Potential Mechanisms of Muscle Mitochondrial Dysfunction in Aging and Obesity and Cellular Consequences

{kind=link}
{kind=link}
Abstract
:1. Introduction
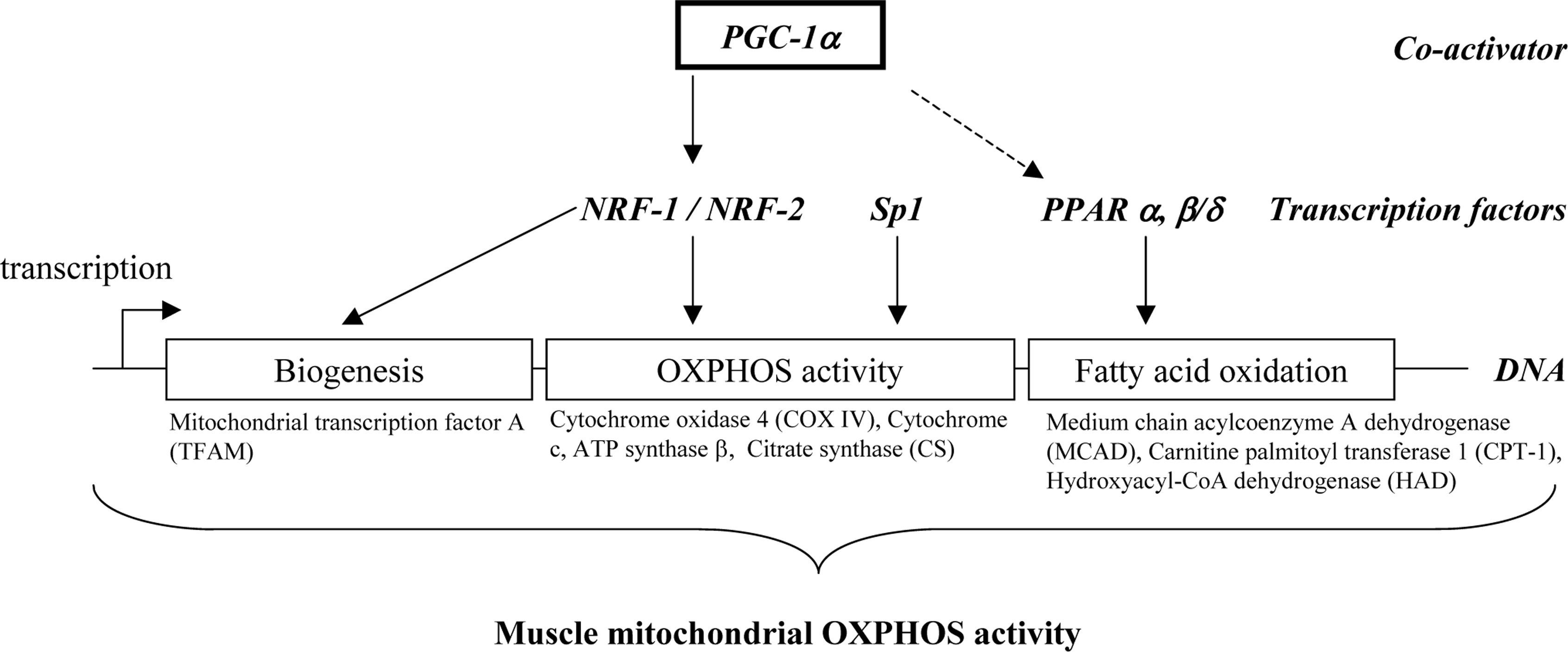
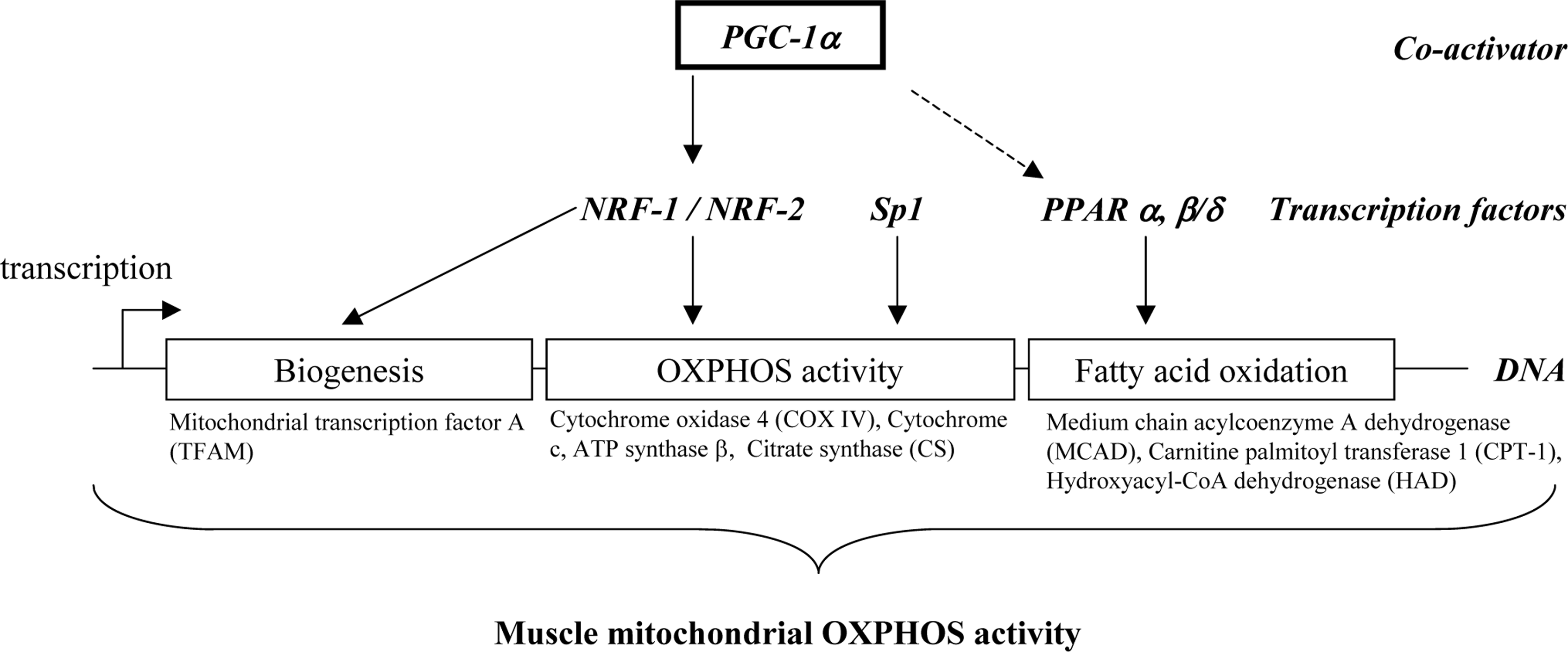
2. Transcriptional regulation of muscle mitochondrial oxidative capacity
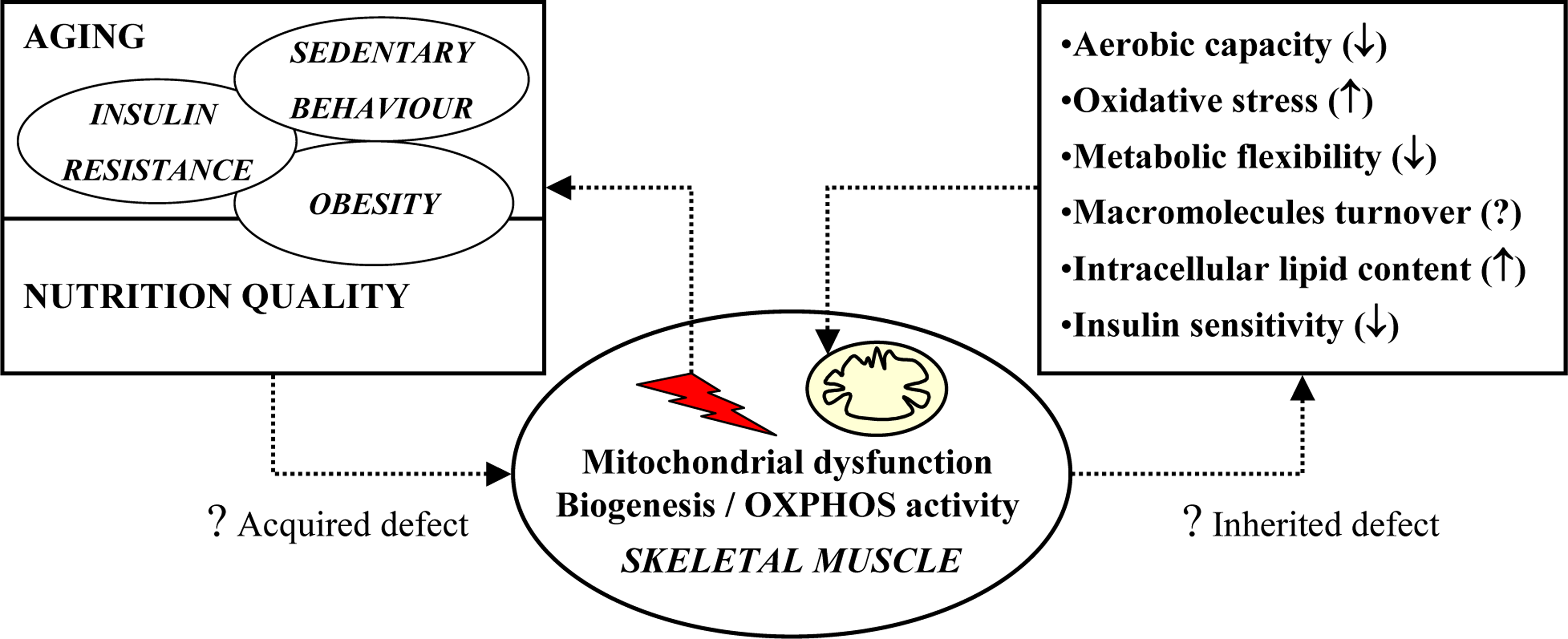
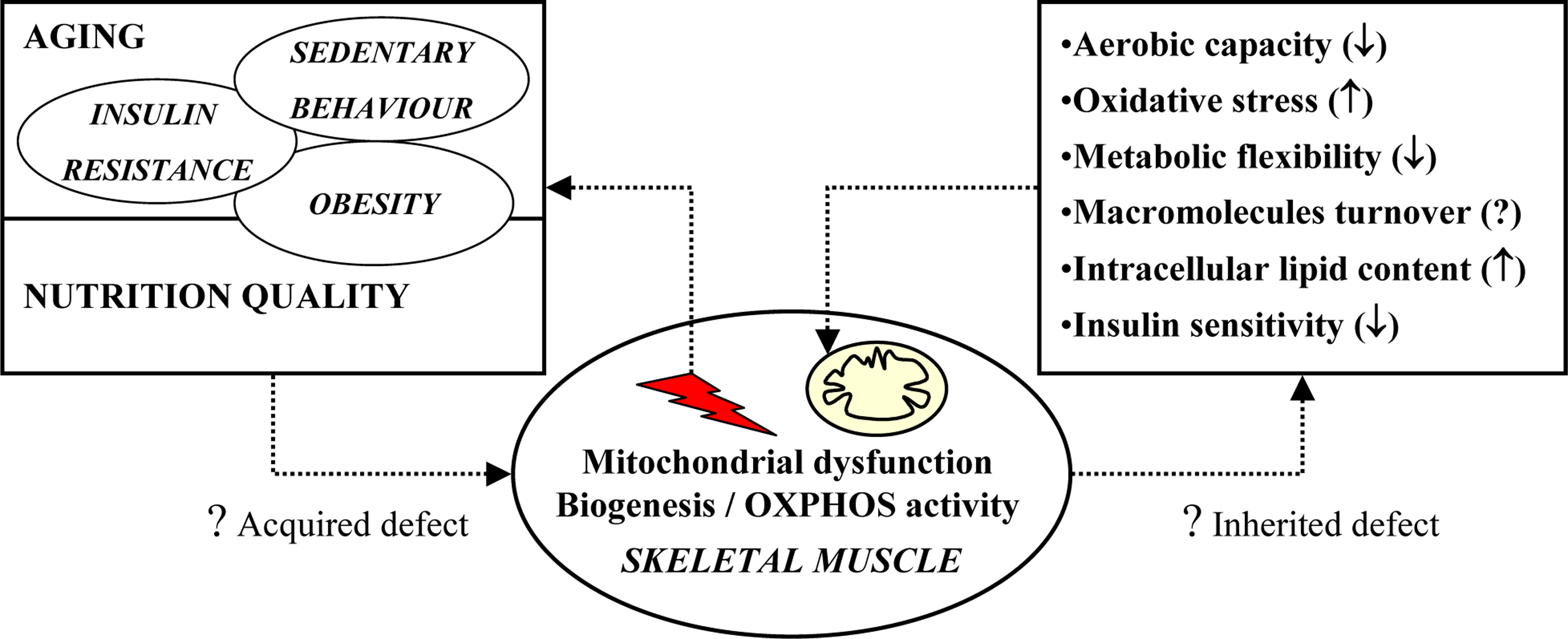
3. Potential causes of impaired mitochondrial oxidative capacity
3.1. Aging
3.2. Insulin resistance
3.3. Sedentary behavior
3.4. Nutrition quality and diet-induced obesity
4. Potential mechanisms: intrinsic factors
5. Cellular consequences of impaired mitochondrial oxidative capacity
5.1. Aerobic capacity
5.2. Oxidative stress
5.3. Metabolic flexibility
5.4. Intracellular lipid content and insulin sensitivity
6. Conclusions
Acknowledgments
References
- Fredenrich, A; Grimaldi, PA. Roles of peroxisome proliferator-activated receptor delta in skeletal muscle function and adaptation. Curr. Opin. Clin. Nutr. Metab. Care 2004, 7, 377–381. [Google Scholar]
- Grimaldi, PA. Regulatory role of peroxisome proliferator-activated receptor delta (PPAR delta) in muscle metabolism. A new target for metabolic syndrome treatment? Biochimie 2005a, 87, 5–8. [Google Scholar]
- Grimaldi, PA. Roles of PPARdelta in skeletal muscle physiology. Med. Sci. (Paris) 2005b, 21, 239–240. [Google Scholar]
- Kliewer, SA; Sundseth, SS; Jones, SA; Brown, PJ; Wisely, GB; Koble, CS; Devchand, P; Wahli, W; Willson, TM; Lenhard, JM; Lehmann, JM. Fatty acids and eicosanoids regulate gene expression through direct interactions with peroxisome proliferator-activated receptors alpha and gamma. Proc. Natl. Acad. Sci. USA 1997, 94, 4318–4323. [Google Scholar]
- Lapsys, NM; Kriketos, AD; Lim-Fraser, M; Poynten, AM; Lowy, A; Furler, SM; Chisholm, DJ; Cooney, GJ. Expression of genes involved in lipid metabolism correlate with peroxisome proliferator-activated receptor gamma expression in human skeletal muscle. J. Clin. Endocrinol. Metab 2000, 85, 4293–4297. [Google Scholar]
- Lazennec, G; Canaple, L; Saugy, D; Wahli, W. Activation of peroxisome proliferator-activated receptors (PPARs) by their ligands and protein kinase A activators. Mol. Endocrinol 2000, 14, 1962–1975. [Google Scholar]
- Patti, ME; Butte, AJ; Crunkhorn, S; Cusi, K; Berria, R; Kashyap, S; Miyazaki, Y; Kohane, I; Costello, M; Saccone, R; Landaker, EJ; Goldfine, AB; Mun, E; DeFronzo, R; Finlayson, J; Kahn, CR; Mandarino, LJ. Coordinated reduction of genes of oxidative metabolism in humans with insulin resistance and diabetes: Potential role of PGC1 and NRF1. Proc. Natl. Acad. Sci. USA 2003, 100, 8466–8471. [Google Scholar]
- Sparks, LM; Xie, H; Koza, RA; Mynatt, R; Hulver, MW; Bray, GA; Smith, SR. A high-fat diet coordinately downregulates genes required for mitochondrial oxidative phosphorylation in skeletal muscle. Diabetes 2005, 54, 1926–1933. [Google Scholar]
- Tanaka, T; Yamamoto, J; Iwasaki, S; Asaba, H; Hamura, H; Ikeda, Y; Watanabe, M; Magoori, K; Ioka, RX; Tachibana, K; Watanabe, Y; Uchiyama, Y; Sumi, K; Iguchi, H; Ito, S; Doi, T; Hamakubo, T; Naito, M; Auwerx, J; Yanagisawa, M; Kodama, T; Sakai, J. Activation of peroxisome proliferator-activated receptor delta induces fatty acid beta-oxidation in skeletal muscle and attenuates metabolic syndrome. Proc. Natl. Acad. Sci. USA 2003, 100, 15924–15929. [Google Scholar]
- Zaid, A; Li, R; Luciakova, K; Barath, P; Nery, S; Nelson, BD. On the role of the general transcription factor Sp1 in the activation and repression of diverse mammalian oxidative phosphorylation genes. J. Bioenerg. Biomembr 1999, 31, 129–135. [Google Scholar]
- Yang, X; Su, K; Roos, MD; Chang, Q; Paterson, AJ; Kudlow, JE. O-linkage of N-acetylglucosamine to Sp1 activation domain inhibits its transcriptional capability. Proc. Natl. Acad. Sci. USA 2001, 98, 6611–661. [Google Scholar]
- Moyes, CD. Controlling muscle mitochondrial content. J. Exp. Biol 2003, 206, 4385–4391. [Google Scholar]
- Lin, J; Wu, H; Tarr, PT; Zhang, CY; Wu, Z; Boss, O; Michael, LF; Puigserver, P; Isotani, E; Olson, EN; Lowell, BB; Bassel-Duby, R; Spiegelman, BM. Transcriptional co-activator PGC-1 alpha drives the formation of slow-twitch muscle fibres. Nature 2002a, 418, 797–801. [Google Scholar]
- Puigserver, P; Spiegelman, BM. Peroxisome proliferator-activated receptor-gamma coactivator 1 alpha (PGC-1 alpha): Transcriptional coactivator and metabolic regulator. Endocr. Rev 2003, 24, 78–90. [Google Scholar]
- Chabi, B; Adhihetty, PJ; Ljubicic, V; Hood, DA. How is mitochondrial biogenesis affected in mitochondrial disease? Med. Sci. Sports Exerc 2005, 37, 2102–2110. [Google Scholar]
- Hood, DA; Irrcher, I; Ljubicic, V; Joseph, AM. Coordination of metabolic plasticity in skeletal muscle. J. Exp. Biol 2006, 209, 2265–2275. [Google Scholar]
- Luquet, S; Lopez-Soriano, J; Holst, D; Fredenrich, A; Melki, J; Rassoulzadegan, M; Grimaldi, PA. Peroxisome proliferator-activated receptor delta controls muscle development and oxidative capability. FASEB J 2003, 17, 2299–22301. [Google Scholar]
- Befroy, DE; Falk Petersen, K; Dufour, S; Mason, GF; de Graaf, RA; Rothman, DL; Shulman, GI. Impaired Mitochondrial Substrate Oxidation in Muscle of Insulin-Resistant Offspring of Type 2 Diabetic Patients. Diabetes 2007, 57, 1376–1381. [Google Scholar]
- Petersen, KF; Dufour, S; Befroy, D; Garcia, R; Shulman, GI. Impaired mitochondrial activity in the insulin-resistant offspring of patients with type 2 diabetes. N. Engl. J. Med 2004, 350, 664–671. [Google Scholar]
- Petersen, KF; Dufour, S; Shulman, GI. Decreased Insulin-Stimulated ATP Synthesis and Phosphate Transport in Muscle of Insulin-Resistant Offspring of Type 2 Diabetic Parents. PLoS Med 2005, 2, e233. [Google Scholar] [Green Version]
- Petersen, KF; Shulman, GI. Etiology of insulin resistance. Am. J. Med 2006, 119, S10–S16. [Google Scholar]
- Drew, B; Phaneuf, S; Dirks, A; Selman, C; Gredilla, R; Lezza, A; Barja, G; Leeuwenburgh, C. Effects of aging and caloric restriction on mitochondrial energy production in gastrocnemius muscle and heart. Am. J. Physiol. Regul. Integr. Comp. Physiol 2003, 284, R474–R480. [Google Scholar]
- Kwong, LK; Sohal, RS. Age-related changes in activities of mitochondrial electron transport complexes in various tissues of the mouse. Arch. Biochem. Biophys 2000, 373, 16–22. [Google Scholar]
- Barrientos, A; Casademont, J; Rotig, A; Miro, O; Urbano-Marquez, A; Rustin, P; Cardellach, F. Absence of relationship between the level of electron transport chain activities and aging in human skeletal muscle. Biochem. Biophys. Res. Commun 1996, 229, 536–539. [Google Scholar]
- Brierley, EJ; Johnson, MA; James, OF; Turnbull, DM. Effects of physical activity and age on mitochondrial function. QJM 1996, 89, 251–258. [Google Scholar]
- Conley, KE; Jubrias, SA; Esselman, PC. Oxidative capacity and ageing in human muscle. J. Physiol 2000, 526, 203–210. [Google Scholar]
- Petersen, KF; Befroy, D; Dufour, S; Dziura, J; Ariyan, C; Rothman, DL; DiPietro, L; Cline, GW; Shulman, GI. Mitochondrial dysfunction in the elderly: Possible role in insulin resistance. Science 2003, 300, 1140–1142. [Google Scholar]
- Rasmussen, UF; Krustrup, P; Kjaer, M; Rasmussen, HN. Human skeletal muscle mitochondrial metabolism in youth and senescence: no signs of functional changes in ATP formation and mitochondrial oxidative capacity. Pflugers Arch 2003, 446, 270–278. [Google Scholar]
- Rimbert, V; Boirie, Y; Bedu, M; Hocquette, JF; Ritz, P; Morio, B. Muscle fat oxidative capacity is not impaired by age but by physical inactivity: association with insulin sensitivity. FASEB J 2004, 18, 737–739. [Google Scholar]
- Kelley, D; He, J; Menshikova, E; Ritov, V. Dysfunction of mitochondria in human skeletal muscle in type 2 diabetes. Diabetes 2002, 51, 2944–2950. [Google Scholar]
- Simoneau, JA; Kelley, DE. Altered glycolytic and oxidative capacities of skeletal muscle contribute to insulin resistance in NIDDM. J. Appl. Physiol 1997, 83, 166–171. [Google Scholar]
- Petersen, KF; Dufour, S; Shulman, GI. Decreased insulin-stimulated ATP synthesis and phosphate transport in muscle of insulin-resistant offspring of Type 2 Diabetic Parents. PLoS Med 2005a, 2, e233. [Google Scholar]
- Hojlund, K; Wrzesinski, K; Larsen, PM; Fey, SJ; Roepstorff, P; Handberg, A; Dela, F; Vinten, J; McCormack, JG; Reynet, C; Beck-Nielsen, H. Proteome analysis reveals phosphorylation of ATP synthase beta -subunit in human skeletal muscle and proteins with potential roles in type 2 diabetes. J. Biol. Chem 2003, 278, 10436–10442. [Google Scholar]
- Liang, P; Hughes, V; Fukagawa, NK. Increased prevalence of mitochondrial DNA deletions in skeletal muscle of older individuals with impaired glucose tolerance: possible marker of glycemic stress. Diabetes 1997, 46, 920–923. [Google Scholar]
- Guillet, C; Prod’homme, M; Balage, M; Gachon, P; Giraudet, C; Morin, L; Grizard, J; Boirie, Y. Impaired anabolic response of muscle protein synthesis is associated with S6K1 dysregulation in elderly humans. Faseb J 2004, 18, 1586–7. [Google Scholar]
- Rooyackers, OE; Adey, DB; Ades, PA; Nair, KS. Effect of age on in vivo rates of mitochondrial protein synthesis in human skeletal muscle. Proc Natl Acad Sci USA 1996a, 93, 15364–15369. [Google Scholar]
- Balagopal, P; Rooyackers, OE; Adey, DB; Ades, PA; Nair, KS. Effects of aging on in vivo synthesis of skeletal muscle myosin heavy-chain and sarcoplasmic protein in humans. Am. J. Physiol 1997, 273, E790–E800. [Google Scholar]
- Boirie, Y; Short, KR; Ahlman, B; Charlton, M; Nair, KS. Tissue-specific regulation of mitochondrial and cytoplasmic protein synthesis rates by insulin. Diabetes 2001, 50, 2652–2658. [Google Scholar]
- Rooyackers, OE; Adey, DB; Ades, PA; Nair, KS. Effect of age on in vivo rates of mitochondrial protein synthesis in human skeletal muscle. Proc. Natl. Acad. Sci. USA 1996, 93, 15364–15369. [Google Scholar]
- Halvatsiotis, P; Short, KR; Bigelow, M; Nair, KS. Synthesis rate of muscle proteins, muscle functions, and amino acid kinetics in type 2 diabetes. Diabetes 2002, 51, 2395–2404. [Google Scholar]
- Stump, CS; Short, KR; Bigelow, ML; Schimke, JM; Nair, KS. Effect of insulin on human skeletal muscle mitochondrial ATP production, protein synthesis, and mRNA transcripts. Proc. Natl. Acad. Sci. USA 2003, 100, 7996–8001. [Google Scholar]
- Boirie, Y. Insulin regulation of mitochondrial proteins and oxidative phosphorylation in human muscle. Trends Endocrinol. Metab 2003, 14, 393–394. [Google Scholar]
- Southgate, RJ; Bruce, CR; Carey, AL; Steinberg, GR; Walder, K; Monks, R; Watt, MJ; Hawley, JA; Birnbaum, MJ; Febbraio, MA. PGC-1alpha gene expression is down-regulated by Akt- mediated phosphorylation and nuclear exclusion of FoxO1 in insulin-stimulated skeletal muscle. Faseb J 2005, 19, 2072–2074. [Google Scholar]
- Short, KR; Vittone, JL; Bigelow, ML; Proctor, DN; Rizza, RA; Coenen-Schimke, JM; Nair, KS. Impact of aerobic exercise training on age-related changes in insulin sensitivity and muscle oxidative capacity. Diabetes 2003, 52, 1888–1896. [Google Scholar]
- Chanseaume, E; Malpuech-Brugere, C; Patrac, V; Bielicki, G; Rousset, P; Couturier, K; Salles, J; Renou, JP; Boirie, Y; Morio, B. Diets high in sugar, fat, and energy induce muscle type-specific adaptations in mitochondrial functions in rats. J. Nutr 2006, 136, 2194–2200. [Google Scholar]
- Iossa, S; Lionetti, L; Mollica, MP; Crescenzo, R; Botta, M; Barletta, A; Liverini, G. Effect of high-fat feeding on metabolic efficiency and mitochondrial oxidative capacity in adult rats. Br. J. Nutr 2003, 90, 953–960. [Google Scholar] [Green Version]
- Obici, S; Wang, J; Chowdury, R; Feng, Z; Siddhanta, U; Morgan, K; Rossetti, L. Identification of a biochemical link between energy intake and energy expenditure. J. Clin. Invest 2002, 109, 1599–1605. [Google Scholar]
- Mootha, VK; Lindgren, CM; Eriksson, KF; Subramanian, A; Sihag, S; Lehar, J; Puigserver, P; Carlsson, E; Ridderstrale, M; Laurila, E; Houstis, N; Daly, MJ; Patterson, N; Mesirov, JP; Golub, TR; Tamayo, P; Spiegelman, B; Lander, ES; Hirschhorn, JN; Altshuler, D; Groop, LC. PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat. Genet 2003, 34, 267–273. [Google Scholar]
- Chavez, JA; Holland, WL; Bar, J; Sandhoff, K; Summers, SA. Acid ceramidase overexpression prevents the inhibitory effects of saturated fatty acids on insulin signaling. J. Biol. Chem 2005, 280, 20148–20153. [Google Scholar]
- Shulman, GI. Cellular mechanisms of insulin resistance. J. Clin. Invest 2000, 106, 171–176. [Google Scholar]
- Staiger, H; Staiger, K; Haas, C; Weisser, M; Machicao, F; Haring, HU. Fatty acid-induced differential regulation of the genes encoding peroxisome proliferator-activated receptor-gamma coactivator-1alpha and -1beta in human skeletal muscle cells that have been differentiated in vitro. Diabetologia 2005, 48, 2115–2118. [Google Scholar]
- Simoneau, JA; Veerkamp, JH; Turcotte, LP; Kelley, DE. Markers of capacity to utilize fatty acids in human skeletal muscle: relation to insulin resistance and obesity and effects of weight loss. Faseb J 1999, 13, 2051–2060. [Google Scholar]
- Coll, T; Jove, M; Rodriguez-Calvo, R; Eyre, E; Palomer, X; Sanchez, RM; Merlos, M; Laguna, JC; Vazquez-Carrera, M. Palmitate-mediated downregulation of peroxisome proliferator-activated receptor-gamma coactivator 1alpha in skeletal muscle cells involves MEK1/2 and nuclear factor-kappaB activation. Diabetes 2006, 55, 2779–2787. [Google Scholar]
- Crunkhorn, S; Dearie, F; Mantzoros, C; Gami, H; da Silva, WS; Espinoza, D; Faucette, R; Barry, K; Bianco, AC; Patti, ME. Peroxisome proliferator activator receptor gamma coactivator-1 expression is reduced in obesity: Potential pathogenic role of saturated fatty acids and p38 mitogen-activated protein kinase activation. J. Biol. Chem 2007, 282, 15439–15450. [Google Scholar]
- Benton, CR; Han, XX; Febbraio, M; Graham, TE; Bonen, A. Inverse relationship between PGC-1alpha protein expression and triacylglycerol accumulation in rodent skeletal muscle. J. Appl. Physiol 2006, 100, 377–383. [Google Scholar]
- Lagouge, M; Argmann, C; Gerhart-Hines, Z; Meziane, H; Lerin, C; Daussin, F; Messadeq, N; Milne, J; Lambert, P; Elliott, P; Geny, B; Laakso, M; Puigserver, P; Auwerx, J. Resveratrol improves mitochondrial function and protects against metabolic disease by activating SIRT1 and PGC-1alpha. Cell 2006, 127, 1109–1122. [Google Scholar]
- Richardson, DK; Kashyap, S; Bajaj, M; Cusi, K; Mandarino, SJ; Finlayson, J; DeFronzo, RA; Jenkinson, CP; Mandarino, LJ. Lipid infusion decreases the expression of nuclear encoded mitochondrial genes and increases the expression of extracellular matrix genes in human skeletal muscle. J. Biol. Chem 2005, 280, 10290–10297. [Google Scholar]
- Brindley, DN; Wang, CN; Mei, J; Xu, J; Hanna, AN. Tumor necrosis factor-alpha and ceramides in insulin resistance. Lipids 1999, (34 Suppl), S85–S88. [Google Scholar]
- Kern, PA; Di Gregorio, GB; Lu, T; Rassouli, N; Ranganathan, G. Adiponectin expression from human adipose tissue: relation to obesity, insulin resistance, and tumor necrosis factor-alpha expression. Diabetes 2003, 52, 1779–1785. [Google Scholar]
- Saghizadeh, M; Ong, JM; Garvey, WT; Henry, RR; Kern, PA. The expression of TNF alpha by human muscle. Relationship to insulin resistance. J. Clin. Invest 1996, 97, 1111–1116. [Google Scholar]
- Neels, JG; Pandey, M; Hotamisligil, GS; Samad, F. Autoamplification of tumor necrosis factor-alpha: A potential mechanism for the maintenance of elevated tumor necrosis factor-alpha in male but not female obese mice. Am. J. Pathol 2006, 168, 435–444. [Google Scholar]
- Halse, R; Pearson, SL; McCormack, JG; Yeaman, SJ; Taylor, R. Effects of tumor necrosis factor-alpha on insulin action in cultured human muscle cells. Diabetes 2001, 50, 1102–1109. [Google Scholar]
- Rieusset, J; Bouzakri, K; Chevillotte, E; Ricard, N; Jacquet, D; Bastard, JP; Laville, M; Vidal, H. Suppressor of cytokine signaling 3 expression and insulin resistance in skeletal muscle of obese and type 2 diabetic patients. Diabetes 2004, 53, 2232–2241. [Google Scholar]
- Jove, M; Planavila, A; Sanchez, RM; Merlos, M; Laguna, JC; Vazquez-Carrera, M. Palmitate induces tumor necrosis factor-alpha expression in C2C12 skeletal muscle cells by a mechanism involving protein kinase C and nuclear factor-kappaB activation. Endocrinology 2006, 147, 552–561. [Google Scholar]
- Steinberg, GR; Michell, BJ; van Denderen, BJ; Watt, MJ; Carey, AL; Fam, BC; Andrikopoulos, S; Proietto, J; Gorgun, CZ; Carling, D; Hotamisligil, GS; Febbraio, MA; Kay, TW; Kemp, BE. Tumor necrosis factor alpha-induced skeletal muscle insulin resistance involves suppression of AMP-kinase signaling. Cell Metab 2006, 4, 465–474. [Google Scholar]
- Munoz-Fernandez, MA; Fresno, M. The role of tumour necrosis factor, interleukin 6, interferon-gamma and inducible nitric oxide synthase in the development and pathology of the nervous system. Prog. Neurobiol 1998, 56, 307–340. [Google Scholar]
- Valerio, A; Cardile, A; Cozzi, V; Bracale, R; Tedesco, L; Pisconti, A; Palomba, L; Cantoni, O; Clementi, E; Moncada, S; Carruba, MO; Nisoli, E. TNF-alpha downregulates eNOS expression and mitochondrial biogenesis in fat and muscle of obese rodents. J. Clin. Invest 2006, 116, 2791–2798. [Google Scholar]
- Perreault, M; Dombrowski, L; Marette, A. Mechanism of impaired nitric oxide synthase activity in skeletal muscle of streptozotocin-induced diabetic rats. Diabetologia 2000, 43, 427–437. [Google Scholar]
- Alderton, WK; Cooper, CE; Knowles, RG. Nitric oxide synthases: structure, function and inhibition. Biochem. J 2001, 357, 593–615. [Google Scholar]
- Nisoli, E; Clementi, E; Paolucci, C; Cozzi, V; Tonello, C; Sciorati, C; Bracale, R; Valerio, A; Francolini, M; Moncada, S; Carruba, MO. Mitochondrial biogenesis in mammals: the role of endogenous nitric oxide. Science 2003, 299, 896–899. [Google Scholar]
- Nisoli, E; Falcone, S; Tonello, C; Cozzi, V; Palomba, L; Fiorani, M; Pisconti, A; Brunelli, S; Cardile, A; Francolini, M; Cantoni, O; Carruba, MO; Moncada, S; Clementi, E. Mitochondrial biogenesis by NO yields functionally active mitochondria in mammals. Proc. Natl. Acad. Sci. USA 2004, 101, 16507–16512. [Google Scholar]
- Hickner, RC; Kemeny, G; Stallings, HW; Manning, SM; McIver, KL. Relationship between body composition and skeletal muscle eNOS. Int. J. Obes. (Lond) 2006, 30, 308–312. [Google Scholar]
- Almendro, V; Busquets, S; Ametller, E; Carbo, N; Figueras, M; Fuster, G; Argiles, JM; Lopez-Soriano, FJ. Effects of interleukin-15 on lipid oxidation: disposal of an oral [(14)C]-triolein load. Biochim. Biophys. Acta 2006, 1761, 37–42. [Google Scholar]
- Busquets, S; Figueras, M; Almendro, V; Lopez-Soriano, FJ; Argiles, JM. Interleukin-15 increases glucose uptake in skeletal muscle. An antidiabetogenic effect of the cytokine. Biochim. Biophys. Acta 2006, 1760, 1613–1617. [Google Scholar]
- Zong, H; Ren, JM; Young, LH; Pypaert, M; Mu, J; Birnbaum, MJ; Shulman, GI. AMP kinase is required for mitochondrial biogenesis in skeletal muscle in response to chronic energy deprivation. Proc. Natl. Acad. Sci. USA 2002, 99, 15983–15987. [Google Scholar]
- Turko, IV; Li, L; Aulak, KS; Stuehr, DJ; Chang, JY; Murad, F. Protein tyrosine nitration in the mitochondria from diabetic mouse heart. Implications to dysfunctional mitochondria in diabetes. J. Biol. Chem 2003, 278, 33972–33977. [Google Scholar]
- Brownlee, M. A radical explanation for glucose-induced beta cell dysfunction. J. Clin. Invest 2003, 112, 1788–1790. [Google Scholar]
- Lowell, BB; Shulman, GI. Mitochondrial dysfunction and type 2 diabetes. Science 2005, 307, 384–387. [Google Scholar]
- Bonnard, C; Durand, A; Peyrol, S; Chanseaume, E; Chauvin, MA; Morio, B; Vidal, H; Rieusset, J. Mitochondrial dysfunction results from oxidative stress in the skeletal muscle of diet-induced nsulin-resistant mice. J. Clin. Invest 2008, 118, 789–800. [Google Scholar]
- Nishikawa, T; Edelstein, D; Du, XL; Yamagishi, S; Matsumura, T; Kaneda, Y; Yorek, MA; Beebe, D; Oates, PJ; Hammes, HP; Giardino, I; Brownlee, M. Normalizing mitochondrial superoxide production blocks three pathways of hyperglycaemic damage. Nature 2000, 404, 787–790. [Google Scholar]
- Zachara, NE; Hart, GW. O-GlcNAc a sensor of cellular state: the role of nucleocytoplasmic glycosylation in modulating cellular function in response to nutrition and stress. Biochim. Biophys. Acta 2004, 1673, 13–28. [Google Scholar]
- Bassett, DR, Jr; Howley, ET. Limiting factors for maximum oxygen uptake and determinants of endurance performance. Med. Sci. Sports Exerc 2000, 32, 70–84. [Google Scholar]
- Haseler, LJ; Lin, AP; Richardson, RS. Skeletal muscle oxidative metabolism in sedentary humans: 31P-MRS assessment of O2 supply and demand limitations. J. Appl. Physiol 2004, 97, 1077–1081. [Google Scholar]
- Arenas, J; Martin, MA. Metabolic intolerance to exercise. Neurologia 2003, 18, 291–302. [Google Scholar]
- Coggan, AR; Abduljalil, AM; Swanson, SC; Earle, MS; Farris, JW; Mendenhall, LA; Robitaille, PM. Muscle metabolism during exercise in young and older untrained and endurance-trained men. J. Appl. Physiol 1993, 75, 2125–2133. [Google Scholar]
- Mousson, B; Collombet, JM; Dumoulin, R; Carrier, H; Flocard, F; Bouzidi, M; Godinot, C; Maire, I; Mathieu, M; Quard, S. An abnormal exercise test response revealing a respiratory chain complex III deficiency. Acta Neurol. Scand 1995, 91, 488–493. [Google Scholar]
- Haller, RG; Lewis, SF; Estabrook, RW; DiMauro, S; Servidei, S; Foster, DW. Exercise intolerance, lactic acidosis, and abnormal cardiopulmonary regulation in exercise associated with adult skeletal muscle cytochrome c oxidase deficiency. J. Clin. Invest 1989, 84, 155–161. [Google Scholar]
- Russell, AP; Gastaldi, G; Bobbioni-Harsch, E; Arboit, P; Gobelet, C; Deriaz, O; Golay, A; Witztum, JL; Giacobino, JP. Lipid peroxidation in skeletal muscle of obese as compared to endurance-trained humans: a case of good vs. bad lipids? FEBS Lett 2003, 551, 104–106. [Google Scholar]
- Nakagawa, Y. Initiation of apoptotic signal by the peroxidation of cardiolipin of mitochondria. Ann. NY Acad. Sci 2004, 1011, 177–184. [Google Scholar]
- Bulteau, AL; Szweda, LI; Friguet, B. Mitochondrial protein oxidation and degradation in response to oxidative stress and aging. Exp. Gerontol 2006, 41, 653–657. [Google Scholar]
- Fridlyand, LE; Philipson, LH. Reactive species and early manifestation of insulin resistance in type 2 diabetes. Diabetes Obes. Metab 2006, 8, 136–145. [Google Scholar]
- Jezek, P; Hlavata, L. Mitochondria in homeostasis of reactive oxygen species in cell, tissues, and organism. Int. J. Biochem. Cell Biol 2005, 37, 2478–2503. [Google Scholar]
- Lenaz, G; D’Aurelio, M; Merlo Pich, M; Genova, ML; Ventura, B; Bovina, C; Formiggini, G; Parenti Castelli, G. Mitochondrial bioenergetics in aging. Biochim. Biophys. Acta 2000, 1459, 397–404. [Google Scholar]
- Kayali, R; Cakatay, U; Telci, A; Akcay, T; Sivas, A; Altug, T. Decrease in mitochondrial oxidative protein damage parameters in the streptozotocin-diabetic rat. Diabetes Metab. Res. Rev 2004, 20, 315–321. [Google Scholar]
- Kelley, DE; Mandarino, LJ. Fuel selection in human skeletal muscle in insulin resistance: A reexamination. Diabetes 2000, 49, 677–683. [Google Scholar]
- Storlien, L; Oakes, ND; Kelley, DE. Metabolic flexibility. Proc. Nutr. Soc 2004, 63, 363–368. [Google Scholar]
- Kraegen, EW; Cooney, GJ; Turner, N. Muscle insulin resistance: A case of fat overconsumption, not mitochondrial dysfunction. Proc. Natl Acad. Sci. USA 2008, 105, 7627–7628. [Google Scholar]
- Holloszy, JO. Skeletal muscle “mitochondrial deficiency” does not mediate insulin resistance. Am. J. Clin. Nutr. 2008. [Google Scholar]
- Bonen, A; Parolin, ML; Steinberg, GR; Calles-Escandon, J; Tandon, NN; Glatz, JF; Luiken, JJ; Heigenhauser, GJ; Dyck, DJ. Triacylglycerol accumulation in human obesity and type 2 diabetes is associated with increased rates of skeletal muscle fatty acid transport and increased sarcolemmal FAT/CD36. Faseb J 2004, 18, 1144–1146. [Google Scholar]
- He, J; Watkins, S; Kelley, D. Skeletal muscle lipid content and oxidative enzyme activity in relation to muscle fiber type in type 2 diabetes and obesity. Diabetes 2001, 50, 817–823. [Google Scholar]
- Schrauwen, P; Hesselink, MK. Oxidative capacity, lipotoxicity, and mitochondrial damage in type 2 diabetes. Diabetes 2004, 53, 1412–1417. [Google Scholar]
- Bruce, CR; Anderson, MJ; Carey, AL; Newman, DG; Bonen, A; Kriketos, AD; Cooney, GJ; Hawley, JA. Muscle oxidative capacity is a better predictor of insulin sensitivity than lipid status. J. Clin. Endocrinol. Metab 2003, 88, 5444–5451. [Google Scholar]
- Morino, K; Petersen, KF; Dufour, S; Befroy, D; Frattini, J; Shatzkes, N; Neschen, S; White, MF; Bilz, S; Sono, S; Pypaert, M; Shulman, GI. Reduced mitochondrial density and increased IRS-1 serine phosphorylation in muscle of insulin-resistant offspring of type 2 diabetic parents. J. Clin. Invest 2005, 115, 3587–3593. [Google Scholar]
- Ritov, VB; Menshikova, EV; He, J; Ferrell, RE; Goodpaster, BH; Kelley, DE. Deficiency of subsarcolemmal mitochondria in obesity and type 2 diabetes. Diabetes 2005, 54, 8–14. [Google Scholar]
- Petersen, KF; Shulman, GI. Cellular mechanism of insulin resistance in skeletal muscle. J. R. Soc. Med. 2002, 95(Suppl 42), 8–13. [Google Scholar]
- Forouhi, NG; Jenkinson, G; Thomas, EL; Mullick, S; Mierisova, S; Bhonsle, U; McKeigue, PM; Bell, JD. Relation of triglyceride stores in skeletal muscle cells to central obesity and insulin sensitivity in European and South Asian men. Diabetologia 1999, 42, 932–935. [Google Scholar]
- Krssak, M; Falk Petersen, K; Dresner, A; DiPietro, L; Vogel, SM; Rothman, DL; Roden, M; Shulman, GI. Intramyocellular lipid concentrations are correlated with insulin sensitivity in humans: A 1H NMR spectroscopy study. Diabetologia 1999, 42, 113–116. [Google Scholar]
- Perseghin, G; Scifo, P; De Cobelli, F; Pagliato, E; Battezzati, A; Arcelloni, C; Vanzulli, A; Testolin, G; Pozza, G; Del Maschio, A; Luzi, L. Intramyocellular triglyceride content is a determinant of in vivo insulin resistance in humans: A 1H-13C nuclear magnetic resonance spectroscopy assessment in offspring of type 2 diabetic parents. Diabetes 1999, 48, 1600–1606. [Google Scholar]
- Chanseaume, E; Tardy, AL; Salles, J; Giraudet, C; Rousset, P; Tissandier, A; Boirie, Y; Morio, B. Chronological approach of diet-induced alterations in muscle mitochondrial functions in rats. Obesity (Silver Spring) 2007, 15, 50–59. [Google Scholar]
© 2009 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/). This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Chanséaume, E.; Morio, B. Potential Mechanisms of Muscle Mitochondrial Dysfunction in Aging and Obesity and Cellular Consequences. Int. J. Mol. Sci. 2009, 10, 306-324. https://doi.org/10.3390/ijms10010306
Chanséaume E, Morio B. Potential Mechanisms of Muscle Mitochondrial Dysfunction in Aging and Obesity and Cellular Consequences. International Journal of Molecular Sciences. 2009; 10(1):306-324. https://doi.org/10.3390/ijms10010306
Chicago/Turabian StyleChanséaume, Emilie, and Béatrice Morio. 2009. "Potential Mechanisms of Muscle Mitochondrial Dysfunction in Aging and Obesity and Cellular Consequences" International Journal of Molecular Sciences 10, no. 1: 306-324. https://doi.org/10.3390/ijms10010306
APA StyleChanséaume, E., & Morio, B. (2009). Potential Mechanisms of Muscle Mitochondrial Dysfunction in Aging and Obesity and Cellular Consequences. International Journal of Molecular Sciences, 10(1), 306-324. https://doi.org/10.3390/ijms10010306