The Cladistic Basis for the Phylogenetic Diversity (PD) Measure Links Evolutionary Features to Environmental Gradients and Supports Broad Applications of Microbial Ecology’s “Phylogenetic Beta Diversity” Framework


Abstract
:
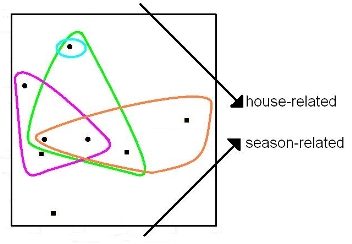{kind=link}
{kind=link}
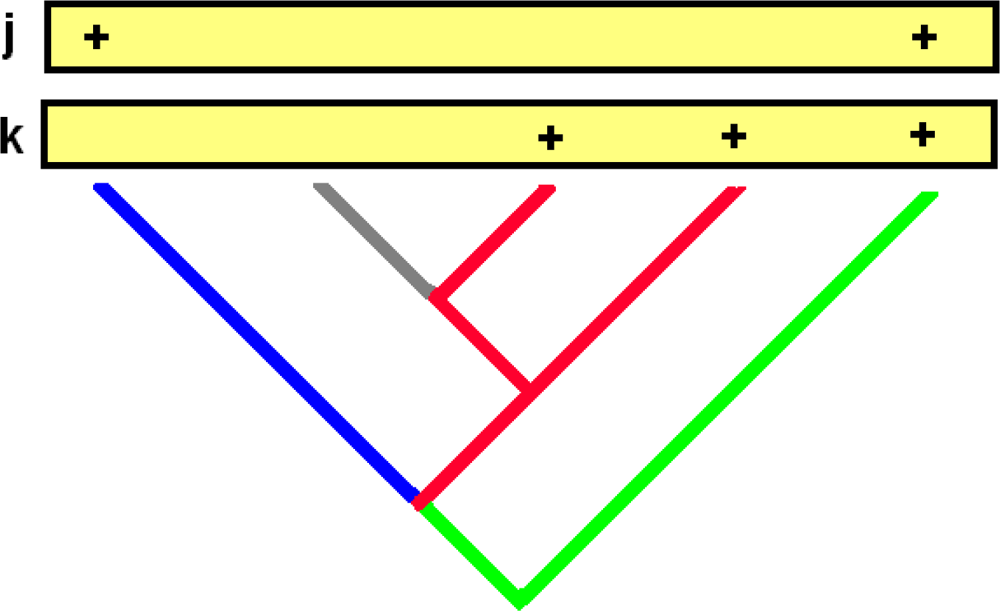{kind=link}
{kind=link}
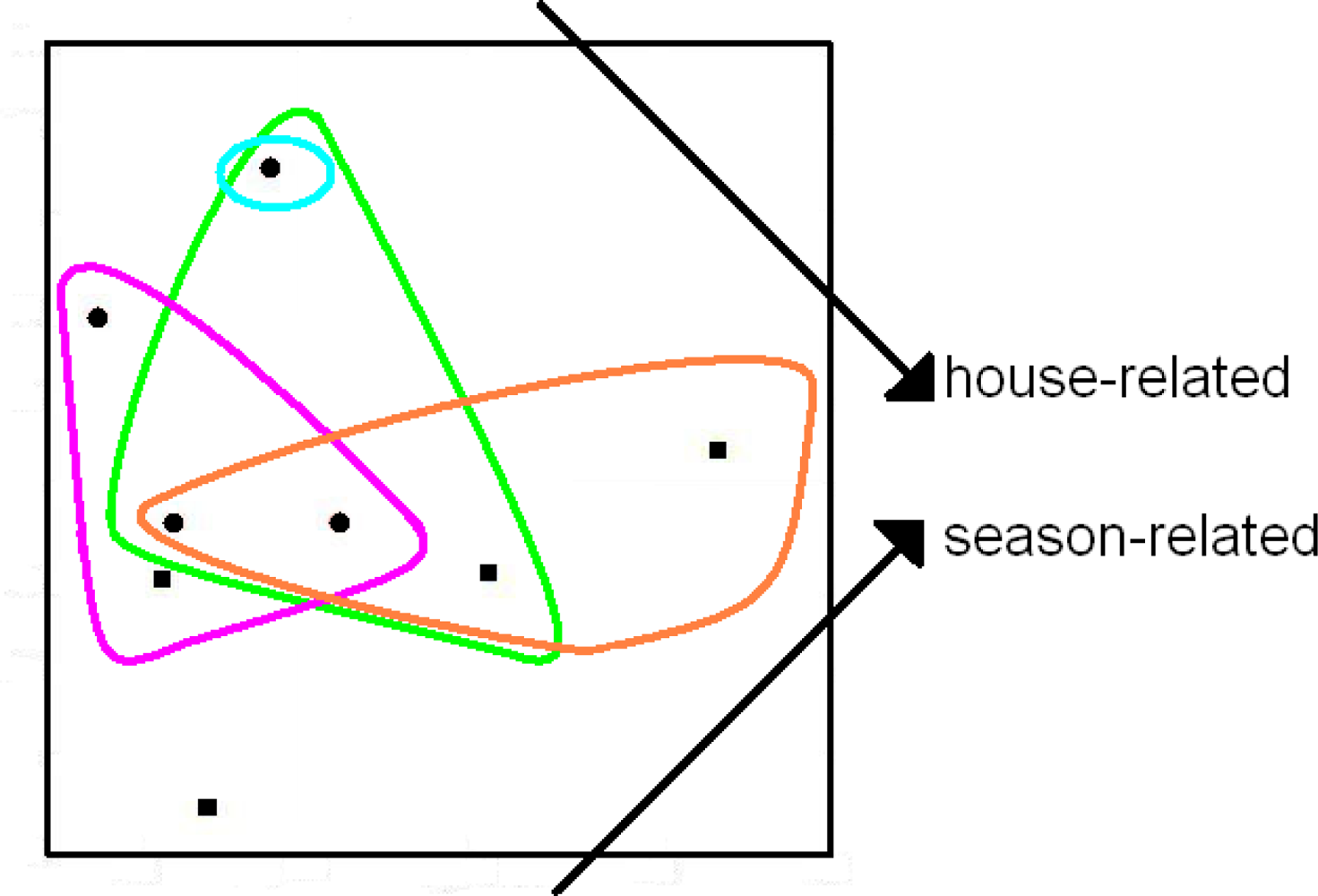{kind=link}
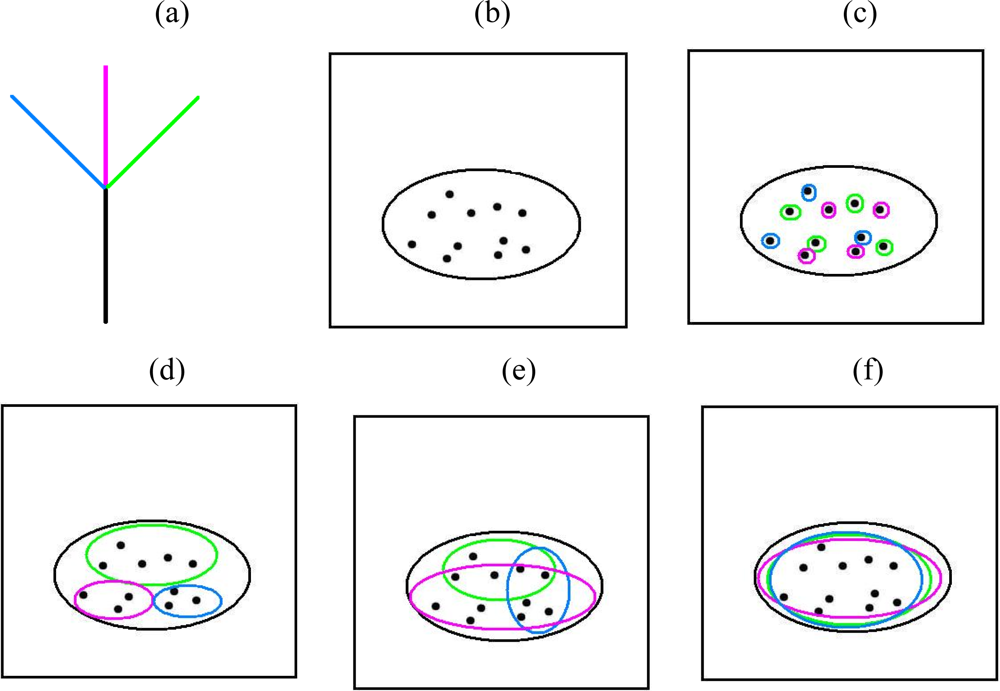{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Microbial Ecology and PD-Dissimilarities
3. The Unimodal Response Model for Evolutionary Features
4. Some Implications for Choice of Methods Used to Explore Patterns of Diversity
5. Some Implications for Methods Used to Explore Processes Driving Community Composition
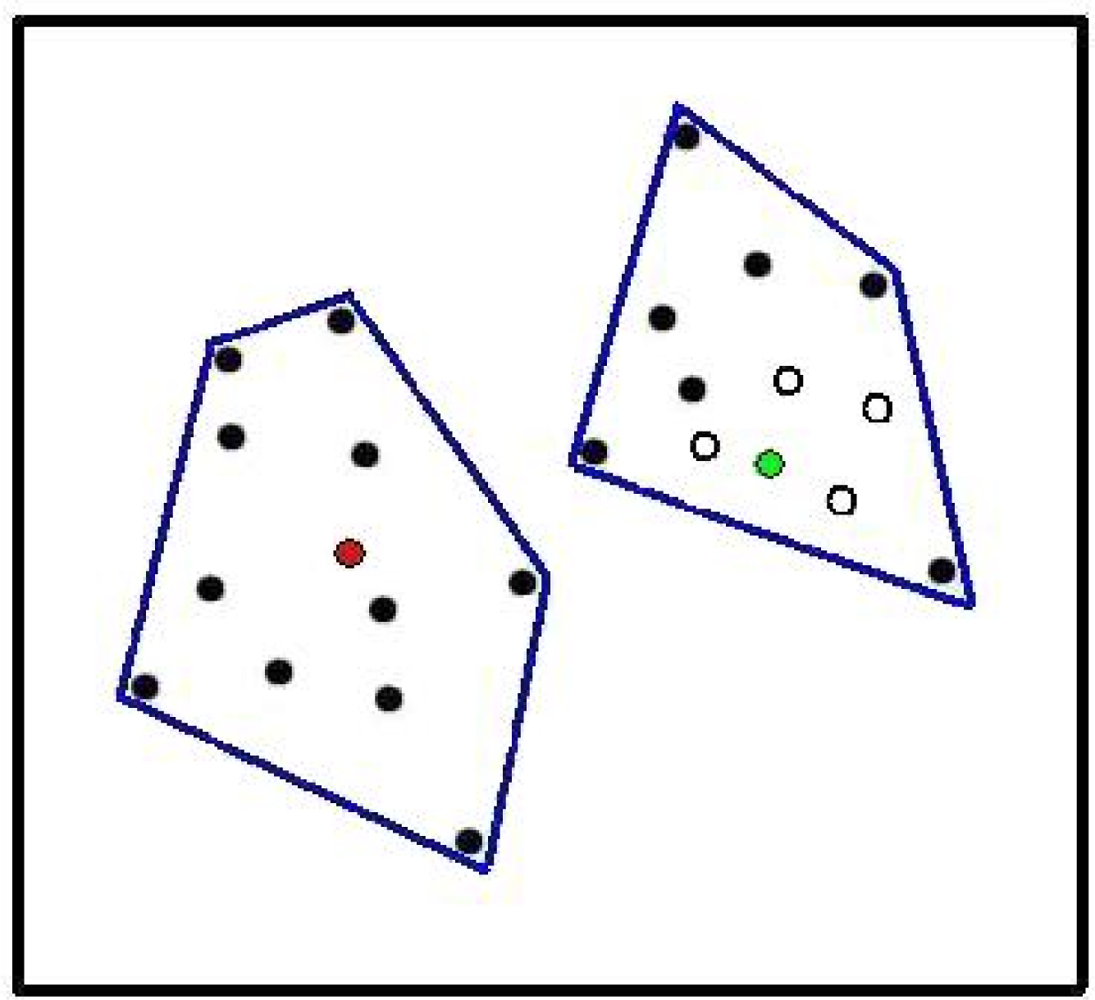
- Co-occurrence in sites of the lineages within that larger clade is significantly low, as indicated by an appropriate randomization test.
- The unimodal response pattern for these lineages (clumping of those sites having the lineage) is absent, suggesting that distribution of the lineage among sites is not simply determined by further specialization within environmental space.
- The GDM residuals for specific pairs of sample sites (where both sites contain members of the larger clade) correspond to higher PD–dissimilarity values than expected.
6. Extending Phylogenetic Beta Diversity Applications
6.1. The Search for New Commercial Products
6.2. Monitoring Human Impacts
7. Conclusions
Acknowledgments
References and Notes
- Faith, DP. Conservation evaluation and phylogenetic diversity. Biol. Cons 1992, 61, 1–10. [Google Scholar]
- Faith, DP. Systematics and conservation—on predicting the feature diversity of subsets of taxa. Cladistics 1992, 8, 361–373. [Google Scholar]
- Faith, DP. Properties of different community-level phylogenetic indices. Annual Meetings of the Ecological Society of Japan (ESJ series), Abstracts 55th Annual Meeting of the Ecological Society of Japan, Kyushu, Japan; 2008.
- Faith, DP; Reid, CAM; Hunter, J. Integrating phylogenetic diversity, complementarity, and endemism. Cons. Biol 2004, 18, 255–261. [Google Scholar]
- Lozupone, C; Knight, R. UniFrac: A new phylogenetic method for comparing microbial communities. Appl. Envir. Microbiol 2005, 71, 8228–8235. [Google Scholar]
- Lozupone, C; Knight, R. Global patterns in bacterial diversity. Proc. Natl. Acad. Sci. USA 2007, 104, 11436–11440. [Google Scholar]
- Lozupone, CA; Hamady, M; Kelley, ST; Knight, R. Quantitative and qualitative beta diversity measures lead to different insights into factors that structure microbial communities. Appl. Environ. Microbiol 2007, 73, 1576–1585. [Google Scholar]
- Lozupone, C; Knight, R. Species divergence and the measurement of microbial diversity. FEMS Microbiol. Rev 2008, 32, 557–578. [Google Scholar]
- Whittaker, RH. Evolution and measurement of species diversity. Taxon 1972, 21, 213–251. [Google Scholar]
- Ferrier, S; Manion, G; Elith, J; Richardson, KS. Using generalized dissimilarity modeling to analyze and predict patterns of beta diversity in regional biodiversity assessment. Div. Distrib 2007, 13, 252–264. [Google Scholar]
- Bryant, JA; Lamanna, C; Morlon, H; Kerkhoff, AJ; Enquist, BJ; Green, JL. Microbes on mountainsides: Contrasting elevational patterns of bacterial and plant diversity. Proc. Natl. Acad. Sci. USA 2008, 105, 11505–11511. [Google Scholar]
- Faith, DP; Minchin, PR; Belbin, L. Compositional Dissimilarity as a robust measure of ecological distance. Vegetatio 1987, 69, 57–68. [Google Scholar]
- Ley, RE; Harris, JK; Wilcox, J; Spear, JR; Miller, SR; Bebout, BM; Maresca, JA; Bryant, DA; Sogin, ML; Pace, NR. Unexpected diversity and complexity of the Guerrero Negro hypersaline microbial mat. Appl. Environ. Microbiol 2006, 72, 3685–3695. [Google Scholar]
- Rintala, H; Pitkäranta, M; Toivola, M; Paulin, L; Nevalainen, A. Diversity and seasonal dynamics of bacterial community in indoor environment. BMC Microbiol 2008, 8, 56. [Google Scholar]
- Harrison, BK; Zhang, H; Berelson, W; Orphan, VJ. Variations in archaeal and bacterial diversity associated with the sulfate-methane transition zone in continental margin sediments (Santa Barbara Basin, California). Appl. Environ. Microbiol 2009, 75, 1487–1499. [Google Scholar]
- Porter, TM; Skillman, JE; Moncalvo, JM. Fruiting body and soil rDNA sampling detects complementary assemblage of Agaricomycotina (Basidiomycota, Fungi) in a hemlock-dominated forest plot in southern Ontario. Mol. Ecol 2008, 17, 3037–3050. [Google Scholar]
- Alexander, E; Stock, A; Breiner, HW; Behnke, A; Bunge, J; Yakimov, MM; Stoeck, T. Microbial eukaryotes in the hypersaline anoxic L’Atalante deep-sea basin. Environ. Microbiol 2009, 11, 360–381. [Google Scholar]
- Desnues, C; Rodriguez-Brito, B; Rayhawk, S; Kelley, S; Tran, T; Haynes, M; Liu, H; Furlan, M; Wegley, L; Chau, B; Ruan, Y; Hall, D; Angly, FE; Edwards, RA; Li, L; Thurber, RV; Reid, RP; Siefert, J; Souza, V; Valentine, DL; Swan, BK; Breitbart, M; Rohwer, F. Biodiversity and biogeography of phages in modern stromatolites and thrombolites. Nature 2008, 452, 340–343. [Google Scholar]
- Marhaver, KL; Edwards, RA; Rohwer, F. Viral communities associated with healthy and bleaching corals. Environ. Microbiol 2008, 10, 2277–2286. [Google Scholar]
- Turnbaugh, PJ; Ley, RE; Mahowald, MA; Magrini, V; Mardis, ER; Gordon, JI. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature 2006, 444, 1027–1031. [Google Scholar]
- Frank, DN; St Amand, AL; Feldman, RA; Boedeker, EC; Harpaz, N; Pace, NR. Molecular-phylogenetic characterization of microbial community imbalances in human inflammatory bowel diseases. Proc. Natl. Acad. Sci. USA 2007, 104, 13780–13785. [Google Scholar]
- Li, M; Wang, B; Zhang, M; Rantalainen, M; Wang, S; Zhou, H; Zhang, Y; Shen, J; Pang, X; Zhang, M; Wei, H; Chen, Y; Lu, H; Zuo, J; Su, M; Qiu, Y; Jia, W; Xiao, C; Smith, LM; Yang, S; Holmes, E; Tang, H; Zhao, G; Nicholson, JK; Li, L; Zhao, L. Symbiotic gut microbes modulate human metabolic phenotypes. Proc. Natl. Acad. Sci. USA 2008, 105, 2117–2122. [Google Scholar]
- Osman, S; La Duc, MT; Dekas, A; Newcombe, D; Venkateswaran, K. Microbial burden and diversity of commercial airline cabin air during short and long durations of travel. ISME: Multidisc. J. Microb. Ecol 2008, 2, 482–497. [Google Scholar]
- Wen, L; Ley, RE; Volchkov, PY; Stranges, PB; Avanesyan, L; Stonebraker, AC; Hu, C; Wong, FS; Szot, GL; Bluestone, JA; Gordon, JI; Chervonsky, AV. Innate immunity and intestinal microbiota in the development of Type 1 diabetes. Nature 2008, 455, 1109–1113. [Google Scholar]
- Hiibel, SR; Pereyra, LP; Inman, LY; Tischer, A; Reisman, DJ; Reardon, KF; Pruden, A. Microbial community analysis of two field-scale sulfate-reducing bioreactors treating mine drainage. Environ. Microb 2008, 10, 2087–2097. [Google Scholar]
- Fraune, S; Bosch, TC. Long-term maintenance of species-specific bacterial microbiota in the basal metazoan Hydra. Proc. Natl. Acad. Sci. USA 2007, 104, 13146–13151. [Google Scholar]
- Balakirev, ES; Pavlyuchkov, VA; Ayala, FJ. DNA variation and symbiotic associations in phenotypically diverse sea urchin Strongylocentrotus intermedius. Proc. Natl. Acad. Sci. USA 2008, 105, 16218–16223. [Google Scholar]
- Ley, RE; Hamady, M; Lozupone, CA; Turnbaugh, PJ; Ramey, RR; Bircher, JS; Schlegel, ML; Tucker, TA; Schrenzel, MD; Knight, R; Gordon, JI. Evolution of mammals and their gut microbes. Science 2009, 320, 1647–1651. [Google Scholar]
- Jones, RT; Robeson, MS; Lauber, CL; Hamady, M; Knight, R; Fierer, N. A comprehensive survey of soil acidobacterial diversity using pyrosequencing and clone library analyses. ISME: Multidisc. J. Microb. Ecol 2009, 2009, 1–12. [Google Scholar]
- Lozupone, CA; Hamaday, M; Canare, BL; Coutinho, PM; Henrissat, B; Gordon, JI; Knight, R. The convergence of carbohydrate active gene repertoires in human gut microbes. PNAS 2008, 105, 15076–15081. [Google Scholar]
- Graham, CH; Fine, PVA. Phylogenetic beta diversity: Linking ecological and evolutionary processes across space in time. Ecol. Lett 2008, 11, 1265–1277. [Google Scholar]
- Cavender-Bares, J; Kozak, KH; Fine, PVA; Kembel, SW. The merging of community ecology and phylogenetic biology. Ecol. Lett 2009, 12, 693–715. [Google Scholar]
- Faith, DP. Phylogenetic triage, efficiency and risk aversion. Tr. Ecol. Evol 2009, 24, 182. [Google Scholar]
- Webb, CO; Ackerly, DD; Kembel, SW. Phylocom: Software for the analysis of phylogenetic community structure and trait evolution. Bioinformatics 2008, 24, 2098–2100. [Google Scholar]
- Minchin, PR. An evaluation of the relative robustmess of techniques for ecological ordination. Vegetatio 1987, 69, 89–107. [Google Scholar]
- Faith, DP. Homoplasy as pattern: Multivariate analysis of morphological convergence in Anseriformes. Cladistics 1989, 5, 235–258. [Google Scholar]
- Kraft, NJB; Cornwell, WK; Webb, CO; Ackerly, DD. Trait evolution, community assembly, and the phylogenetic structure of ecological communities. Am. Nat 2007, 170, 271–283. [Google Scholar]
- Chave, J; Chust, G; Thebaud, C. The importance of phylogenetic structure in biodiversity studies. In Scaling Biodiversity; Storch, D, Marquet, PL, Brown, JH, Eds.; Cambridge University Press: Cambridge, UK, 2007; pp. 150–167. [Google Scholar]
- Ramette, A. Multivariate analyses in microbial ecology. FEMS Microbiol. Ecol 2007, 62, 142–160. [Google Scholar]
- Austin, MP. Continuum concept, ordination methods, and niche theory. Ann. Rev. Ecol. Syst 1985, 16, 39–61. [Google Scholar]
- Terahara, T; Ikeda, S; Noritake, C; Minamisawa, K; Ando, K; Tsuneda, S; Harayama, S. Molecular diversity of bacterial chitinases in arable soils and the effects of environmental factors on the chitinolytic bacterial community. Soil Biol. Biochem 2009, 41, 473–480. [Google Scholar]
- Belbin, L; Faith, DP; Milligan, GW. A comparison of two approaches to beta-flexible clustering. Multiv. Behav. Res 1992, 27, 417–433. [Google Scholar]
- Green, JL; Bohannan, BJM; Whitaker, RJ. Microbial biogeography: From taxonomy to traits. Science 2008, 320, 1039–1043. [Google Scholar]
- Koleff, P; Gaston, KJ; Lennon, JJ. Measuring beta diversity for presence-absence data. J. Anim. Ecol 2003, 72, 367–382. [Google Scholar]
- Horner-Devine, MC; Bohannan, BJM. Phylogenetic clustering and overdispersion in bacterial communities. Ecology 2006, 87, S100–S108. [Google Scholar]
- Webb, CO; Ackerly, DD; McPeek, MA; Donoghue, MJ. Phylogenies and community ecology. Annu. Rev. Ecol. Syst 2002, 33, 475–505. [Google Scholar]
- Lovette, IJ; Hochachka, WM. Simultaneous effects of phylogenetic niche conservatism and competition on avian community structure. Ecology 2006, 87, S14–S28. [Google Scholar]
- Helmus, MR; Savage, K; Diebel, MW; Maxted, JT; Ives, AR. Separating the determinants of phylogenetic community structure. Ecol. Lett 2007, 10, 917–925. [Google Scholar]
- Emerson, BC; Gillespie, RG. Phylogenetic analysis of community assembly and structure over space and time. Tr. Ecol. Evol 2008, 23, 619–630. [Google Scholar]
- Faith, DP; Walker, PA. Environmental diversity: On the best-possible use of surrogate data for assessing the relative biodiversity of sets of areas. Biodiv. Cons 1996, 5, 399–415. [Google Scholar]
- Faith, DP; Ferrier, S; Walker, PA. The ED strategy: How species-level surrogates indicate general biodiversity patterns through an “environmental diversity” perspective. J. Biogeog 2004, 31, 1207–1217. [Google Scholar]
- Forest, F; Grenyer, R; Rouget, M; Davies, TJ; Cowling, RM; Faith, DP; Balmford, A; Manning, JC; Proches, S; van der Bank, M; Reeves, G; Hedderson, TAJ; Savolainen, V. Preserving the evolutionary potential of floras in biodiversity hotspots. Nature 2007, 445, 757–760. [Google Scholar]
- Pacharawongsakda, E; Yokwai, S; Ingsriswang, S. Potential natural product discovery from microbes through a diversity-guided computational framework. Appl. Microbiol. Biotechnol 2009, 82, 579–586. [Google Scholar]
- Linke, S; Norris, RH; Faith, DP; Stockwell, D. ANNA: A new prediction method for bioassessment programs. Freshw. Biol 2004, 50, 147–158. [Google Scholar]
- Downes, BJ; Barmuta, LA; Fairweather, PG; Faith, DP; Keough, MJ; Lake, PS; Mapstone, BD; Quinn, GP. Assessing Ecological Impacts: Applications in flowing waters; Cambridge University Press: Cambridge, UK, 2002. [Google Scholar]
- Andrefouet, S; Costello, MJ; Faith, DP; Ferrier, S; Geller, GN; Höft, R; Jürgens, N; Lane, MA; Larigauderie, A; Mace, G; Miazza, S; Muchoney, D; Parr, T; Pereira, HM; Sayre, R; Scholes, RJ; Stiassny, MLJ; Turner, W; Walther, BA; Yahara, T. The GEO Biodiversity Observation Network: Concept Document; GEO—Group on Earth Observations: Geneva, Switzerland, 2008.
- Faith, DP. Phylogenetic diversity and conservation. In Conservation Biology: Evolution in Action; Carroll, SP, Fox, C, Eds.; Oxford University Press: New York, NY, USA, 2008; pp. 99–115. [Google Scholar]






© 2009 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Faith, D.P.; Lozupone, C.A.; Nipperess, D.; Knight, R. The Cladistic Basis for the Phylogenetic Diversity (PD) Measure Links Evolutionary Features to Environmental Gradients and Supports Broad Applications of Microbial Ecology’s “Phylogenetic Beta Diversity” Framework. Int. J. Mol. Sci. 2009, 10, 4723-4741. https://doi.org/10.3390/ijms10114723
Faith DP, Lozupone CA, Nipperess D, Knight R. The Cladistic Basis for the Phylogenetic Diversity (PD) Measure Links Evolutionary Features to Environmental Gradients and Supports Broad Applications of Microbial Ecology’s “Phylogenetic Beta Diversity” Framework. International Journal of Molecular Sciences. 2009; 10(11):4723-4741. https://doi.org/10.3390/ijms10114723
Chicago/Turabian StyleFaith, Daniel P., Catherine A. Lozupone, David Nipperess, and Rob Knight. 2009. "The Cladistic Basis for the Phylogenetic Diversity (PD) Measure Links Evolutionary Features to Environmental Gradients and Supports Broad Applications of Microbial Ecology’s “Phylogenetic Beta Diversity” Framework" International Journal of Molecular Sciences 10, no. 11: 4723-4741. https://doi.org/10.3390/ijms10114723
APA StyleFaith, D. P., Lozupone, C. A., Nipperess, D., & Knight, R. (2009). The Cladistic Basis for the Phylogenetic Diversity (PD) Measure Links Evolutionary Features to Environmental Gradients and Supports Broad Applications of Microbial Ecology’s “Phylogenetic Beta Diversity” Framework. International Journal of Molecular Sciences, 10(11), 4723-4741. https://doi.org/10.3390/ijms10114723