Phenolic Compounds Protect Cultured Hippocampal Neurons against Ethanol-Withdrawal Induced Oxidative Stress

Abstract
:1. Introduction
2. Results and Discussion
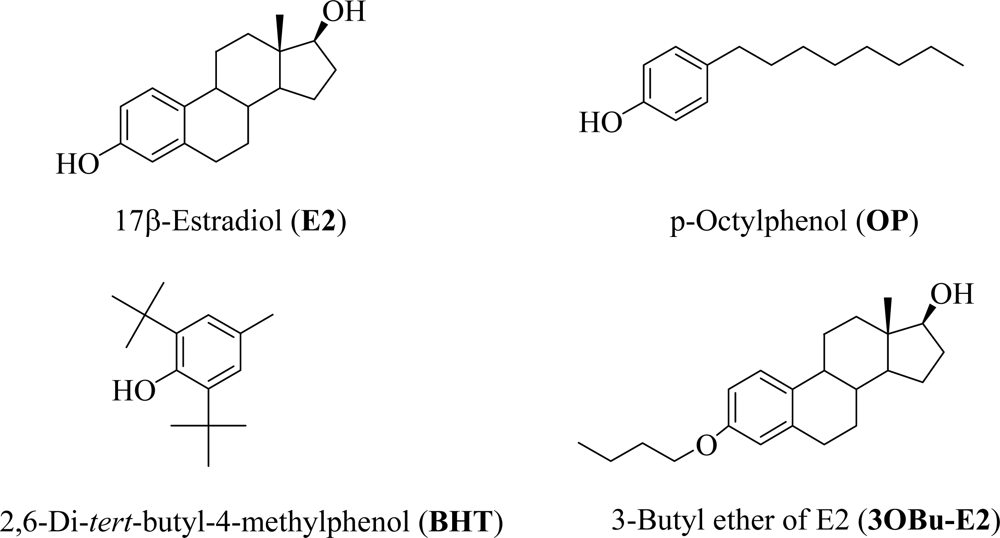
2.1. Computational and cell-free inhibition of LPO studies
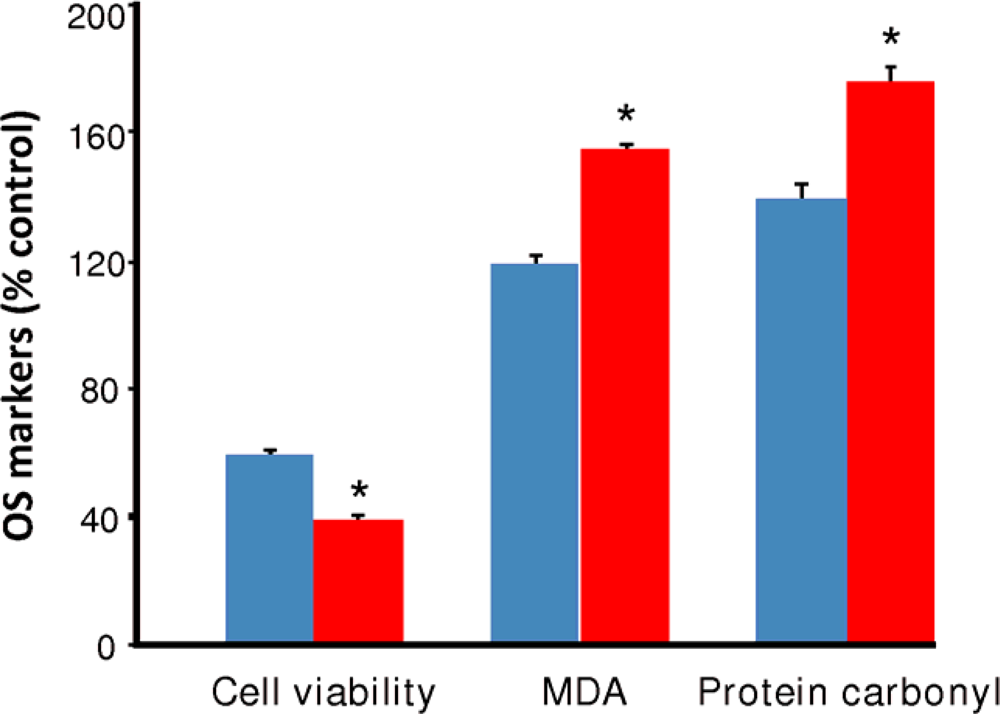
2.2. Oxidative stress by continuous ethanol exposure and its subsequent removal on HT-22 cells
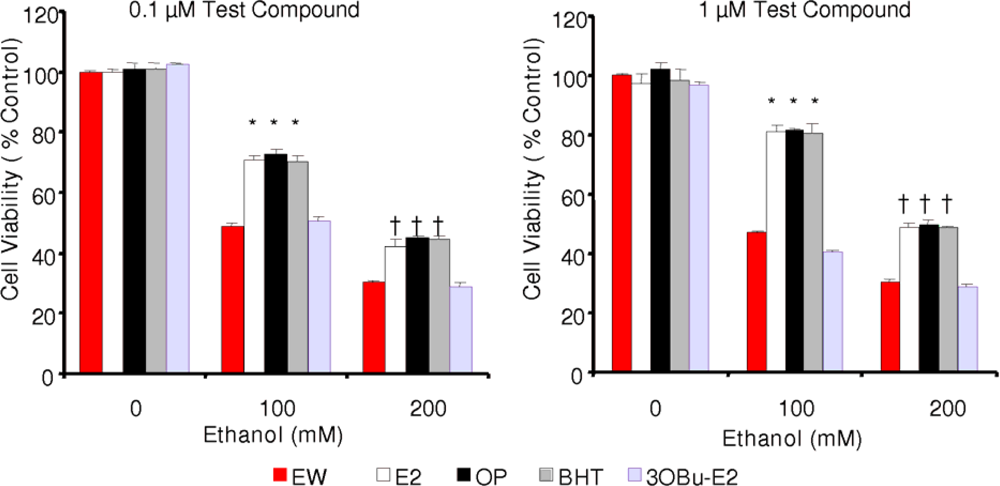
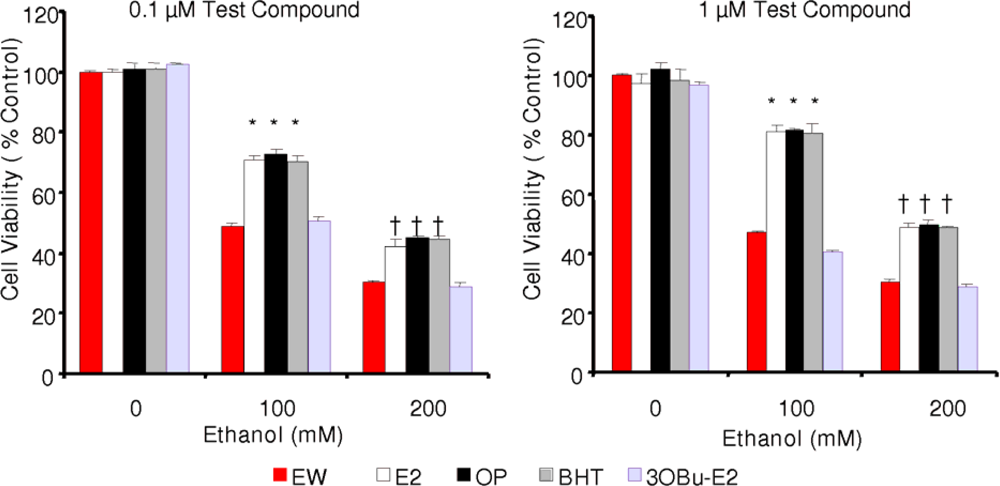
2.3. Effect of phenolic antioxidants on cell survival of EW-subjected HT-22 cells
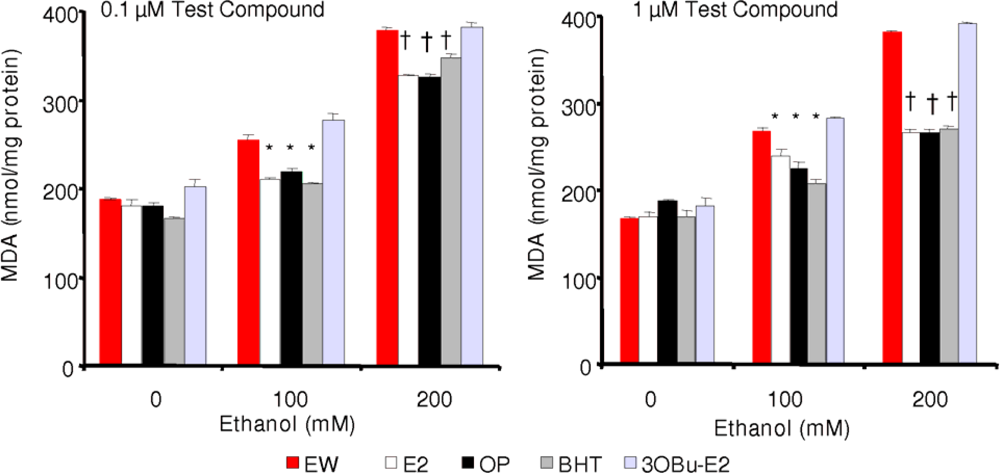
2.4. Effect of phenolic antioxidants on EW-induced lipid peroxidation
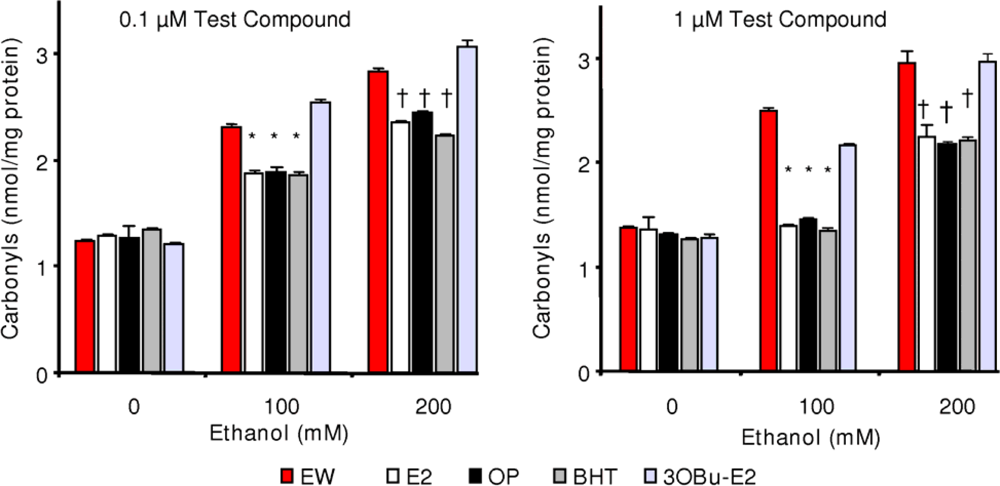
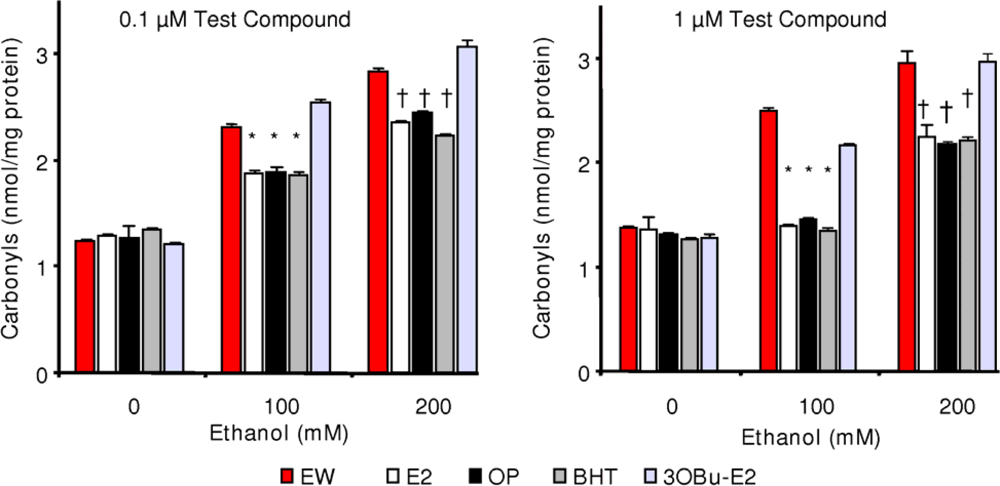
2.5. Effect of phenolic antioxidants on protein carbonylation
3. Experimental Section
3.1. Materials
3.2. Computational studies
3.3. Inhibition of lipid peroxidation
3.4. Cell culture
3.5. HT-22 cell treatments [24,25]
3.6. Calcein-AM viability assay
3.7. MDA assay
3.8. Measurement of protein carbonyl formation
3.9. Statistical analysis
4. Conclusions
Acknowledgments
References
- Sun, AY; Sun, GY. Ethanol and oxidative mechanisms in the brain. J. Biomed. Sci 2001, 8, 37–43. [Google Scholar]
- Gauvin, V. Free radical production during ethanol intoxication, dependence, and withdrawal. Alcohol. Clin. Exp. Res 1997, 2, 275–285. [Google Scholar]
- Shulman, RG; Rothman, D; Behar, KL; Hyder, F. Energetic basis of brain activity: Implications for neuroimaging. Trends Neurosci 2004, 27, 489–495. [Google Scholar]
- Bush, AI. Metals and neuroscience. Curr. Opin. Chem. Biol 2000, 4, 184–191. [Google Scholar]
- Prokai, L; Prokai-Tatrai, K; Perjesi, P; Zharikova, AD; Perez, EJ; Liu, R; Simpkins, JW. Quinol-based cyclic antioxidant mechanism in estrogen neuroprotection. Proc Natl Acad Sci USA 2003, 100, 11741–11746. [Google Scholar]
- Wagner, BA; Buettner, GR; Burns, CP. Free radical-mediated lipid peroxidation in cells: Oxidizability is a function of cell lipid bis-allylic hydrogen content. Biochemistry 1995, 33, 4449–4453. [Google Scholar]
- Janero, DR. Malondialdehyde and thiobarbituric acid-reactivity as diagnostic indices of lipid peroxidation and peroxidative tissue injury. Free Radical Biol. Med 1990, 9, 515–540. [Google Scholar]
- Prokai, L; Jun Yan, L; Vera-Serrano, JL; Stevens, SM, Jr; Forster, MJ. Mass spectrometry-based survey of age-associated protein carbonylation in rat brain mitochondria. J. Mass Spectrom 2007, 42, 1583–1589. [Google Scholar]
- Rauniyar, N; Stevens, SM; Prokai-Tatrai, K; Prokai, L. Characterization of 4-hydroxy-2-nonenal-modified peptides by liquid chromatography–tandem mass spectrometry using data-dependent acquisition: Neutral Loss-Driven MS3 versus Neutral Loss-Driven electron capture dissociation. Anal. Chem 2009, 81, 782–789. [Google Scholar]
- Nagy, J; Müller, F; László, L. Cytotoxic effect of alcohol-withdrawal on primary cultures of cortical neurons. Drug Alcohol. Depend 2001, 61, 155–162. [Google Scholar]
- Nagy, J; Horváth, C; Farkas, S; Kolok, S; Szombathely, Z. NR2B subunit selective NMDA antagonists inhibit neurotoxic effect of alcohol-withdrawal in primary cultures of rat cortical neurons. Neurochem. Int 2004, 44, 17–23. [Google Scholar]
- Crews, FT; Morrow, AL; Criswell, H; Breese, G. Effects of ethanol on ion channels. Int. Rev. Neurobiol 1996, 39, 283–367. [Google Scholar]
- Mantle, D; Preedy, VR. Free radicals as mediators of alcohol toxicity. Adverse Drug React. Toxicol. Rev 1999, 18, 235–252. [Google Scholar]
- N’Gouemo, P; Morad, M. Ethanol withdrawal seizure susceptibility is associated with upregulation of L- and P-type Ca2+ channel currents in rat inferior colliculus neurons. Neuropharmacology 2003, 45, 429–437. [Google Scholar]
- Tsai, GE; Ragan, P; Chang, R; Chen, S; Linnoila, VM; Coyle, JT. Increased glutamatergic neurotransmission and oxidative stress after alcohol withdrawal. Am. J. Psychiatry 1998, 155, 726–732. [Google Scholar]
- Rossett, ZL; Carboni, S. Ethanol withdrawal is associated with increased extracellular glutamate in the rat striatum. Eur. J. Pharmacol 1995, 283, 177–183. [Google Scholar]
- Hu, XJ; Ticku, MK. Functional characterization of a kindling-like model of ethanol withdrawal in cortical cultured neurons after chronic intermittent ethanol exposure. Brain Res 1997, 767, 228–234. [Google Scholar]
- Kumar, S; Fleming, RL; Morrow, AL. Ethanol regulation of gammaaminobutyric acid A receptors: genomic and nongenomic mechanisms. Pharmacol. Ther 2004, 101, 211–226. [Google Scholar]
- Girouard, H; Wang, G; Gallo, EF; Anrather, J; Zhou, P; Pickel, VM; Iadecola, C. NMDA receptor activation increases free radical production through nitric oxide and NOX2. J. Neurosci 2009, 29, 2545–2552. [Google Scholar]
- Prokai, L; Prokai-Tatrai, K; Perjesi, P; Simpkins, JW. Mechanistic insights into the direct antioxidant effects of estrogens. Drug Dev Res 2006, 66, 118–125. [Google Scholar]
- Jung, ME; Yang, SH; Brun-Zinkernagel, AM; Simpkins, JW. Estradiol protects against cerebellar damage and motor deficit in ethanol-withdrawn rats. Alcohol 2002, 26, 83–93. [Google Scholar]
- Jung, ME; Rewal, M; Perez, E; Wen, Y; Simpkins, JW. Estrogen protects against brain lipid peroxidation in ethanol-withdrawn rats. Pharmacol. Biochem. Behav 2004, 79, 573–586. [Google Scholar]
- Jung, ME; Gatch, BM; Simpkins, JW. The role of female sex steroids in chronic ethanol/ethanol withdrawal toxicity: potential mechanisms of estrogenic protection: review article. Exp. Biol. Med 2005, 230, 8–22. [Google Scholar]
- Jung, ME; Wilson, AM; Simpkins, JW. A nonfeminizing estrogen analog protects against ethanol withdrawal toxicity in immortalized hippocampal cells. J. Pharmacol. Exp. Ther 2006, 319, 543–550. [Google Scholar]
- Jung, ME; Wilson, AM; Ju, X; Wen, Y; Metzger, DB; Simpkins, JF. Ethanol withdrawal provokes opening of the mitochondrial membrane permeability transition pore in an estrogen preventable manner. J. Pharmacol. Exp. Ther 2009, 328, 692–698. [Google Scholar]
- Prokai, L; Oon, SM; Prokai-Tatrai, K; Abboud, K; Simpkins, JW. Synthesis and biological evaluation of 17β-alkoxyestra-1,3,5(10)-trienes as potential neuroprotectants against oxidative stress. J Med Chem 2001, 44, 110–114. [Google Scholar]
- Prokai, L; Simpkins, JW. Structure–nongenomic neuroprotection relationship of estrogens and estrogen-derived compounds. Pharmacol. Ther 2007, 114, 1–12. [Google Scholar]
- Koch, RB; Stern, B; Ferrari, CG. Linoleic acid and trilinolein as substrates for soybean lipoxidase(s). Arch. Biochem. Biophys 1958, 78, 165–179. [Google Scholar]
- Huang, D; Ou, B; Prior, RL. The chemistry behind antioxidant capacity assays. J. Agric. Food Chem 2005, 23, 1841–1856. [Google Scholar]
- Lucarini, M; Pedulli, GF. Bond dissociation enthalpy of α-tocopherol and other phenolic antioxidants. J. Org. Chem 1994, 59, 5063–5070. [Google Scholar]
- Zhang, HY; Sun, YM; Chen, DZ. O–H bond dissociation energies of phenolic compounds are determined by field/inductive effect or resonance effects? A DTF study and its implications. Quant. Struct.-Act. Relat 2001, 20, 148–152. [Google Scholar]
- Wright, JS; Johnson, ER; DiLabio, GA. Predicting the activity of phenolic antioxidants: theoretical method, analysis of substituent effects, and application to major families of antioxidants. J. Am. Chem. Soc 2001, 123, 11730–1183. [Google Scholar]
- Aĉanski, MM. Chromatographic behavior and lipophilicity of estradiol derivatives. Chromatographia 2005, 62, 475–482. [Google Scholar]
- Bizzozero, OA; Reyes, S; Ziegler, J; Smerjac, S. Lipid peroxidation scavengers prevent the carbonylation of cytoskeletal brain proteins induced by glutathione depletion. Neurochem. Res 2007, 32, 2114–2122. [Google Scholar]
- Jung, YJ; Kiso, Y; Park, HJ; Nishioka, K; Min, KS. Rejection properties of NF membranes for alkylphenols. Desalination 2006, 202, 278–285. [Google Scholar]
- Kikuzaki, H; Nakatani, N. Antioxidant effects of some ginger constituents. J. Food Sci 1993, 58, 1407–1410. [Google Scholar]
- Du, Z; Bramlage, WJ. Modified thiobarbituric acid assay for measuring lipid oxidation in sugar-rich plant tissue extracts. J. Agric. Food Chem 1992, 40, 1566–1570. [Google Scholar]
- Miller, CP; Jirkovsky, I; Hayhurst, DA; Adelman, SJ. In vitro antioxidant effects of estrogens with a hindered 3-OH function on the copper-induced oxidation of low density lipoprotein. Steroids 1991, 65, 305–308. [Google Scholar]
- Smothers, CT; Mrotek, JJ; Lovinger, DMN. Chronic ethanol exposure leads to a selective enhancement of N-methyl-D-aspartate receptor function in cultured hippocampal neurons. J. Pharmacol. Exp. Ther 1997, 293, 1214–1222. [Google Scholar]
- Ramarathnam, N; Osawa, T; Namiki, M; Kawakishi, S. Chemical studies on novel rice hull antioxidants. 1. Isolation, fractionation, and partial characterization. J. Agric. Food Chem 1988, 36, 732–737. [Google Scholar]
- Morimoto, BH; Koshland, DE, Jr. Induction and expression of long- and short-term neurosecretory potentiation in a neural cell line. Neuron 1990, 5, 875–880. [Google Scholar]
- Bradford, MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem 1976, 72, 248–254. [Google Scholar]
- Mostallino, MC; Mascia, MP; Pisu, MG; Busonero, F; Talani, G; Biggio, G. Inhibition by miltirone of up-regulation of GABAA receptor alpha4 subunit mRNA by ethanol withdrawal in hippocampal neurons. Eur. J. Pharmacol 2004, 494, 83–90. [Google Scholar]
- Li, HF; Kendig, JJ. Ethanol withdrawal hyper-responsiveness mediated by NMDA receptors in spinal cord motor neurons. Br. J. Pharmacol 2003, 139, 73–80. [Google Scholar]
- Levine, RL; Garland, D; Oliver, CN; Amici, A; Climent, I; Lenz, AG; Ahn, BW; Shaltiel, S; Stadtman, ER. Determination of carbonyl content in oxidatively modified proteins. Methods Enzymol 1990, 186, 464–478. [Google Scholar]
- Behl, C; Widmann, M; Trapp, T; Holsboer, F. 17-Beta estradiol protects neurons from oxidative stress-induced cell death in vitro. Biochem. Biophys. Res. Commun 1995, 216, 473–482. [Google Scholar]
- Deecher, DC; Daoud, P; Bhat, RA; O’Connor, LT. Endogenously expressed estrogen receptors mediate neuroprotection in hippocampal cells (HT22). J. Cell. Biochem 2005, 95, 302–312. [Google Scholar]
- Laws, SC; Carey, SA; Ferrell, JM; Bodman, GJ; Cooper, RL. Estrogenic activity of octylphenol, nonylphenol, bisphenol A and methoxychlor in rats. Toxicol. Sci 2000, 54, 154–167. [Google Scholar]
- Johansson, T; Frändberg, PA; Nyberg, F; Le Grevès, P. Molecular mechanisms for nanomolar concentrations of neurosteroids at NR1/NR2B receptors. J Pharmacol Exp Ther 2008, 324, 759–768. [Google Scholar]
- Zaulyanov, LL; Green, PS; Simpkins, JW. Glutamate receptor requirement for neuronal death from anoxia-reoxygenation: an in Vitro model for assessment of the neuroprotective effects of estrogens. Cell. Mol. Neurobiol 1999, 19, 705–718. [Google Scholar]
- Martin, AM; Kuhlmann, C; Trossbach, S; Jaeger, S; Waldron, E; Roebroek, A; Luhmann, HJ; Laatsch, A; Weggen, S; Lessmann, V; Pietrzik, CU. The functional role of the second NPXY motif of the LRP1 beta-chain in tissue-type plasminogen activator-mediated activation of Nmethyl-D-aspartate receptors. J. Biol. Chem 2008, 283, 12004–12013. [Google Scholar]
 ) and EW (
) and EW (
 ), produced a statistically significant difference (p<0.001) in OS markers from those measured in control experiments. Asterisk indicates a significant difference (p<0.01) between EW vs EtOH exposure only.
) and EW (
), produced a statistically significant difference (p<0.001) in OS markers from those measured in control experiments. Asterisk indicates a significant difference (p<0.01) between EW vs EtOH exposure only.
), produced a statistically significant difference (p<0.001) in OS markers from those measured in control experiments. Asterisk indicates a significant difference (p<0.01) between EW vs EtOH exposure only.
) and EW (
), produced a statistically significant difference (p<0.001) in OS markers from those measured in control experiments. Asterisk indicates a significant difference (p<0.01) between EW vs EtOH exposure only.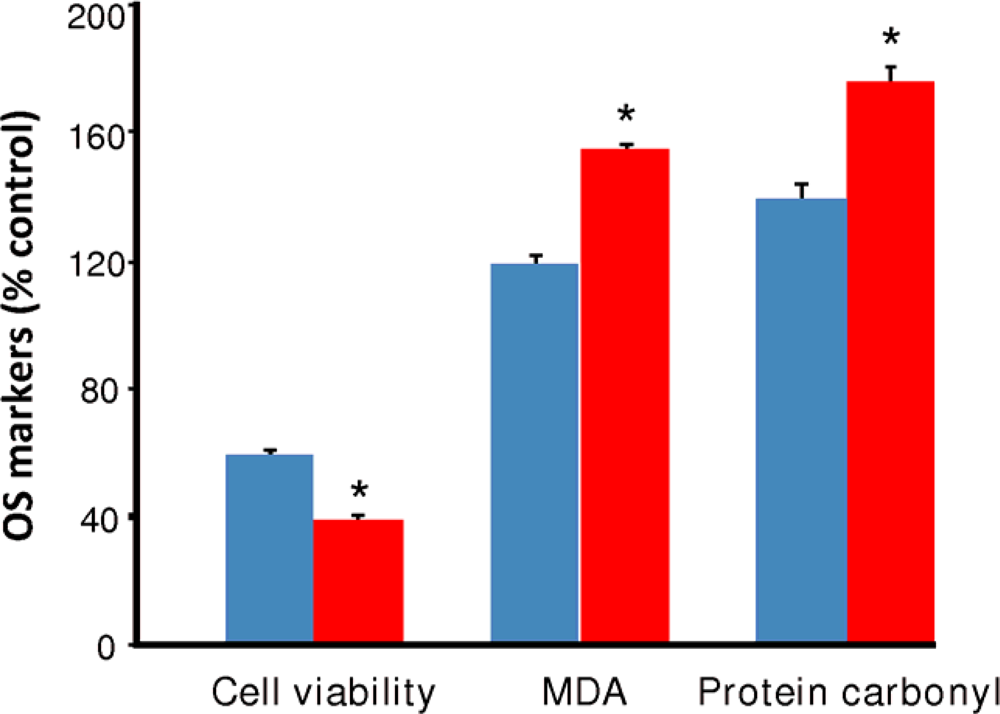

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | BDE (kcal/mol) | IC50 (μM)
| |
|---|---|---|---|
| FTC | TBARS | ||
| E2 | 81.1 | 11.8 ± 1.6 | 3.9 ± 0.4 |
| OP | 80.7 | 130.0 ± 18 | 64.8 ± 5.3 |
| BHT | 77.0 | 4.4 ± 0.6 | 3.5 ± 0.5 |
© 2009 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Prokai-Tatrai, K.; Prokai, L.; Simpkins, J.W.; Jung, M.E. Phenolic Compounds Protect Cultured Hippocampal Neurons against Ethanol-Withdrawal Induced Oxidative Stress. Int. J. Mol. Sci. 2009, 10, 1773-1787. https://doi.org/10.3390/ijms10041773
Prokai-Tatrai K, Prokai L, Simpkins JW, Jung ME. Phenolic Compounds Protect Cultured Hippocampal Neurons against Ethanol-Withdrawal Induced Oxidative Stress. International Journal of Molecular Sciences. 2009; 10(4):1773-1787. https://doi.org/10.3390/ijms10041773
Chicago/Turabian StyleProkai-Tatrai, Katalin, Laszlo Prokai, James W. Simpkins, and Marianna E. Jung. 2009. "Phenolic Compounds Protect Cultured Hippocampal Neurons against Ethanol-Withdrawal Induced Oxidative Stress" International Journal of Molecular Sciences 10, no. 4: 1773-1787. https://doi.org/10.3390/ijms10041773
APA StyleProkai-Tatrai, K., Prokai, L., Simpkins, J. W., & Jung, M. E. (2009). Phenolic Compounds Protect Cultured Hippocampal Neurons against Ethanol-Withdrawal Induced Oxidative Stress. International Journal of Molecular Sciences, 10(4), 1773-1787. https://doi.org/10.3390/ijms10041773