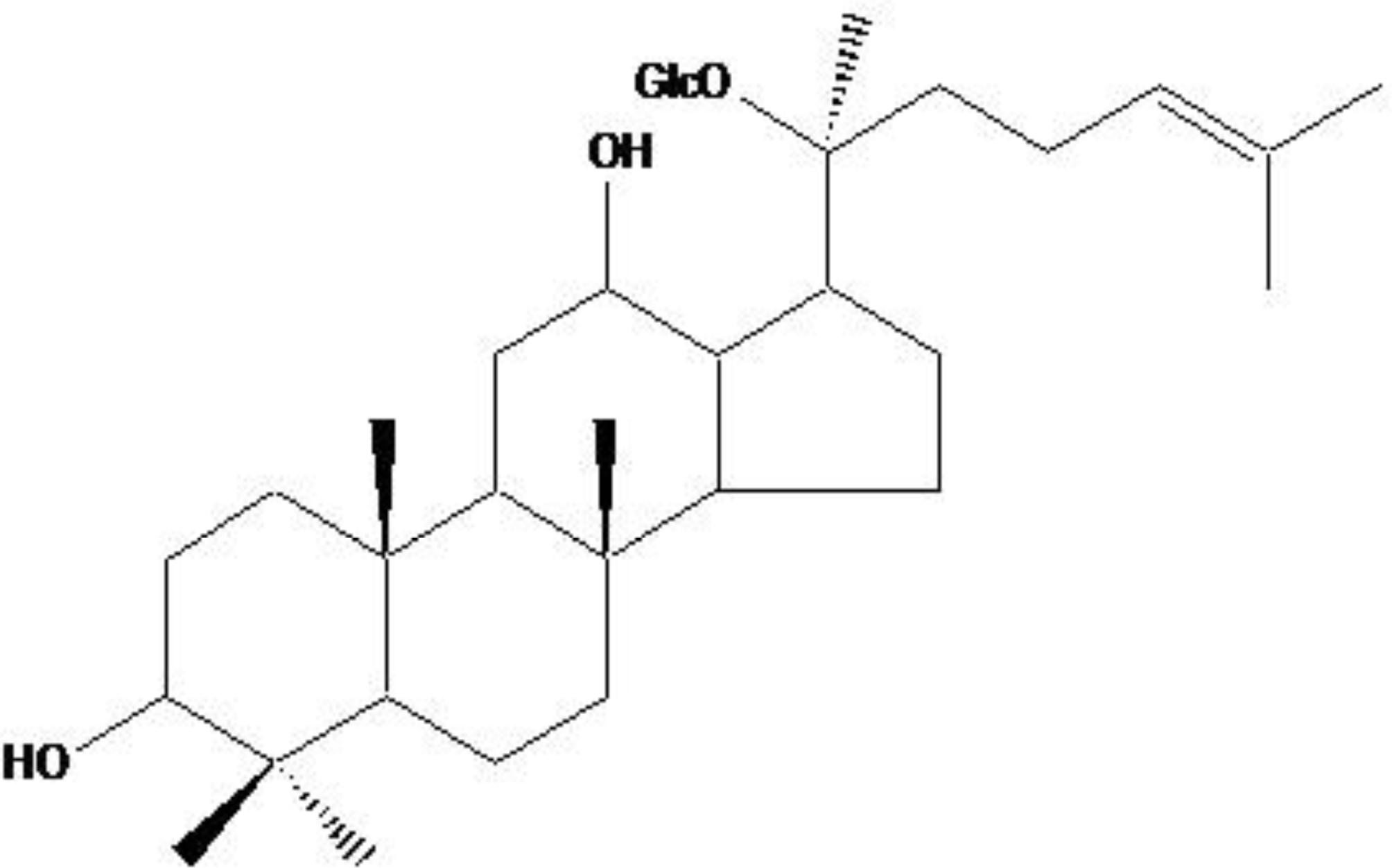
Compound K, a Metabolite of Ginseng Saponin, Induces Mitochondria-Dependent and Caspase-Dependent Apoptosis via the Generation of Reactive Oxygen Species in Human Colon Cancer Cells

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
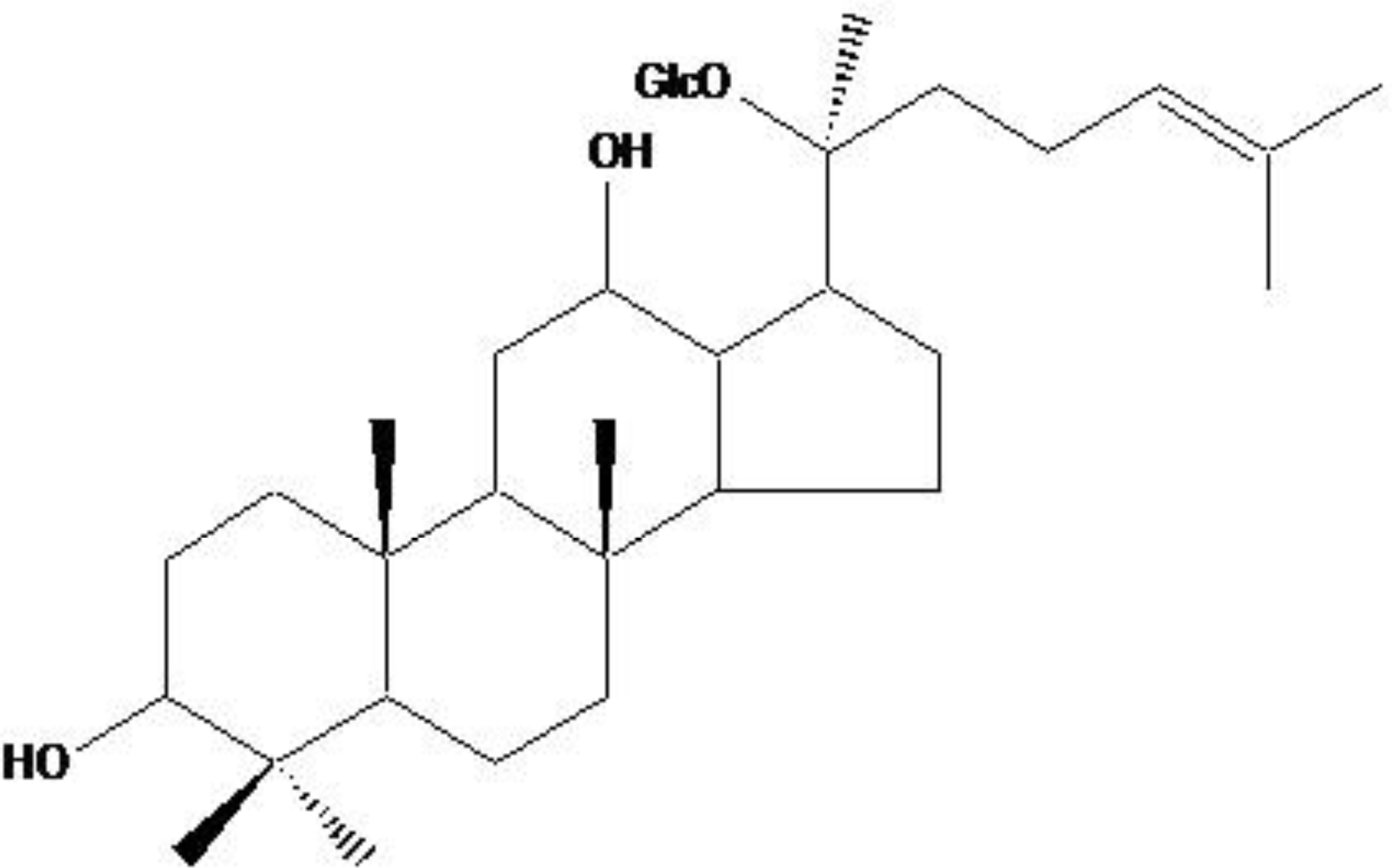
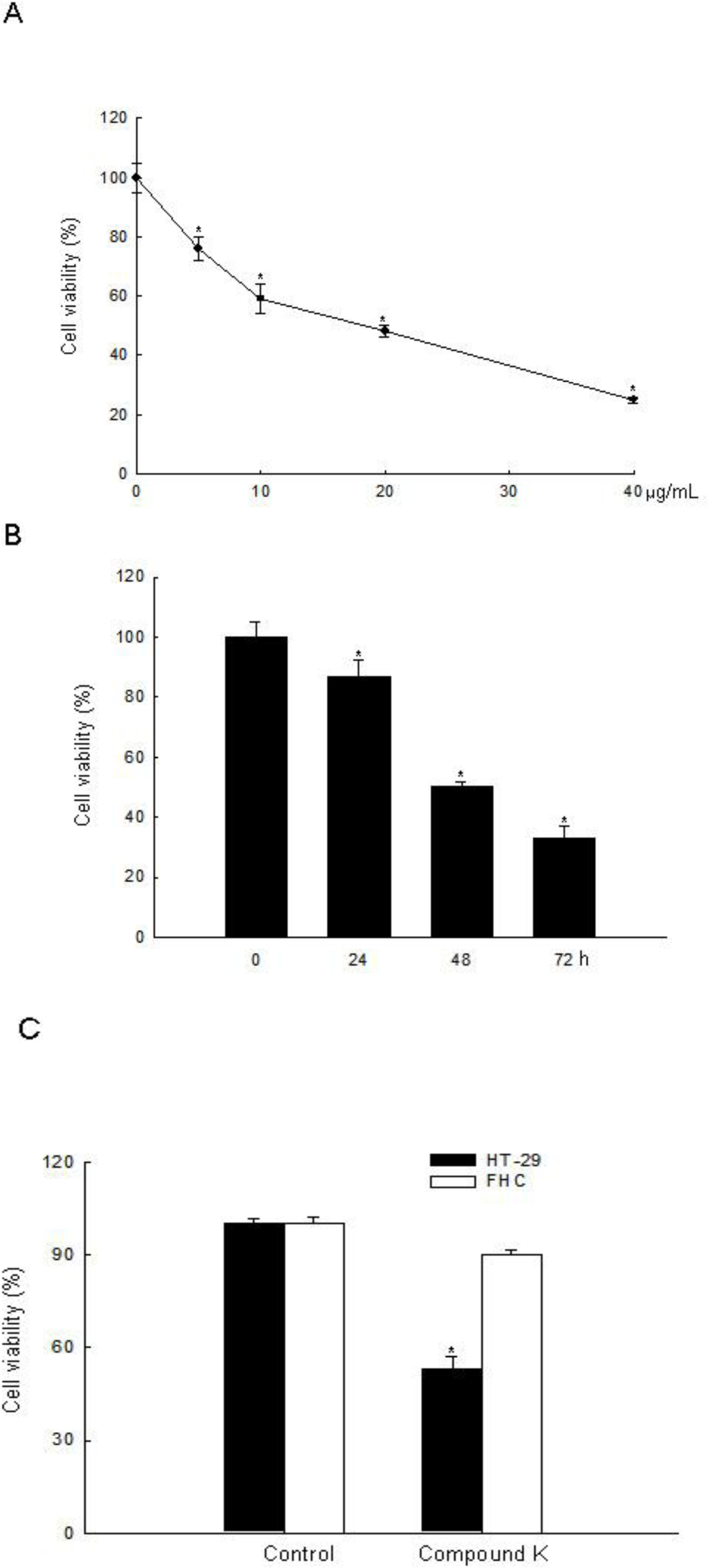
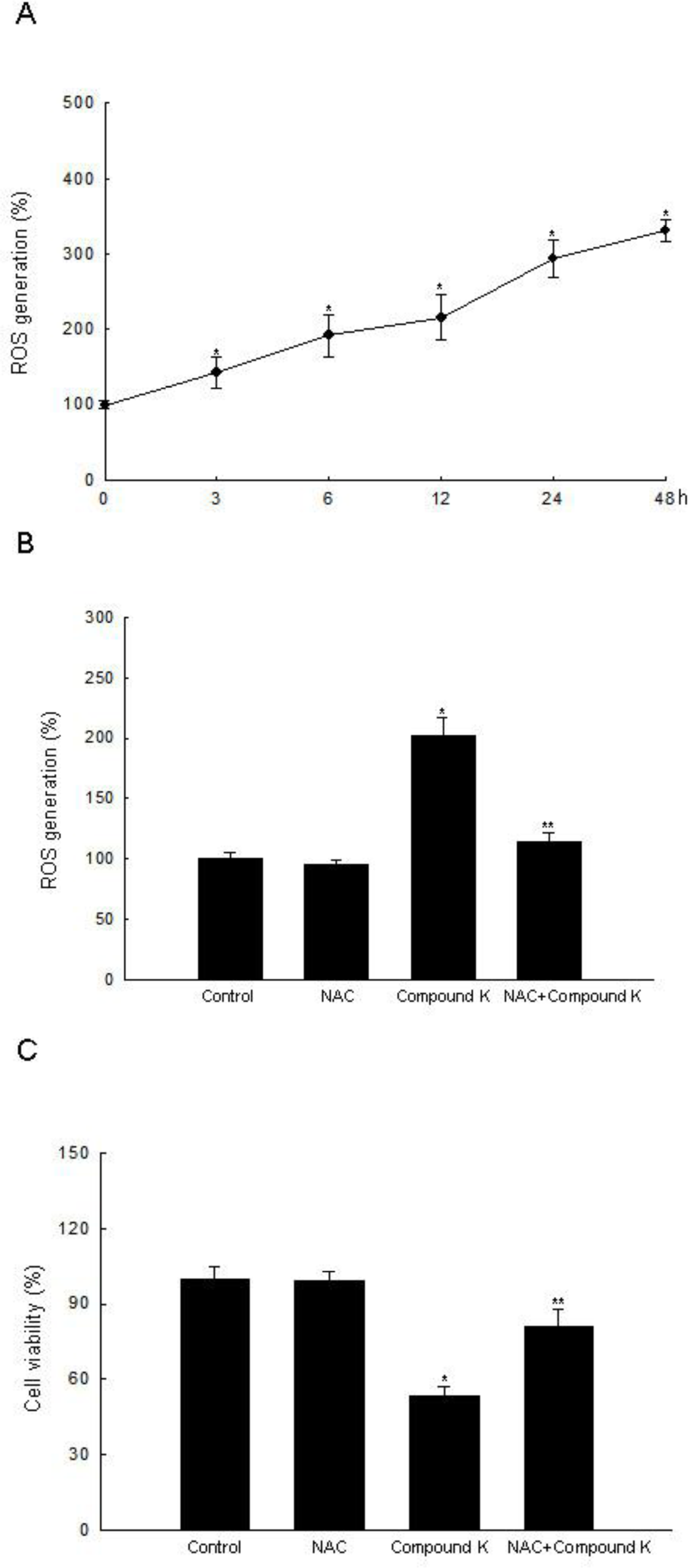
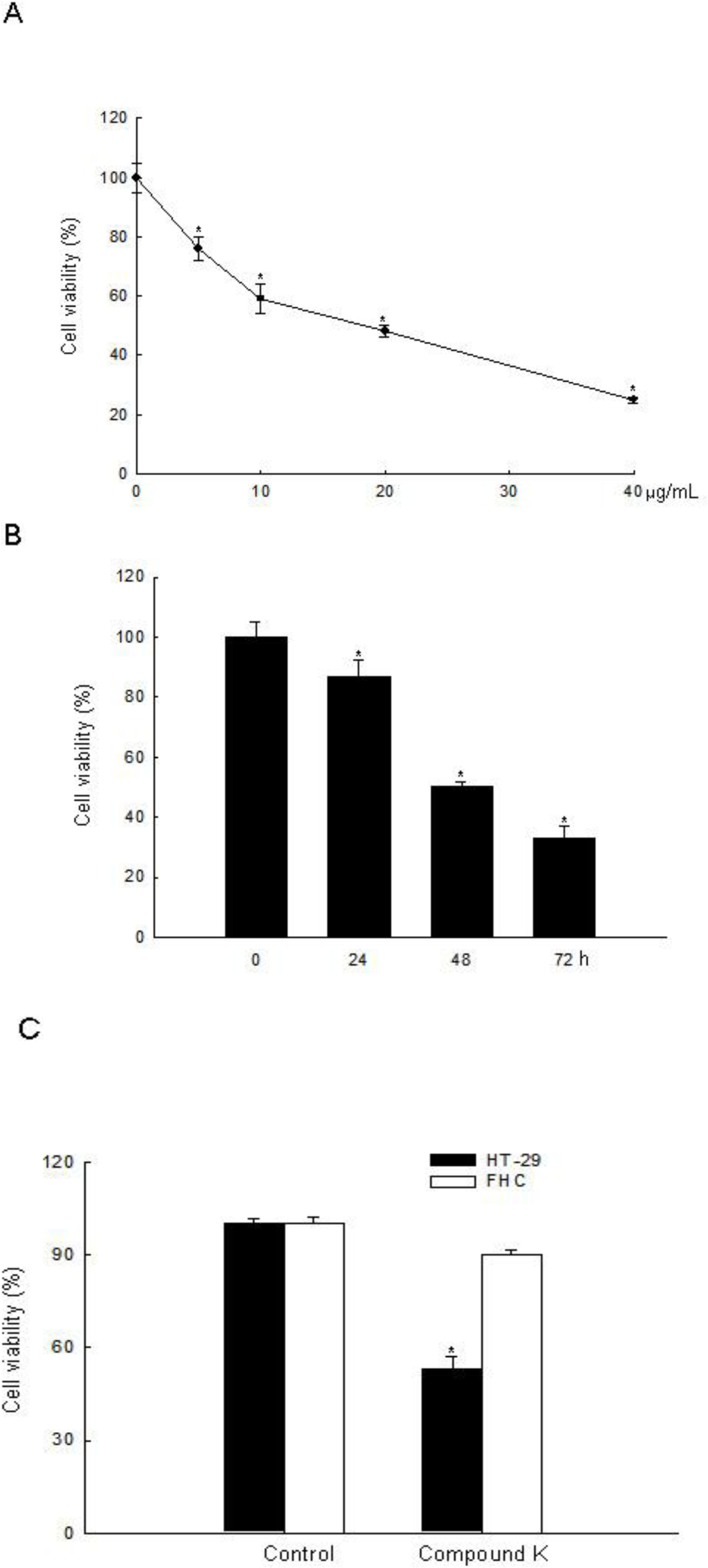
2.1. ROS-Induced Cytotoxic Effect of Compound K on HT-29 Colon Cancer Cells
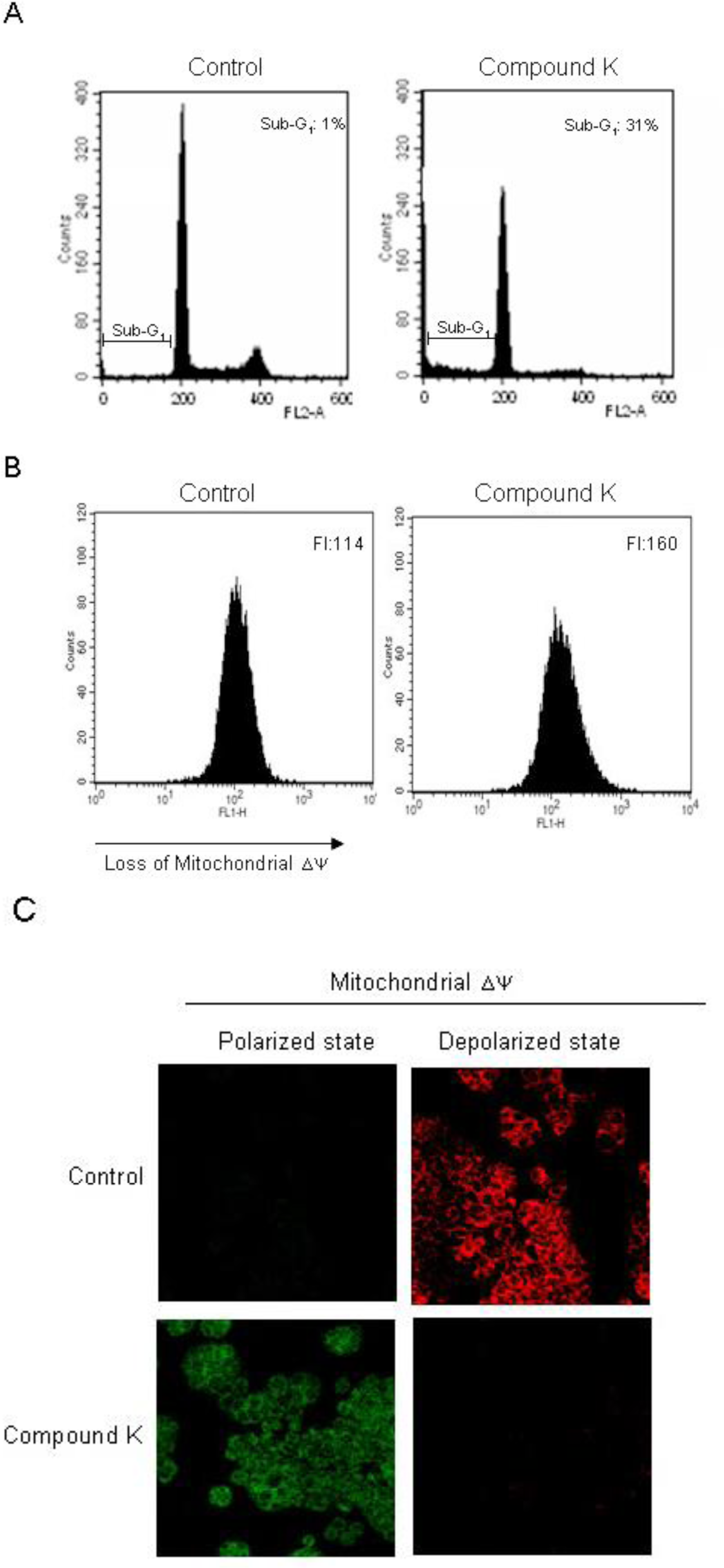
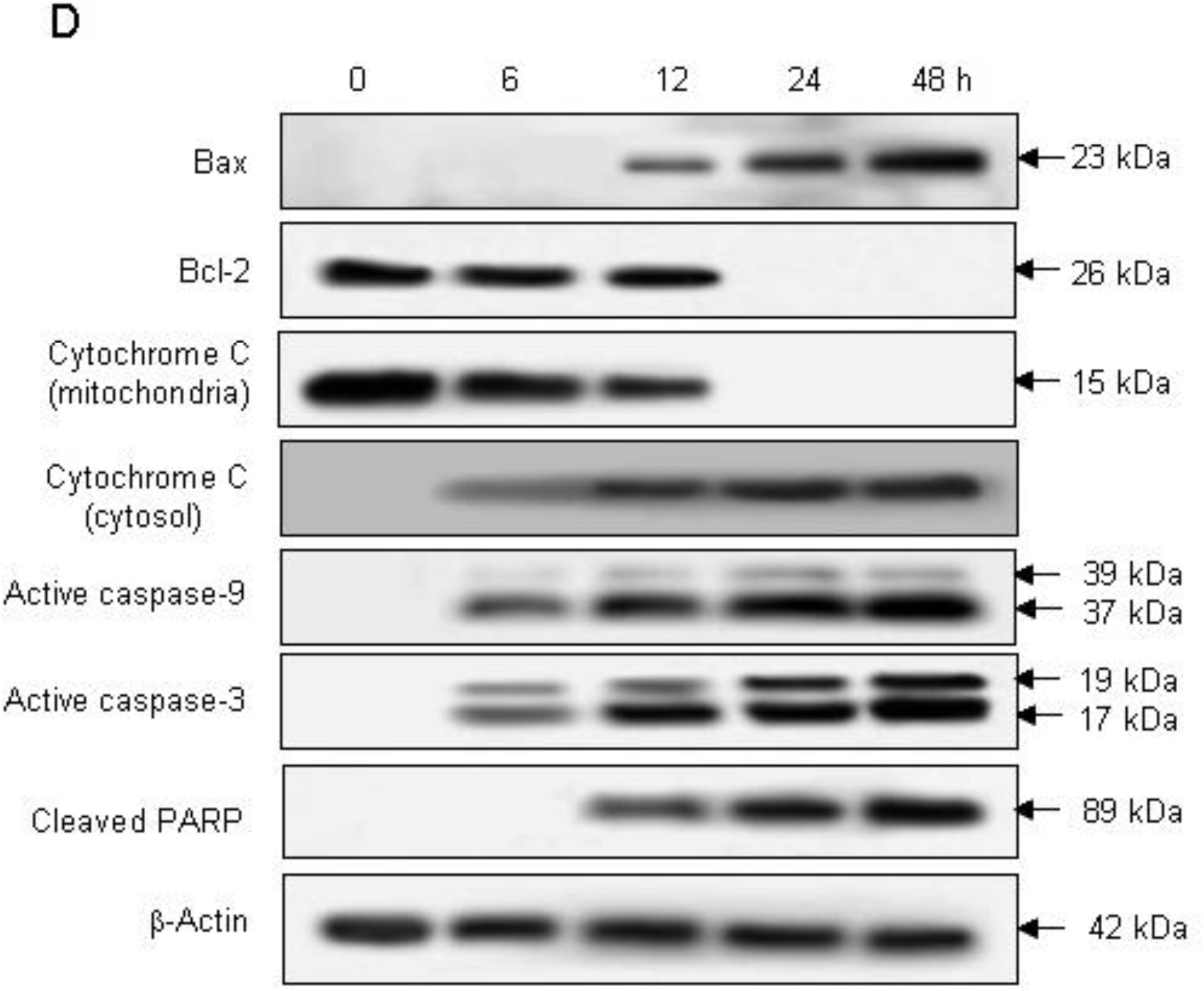
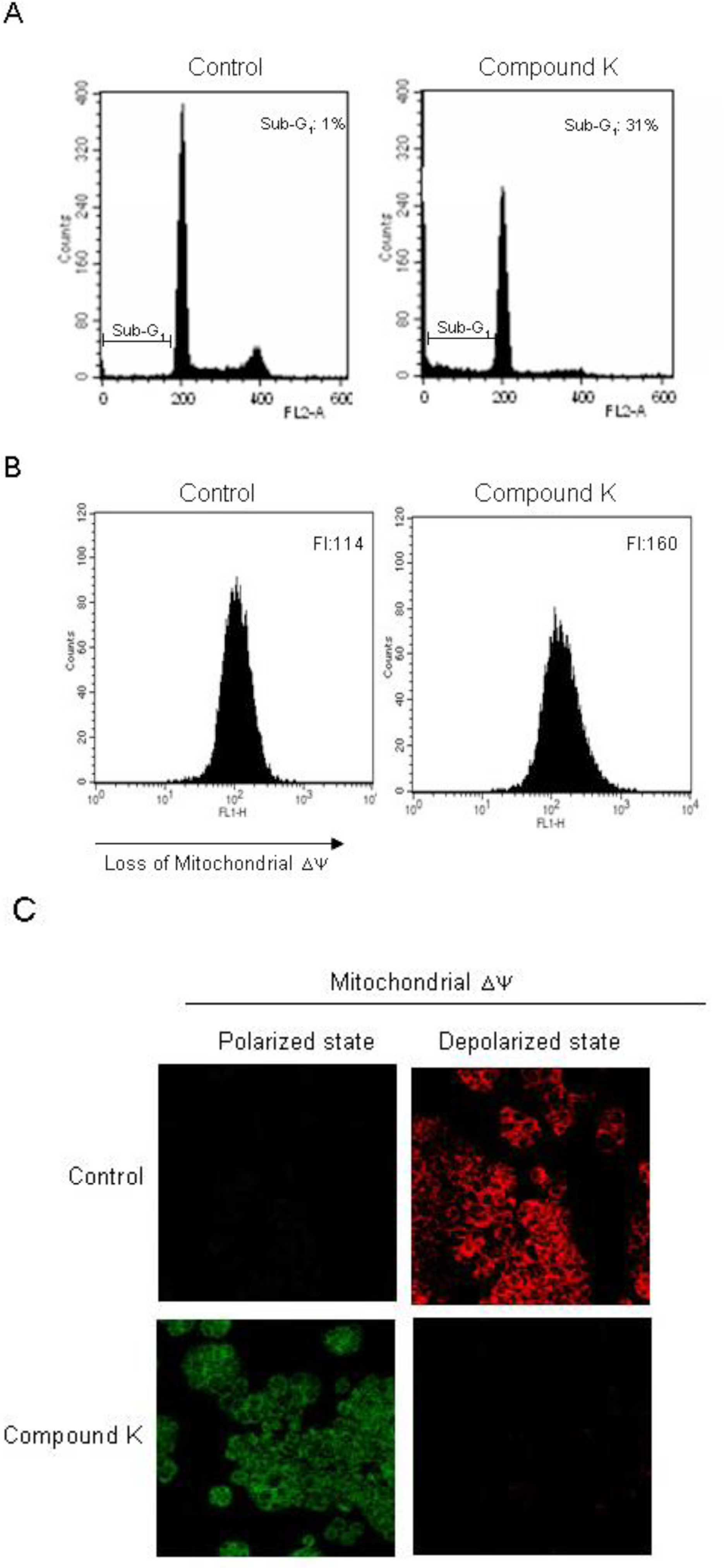
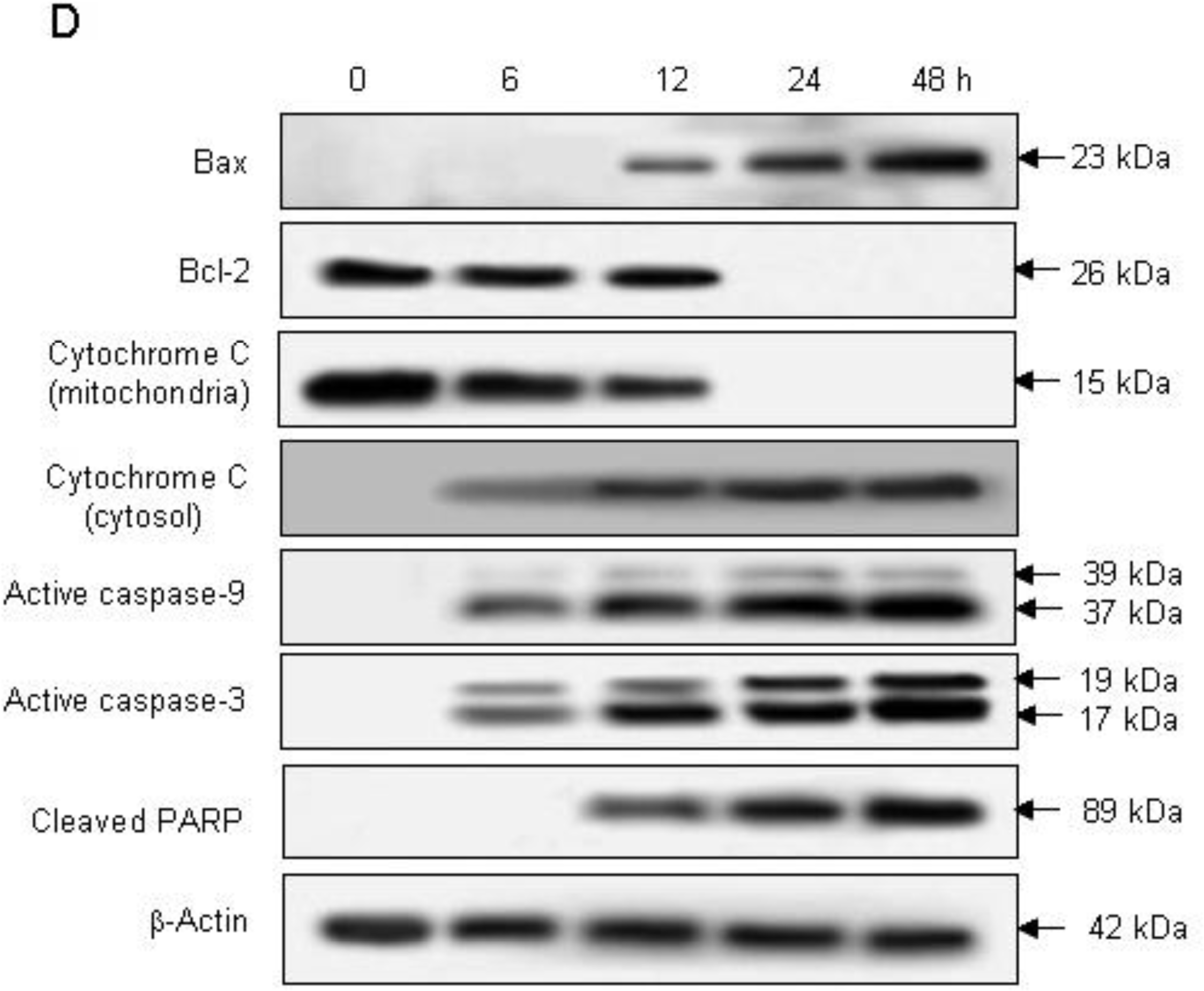
2.2. Induction of Apoptosis by Compound K via a Mitochondria-Dependent Pathway
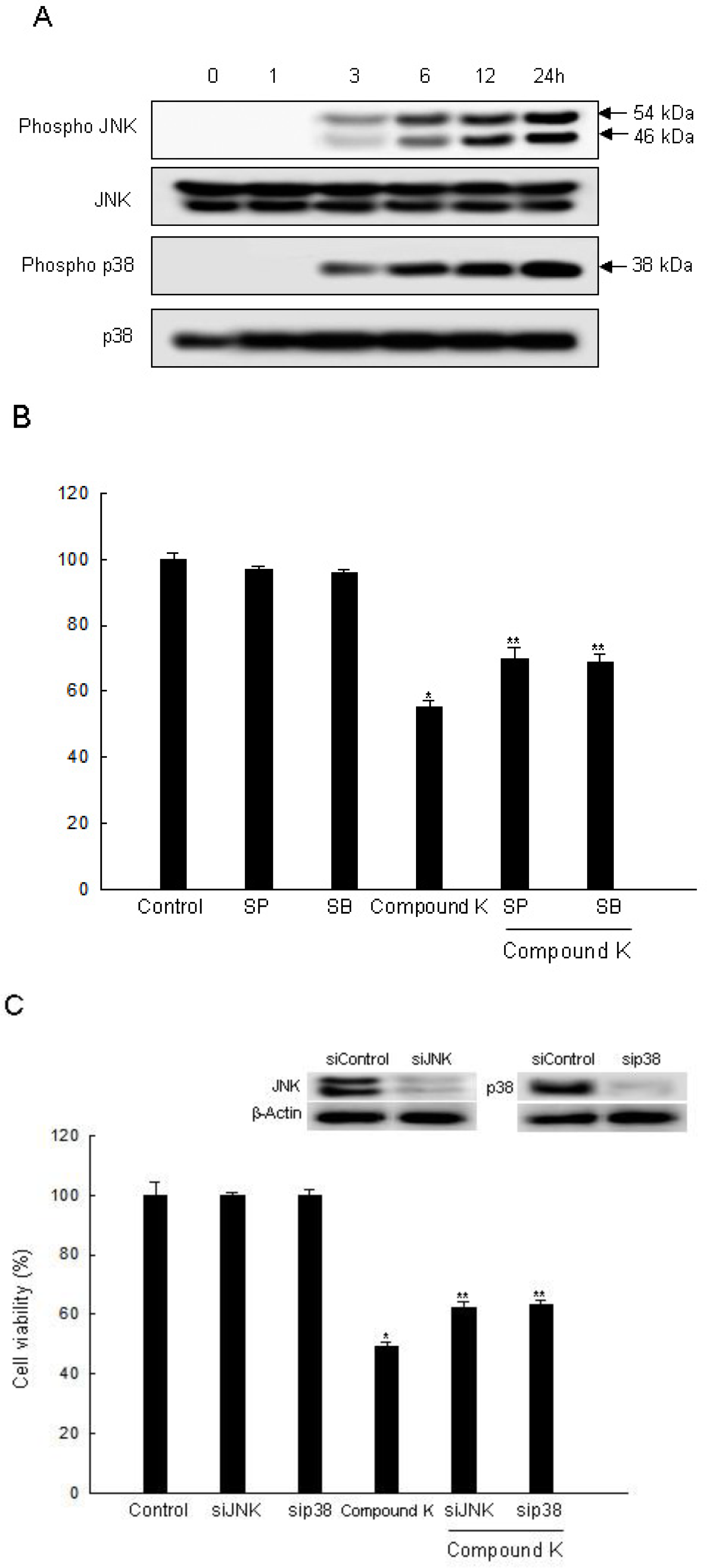
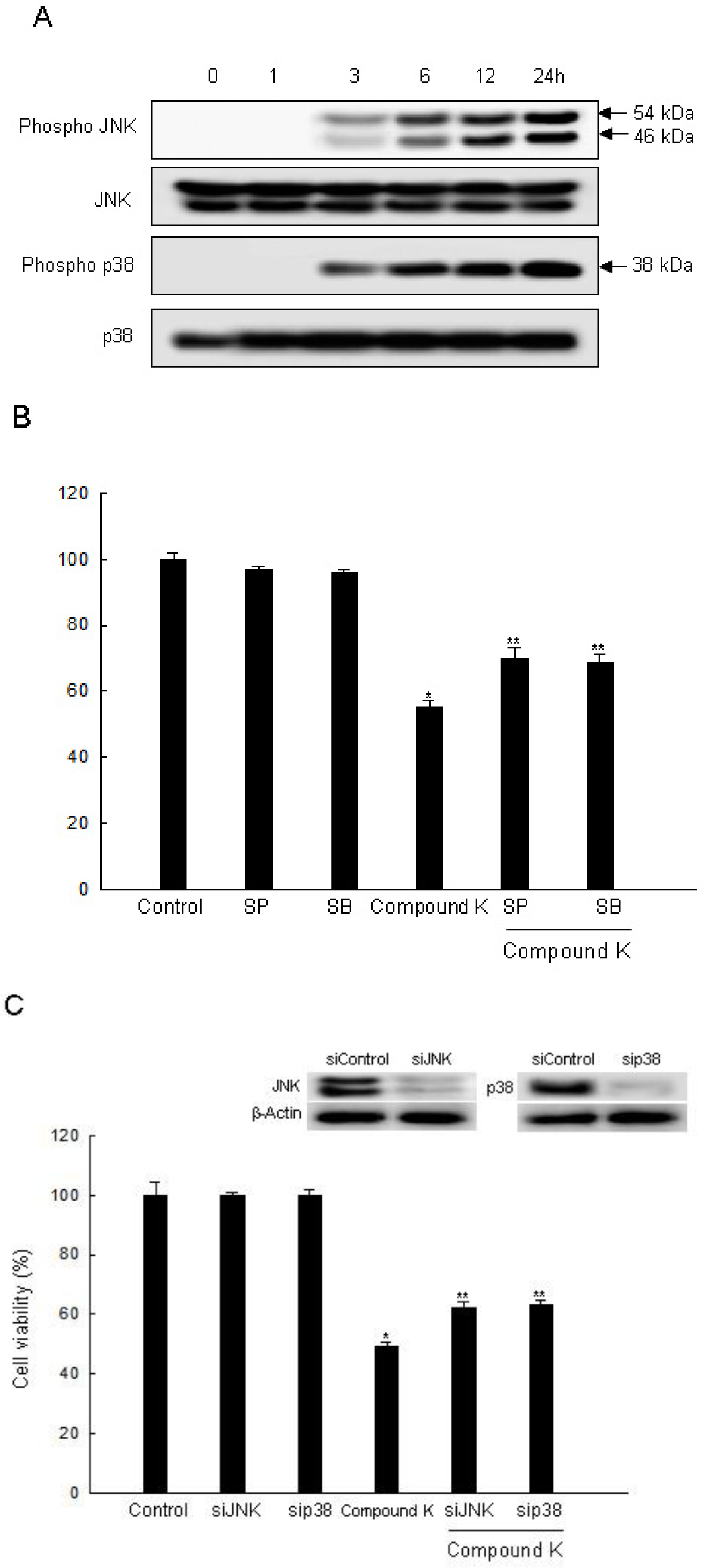
2.3. Induction of Apoptosis by Compound K via JNK and p38 MAPK Activation
3. Experimental Section
3.1. Chemicals
3.2. Cells and Cell Culture
3.3. Cell Viability Assay
3.4. Measurement of Intracellular Reactive Oxygen Species (ROS)
3.5. Detection of Sub-G1 Hypodiploid Cells
3.6. Analysis of Mitochondrial Membrane Potential (Δψm)
3.7. Western Blot Analysis
3.8. Transient Transfection of Small RNA Interference (siRNA)
3.9. Statistical Analysis
4. Conclusions
Acknowledgments
References
- Suzuki, YJ; Forman, HJ; Sevanian, A. Oxidants as stimulators of signal transduction. Free Radic. Biol. Med 1997, 22, 269–285. [Google Scholar]
- Kannan, K; Jain, SK. Oxidative stress and apoptosis. Pathophysilogy 2000, 7, 153–163. [Google Scholar]
- Dröge, W. Free radicals in the physiological control of cell function. Physiol. Rev 2002, 82, 47–95. [Google Scholar]
- Green, DR; Reed, JC. Mitochondria and apoptosis. Science 1998, 281, 1309–1312. [Google Scholar]
- Li, J; Huang, CY; Zheng, RL; Cui, KR; Li, JF. Hydrogen peroxide induces apoptosis in human hepatoma cells and alters cell redox status. Cell Biol. Int 2000, 24, 9–23. [Google Scholar]
- Shih, CM; Ko, WC; Wu, JS; Wei, YH; Wang, LF; Chang, EE; Lo, TY; Cheng, HH; Chen, CT. Mediating of caspase-independent apoptosis by cadmium through the mitochondria-ROS pathway in MRC-5 fibroblasts. J. Cell. Biochem 2004, 91, 384–397. [Google Scholar]
- Thompson, CB. Apoptosis in the pathogenesis and treatment of disease. Science 1995, 267, 1456–1462. [Google Scholar]
- Cobb, JP; Hotchkiss, RS; Karl, IE; Buchman, TG. Mechanisms of cell injury and death. Br. J. Anaesth 1996, 77, 3–10. [Google Scholar]
- Ziegler, U; Groscurth, P. Morphological features of cell death. News Physiol. Sci 2004, 19, 124–128. [Google Scholar]
- Thornberry, NA; Lazebnik, Y. Caspases: Enemies within. Science 1998, 281, 1312–1316. [Google Scholar]
- Budihardjo, I; Oliver, H; Lutter, M; Luo, X; Wang, X. Biochemical pathways of caspase activation during apoptosis. Annu. Rev. Cell Dev. Biol 1999, 15, 269–290. [Google Scholar]
- Earnshaw, WC; Martins, LM; Kaufmann, SH. Mammalian caspases: Structure, activation, substrates, and functions during apoptosis. Annu. Rev. Biochem 1999, 68, 383–424. [Google Scholar]
- Izban, KF; Wrone-Smith, T; Hsi, ED; Schnitzer, B; Quevedo, ME; Alkan, S. Characterization of the interleukin-1beta-converting enzyme/ced-3-family protease, caspase-3/CPP32, in Hodgkin’s disease: Lack of caspase-3 expression in nodular lymphocyte predominance Hodgkin’s disease. Am. J. Pathol 1999, 154, 1439–1447. [Google Scholar]
- Chen, M; Wang, J. Initiator caspases in apoptosis signaling pathways. Apoptosis 2002, 7, 313–319. [Google Scholar]
- Akao, T; Kida, H; Kanaoka, M; Hattori, M; Kobashi, K. Intestinal bacterial hydrolysis is required for the appearance of compound K in rat plasma after oral administration of ginsenoside Rb1 from Panax ginseng. J. Pharm. Pharmacol 1998, 50, 1155–1160. [Google Scholar]
- Hasegawa, H; Sung, JH; Huh, JH. Ginseng intestinal bacterial metabolite IH901 as a new anti-metastatic agent. Arch. Pharm. Res 1997, 20, 539–544. [Google Scholar]
- Hasegawa, H; Sung, JH; Benno, Y. Role of human intestinal Prevotella oris in hydrolyzing ginseng saponins. Planta Med 1997, 63, 436–440. [Google Scholar]
- Akao, T; Kanaoka, M; Kobashi, K. Appearance of compound K, a major metabolite of ginsenoside Rb1 by intestinal bacteria, in rat plasma after oral administration--measurement of compound K by enzyme immunoassay. Biol. Pharm. Bull 1998, 21, 245–249. [Google Scholar]
- Kang, KA; Kim, YW; Kim, SU; Chae, S; Koh, YS; Kim, HS; Choo, MK; Kim, DH; Hyun, JW. G1 phase arrest of the cell cycle by a ginseng metabolite, compound K, in U937 human monocytic leukamia cells. Arch. Pharm. Res 2005, 28, 685–690. [Google Scholar]
- Kang, KA; Lim, HK; Kim, SU; Kim, YW; Kim, WT; Chung, HS; Choo, MK; Kim, DH; Kim, HS; Shim, MJ; Chung, MH; Hyun, JW. Induction of apoptosis by ginseng saponin metabolite in U937 human monocytic leukemia cells. J. Food Biochem 2005, 29, 27–40. [Google Scholar]
- Kang, KA; Lim, HK; Chae, S; Kim, JK; Seo, JY; Ham, YH; Lee, KH; Kim, BJ; Kim, HS; Kim, DH; Hyun, JW. Inhibition of telomerase activity in U937 human monocytic leukemia cells by compound K, a ginseng saponin metabolite. Biotechnol. Bioprocess Eng 2005, 11, 7–12. [Google Scholar]
- Chae, S; Kang, KA; Chang, WY; Kim, MJ; Lee, SJ; Lee, YS; Kim, HS; Kim, DH; Hyun, JW. Effect of compound K, a metabolite of ginseng saponin, combined with gamma-ray radiation in human lung cancer cells in vitro and in vivo. J. Agric. Food Chem 2009, 57, 5777–5782. [Google Scholar]
- Kim, AD; Kang, KA; Zhang, R; Lim, CM; Kim, HS; Kim, DH; Jeon, YJ; Lee, CH; Park, J; Chang, WY; Hyun, JW. Ginseng saponin metabolite induces apoptosis in MCF-7 breast cancer cells through the modulation of AMP-activated protein kinase. Environ. Toxicol. Pharmacol 2010, 30, 134–140. [Google Scholar]
- Blau, S; Rubinstein, A; Bass, P; Singaram, C; Kohen, R. Differences in the reducing power along the rat GI tract: Lower antioxidant capacity of the colon. Mol. Cell. Biochem 1999, 194, 185–191. [Google Scholar]
- Rainis, T; Keren, D; Goldstein, O; Stermer, E; Lavy, A. Diagnostic yield and safety of colonoscopy in Israeli patients in an open access referral system. J. Clin. Gastroenterol 2007, 41, 394–399. [Google Scholar]
- Gushima, M; Hirahashi, M; Matsumoto, T; Fujita, K; Fujisawa, R; Mizumoto, K; Nakabeppu, Y; Iida, M; Yao, T; Tsuneyoshi, M. Altered expression of MUTYH and an increase in 8-hydroxydeoxyguanosine are early events in ulcerative colitis-associated carcinogenesis. J. Pathol 2009, 219, 77–86. [Google Scholar]
- Meira, LB; Bugni, JM; Green, SL; Lee, CW; Pang, B; Borenshtein, D; Rickman, BH; Rogers, AB; Moroski-Erkul, CA; McFaline, JL; Schauer, DB; Dedon, PC; Fox, JG; Samson, LD. DNA damage induced by chronic inflammation contributes to colon carcinogenesis in mice. J. Clin. Invest 2008, 118, 2516–2525. [Google Scholar] [Green Version]
- Choo, MK; Sakurai, H; Kim, DH; Saiki, I. A ginseng saponin metabolite suppresses tumor necrosis factor-alpha-promoted metastasis by suppressing nuclear factor-kappaB signaling in murine colon cancer cells. Oncol. Rep 2008, 19, 595–600. [Google Scholar]
- Kim, do Y; Yuan, HD; Chung, IK; Chung, SH. Compound K, intestinal metabolite of ginsenoside, attenuates hepatic lipid accumulation via AMPK activation in human hepatoma cells. J. Agric. Food Chem 2009, 57, 1532–1537. [Google Scholar]
- Kim, do Y; Park, MW; Yuan, HD; Lee, HJ; Kim, SH; Chung, SH. Compound K induces apoptosis via CAMK-IV/AMPK pathways in HT-29 colon cancer cells. J. Agric. Food Chem 2009, 57, 10573–10578. [Google Scholar]
- Hasegawa, H; Matsumiya, S; Uchiyama, M; Kurokawa, T; Inouye, Y; Kasai, R; Ishibashi, S; Yamasaki, K. Inhibitory effect of some triterpenoid saponins on glucose transport in tumor cells and its application to in vitro cytotoxic and antiviral activities. Planta Med 1994, 60, 240–243. [Google Scholar]
- Lee, BH; Lee, SJ; Hur, JH; Lee, S; Sung, JH; Huh, JD; Moon, CK. In vitro antigenotoxic activity of novel ginseng saponin metabolites formed by intestinal bacteria. Planta Med 1998, 64, 500–503. [Google Scholar]
- Lee, JY; Shin, JW; Chun, KS; Park, KK; Chung, WY; Bang, YJ; Sung, JH; Surh, YJ. Antitumor promotional effects of a novel intestinal bacterial metabolite (IH-901) derived from the protopanaxadiol-type ginsenosides in mouse skin. Carcinogenesis 2005, 26, 359–367. [Google Scholar]
- Yim, HW; Jong, HS; Kim, TY; Choi, HH; Kim, SG; Song, SH; Kim, J; Ko, SG; Lee, JW; Kim, TY; Bang, YJ. Cyclooxygenase-2 inhibits novel ginseng metabolite-mediated apoptosis. Cancer Res 2005, 65, 1952–1960. [Google Scholar]
- Kim, YS; Kim, JJ; Cho, KH; Jung, WS; Moon, SK; Park, EK; Kim, DH. Biotransformation of ginsenoside Rb1, crocin, amygdalin, geniposide, puerarin, ginsenoside Re, hesperidin, poncirin, glycyrrhizin, and baicalin by human fecal microflora and its relation to cytotoxicity against tumor cells. J. Microbiol. Biotechnol 2008, 18, 1109–1114. [Google Scholar]
- Choi, K; Choi, C. Proapoptotic ginsenosides compound K and Rh enhance Fas-induced cell death of human astrocytoma cells through distinct apoptotic signaling pathways. Cancer Res. Treat 2009, 41, 36–44. [Google Scholar]
- Pelicano, H; Carney, D; Huang, P. ROS stress in cancer cells and therapeutic implications. Drug Resist. Updat 2004, 7, 97–110. [Google Scholar]
- Hwang, PM; Bunz, F; Yu, J; Rago, C; Chan, TA; Murphy, MP; Kelso, GF; Smith, RA; Kinzler, KW. Vogelstein, B. Ferredoxin reductase affects p53-dependent, 5-fluorouracil-induced apoptosis in colorectal cancer cells. Nat. Med 2001, 7, 1111–1117. [Google Scholar]
- Alexandre, J; Hu, Y; Lu, W; Pelicano, H; Huang, P. Novel action of paclitaxel against cancer cells: Bystander effect mediated by reactive oxygen species. Cancer Res 2007, 67, 3512–3517. [Google Scholar]
- Qanungo, S; Das, M; Haldar, S; Basu, A. Epigallocatechin-3-gallate induces mitochondrial membrane depolarization and caspase-dependent apoptosis in pancreatic cancer cells. Carcinogenesis 2005, 26, 958–967. [Google Scholar]
- Cao, XH; Wang, AH; Wang, CL; Mao, DZ; Lu, MF; Cui, YQ; Jiao, RZ. Surfactin induces apoptosis in human breast cancer MCF-7 cells through a ROS/JNK-mediated mitochondrial/caspase pathway. Chem. Biol. Interact 2010, 183, 357–362. [Google Scholar]
- Rosenkranz, AR; Schmaldienst, S; Stuhlmeier, KM; Chen, W; Knapp, W; Zlabinger, GJ. A microplate assay for the detection of oxidative products using 2′,7′-dichlorofluorescin-diacetate. J. Immunol. Methods 1992, 156, 39–45. [Google Scholar]
- Zamzami, N; Marchetti, P; Castedo, M; Zanin, C; Vayssière, JL; Petit, PX; Kroemer, G. Reduction in mitochondrial potential constitutes an early irreversible step of programmed lymphocyte death in vivo. J. Exp. Med 1995, 181, 1661–1672. [Google Scholar]
- Zamzami, N; Susin, SA; Marchetti, P; Hirsch, T; Gómez-Monterrey, I; Castedo, M; Kroemer, G. Mitochondrial control of nuclear apoptosis. J. Exp. Med 1996, 183, 1533–1544. [Google Scholar]
- Cai, J; Yang, J; Jones, DP. Mitochondrial control of apoptosis: The role of cytochrome c. Biochim. Biophys. Acta 1998, 1366, 139–149. [Google Scholar]
- Petrosillo, G; Ruggiero, FM; Pistolese, M; Paradies, G. Reactive oxygen species generated from the mitochondrial electron transport chain induce cytochrome c dissociation from beef-heart submitochondrial particles via cardiolipin peroxidation. Possible role in the apoptosis. FEBS Lett 2001, 509, 435–438. [Google Scholar]
- Petrosillo, G; Ruggiero, FM; Paradies, G. Role of reactive oxygen species and cardiolipin in the release of cytochrome c from mitochondria. FASEB J 2003, 17, 2202–2208. [Google Scholar]
- Chen, KC; Chang, LS. Arachidonic acid-induced apoptosis of human neuroblastoma SK-N-SH cells is mediated through mitochondrial alteration elicited by ROS and Ca(2+)-evoked activation of p38alpha MAPK and JNK1. Toxicology 2009, 262, 199–206. [Google Scholar]
- Chen, HL; Chang, CY; Lee, HT; Lin, HH; Lu, PJ; Yang, CN; Shiau, CW; Shaw, AY. Synthesis and pharmacological exploitation of clioquinol-derived copper-binding apoptosis inducers triggering reactive oxygen species generation and MAPK pathway activation. Bioorg. Med. Chem 2009, 17, 7239–7247. [Google Scholar]
- Sahu, RP; Zhang, R; Batra, S; Shi, Y; Srivastava, SK. Benzyl isothiocyanate-mediated generation of reactive oxygen species causes cell cycle arrest and induces apoptosis via activation of MAPK in human pancreatic cancer cells. Carcinogenesis 2009, 30, 1744–1753. [Google Scholar]
- El-Najjar, N; Chatila, M; Moukadem, H; Vuorela, H; Ocker, M; Gandesiri, M; Schneider-Stock, R; Gali-Muhtasib, H. Reactive oxygen species mediate thymoquinone-induced apoptosis and activate ERK and JNK signaling. Apoptosis 2010, 15, 183–195. [Google Scholar]
- Chen, YJ; Liu, WH; Kao, PH; Wang, JJ; Chang, LS. Involvement of p38 MAPK- and JNK-modulated expression of Bcl-2 and Bax in Naja nigricollis CMS-9-induced apoptosis of human leukemia K562 cells. Toxicon 2010, 55, 1306–1316. [Google Scholar]
- Deng, YT; Huang, HC; Lin, JK. Rotenone induces apoptosis in MCF-7 human breast cancer cell-mediated ROS through JNK and p38 signaling. Mol. Carcinog 2010, 49, 141–151. [Google Scholar]
- Choi, SL; Kim, SJ; Lee, KT; Kim, J; Mu, J; Birnbaum, MJ; Kim, SS; Ha, J. The regulation of AMP-activated protein kinase by H2O2. Biochem. Biophys. Res. Commun 2001, 287, 92–97. [Google Scholar]
- Yun, H; Lee, M; Kim, SS. Glucose deprivation increases mRNA stability of vascular endothelial growth factor through activation of AMP-activated protein kinase in DU145 prostate carcinoma. J. Biol. Chem 2005, 280, 9963–9972. [Google Scholar]
- Carmichael, J; DeGraff, WG; Gazdar, AF; Minna, JD; Mitchell, JB. Evaluation of a tetrazolium-based semiautomated colorimetric assay: Assessment of chemosensitivity testing. Cancer Res 1987, 47, 936–942. [Google Scholar]
- Nicoletti, I; Migliorati, G; Pagliacci, MC; Grignani, F; Riccardi, C. A rapid and simple method for measuring thymocyte apoptosis by propidium iodide staining and flow cytometry. J. Immunol. Methods 1991, 139, 271–279. [Google Scholar]
- Troiano, L; Ferraresi, R; Lugli, E; Nemes, E; Roat, E; Nasi, M; Pinti, M; Cossarizza, A. Multiparametric analysis of cells with different mitochondrial membrane potential during apoptosis by polychromatic flow cytometry. Nat. Protoc 2007, 2, 2719–2727. [Google Scholar]

© 2010 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Lee, I.K.; Kang, K.A.; Lim, C.M.; Kim, K.C.; Kim, H.S.; Kim, D.H.; Kim, B.J.; Chang, W.Y.; Choi, J.H.; Hyun, J.W. Compound K, a Metabolite of Ginseng Saponin, Induces Mitochondria-Dependent and Caspase-Dependent Apoptosis via the Generation of Reactive Oxygen Species in Human Colon Cancer Cells. Int. J. Mol. Sci. 2010, 11, 4916-4931. https://doi.org/10.3390/ijms11124916
Lee IK, Kang KA, Lim CM, Kim KC, Kim HS, Kim DH, Kim BJ, Chang WY, Choi JH, Hyun JW. Compound K, a Metabolite of Ginseng Saponin, Induces Mitochondria-Dependent and Caspase-Dependent Apoptosis via the Generation of Reactive Oxygen Species in Human Colon Cancer Cells. International Journal of Molecular Sciences. 2010; 11(12):4916-4931. https://doi.org/10.3390/ijms11124916
Chicago/Turabian StyleLee, In Kyung, Kyoung Ah Kang, Chae Moon Lim, Ki Cheon Kim, Hee Sun Kim, Dong Hyun Kim, Bum Joon Kim, Weon Young Chang, Jae Hyuck Choi, and Jin Won Hyun. 2010. "Compound K, a Metabolite of Ginseng Saponin, Induces Mitochondria-Dependent and Caspase-Dependent Apoptosis via the Generation of Reactive Oxygen Species in Human Colon Cancer Cells" International Journal of Molecular Sciences 11, no. 12: 4916-4931. https://doi.org/10.3390/ijms11124916
APA StyleLee, I. K., Kang, K. A., Lim, C. M., Kim, K. C., Kim, H. S., Kim, D. H., Kim, B. J., Chang, W. Y., Choi, J. H., & Hyun, J. W. (2010). Compound K, a Metabolite of Ginseng Saponin, Induces Mitochondria-Dependent and Caspase-Dependent Apoptosis via the Generation of Reactive Oxygen Species in Human Colon Cancer Cells. International Journal of Molecular Sciences, 11(12), 4916-4931. https://doi.org/10.3390/ijms11124916