rozgaj, R; Cepelak, I; Pepeljnjak, S; Grubisic, TZ. Cytotoxic and genotoxic effects of fumonisin B1 on rabbit kidney RK13 cell line. Arch. Toxicol 2002, 76, 55–61. [Google Scholar]
- Dresden-Osborne, C; Noblet, GP. Fumonisin B1 affects viability and alters nitric oxide production of a murine macrophage cell line. Int Immunopharmacol 2002, 2, 1087–1093. [Google Scholar]
- Yoo, HS; Norred, WP; Showker, J; Riley, RT. Elevated sphingoid bases and complex sphingolipid depletion as contributing factors in fumonisin-iduced cytotoxicity. Toxicol. Appl. Pharmacol 1996, 138, 211–218. [Google Scholar]
- Mobio, TA; Anane, R; Baudrimont, I; Carratu, MR; Shier, TW; Dano, SD; Ueno, Y; Creppy, EE. Epigenetic properties of fumonisin B1: Cell cycle arrest and DNA base modification in C6 glioma cells. Toxicol. Appl. Pharmacol 2000, 164, 91–96. [Google Scholar]
- Schmelz, EM; Dobrink-Kurtzman, MA; Roberts, PC; Kozutsumi, Y; Kawasaki, T; Merril, AH, Jr. Induction of apoptosis by fumonisin B1 in HT29 cells in mediated by the acumulation of endogenous free sphingoid bases. Toxicol. Appl. Pharmacol 1998, 148, 252–260. [Google Scholar]
- Bouhet, S; Hourcade, E; Loiseau, N; Fikry, A; Martinez, S; Roselli, M; Galtier, P; Mengheri, E; Oswald, IP. The mycotoxin fumonisin B1 alters the proliferation and the barrier function of porcine intestinal epithelial cells. Toxicol. Sci 2004, 77, 165–171. [Google Scholar]
- Ciacci-Zanela, JR; Jones, C. Fumonisin B1, a mycotoxin contaminant of cereal grains, and inducer of apoptosis via the tumour necrosis factor pathway and caspase activation. Food Chem. Toxicol 1999, 37, 703–712. [Google Scholar]
- Ramljak, D; Calvert, RJ; Wiesenfeld, PW; Diwan, BA; Catipovic, B; Marasas, WF; Victor, TC; Anderson, LM; Gelderblom, WC. Potential mechanism for fumonisin B(1)-mediated hepatocarcinogenesis: ciclin D1 stabilization associated with activation of AKT and inibition of GSK-3beta activity. Carcinogenesis 2000, 21, 1537–1546. [Google Scholar]
- Seegers, JC; Joubert, AM; Panzer, A; Lottering, ML; Jordan, CA; Joubert, F; Maree, JL; Bianchi, P; Kock, M; Gelderblom, WCA. Fumonisin B1 influenced the effects of arachidonic acid, prostaglandis E2 and A2 on cell cycle progression, apoptosis induction, tyrosine- and CDC2-kinase activity in oesophageal cancer cells. Prostaglandins Leukot. Essent. Fatty Acids 2000, 62, 75–84. [Google Scholar]
- Mello, MLS; Vidal, BC; Maria, SS. Morte celular. In A Célula 2001, 1st ed; Manole: Barueri, Sao Paulo, Brazil, 2001; pp. 275–281. [Google Scholar]
- Tolleson, WH; Couch, LH; Melchior, WB, Jr; Jenkins, GR; Muskhelishvili, M; Muskhelishvili, L; Mcgarrity, LJ; Domon, O; Morris, SM; Howard, PC. Fumonisin B1 induces apoptosis in cultured human keratinocytes through sphinganine accumulation and ceramide depletion. Int. J. Oncol 1999, 14, 833–843. [Google Scholar]
- van Cruchten, S; van den Broeck, W. Morphological and biochemical aspects of apoptosis, oncosis and necrosis. Anat. Histol. Embryol 2002, 31, 214–223. [Google Scholar]
- Gelderblom, WCA; Abel, S; Smut, CM; Marnewick, J; Marasas, WFO; Lemmer, ER; Ramljak, D. Fumonisin-Induced hepatocarcinogenesis: mechanism related to cancwer initiation and promotion. Environ. Health Pespect 2001, 109, 291–300. [Google Scholar]
- Theumer, MG; Lopez, AG; Masih, DT; Chulze, SN; Rubinstein, HR. Immunobiological effects of AFB1 and AFB1–FB1 mixture in experimental subchronic mycotoxicoses in rats. Toxicology 2003, 186, 159–170. [Google Scholar]
- Theumer, MG; López, AG; Aoki, MP; Cánepa, MC; Rubinstein, HR. Subchronic mycotoxicoses in rats. Histopathological changes and modulation of the sphinganine to sphingosine (Sa/So) ratio imbalance induced by Fusarium verticillioides culture material, due to the coexistence of aflatoxin B1 in the diet. Food Chem. Toxicol 2008, 46, 967–977. [Google Scholar]
© 2010 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Ribeiro, D.H.B.; Ferreira, F.L.; Da Silva, V.N.; Aquino, S.; Corrêa, B. Effects of Aflatoxin B1 and Fumonisin B1 on the Viability and Induction of Apoptosis in Rat Primary Hepatocytes. Int. J. Mol. Sci. 2010, 11, 1944-1955. https://doi.org/10.3390/ijms11041944
Ribeiro DHB, Ferreira FL, Da Silva VN, Aquino S, Corrêa B. Effects of Aflatoxin B1 and Fumonisin B1 on the Viability and Induction of Apoptosis in Rat Primary Hepatocytes. International Journal of Molecular Sciences. 2010; 11(4):1944-1955. https://doi.org/10.3390/ijms11041944
Chicago/Turabian StyleRibeiro, Deise H. B., Fabiane L. Ferreira, Valéria N. Da Silva, Simone Aquino, and Benedito Corrêa. 2010. "Effects of Aflatoxin B1 and Fumonisin B1 on the Viability and Induction of Apoptosis in Rat Primary Hepatocytes" International Journal of Molecular Sciences 11, no. 4: 1944-1955. https://doi.org/10.3390/ijms11041944
APA StyleRibeiro, D. H. B., Ferreira, F. L., Da Silva, V. N., Aquino, S., & Corrêa, B. (2010). Effects of Aflatoxin B1 and Fumonisin B1 on the Viability and Induction of Apoptosis in Rat Primary Hepatocytes. International Journal of Molecular Sciences, 11(4), 1944-1955. https://doi.org/10.3390/ijms11041944